

Abstract
Multiple sclerosis (MS) is a chronic immunological disease of the central nervous system characterized by early inflammatory demyelination and subsequent neurodegeneration. Major therapeutic progress has occurred during the past decade, in particular since the introduction of immunomodulatory agents, however, MS is still an incurable disease. In addition, parenteral application of the currently licensed drugs is associated with injection-related adverse events (AEs) and low patient compliance. Thus, there remains an unmet need for the development of more effective and well tolerated oral therapies for the treatment of MS. A number of new orally administered agents including fingolimod, laquinimod, teriflunomide, cladribine, and BG-12 have been licensed recently or are currently under investigation in relapsing remitting MS patients. In multi-center, randomized, placebo-controlled phase III clinical studies, all of these agents have already shown their efficacy on both clinical disease parameters and magnetic resonance imaging-based measures of disease activity in patients with relapsing remitting MS. However, there are essential differences concerning their clinical efficacy and side-effect profiles. Additionally, the mechanisms by which these substances exert clinical efficacy have not been fully elucidated. In this article, we review the pharmaceutical properties of fingolimod, laquinimod, teriflunomide, cladribine, and BG-12; and their suggested mechanisms of action, clinical efficacy, and side-effect profiles.
Keywords: cladribine, fingolimod (FTY), fumaric acid esters (BG-12), laquinimod, teriflunomide
Introduction
Although great efforts have been made towards improving multiple sclerosis (MS) therapy and understanding the underlying pathophysiological mechanisms, MS is still an incurable disease and the most prevalent cause of sustained disability in young adults.1 Symptoms at disease onset are often variable, including visual and sensory abnormalities, motor dysfunction, ataxia, fatigue, and disturbances of the bladder and bowel function.1 In the majority of patients, the initial disease course shows complete remission after an acute episode, called relapsing remitting MS (RRMS) followed by a slow accumulation of persistent symptoms. Eventually, the natural disease course enters a stage referred to as secondary progressive MS (SPMS). The pathophysiological pattern of MS is characterized by inflammatory cell infiltration, demyelination, axonal damage, astrogliosis, and neurodegenerative processes.1
The arsenal for MS therapy involves treatment of acute relapses with corticosteroids, symptomatic treatment with appropriate agents and disease modification with immunomodulatory agents. Currently, approved disease modifying therapies for the treatment of RRMS – including interferon beta (IFN-β) preparations (Betaferon®, Extavia®, Avonex®, and Rebif®), glatiramer acetate (Copaxone®), mitoxantrone (Ralenova®), natalizumab (Tysabri®), fingolimod (Gilenya®), and cladribine – modify the immune response that occurs in MS through various immunomodulatory or immunosuppressive effects. However, except for fingolimod and cladribine, all currently approved agents require parenteral administration. Yet, the majority of RRMS patients would prefer safe and effective oral therapies. At this stage, different available oral disease modifying therapies including laquinimod, teriflunomide, and BG-12 have entered or completed phase III clinical trials in patients with MS.
In this article, we review the pharmaceutical properties of different promising available oral agents for the treatment of RRMS as well as their effects in clinical studies and in particular possible adverse profiles.
Laquinimod
Laquinimod, also known as ABR-215062 sodium salt ( Figure 1) is a new orally administered synthetic drug designed for the treatment of RRMS and relates to the antecessor substance roquinimex (Linomide®). Roquinimex has demonstrated beneficial effects in MS, however, due to its serious adverse profile including cardiopulmonary toxicity, further development was stopped.
Figure 1.
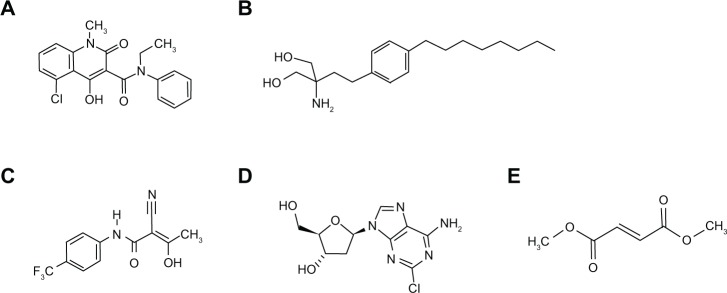
Chemical structure of oral agents. (A) Laquinimod. Molecular formula: C19H16O3N2ClNa; relative molar weight salt: 378,78; relative molar weight corresponding acid: 356.803 g/mol. (B) Fingolimod. Molecular formula: C19H33NO2 • HCl; relative molar weight: 343.93. (C) teriflunomide. Molecular formula: C12H9F3N2O2; relative molar weight: 270.2. (D) Cladribine. Molecular formula: C10H12ClN5O3; relative molar weight 285.68. (E) Dimethyl fumarate. Molecular formula: C6H8O4; relative molar weight: 144.12.
Laquinimod shows a clearly superior safety profile. Its exact mechanism of action has not been fully elucidated. Different studies conducted in the experimental autoimmune encephalomyelitis (EAE) model, which represents the major animal model of MS, clearly demonstrate the capacity of laquinimod to ameliorate clinical EAE disease course.2,3 Histopathological analyses elucidate reduced infiltration of both CD4+ T cells and macrophages into the central nervous system (CNS) and less axonal damage.4–7 Laquinimod acts via modulation of the brain-derived neurotrophic factor and targets the distribution of monocyte subsets towards an anti-inflammatory type II phenotype.5–8 Interestingly, its beneficial effect in EAE seems to be independent of endogenous IFN-β.4 Thus, laquinimod might be an alternative in RRMS patients not responding to IFN-β treatments.
In summary, the data available to date suggest both immunomodulatory and neuroprotective mechanisms of action of laquinimod (Table 1).
Table 1.
Oral agents in RRMS: Overview of mechanisms of action and efficiency.
| Drug | MOA | MRI Outcome | Clinical outcome |
|---|---|---|---|
| Laquinimod | – Induction of type ii myeloid cells. – Induction of Th2 cytokine balance. – Increase of BDNF and suppression of TH17 responses. – Attenuation of astrocytic NF-κB activation. |
– Reduction of enlarging or new T2 hyperintense lesions. – Reduction of gadolinium enhancing lesions. – Reduction in the loss of brain volume. |
– Reduction of ARR. – Modest reduction in the risk of confirmed disability progression. |
| Fingolimod | – Modulation of S1PR 1, 3–5. – Inhibits egress of lymphocytes. from lymph nodes towards CNS. – Retains Th17 cells. – May reduce astrogliosis and favor remyelination. |
– Reduction of enlarging or new T2 hyperintense lesions. – Reduction of gadolinium enhancing lesions. – Reduction of brain atrophy. |
– Reduction of ARR. – Reduces the risk of disability progression. – Superior efficacy with respect to ARR compared to IFN-β-1a. |
| Teriflunomide | – Downregulation of T- and B-cell proliferation by suppression of pyrimidine synthesis. | – Reduction of enlarging or new T2 hyperintense lesions. – Reduction of gadolinium enhancing lesions. |
– Reduction of ARR. – Reduces the risk of disability progression. |
| Cladribine | – Induction of DNA damage via accumulation in monocytes and lymphocytes. | – Reduction of active T2 lesions. – Reduction of gadolinium enhancing lesions. |
– Reduction of ARR. – Reduces the risk for disability progression. |
| BG-12 | – Induction of Th2-like cytokines and apoptosis in activated T cell. – Activation of Nrf2 pathway and antioxidant response elements. – Decrease of vascular and intracellular adhesion molecules. |
– Reduction of enlarging or new T2 hyperintense lesions. – Reduction of gadolinium enhancing lesions. – Reduction of brain atrophy. – Reduces the number of newhypointense T1-lesions. |
– Reduction of ARR and the proportion of patients relapsing. – Reduces the risk of disability progression. – Superior efficacy with respect to ARR compared to GA. |
Abbreviations: ARR, annual relapse rate; GA, glatiramer acetate; Ian; interferon, MOA; mechanisms of action, MRI; magnetic resonance imaging.
Studies and clinical efficacy
To date, the efficacy of oral laquinimod in adult patients with RRMS has been studied in one phase II, one phase IIb (LAQ/5062), and one phase III (ALLEGRO) randomized, double-blind clinical trial of 6–24 months duration.9–12 In addition, one global phase III (BRAVO) study has been completed and the results will be published soon.
Phase II studies, including one double-blind active extension, investigated the beneficial effect of different doses of laquinimod (0.1 mg/day, 0.3 mg/day, and 0.6 mg/day) vs a placebo in RRMS.9–11 Studies were scheduled for 24 and 36 weeks. The active extension study was scheduled for 36 weeks. Magnetic resonance imaging (MRI) measures of disease activity were the basis for statistical power and sample size calculations in all studies. In summary, laquinimod (0.3 mg/day and 0.6 mg/day) demonstrated sustained beneficial effects on MRI surrogate parameters, but failed to detect statistically significant effects on the relapse rate in these phase II studies, where clinical effects were only exploratory/tertiary endpoints (Table 1).9–11
To further evaluate the clinical effects of laquinimod in RRMS and its safety profile two global, multicenter, randomized, parallel-group, double-blind phase III studies have been initiated. The ALLEGRO study (MS-LAQ-301) is a placebo-controlled study, encompassing over 1100 participants, designed to evaluate the efficacy, tolerability, and safety of laquinimod in a dosage of 0.6 mg/day in patients with RRMS. The BRAVO study (MS-LAQ-302) encompasses over 1300 participants and was designed to assess the efficacy, tolerability, and safety of laquinimod in a dosage of 0.6 mg/day in comparison to a placebo and IFN-β-1a (Avonex®) in patients with RRMS in a rater blinded-fashion. Both studies were scheduled for 24 months and the primary endpoint was defined as the number of confirmed relapses during the treatment phase.
Results of the ALLEGRO study were published recently.12 Treatment with laquinimod resulted in a 23% reduction of the mean annualized relapse rate (ARR) compared to the placebo (0.30 ± 0.02 vs 0.39 ± 0.03; P = 0.002) (Table 1). Furthermore, there was a modest reduction in the risk of confirmed disability progression (11.1% vs 15.7%, P = 0.01). Similar to phase II studies, laquinimod reduces the mean number of gadolinium (Gd)-enhancing lesions and new or enlarging MRI lesions on T2-weighted images (P < 0.001 for both comparisons) (Table 1).12
First preliminary results of the BRAVO study have been announced.13 No statistical superiority was observed between the IFN-β-1a and laquinimod arm. Interestingly, participants who were randomized to receive laquinimod showed a significant reduction in the loss of brain volume and Expanded Disability Status Scale progression compared to intramuscular IFN-β-1a.13 At this point in time, results of the BRAVO study have to be interpreted with caution since data have not been published in a peer-reviewed publication.
Specific safety and tolerability
So far, different doses of laquinimod (0.1 mg/day vs 0.3 mg/day vs 0.6 mg/day) have been studied in placebo-controlled clinical trials in patients with RRMS.9–12 Based on the clinical data available to date, laquinimod is well tolerated in patients with RRMS. The drop-out rate in the Phase II study in the 0.6 mg/day treatment group was only 5%. The favorable safety profile was confirmed in the ALLEGRO trial. In the ALLEGRO study, serious adverse reactions occurred in 16.2% of placebo participants and 22.2% of laquinimod participants.13
The most common AE associated with the laquinimod 0.6 mg/day dosage was a dose dependent elevation of alanine aminotransferase. In the majority of cases elevated liver enzymes occurred within the first month of treatment and normalized without discontinuation of the drug. In only a few cases, an increase of liver enzymes caused termination of laquinimod. In those subjects, withdrawal of therapy was followed by rapid normalization of liver enzymes and no clinical sequelae was noted. In the ALLEGRO study, there were no cases of fatal liver failure, concomitant elevation of bilirubin, or coagulation values.13
Aside from the elevation of liver enzymes, laboratory examination revealed a shift of fibrinogen and C-reactive protein level more frequently in the laquinimod group in comparison to the placebo group.4–7 Other common AEs in the laquinimod group included abdominal pain, back pain, cough, respiratory tract infections, headache, asthenic conditions, insomnia, nausea and vomiting, dizziness, arthralgia, and diarrhea (Table 2).9–13 Based on current clinical data, there seems to be no evidence for cardiac AEs due to laquinimod.9–13 So far, three death cases were reported, however, all were assessed by the investigator as unrelated to the study medication. In the ALLEGRO study, there was no increased likelihood of herpes virus infections or cancer.13
Table 2.
Oral agents in RRMS: Overview of safety issues.
| Drug | Side Effects | Saftey Precautions | Risk in pregnancy |
|---|---|---|---|
| Laquinimod | – Elevation of liver enzymes. – Abdominal and back pain, cough, dizziness, headache, diarrhea, respiratory tract infections. |
– Monitoring for liver enzyme elevation and signs of infection, | – No data available |
| Fingolimod | – Skin malignancies. – Herpes infections. – Cardiac arrhythmia. – Macula edema. – Lymphopenia, influenza, nausea, headaches, increased liver enzymes, back pain, diarrhea. |
– Close of vaccination gaps before treatment initiation => VZV!– During initiation of treatment monitoring for alterations of cardiac rhythm. – Monitor for macula edema and dermatological changes. – Monitoring of signs of infection. – Regular blood monitoring, including liver enzymes. |
– Results of animal studies suggest teratogenic effects. |
| Teriflunomide | – Neutro- and lymphopenia. – Urinary tract infections and pyelonephritis. – increased liver enzymes, nausea, alopecia, nasopharyngitis, back pain, paresthesia, diarrhea, arthralgia. |
– Regular blood monitoring, including liver enzymes. – Monitoring of signs of infection. |
– Studies suggest an increased risk of fetal abnormalities. |
| Cladribine | – isolated malignancies. – Herpes zoster. – Lymphopenia, nausea, fatigue, respiratory tract infections, rash, depression, headache, nasopharyngitis. |
– Regular blood monitoring. – Monitoring for signs of infection. – Monitoring for dermatological changes. |
– Studies showed an increased teratogenic risk and frequency of fetal abnormalities. |
| BG-12 | – Lymphopenia. – Abdominal pain, diarrhea, nausea, vomiting, proteinuria, pruritus, flushing. |
– Regular blood monitoring. – Monitoring for signs of infection. |
– No data in RRMS available. Experiences from psoriasis use showed no evidence for teratogenic effects. |
Abbreviations: RRMS, relapsing remitting multiple sclerosis; VZV, varizella zoster virus.
As the cytochrome isoenzyme CYP 3A4 was found to be the primary catalyst of laquinimod, a concomitant systemic use of CYP 3A4 inhibitors or inducers should be avoided. However, this point is still under investigation.
So far no reliable data exist concerning potential teratogenic effects in humans. Therefore in female patients of child bearing age, a consequent use of contraceptives is mandatory and regular pregnancy tests are recommended. In parallel, breastfeeding women should not be exposed to laquinimod (Table 2).
Place in therapy
Published studies demonstrate beneficial effects of laquinimod (0.6 mg/day) on clinical and neuroimaging surrogate markers in adult patients with RRMS. In parallel, laquinimod shows a favorable risk-benefit profile. In particular, there is no evidence for an increased risk of cardiac AEs. Interestingly, neuroimaging and EAE data suggest neuroprotective effects of laquinimod. However, further studies are required to evaluate the efficacy of laquinimod with respect to established disease modifying therapies.
Fingolimod
Fingolimod (Gilenya®) is an oral sphingosine 1-phosphate receptor (S1PR) modulator (Figure 1). In 2010 and 2011 fingolimod was approved as the first available oral agent for the treatment of highly active RRMS in North America and Europe. Subsequent to its phosphorylation, fingolimod binds with high affinity to S1PR, which in turn leads to an internalization and degradation of the receptor on different tissues and cell types, including lymphocytes. As a consequence, fingolimod inhibits the ability of autoaggressive lymphocytes to egress from the lymph nodes towards the CNS, thereby limiting inflammatory and neurodegenerative processes in MS.14 Furthermore, fingolimod reduces the circulation of Th17 cells in MS patients and may show positive effects on both astrocytosis and remyelination via direct interaction with S1PR on astrocytes and oligodendrocytes and the corresponding precursor cells, respectively (Table 1).
Studies and clinical efficacy
One phase II study, followed by an active extension and two phase III (FREEDOMS and TRANSFORMS) placebo-controlled or active comparator-controlled clinical trials have been published in adults with RRMS (Table 1).15–20 The phase II study was scheduled for 36 weeks and the primary endpoint was defined as the total number of Gd-enhanced MRI lesions. In comparison to the placebo, fingolimod (1.25 mg/day or 5 mg/day) significantly reduced the absolute number of Gd-enhanced lesions, evident from 8 weeks onwards. Furthermore, there was a significant effect on clinical outcomes such as ARR and time to first confirmed relapse in both fingolimod treatment groups.15 The following active extension study was scheduled for a further 36 months. Patients treated with placebo were switched to either dose of fingolimod. Results of the extension study demonstrated sustained beneficial effects on MRI activity and ARR.16
FREEDOMS was scheduled for 24 months and included a total of 1272 patients with RRMS. Patients were randomized either to fingolimod 0.5 mg/day, fingolimod 1.25 mg/day, or placebo.17 The primary endpoint was defined as the ARR. In comparison to the placebo, treatment with both dosages of fingolimod resulted in a significant reduction of the ARR by 55% (fingolimod 0.5 mg/day group) and by 60% (fingolimod 1.25 mg/day group), respectively.17 Secondary clinical endpoints and MRI measures of disease activity favored treatment with fingolimod over the placebo as well. Both dosages significantly decreased the risk of disease progression, decreased the risk of relapse, and increased the number of relapse-free patients in comparison to the placebo. MRI measurements demonstrated significantly less Gd-enhancing lesions, less new or enlarged T2 lesions, and reduction of brain volume loss in both fingolimod groups compared to the placebo treatment group.17,18
The 12-month TRANSFORMS study enrolled 1292 patients and investigated two dosages of fingolimod (0.5 mg/day, 1.25 mg/day) vs treatment with intramuscular IFN-β-1a (Avonex®).19 The primary endpoint was defined as the ARR. Both dosages reduced the ARR (40%–50%) significantly compared to IFN-β-1a treatment.19 Similar to FREEDOMS, secondary endpoints, including risk of disease progression, risk of relapse, number of relapse-free patients, and MRI measures of disease activity, favored treatment with both dosages of fingolimod as well. Subsequent to completion of the TRANSFORMS trial subjects were enrolled in a 1 year extension study. Subjects initially treated with IFN-β-1a were switched to fingolimod at a dosage of either 0.5 mg/day or 1.25 mg/day, while actively treated subjects continued their medication. In general, the extension study corroborated the beneficial effects of both dosages in terms of MRI measures of disease activity (Table 1).20
Specific safety and tolerability
In FREEDOMS and TRANSFORMS the majority of AEs were characterized as mild to moderate and were resolved without discontinuation of the study drug. Common AEs in patients treated with fingolimod included flu infections, upper respiratory tract infection, headache, cough, diarrhea, and back pain. Furthermore, incidence of basal cell carcinoma was higher in patients receiving fingolimod. Laboratory examination revealed abnormal liver function tests and lymphopenia more frequently in either of the fingolimod groups in comparison to the placebo group (Table 2).17–19
FREEDOMS reported on serious AEs in 13.4% of subjects treated with the placebo vs 10.1% and 11.9% of subjects receiving fingolimod in a dosage of 0.5 mg/day or fingolimod in a dosage of 1.25 mg/day, respectively. Serious AEs related to either of the fingolimod groups include bradycardia, basal cell carcinoma, chest pain, and macular edema. In total, seven cases of serious bradycardia were noted in the fingolimod groups. Remarkably, all episodes appeared after the first dose of fingolimod. No cases of skin cancer or macular edema appeared during FREEDOMS.17
TRANSFORMS described serious AEs in 5.8% of subjects treated with IFN-β-1a vs 7.0% and 10.7% of subjects receiving fingolimod in a dosage of 0.5 mg/day or fingolimod in a dosage of 1.25 mg/day, respectively. Similar to FREEDOMS, serious AEs that were observed included bradycardia and basal cell carcinoma. Additionally, atrioventricular block, herpes virus infection and melanoma were reported. In TRANSFORMS two deaths occurred in patients treated with fingolimod in a dosage of 1.25 mg/day: one due to disseminated primary varicella zoster infection and one due to herpes simplex encephalitis.19
Given its immunosuppressive action fingolimod should be avoided in subjects at higher risk of infections or known chronic infection. Vaccination gaps should be closed before initiation of fingolimod and monitoring for macular edema; investigation of dermatological changes as well as regular blood examinations are essential subsequent to treatment initiation. Due to its effect on heart rate, fingolimod is contraindicated in patients with known cardiac arrhythmia or history of cardiovascular disease. In parallel, concomitant medication of agents that lower the heart rate should be avoided. Usually, the maximum heart rate lowering effect of fingolimod occurred within 6 hours of the first dose, however effects may also have occurred as late as 20 hours after the first dose. As a consequence, the US Food and Drug Administration (FDA) recommend that prior to treatment, initiation of electrocardiogram testing is necessary and subsequent to the first dose of fingolimod all patients must be monitored for signs of bradycardia for at least 6 hours. In patients who are at higher risk the period of cardiovascular monitoring must be extended (Table 2).
There are no adequate clinical trials in patients with MS which have studied AEs of fingolimod in pregnancy. However, experimental studies in rabbits and rats suggest teratogenicity and embryo lethality subsequent to medication with fingolimod in pregnant animals. Furthermore, the S1PR is critical for cardiovascular development and loss of S1PR expression leads to malformed embryonic hearts.21
The FDA has given fingolimod a pregnancy category C (animal studies have shown an AE on the fetus and there are no adequate studies in humans) and the US prescribing information advises women of childbearing age to have a strict use of contraceptives to avoid pregnancies during and for the 8 weeks after discontinuation of fingolimod. In parallel, breastfeeding women should not be exposed to fingolimod (Table 2).
Place in therapy
There is clear evidence of the benefit of fingolimod in adults with RRMS and fingolimod has demonstrated a superior efficacy than IFN-β-1a. Furthermore, fingolimod in a dosage of 1.25 mg/day improves health-related quality of life and depression compared with a placebo in RRMS.22 However, due to its safety profile, treatment with fingolimod requires intensive monitoring both before the first dose and in the course of treatment. Thus, fingolimod should be used as a therapeutic option only in MS patients not responding to established injectable agents or because of a rapid and severe disease course.
Teriflunomide
Teriflunomide is an orally available anti-inflammatory drug being developed for the treatment of MS. It is the active metabolite of leflunomide, a disease modifying agent used in the treatment of rheumatoid arthritis (Figure 1). The exact mechanism of action of teriflunomide remains unclear. Currently, both anti-proliferative and anti-inflammatory mechanisms of action have been shown.23,24 Subsequent to its metabolization in the liver, teriflunomide inhibits the mitochondrial dihydroorotate dehydrogenase which in turn influences the pyrimidine synthesis resulting in a reduction of lymphocyte proliferation.23,24 Furthermore, teriflunomide might influence the NF-κB pathway and shifts T-cells and macrophages towards an anti-inflammatory cytokine profile (Table 1).23,24
Studies and clinical efficacy
Teriflunomide has been studied primarily in RRMS. Results of a 36 week, placebo-controlled phase II study of the safety and efficacy of teriflunomide in patients with RRMS and SPMS still experiencing relapses was published in 2006.25 Treatment with teriflunomide (7 mg/day and 14 mg/day) resulted in a significant reduction of MRI measures of disease activity (Table 1). Apart from that, teriflunomide showed beneficial effects on disease progression at 36 weeks. Further phase II studies investigated the safety and efficacy of teriflunomide (7 or 14 mg/day) as adjunctive therapy in RRMS.26,27 teriflunomide (7 or 14 mg/day) added to ongoing stable-dosed IFN-β in RRMS resulted in a significant reduction of MRI disease activity in comparison to treatment with IFN-β alone after a 48 week treatment period. Another phase II study designed to evaluate the safety and efficacy of teriflunomide (7 or 14 mg/day) added to ongoing stable-dosed treatment with glatiramer acetate has not yet been published in a peer-reviewed publication. However, first data presented at conferences showed that adjunctive treatment with teriflunomide is superior to glatiramer acetate monotherapy with regard to MRI disease activity.27
So far, one phase III study encompassing a total of 1088 patients with RRMS evaluated the efficacy and safety of oral teriflunomide. The primary endpoint was the ARR after treatment with teriflunimode (7 mg/day and 14 mg/day) versus a placebo for 108 weeks.28 Secondary endpoints included progression of disability as well as the progression of MRI activity. There was a significant difference of the ARR within the groups (teriflunomide at a dosage of 7 mg/day or 14 mg/day versus placebo; P < 0.001). The equivalent relative reduction of ARR for patients using teriflunomide was 31.2% (7 mg/day) and 31.5% (14 mg/day) lower than for those taking placebo. Additionally, there was a significant reduction of the MRI disease burden (total area, number of lesions, volume of T2 lesions, and number of Gd-enhancing T1 lesions) in both teriflunomide groups and teriflunomide (14 mg/day) was superior to the placebo with regard to disease progression (P = 0.03).
In addition to TEMSO, results of the phase III trial TENERE (ClinicalTrials.gov identifier: NCT00883337) are expected soon. TENERE enrolled 324 participants, was scheduled for 48 weeks, and was designed to compare the efficacy and safety of teriflunomide (7 mg/day and 14 mg/day) and IFN-β-1a (Rebif®; 44 mg subcutaneously three times per week) in RRMS. Preliminary results were announced in 2011.29 Both treatment regimens failed to demonstrate a statistical superiority in comparison to treatment with IFN-β-1a. However, the data have not yet been published in a peer-reviewed publication and must be interpreted with caution.
The TOWER study is a further placebo-controlled phase III study evaluating the effect of two doses of teriflunomide (7 mg/day and 14 mg/day) in patients with RRMS. The primary outcome measure is the ARR, and the treatment period is scheduled for 24 months. Currently the study is ongoing but not recruiting participants (ClinicalTrials.gov identifier: NCT00751881). Another active phase III study, TOPIC (ClinicalTrials.gov identifier: NCT00622700), is underway in patients with early MS or clinically isolated syndrome.
Specific safety and tolerability
In comparison to a placebo, teriflunomide (7 mg/day and 14 mg/day) was well tolerated. In TEMSO, the proportion of AEs (87.5%, 89.1%, and 90.8%) and severe AEs (12.8%, 14.1%, and 15.9%) was similar across groups. In parallel, the frequency of AEs leading to treatment discontinuation was similar in all study groups (8.1%, 9.8%, and 10.8%).27 Diarrhea, dizziness, nausea, paresthesia, alopecia, and elevated alanine amino-transferase (ALT) were more common in patients treated with teriflunomide as compared with those taking the placebo (Table 2).27 Usually, AEs were mild to moderate and normalized without discontinuation of the drug. Aside from elevation of ALT, laboratory examination revealed a mean reduction of neutrophil and lymphocyte counts from baseline more often in the teriflunomide 14 mg/day group than the teriflunomide 7 mg/day or placebo groups. In the majority of cases reductions occurred during the first 12 weeks and stabilized within the course.27 Serious AEs included pathological liver function, neutropenia, and trigeminal neuralgia as well as one case of progressive multifocal leukoencephalopathy in a patient with systemic lupus erythematosus.27 No opportunistic infections and cases of death related to the study drug were reported. In all groups the incidence of serious infections was similar (2.2%, 1.6%, and 2.5%) (Table 2).
Leflunomide is contraindicated in pregnant women. Animal studies provided positive evidence for fetal malformations. The FDA classified leflunomide as pregnancy category X (animal or human studies have shown fetal abnormalities or toxicity, and the risk outweighs the benefits). Thus, leflunomide is contraindicated in women who plan to become pregnant. In parallel, male patients are recommended to stop treatment with leflunomide if they plan to father a child. A total of 45 pregnancies in female study patients were identified. No structural defects or functional deficits were reported in newborns whereas there were 21 induced abortions and 9 spontaneous abortions.22,30
Place in therapy
Teriflunomide is a promising available oral agent. Trial data underline the efficacy of teriflunomide in RRMS although preliminary data showed no superiority to IFN-β-1a. Due to its favorable risk benefit profile, teriflunomide might provide an alternative therapeutic option to established injectable agents. It may replace IFN-β and glatiramer acetate or supplement established therapies. Final statements concerning beneficial effects and AEs as well as the time point of licensing are possible after finishing and analyzing ongoing studies.
Cladribine
Cladribine (2-chloro-2′-deoxyadenosine) is a deaminaseresistant deoxyadenosine analogue (Figure 1). It is a long-lasting treatment and application is very comfortable without daily intake. Tablets are taken in two cycles per year, each lasting a few days. As the structure of cladribine is similar to adenosine triphosphate, cladribine triphosphate gets incorporated into DNA in dividing cells, leading to DNA damage and subsequent cell death (Table 1).31 The mode of action selectively depletes numbers of circulating peripheral T(CD4+ and CD8+) and B lymphocytes. In contrast, cladribine has only minor effects on natural killer cells. Apart from its effect on lymphocytes, cladribine targets the influx of T cells into the CNS and exerts immunomodulatory effects.31 The drug can penetrate the CNS and therefore affects both the peripheral and central circulation.31 It displays less side effects in other organs.
Studies and clinical efficacy
Cladribine was first tested intravenously in chronic progressive MS patients in two randomized double-blind clinical trials.32,33 Several studies followed and on the basis of various clinical and MRI measurements, the phase III Cladribine Tablets Treating Multiple Sclerosis Orally (CLARITY) study in patients with RRMS was initiated. CLARITY demonstrated that short-course oral treatment with oral cladribine at cumulative dosages of either 3.5 mg/kg or 5.25 mg/kg over 96 weeks were more effective than a placebo (Table 1).34,35 In 2011 the FDA declined approval of cladribine because of severe safety issues. The drug-developing company announced that it will not pursue its efforts to obtain approval but will complete ongoing clinical trials (ONWARD, ORALCE MS, and CLARITY extension study).
Specific safety and tolerability
The therapeutic efficacy and safety of cladribine have been assessed in several autoimmune disorders and the parenteral formulation of cladribine is used as a first-line treatment for hairy cell leukemia.36
Overall AEs in the CLARITY trial were comparable for the placebo and cladribine treated subjects.34 Common AEs in all study groups were upper respiratory tract infection, nasopharyngitis, nausea, fatigue, depression, rash, headache, and lymphopenia (Table 2).34,35 Lymphocyte count decreased with a nadir at week 16 and was a dose-dependent side effect. It is important to note that the immunosuppressive effect is long-lasting and cannot be reversed.34 Other AEs associated with cladribine intake included benign uterine leiomyomas and mild dermal herpes zoster. One patient suffered from hepatitis B and one died after reactivation of latent tuberculosis.34,37 Neoplasms and serious AEs were reported in 3.7% and 12.9% of all cladribine treated subjects compared with 1.7% and 8.1% of placebo-treated subjects.34,35 No cases of malignancies occurred in the placebo group. In contrast, four isolated malignancies (malignant melanoma, pancreatic, cervical, and ovarian cancer) were associated with cladribine intake and one case of choriocarcinoma after study termination.34,35
Animal studies have shown a teratogenic risk to the fetus. Moreover, an increased likelihood of fetal malformations was seen in mice and rabbits. The FDA rated cladribine in its currently approved parenteral form as pregnancy category D (there is positive evidence of human fetal risk, but the benefits may outweigh the risks). US prescribing information advises women of childbearing age to avoid becoming pregnant (Table 2).
Place in therapy
The CLARITY study demonstrated a beneficial effect of oral cladribine. After a 96 week treatment period about 45% of patients in the cladribine group were free from disease activity compared to 16% in the placebo group.34 However, apart from clinical effectiveness, AEs have to be considered as well. Based on the current data, cladribine did not achieve regulatory approval by the FDA or European Medicines Agency due to insufficient data on its safety and benefit risk balance.38–40 Although, cladribine is already accepted for the treatment of MS in Australia and Russia, the drug company does not intend to pursue further worldwide approval of cladribine tablets for the treatment of MS. Therefore, cladribine is not yet relevant in a daily routine of MS therapy.
Fumaric acid esters (BG-12)
Since 1990, fumaric acid esters (FAE) have been licensed under the brand name Fumaderm® for the treatment of psoriasis. Fumaderm® mainly consists of dimethyl fumarate (DMF) and different salts of ethyl hydrogen fumarate. After oral intake, DMF is hydrolyzed to its active metabolite monomethyl fumarate. DMF (study name BG-12) is a novel oral drug, which has shown strong therapeutic activity in two phase III studies in adults with RRMS. The exact mechanisms of action are still under investigation. Experimental studies in EAE suggest immunomodulatory and neuroprotective properties.41,42 In particular, BG-12 and its primary metabolite modulate the secretion of proinflammatory cytokines, downregulate the infiltration of inflammatory cells into the CNS, and influence inflammatory cascades. Additionally, Linker et al demonstrated neuroprotective effects of BG-12 via targeting of the Nrf2 antioxidant pathway.41
Studies and clinical efficacy
The first open-label study of FAE in RRMS patients was published in 2006.43 FAE reduced the number of Gd-enhancing lesions on brain MRI scans significantly. Double-blind, placebo-controlled phase II and phase IIb clinical studies confirmed the beneficial effect of FAE on MRI parameters of disease activity in patients with RRMS (Table 1).
The efficacy and safety of oral BG-12 are being further evaluated in two large phase III studies (DEFINE and CONFIRM) and results were published recently in the New England Journal of Medicine.44,45 Both studies were designed to investigate the clinical efficacy and risk-benefit profile of BG-12, were scheduled for 24 months and enrolled in each case over 1200 adults with RRMS. The DEFINE study investigated the effect of oral BG-12 (240 mg), applied twice or three times daily in comparison to placebo.44 The CONFIRM study evaluated the effect of BG-12 (240 mg) applied twice or three times daily compared with placebo and glatiramer acetate (copaxone®) as an active comparator.45 In comparison to placebo, BG-12 in both treatment regimens reduced the ARR (relative reduction of 44%–53%). The proportion of patients with disease progression was reduced significantly in DEFINE but failed to reach statistical significance in CONFIRM.44,45 Secondary clinical endpoints and MRI measures of disease activity, including the number of Gd-enhancing lesions and T2-weighted hyperintense lesions, favored BG-12 over placebo as well. Furthermore, BG-12 three times daily was superior compared to treatment with glatiramer acetate with regard to the ARR (P < 0.05) and the number of new T1-weighted hypointense lesions (P < 0.05).
Specific safety and tolerability
In phase II and III clinical trials, FAE was well tolerated especially after using BG-12 containing only DMF in enteric-coated microtablets to improve gastrointestinal tolerability.
In both phase III studies the overall incidence of AEs was similar across all treatment groups, highlighting the favorable safety profile of BG-12.44,45 In parallel, the incidence of AEs leading to discontinuation of the study agent did not differ between groups. The majority of AEs were of mild or moderate severity and normalized without discontinuation of the study drug. Common AEs in the BG-12 group involved gastrointestinal events (diarrhea, nausea, vomiting, and abdominal pain), flushing, pruritus, and proteinuria (Table 2). Gastrointestinal side effects and flushing occurred more frequently at the beginning of therapy and often diminished while continuing treatment. Nevertheless, gastrointestinal events and flushing resulted more often in discontinuation of treatment in patients receiving BG-12 compared to the placebo. Serious AEs were rare and similar across all treatment groups. The overall incidence of both malignant neoplasms and opportunistic infections was not increased in patients receiving BG-12. No cases of death, liver failure, or renal failure were assessed by the investigator as related to the study medication.44,45
As expected, the mean white cell count and lymphocyte count decreased in patients treated with BG-12. Usually, the mean white cell count and lymphocyte count decreased over the first 12 months and then plateaued, remaining within normal range. Overwhelming decreases of the white cell count and lymphocyte count (<3.0 × 1 0 9/L and <0.5 × 1 0 9/L, respectively) occurred in 4%–10% of patients treated with BG-12. Decreases of the mean white cell count and lymphocyte count were not associated with an increase of infections. In DEFINE, the overall incidence of infections was balanced between study groups (64% in the placebo group vs 64% and 68% in the BG12 twice daily and thrice daily group, respectively).44 In parallel, the incidence of severe infections was not increased in patients treated with BG-12 compared to the placebo (2% in all study groups). Similar results were found in CONFIRM.45 The most common infections included nasopharyngitis, upper respiratory tract infection, urinary tract infection, and influenza.
Place in therapy
Large studies confirm the effectiveness of BG-12 reducing the ARR by about 50% compared to the placebo. Compared to glatiramer acetate there was a reduction of relapse rate of approximately 30%. Phase III studies underscore the favorable risk-benefit profile. Based on current data, there is clear evidence of the benefit of BG-12. Thus, BG-12 seems to be an appealing alternative therapy in RRMS. Approval is expected for 2013. BG-12 might also be helpful in patients suffering from SPMS, but referring trials have not yet been conducted.
Conclusion
Oral treatments are heralding a new era in MS therapy. They enable an extension of individualized therapies and open new challenges. As many studies are still ongoing and safety profiles are not yet completely developed, fundamental recommendations are not yet possible. Long-term data and comparisons of the efficacy of new oral agents with established treatments are eagerly awaited.
Acknowledgment
JT and GE contributed equally to the review.
Footnotes
Disclosure
JT received funding from Teva Pharmaceuticals Ltd. GE received funding from Biogen Idec and speakers’ fees from Almirall.
References
- 1.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372(9648):1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 2.Thöne J, Gold R. Laquinimod: a promising oral medication for the treatment of relapsing-remitting multiple sclerosis. Expert Opin Drug Metab Toxicol. 2011;7(3):365–370. doi: 10.1517/17425255.2011.556618. [DOI] [PubMed] [Google Scholar]
- 3.Brück W, Zamvill SS. Laquinimod, a once-daily oral drug in development for the treatment of relapsing-remitting multiple sclerosis. Expert Rev Clin Pharmacol. 2012;5(3):245–256. doi: 10.1586/ecp.12.12. [DOI] [PubMed] [Google Scholar]
- 4.Runström A, Leanderson T, Ohlsson L, Axelsson B. Inhibition of the development of chronic experimental autoimmune encephalomyelitis by laquinimod (ABR-215062) in IFN-beta ko and wild type mice. J Neuroimmunol. 2006;173(1–2):69–78. doi: 10.1016/j.jneuroim.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 5.Thöne J, Ellrichmann G, Seubert S, et al. Modulation of autoimmune demyelination by laquinimod via induction of brain-derived neurotrophic factor. Am J Pathol. 2012;180(1):267–274. doi: 10.1016/j.ajpath.2011.09.037. [DOI] [PubMed] [Google Scholar]
- 6.Brück W, Pförtner R, Pham T, et al. Reduced astrocytic NF-κB activation by laquinimod protects from cuprizone-induced demyelination. Acta Neuropathol. 2012;124(3):411–424. doi: 10.1007/s00401-012-1009-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aharoni R, Saada R, Eilam R, Hayardeny L, Sela M, Arnon R. Oral treatment with laquinimod augments regulatory T-cells and brain-derived neurotrophic factor expression and reduces injury in the CNS of mice with experimental autoimmune encephalomyelitis. J Neuroimmunol. 2012;251(1–2):14–24. doi: 10.1016/j.jneuroim.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 8.Mishra MK, Wang J, Silva C, Mack M, Yong VW. Kinetics of proinflammatory monocytes in a model of multiple sclerosis and its perturbation by laquinimod. Am J Pathol. 2012;181(2):642–651. doi: 10.1016/j.ajpath.2012.05.011. [DOI] [PubMed] [Google Scholar]
- 9.Polman C, Barkhof F, Sandberg-Wollheim M, et al. Treatment with laquinimod reduces development of active MRI lesions in relapsing MS. Neurology. 2005;64(6):987–991. doi: 10.1212/01.WNL.0000154520.48391.69. [DOI] [PubMed] [Google Scholar]
- 10.Comi G, Pulizzi A, Rovaris M, et al. Effect of laquinimod on MRI-monitored disease activity in patients with relapsing-remitting multiple sclerosis: a multicentre, randomised, double-blind, placebo-controlled phase IIb study. Lancet. 2008;371(9630):2085–2092. doi: 10.1016/S0140-6736(08)60918-6. [DOI] [PubMed] [Google Scholar]
- 11.Comi G, Abramsky A, Arbizu T, et al. Oral laquinimod in patients with relapsing–remitting multiple sclerosis: 36-week double-blind active extension of the multi-centre, randomized, double-blind, parallel-group placebo-controlled study. Mult Scler. 2010;16(11):1360–1366. doi: 10.1177/1352458510378127. [DOI] [PubMed] [Google Scholar]
- 12.Comi G, Jeffery D, Kappos L, et al. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N Engl J Med. 2012;366(11):1000–1009. doi: 10.1056/NEJMoa1104318. [DOI] [PubMed] [Google Scholar]
- 13.CMSC INForMS: Results of phase III BRAVO trial Reinforce Unique profile of Laquinimod for MS [webpage on the Internet] Hackensack: Consortium of the Multiple Sclerosis Centers; 2011Available from: http://bit.Ly/oAuZD6Accessed February 12, 2012 [Google Scholar]
- 14.Scott LJ. Fingolimod: a review of its use in the management of relapsing-remitting multiple sclerosis. CNS Drugs. 2011;25(8):673–698. doi: 10.2165/11207350-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 15.Kappos L, Antel J, Comi G, et al. Oral fingolimod (FTY720) for relapsing multiple sclerosis. N Engl J Med. 2006;355(11):1124–1140. doi: 10.1056/NEJMoa052643. [DOI] [PubMed] [Google Scholar]
- 16.Comi G, O’Connor P, Montalban X, et al. Phase II study of oral fingolimod (FTY720) in multiple sclerosis: 3-year results. Mult Scler. 2010;16(2):197–207. doi: 10.1177/1352458509357065. [DOI] [PubMed] [Google Scholar]
- 17.Kappos L, Radue EW, O’Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362(5):387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 18.Devonshire V, Havrdova E, Radue E W, et al. Relapse and disability outcomes in patients with multiple sclerosis treated with fingolimod: subgroup analyses of the double-blind, randomised, placebo-controlled FREEDOMS study. Lancet Neurol. 2012;11(5):420–428. doi: 10.1016/S1474-4422(12)70056-X. [DOI] [PubMed] [Google Scholar]
- 19.Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- 20.Khatri B, Barkhof F, Comi G, et al. Comparison of fingolimod with interferon beta-1a in relapsing-remitting multiple sclerosis: a randomised extension of the TRANSFORMS study. Lancet Neurol. 2011;10(6):520–529. doi: 10.1016/S1474-4422(11)70099-0. [DOI] [PubMed] [Google Scholar]
- 21.Poulsen RR, McClaskey CM, Rivkees SA, Wendler CC. The Sphingosine-1-phospate receptor 1 mediates S1P action during cardiac development. BMC Dev Biol. 2011;11:37. doi: 10.1186/1471-213X-11-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Montalban X, Comi G, O’Connor P, et al. Oral fingolimod (FTY720) in relapsing multiple sclerosis: impact on health-related quality of life in a phase II study. Mult Scler. 2011;17(11):1341–1350. doi: 10.1177/1352458511411061. [DOI] [PubMed] [Google Scholar]
- 23.Claussen MC, Korn T. Immune mechanisms of new therapeutic strategies in MS: teriflunomide. Clin Immunol. 2012;142(1):49–56. doi: 10.1016/j.clim.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 24.Gold R, Wolinsky JS. Pathophysiology of multiple sclerosis and the place of teriflunomide. Acta Neurol Scand. 2011;124(2):75–84. doi: 10.1111/j.1600-0404.2010.01444.x. [DOI] [PubMed] [Google Scholar]
- 25.O’Connor P W, Li D, Freedman MS, et al. Teriflunomide Multiple Sclerosis Trial Group. University of British Columbia MS/MRI Research Group A Phase II study of the safety and efficacy of teriflunomide in multiple sclerosis with relapses. Neurology. 2006;66(6):894–900. doi: 10.1212/01.wnl.0000203121.04509.31. [DOI] [PubMed] [Google Scholar]
- 26.Freedman MS, Wolinsky JS. Wamil B, et al; Teriflunomide Multiple Sclerosis Trial Group and the MRI Analysis Center. Teriflunomide added to interferon-β in relapsing multiple sclerosis: a randomized phase II trial. Neurology. 2012;78(23):1877–1885. doi: 10.1212/WNL.0b013e318258f7d4. [DOI] [PubMed] [Google Scholar]
- 27.Freedman MS, Wolinsky JS, Wamil B, et al. Oral teriflunomide plus glatiramer acetate in relapsing multiple sclerosis [abstract P17] Int J MS Care. 2011;13:9. [Google Scholar]
- 28.O’Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365(14):1293–1303. doi: 10.1056/NEJMoa1014656. [DOI] [PubMed] [Google Scholar]
- 29.Genzyme Reports Top-line Results for TENERE Study of Oral Teriflunomide in Relapsing Multiple Sclerosis [webpage on the Internet] Cambridge: Business Wire; 2011Available from: http://www.businesswire.com/news/home/20111219006550/en/Genzyme-Reports-Top-line-Results-TENERE-Study-OralAccessed December 4, 2012 [Google Scholar]
- 30.Stuve O, Benamor M, Benzerdjeb H, Kieseier B. Pregnancy Outcomes from the teriflunomide clinical development program: retrospective analysis of a global pharmacovigilance database [abstract] Neurology. 2012;78(Meeting Abstracts 1):P06.190. [Google Scholar]
- 31.Leist TP, Weissert R. Cladribine: mode of action and implications for treatment of multiple sclerosis. Clin Neuropharmacol. 2011;34(1):28–35. doi: 10.1097/WNF.0b013e318204cd90. [DOI] [PubMed] [Google Scholar]
- 32.Beutler E, Sipe JC, Romine JS, Koziol JA, McMillan R, Zyroff J. The treatment of chronic progressive multiple sclerosis with cladribine. Proc Natl Acad Sci USA. 1996;93(4):1716–1720. doi: 10.1073/pnas.93.4.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sipe JC, Romine JS, Koziol JA, McMillan R, Zyroff J, Beutler E. Cladribine in treatment of chronic progressive multiple sclerosis. Lancet. 1994;34(8914):9–13. doi: 10.1016/s0140-6736(94)91046-4. [DOI] [PubMed] [Google Scholar]
- 34.Giovannoni G, Comi G, Cook S, et al. CLARITY Study Group A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med. 2010;362(5):416–426. doi: 10.1056/NEJMoa0902533. [DOI] [PubMed] [Google Scholar]
- 35.Giovannoni G, Cook S, Rammohan K, et al. CLARITY study group Sustained disease-activity-free status in patients with relapsing-remitting multiple sclerosis treated with cladribine tablets in the CLARITY study: a post-hoc and subgroup analysis. Lancet Neurol. 2011;10(4):329–337. doi: 10.1016/S1474-4422(11)70023-0. [DOI] [PubMed] [Google Scholar]
- 36.Hartung HP, Aktas O, Kieseier B, Giancarlo Comi GC. Development of oral cladribine for the treatment of multiple sclerosis. J Neurol. 2010;257(2):163–170. doi: 10.1007/s00415-009-5359-0. [DOI] [PubMed] [Google Scholar]
- 37.Sipe JC. Cladribine for multiple sclerosis: review and current status. Expert Rev Neurother. 2005;5(6):721–727. doi: 10.1586/14737175.5.6.721. [DOI] [PubMed] [Google Scholar]
- 38.US Food and Drug Administration [homepage on the Internet] Silver Spring: FDA; 2012Available from: http://www.fda.govAccessed December 4, 2012 [Google Scholar]
- 39.European Medicines Agency [homepage on the Internet] London: EMA; 2011Available from: http://www.ema.europa.eu/emaAccessed December 4, 2012 [Google Scholar]
- 40.multiple Sclerosis Resource Center [homepage on the Internet] Colchester: MSRC; 2012Available from: http://www.msrc.co.ukAccessed December 4, 2012 [Google Scholar]
- 41.Linker RA, Lee DH, Ryan S, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134(Pt 3):678–692. doi: 10.1093/brain/awq386. [DOI] [PubMed] [Google Scholar]
- 42.Papadopoulou A, D’Souza M, Kappos L, Yaldizli O. Dimethyl fumarate for multiple sclerosis. Expert Opin Investig Drugs. 2010;19(12):1603–1612. doi: 10.1517/13543784.2010.534778. [DOI] [PubMed] [Google Scholar]
- 43.Schimrigk S, Brune N, Hellwig K, et al. Oral fumaric acid esters for the treatment of active multiple sclerosis: an open-label, baseline-controlled pilot study. Eur J Neurol. 2006;13(6):604–610. doi: 10.1111/j.1468-1331.2006.01292.x. [DOI] [PubMed] [Google Scholar]
- 44.Gold R, Kappos L, Arnold DL, et al. DEFINE Study Investigators Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367(12):1098–1107. doi: 10.1056/NEJMoa1114287. [DOI] [PubMed] [Google Scholar]
- 45.Fox RJ, Miller DH, Phillips JT, et al. CONFIRM Study Investigators Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367(12):1087–1097. doi: 10.1056/NEJMoa1206328. [DOI] [PubMed] [Google Scholar]