

Abstract
Arteries and veins acquire distinct molecular identities prior to the onset of embryonic blood circulation, and their specification is crucial for vascular development. The transcription factor COUP-TFII currently functions at the top of a signaling pathway governing venous fate. It promotes venous identity by inhibiting Notch signaling and subsequent arterialization of endothelial cells, yet nothing is known about what regulates COUP-TFII expression in veins. We now report that the chromatin-remodeling enzyme BRG1 promotes COUP-TFII expression in venous endothelial cells during murine embryonic development. Conditional deletion of Brg1 from vascular endothelial cells resulted in downregulated COUP-TFII expression and aberrant expression of arterial markers on veins. BRG1 promotes COUP-TFII expression by binding conserved regulatory elements within the COUP-TFII promoter and remodeling chromatin to make the promoter accessible to transcriptional machinery. This study provides the first description of a factor promoting COUP-TFII expression in vascular endothelium and highlights a novel role for chromatin remodeling in venous specification.
Keywords: SWI/SNF, Chromatin remodeling, Veins, Mouse, NR2F2
INTRODUCTION
Arteries and veins are structurally and functionally distinct vessels that play crucial roles in circulating blood to and from sources of oxygen. Arteries carry blood from the heart under high pressure and are surrounded by multiple layers of smooth muscle cells and extracellular matrix components that provide strength and elasticity to their vascular walls. Veins, by contrast, have thinner and less elastic vascular walls, and rely on specialized valves to return blood under low pressure to the heart. Arterial-venous identity is specified during embryonic development and is influenced by hemodynamic forces. Moreover, experimental manipulation of blood flow can alter the fate of arteries and veins, indicating that vessel identity and function are plastic properties (Garcia-Cardeña et al., 2001; le Noble et al., 2004).
Despite the important roles that hemodynamic forces play in differentiating arteries from veins, it is also clear that genetic factors contribute significantly to their identity. Molecular markers that distinguish arteries from veins are expressed before the onset of blood circulation during embryonic development (Wang et al., 1998; Zhong et al., 2000; Herzog et al., 2001). In addition, genetic mutations can induce changes in arterial/venous marker expression even when hemodynamic forces are unaltered (Jones et al., 2008).
The first genetic markers shown to be differentially expressed on arteries and veins were the transmembrane ligand ephrin B2 (EFNB2) and its cognate tyrosine kinase receptor EPHB4 (Wang et al., 1998). These markers are respectively expressed on arterial and venous endothelial cells of the developing murine yolk sac prior to blood flow, and their genetic ablation results in failed vascular remodeling and embryonic lethality by embryonic day 11 (E11.0) (Wang et al., 1998; Adams et al., 1999; Gerety et al., 1999). However, EFNB2 and EPHB4 are not determinants of arterial-venous specification, as their deletion does not switch vessel identity (Gerety et al., 1999). Rather, Notch signaling determines arterial specification (Lawson et al., 2001; Zhong et al., 2001; Duarte et al., 2004; Krebs et al., 2004). Loss-of-function mutations in Notch pathway components result in downregulated Efnb2 expression in arteries (Duarte et al., 2004; Fischer et al., 2004; Krebs et al., 2004; Kokubo et al., 2005; Koo et al., 2005), whereas overexpression of Notch signaling components causes veins to ectopically express Efnb2 (Trindade et al., 2008).
Venous specification was originally thought to occur by default in the absence of Notch signaling (Thurston and Yancopoulos, 2001). But it is now recognized that the orphan nuclear receptor chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII, also known as NR2F2) actively promotes venous specification by inhibiting Notch signaling in a subset of endothelial cells (You et al., 2005; Chen et al., 2012). Conditional deletion of COUP-TFII in embryonic endothelial cells upregulates Notch signaling in venous endothelium and leads to aberrant arterialization of veins. COUP-TFII currently functions at the top of the venous specification pathway, and nothing is known about what regulates its expression and activity in veins.
ATP-dependent chromatin-remodeling complexes modulate expression of their target genes by altering accessibility of transcriptional machinery to gene regulatory regions (Hargreaves and Crabtree, 2011). Each complex contains a catalytic ATPase, which generates energy required for altering chromatin accessibility. Mammalian SWITCH/sucrose non-fermentable (SWI/SNF)-like complexes contain one of two mutually exclusive ATPases: brahma (BRM, also known as SMARCA2) or brahma-related gene 1 (BRG1, also known as SMARCA4). Conditional deletion of Brg1 has revealed its importance in a variety of developmental processes, including embryonic vascular development (Curtis et al., 2012).
We now present evidence that the chromatin-remodeling enzyme BRG1 epigenetically regulates COUP-TFII expression in veins during embryonic development. BRG1 remodels chromatin within the COUP-TFII promoter, impacting the ability of transcriptional machinery to access the promoter. Genetic depletion of Brg1 results in downregulated COUP-TFII and aberrant arterial marker expression in developing veins. Our data provide important new insight into an upstream epigenetic regulatory mechanism for the venous specification cascade.
MATERIALS AND METHODS
Mice
Brg1-floxed mice (Brg1fl/fl) (Gebuhr et al., 2003), β-catenin-floxed mice (Catnbfl/fl) (Brault et al., 2001), Tie2-Cre+ transgenic mice (Kisanuki et al., 2001), Efnb2LacZ mice (Wang et al., 1998) and BAT-gal transgenic mice (Maretto et al., 2003) were maintained on a mixed genetic background at the Oklahoma Medical Research Foundation animal facility. All animal use protocols were approved by the Institutional Animal Care and Use Committee.
Genotyping
PCR genotyping of Brg1-floxed, Tie2-Cre+ and BAT-gal transgenic embryos and mice was performed as described previously (Griffin et al., 2011). β-catenin-floxed mice and embryos were PCR genotyped using the following primers: forward (5′-AAGGTAGAGTGATGAAAGTTGTT-3′) and reverse (5′-CACCATGTCCTCTGTCTATTC-3′). These primers amplified a 221 bp fragment of the wild-type allele and a 324 bp fragment of the floxed allele. The PCR was performed at an annealing temperature of 60°C. Efnb2LacZ mice were genotyped by whole-mount X-gal staining for β-galactosidase activity on ear punches.
Primary endothelial cell isolation and maintenance
Endothelial cells from individual murine yolk sacs and embryos were isolated using anti-PECAM-1-conjugated magnetic beads, as described previously (Griffin et al., 2011). Primary human umbilical vein endothelial cells (HUVECs) were isolated and maintained as previously described (Yao et al., 1996). Primary human aortic endothelial cells (ATCC, Manassas, VA, USA; PCS-100-011) were maintained using the Endothelial Cell Growth Kit-BBE (ATCC; PCS 100-040) and Vascular Cell Basal Medium (ATCC; PCS 100-030). HUVECs and primary aortic endothelial cells were passaged no more than twice.
Cell culture and transfections
C166 yolk sac endothelial cells (ATCC; CRL-2581) were maintained and transfected with 100 nM BRG1 siGENOME SMART pool or nonspecific control small interfering RNA (siRNA) oligonucleotides (Dharmacon, Lafayette, CO, USA; M-041135-01 and D-001210-01, respectively), as described previously (Griffin et al., 2011). After 24 hours, protein was harvested in Laemmli buffer [62.5 mM Tris (pH 6.8)/10% glycerol/5% SDS/0.01% bromophenol blue] plus 0.2 M dithiothreitol for western blotting. Alternatively, RNA was harvested after 24 hours in TRIzol (Invitrogen, Grand Island, NY, USA) for transcript analysis. For the BRG1 overexpression studies, a BRG1 expression plasmid was constructed by excising a 5338 bp murine Brg1 cDNA from IMAGE clone 30533489 (ATCC; 10698217) with SalI and XbaI, and inserting it into the multiple cloning site (XhoI-XbaI) of the mammalian expression vector pcDNA3.1 (Invitrogen). The resulting plasmid (or empty pcDNA3.1 vector) was transfected into C166 endothelial cells at various concentrations for 24 hours with Lipofectamine 2000 (Invitrogen). For the COUP-TFII rescue experiment, 200 ng of a COUP-TFII expression plasmid (pCNX-COUP-TFII; obtained from Dr Sophia Tsai, Baylor College of Medicine, Houston, TX, USA) was co-transfected with 100 nM BRG1 siRNA into C166 endothelial cells for 24 hours with Lipofectamine 2000.
Western blot
Total protein was harvested from siRNA-transfected C166 cells after 24 hours knockdown, fractionated in a 9% SDS polyacrylamide gel, and transferred to a PVDF membrane for western blot analysis using antibodies to BRG1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA; sc-17796), COUP-TFII (R&D Systems, Minneapolis, MN, USA; PP-H7147-00) and GAPDH (Sigma, St. Louis, MO, USA; 9545). Band intensity was determined for each protein using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Quantitative real-time PCR (qPCR)
To analyze transcript levels, total RNA from primary yolk sac or embryonic endothelial cells or C166 siRNA-transfected cells was harvested in TRIzol (Invitrogen), and RNA was prepared using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. cDNA was prepared using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA), and qPCR was performed using RT2 Fast SYBR Green qPCR Master Mix (SABioscience, Valencia, CA, USA) and the CFX96 detection system (Bio-Rad) with gene-specific primers.
qPCR primers
qPCR primers are listed in supplementary material Table S1.
qPCR analysis
The relative fold changes in transcript levels were determined using the comparative threshold cycle (CT) method with the β-actin and Gapdh housekeeping genes as internal controls. Data from three independent experiments were combined and are presented as mean±s.e.m. Statistical differences were detected using a two-tailed Student’s t-test.
Whole-mount yolk sac and placental staining
For whole-mount yolk sac immunostaining, E9.5 Brg1fl/fl:Tie2-Cre+ embryos with attached yolk sacs were dissected from maternal tissue, fixed for 10 minutes in 3% paraformaldehyde (PFA), permeabilized, blocked and stained as described (Griffin et al., 2011). Primary anti-PECAM-1 (1:100; BD Biosciences, San Jose, CA, USA; 557355) and secondary donkey-anti-rat-Cy3 IgG (1:250; Jackson ImmunoResearch, West Grove, PA, USA; 9712-165-153) antibodies were used. The tissues were then washed and post-fixed as described previously (Griffin et al., 2011) and transferred to glass slides. On the glass slides, the embryos were dissected away from the yolk sacs while maintaining the orientation of the vitelline arteries and veins within the yolk sacs. The yolk sacs were then flat-mounted and coverslipped with DABCO mounting media [2.5% 1,4-diazabicyclo[2.2.2]octane in 9:1 glycerol/PBS (pH 8.6)] for microscopic examination. Embryos were simultaneously digested for genotyping.
Whole-mount X-gal staining for β-galactosidase activity was performed on BAT-gal and Efnb2LacZ embryos attached to their yolk sacs and placentae. Tissues were fixed in a 2% PFA/0.2% glutaraldehyde solution, washed and X-gal stained as described (Griffin et al., 2008) for 48 hours at room temperature. The stained yolk sacs were dissected away from embryos and placentae and flat-mounted in DABCO mounting media on glass slides as described above.
Immunofluorescence
Brg1fl/fl:Tie2-Cre+ and littermate controls were cryoembedded and sectioned (10-12 μm). Double immunostaining using anti-PECAM-1 (1:500; BD Biosciences, 553370) and anti-BRG1 (1:100; Santa Cruz Biotechnology, SC-17796) antibodies was performed as described previously (Curtis and Griffin, 2012). Double immunostaining for PECAM-1 and αSMA in embryonic sections was performed as follows: cryosections were thawed and blocked in blocking solution [3% normal donkey serum (Jackson ImmunoResearch)/3% BSA/0.3% Triton X-100/PBS] for 2 hours at room temperature. Sections were then co-incubated overnight at 4°C in anti-PECAM-1 (1:500) and Cy3-conjugated anti-αSMA (1:200; Sigma, C6198) diluted in blocking solution. After washing three times (3 minutes each) in 0.1% Triton X-100/PBS, sections were incubated for 1 hour at room temperature in Alexa 488-donkey anti-rat IgG (1:500; Invitrogen) and 1:500 Hoechst (20 μg/ml) diluted in blocking solution. Finally, sections were washed as above and mounted with DABCO mounting media and glass coverslips. Double immunostaining for PECAM-1 and either COUP-TFII, NRP1, DLL4 or EPHB4 was performed similarly with the following exceptions: cryosections were blocked for 1 hour at room temperature and co-incubated overnight at 4°C in anti-PECAM-1 (1:500) and either anti-COUP-TFII (1:100; R&D Systems, PP-H7147-00), anti-NRP1 (1:100; R&D Systems, AF566), anti-DLL4 (1:100; R&D Systems, AF1389) or anti-EPHB4 (1:100; R&D Systems, AF446) diluted in blocking solution. For secondary antibodies, Cy3-donkey anti-rat IgG (1:500; Jackson ImmunoResearch) and Alexa 488-goat anti-mouse IgG or Alexa 488-donkey anti-goat (1:500; Invitrogen) were used.
Microscopy and image acquisition
Fluorescent images were obtained with a Nikon Eclipse 80i microscope using 4× (NA 0.13), 10× (NA 0.3) and 20× (NA 0.5) objectives, an X-cite 120Q light source, and a Nikon DS-Qi1Mc camera. NIS-Elements AR3.0 (Nikon, Melville, NY, USA) software was used for all fluorescent image acquisition and assembly. ImageJ software was used to quantify fluorescence intensity of immunostained tissue sections. Gross yolk sac images were obtained with a Nikon SMZ800 stereomicroscope and Nikon DS-Fi1 camera and monitor.
Chromatin immunoprecipitation (ChIP)
Subconfluent C166 yolk sac endothelial cells, HUVECs or primary human aortic endothelial cells were used for ChIP with the MAGnify Chromatin Immunoprecipitation System (Invitrogen) according to the manufacturer’s instructions. A mixture of BRG1-specific antibodies (Millipore, 07-478; Abcam, ab4081) was used to immunoprecipitate protein-DNA complexes. Isotype-matched IgG antibodies (Invitrogen) were used as a negative control. For total histone H3 ChIP, chromatin was harvested from C166 cells transfected with nonspecific or BRG1-specific siRNAs and immunoprecipitated using the histone H3-specific and negative-control antibodies supplied in the ChIPAb+ Histone H3 kit (Millipore, Billerica, MA, USA; 17-10046). For RNA polymerase II ChIP, chromatin was harvested as described for H3 ChIP using an anti-RNAPolII antibody (Abcam, Cambridge, MA, USA; ab5408) and negative control antibodies from the H3 kit. For H3K9Ac ChIP, chromatin was harvested as described for H3 ChIP using an anti-histone H3K9Ac antibody (Active Motif, Carlsbad, CA, USA; 39137), and isotype-matched IgG antibodies (Invitrogen) were used as a negative control. Real-time quantitative PCR was performed using RT2 Fast SYBR green qPCR master mix (SABiosciences) and the CFX96 detection system (Bio-Rad) with specific primers. Data from three or four independent experiments were combined and presented as n-fold levels of enrichment over the negative control±s.e.m. Statistical differences were detected using a two-tailed Student’s t-test.
ChIP qPCR primers
Primers used are listed in supplementary material Table S2.
RESULTS
Brg1fl/fl:Tie2-Cre+ mutant yolk sac veins are morphologically abnormal
We previously showed that deletion of Brg1 from developing endothelium using a Tie2-Cre transgene results in abnormal angiogenesis in yolk sac vessels owing, in part, to misregulated Wnt signaling (Griffin et al., 2011). To examine angiogenesis defects in Brg1fl/fl:Tie2-Cre+ yolk sacs more thoroughly, we immunostained E9.5 littermate control and Brg1fl/fl:Tie2-Cre+ yolk sacs with an endothelial-specific (anti-PECAM-1) antibody and flat-mounted the yolk sacs on slides while maintaining the orientation of vitelline arteries and veins (Fig. 1). We found that angiogenesis defects were more exaggerated in Brg1fl/fl:Tie2-Cre+ yolk sac veins than in arteries (Fig. 1B). Brg1fl/fl:Tie2-Cre+ yolk sac veins were poorly remodeled, and many veins were thin, blunt-ended or failed to interconnect (Fig. 1D). This venous phenotype was unexpected as BRG1 is expressed in both arterial and venous endothelial cells and is excised efficiently from arteries and veins in Brg1fl/fl:Tie2-Cre+ extra-embryonic vessels (supplementary material Fig. S1). Nevertheless, our observations indicate that Brg1fl/fl:Tie2-Cre+ yolk sac veins exhibit more phenotypic abnormalities than mutant arteries.
Fig. 1.
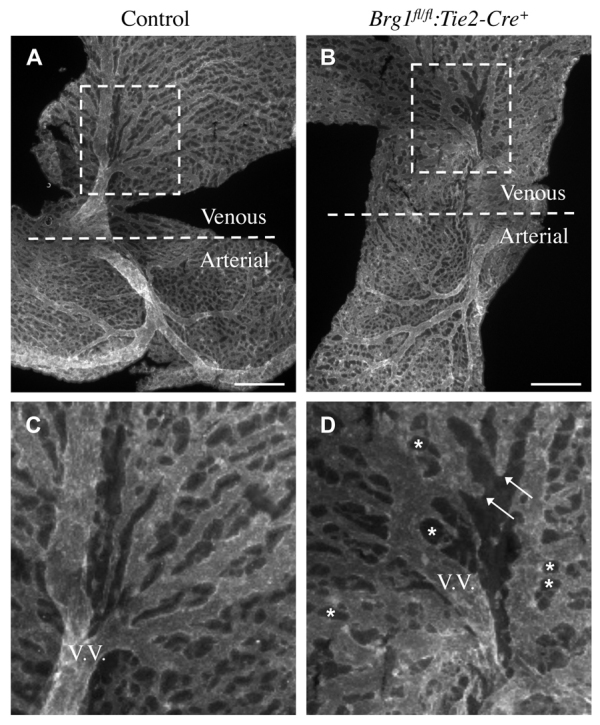
Brg1fl/fl:Tie2-Cre+ yolk sac veins are morphologically abnormal. (A-D) Anti-PECAM1 staining on flat-mounted E9.5 yolk sacs revealed Brg1fl/fl:Tie2-Cre+ veins were more abnormal than arteries. Whereas the arterial sides of control and Brg1fl/fl:Tie2-Cre+ yolk sacs contained branching networks of vessels, the venous side of Brg1fl/fl:Tie2-Cre+ yolk sacs failed to undergo normal branching and development. (C,D) Magnified views of the boxed regions in A,B, respectively, including vitelline veins (V.V.). Arrows in D indicate Brg1fl/fl:Tie2-Cre+ veins that failed to interconnect or underwent aberrant regression. Asterisks indicate round, non-vascular spaces that are characteristic of failed vascular plexus remodeling. Scale bars: 500 μm.
Brg1fl/fl:Tie2-Cre+ veins express arterial markers and characteristics
To examine Brg1fl/fl:Tie2-Cre+ yolk sac venous abnormalities further, we crossed control and Brg1fl/fl:Tie2-Cre+ embryos onto an arterial reporter line in which LacZ is knocked into the Efnb2 locus (Efnb2LacZ) (Wang et al., 1998). Upon whole-mount X-gal staining of E9.5 yolk sacs, we saw Efnb2LacZ reporter activity in control and Brg1fl/fl:Tie2-Cre+ arteries, as expected (Fig. 2A,B). However, Brg1fl/fl:Tie2-Cre+ yolk sacs also showed ectopic Efnb2LacZ reporter activity in veins (Fig. 2B).
Fig. 2.
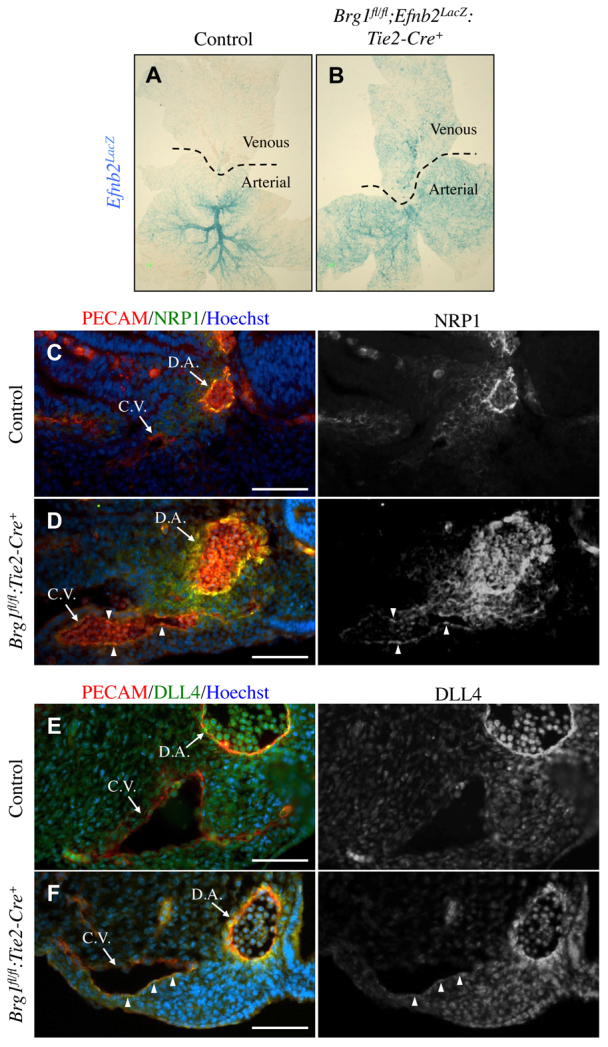
Brg1fl/fl:Tie2-Cre+ veins express arterial markers. (A,B) Control and Brg1fl/fl:Tie2-Cre+ embryos were crossed onto an Efnb2LacZ arterial reporter line and stained with X-gal solution to reveal sites of Efnb2 (LacZ) expression (blue). Flat-mounted E9.5 yolk sacs displayed aberrant Efnb2 expression in Brg1fl/fl;Efnb2LacZ:Tie2-Cre+ veins (B) compared with control veins (A). (C-F) E9.75 littermate control and Brg1fl/fl:Tie2-Cre+ embryos were cross-sectioned, and sections containing a dorsal aorta (D.A.) and cardinal vein (C.V.) were immunostained for arterial markers. (C,D) Sections were stained for the endothelial cell marker PECAM1 (red), the arterial marker NRP1 (green) and the nuclear marker Hoechst (blue). NRP1 was aberrantly upregulated on endothelial cells within Brg1fl/fl:Tie2-Cre+ cardinal veins (arrowheads in D). (E,F) Sections were stained for PECAM1 (red), DLL4 (green) and Hoechst (blue). Arterial DLL4 was likewise upregulated on Brg1fl/fl:Tie2-Cre+ cardinal veins (see arrowheads in F). Scale bars: 100 μm.
We performed immunostaining on cross-sections of E9.75 control and Brg1fl/fl:Tie2-Cre+ embryos to determine whether BRG1 also influences arterial marker expression in the embryo proper. Brg1fl/fl:Tie2-Cre+ cardinal vein endothelial cells aberrantly expressed the arterial markers neuropilin 1 (NRP1) (Fig. 2D) and DLL4 (Fig. 2F) at this developmental timepoint. Therefore, both extra-embryonic and embryonic venous endothelial cells express arterial markers in Brg1fl/fl:Tie2-Cre+ mutants.
To investigate whether Brg1fl/fl:Tie2-Cre+ extra-embryonic and embryonic veins acquire other arterial characteristics, E10.5 control and Brg1fl/fl:Tie2-Cre+ umbilical vessels were cross-sectioned and immunostained with an antibody against the smooth muscle cell marker alpha-smooth muscle actin (αSMA). In control sections, αSMA-positive cells predominantly surrounded umbilical arteries rather than umbilical veins (Fig. 3A). By contrast, Brg1fl/fl:Tie2-Cre+ umbilical veins recruited αSMA-positive cells to a similar extent as umbilical arteries (Fig. 3B). Likewise, evidence of aberrant αSMA-positive cell recruitment to cardinal veins was evident in E10.5 embryos (Fig. 3D). These results indicate that Brg1fl/fl:Tie2-Cre+ extra-embryonic and embryonic veins acquire arterial characteristics at midgestation and represent the first evidence of a role for BRG1 in embryonic blood vessels outside of the developing heart (Curtis et al., 2012).
Fig. 3.
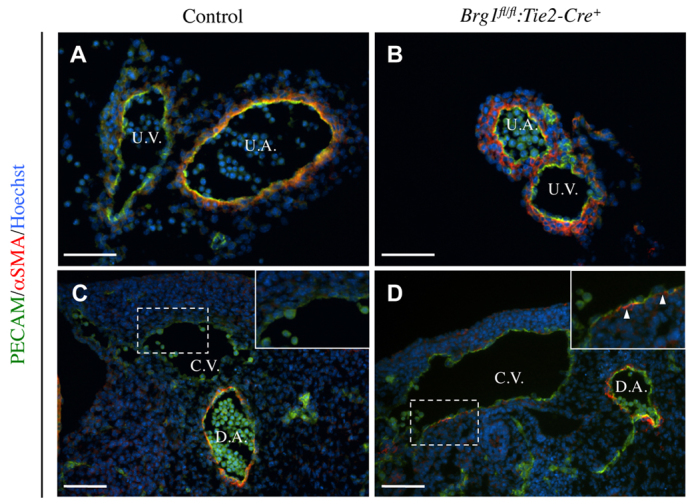
Brg1fl/fl:Tie2-Cre+ veins aberrantly recruit smooth muscle cells. Tissues from E10.5 littermate control and Brg1fl/fl:Tie2-Cre+ mutants were sectioned and immunostained for the endothelial cell marker PECAM1 (green), the smooth muscle cell marker α-smooth muscle actin (αSMA) (red) and the nuclear marker Hoechst (blue). (A,B) Extra-embryonic umbilical vessels, including umbilical veins (U.V.) and umbilical arteries (U.A.), were sectioned and stained. In control vessels, αSMA-positive cells predominantly accumulated around umbilical arteries (A). However, αSMA-positive cells accumulated around both umbilical arteries and veins in Brg1fl/fl:Tie2-Cre+ mutants (B). (C,D) Sections of embryos containing a dorsal aorta (D.A.) and cardinal vein (C.V.) were immunostained. αSMA-positive cells were detected around the Brg1fl/fl:Tie2-Cre+ C.V. (arrowheads in magnified inset of D) but not around the control C.V. (C). Scale bars: 100 μm.
COUP-TFII expression is downregulated in BRG1-deficient endothelial cells
The nuclear receptor and transcription factor COUP-TFII functions at the top of the known venous specification signaling cascade (Lin et al., 2011). In order to determine whether COUP-TFII is properly expressed in Brg1fl/fl:Tie2-Cre+ veins, we immunostained extra-embryonic and embryonic vessels with anti-COUP-TFII antibody. COUP-TFII was downregulated in endothelial cells lining E10.5 Brg1fl/fl:Tie2-Cre+ umbilical veins (Fig. 4B) and E9.75 Brg1fl/fl:Tie2-Cre+ cardinal veins (Fig. 4D). To examine COUP-TFII transcript levels in Brg1fl/fl:Tie2-Cre+ endothelial cells, we isolated RNA from E10.5 yolk sac and embryonic endothelial cells and performed quantitative real-time PCR (qPCR). We found COUP-TFII mRNA levels were significantly reduced in Brg1fl/fl:Tie2-Cre+ endothelial cells compared with control endothelial cells (Fig. 4E,F). We also knocked down BRG1 in the C166 murine yolk sac-derived endothelial cell line (Wang et al., 1996) and found COUP-TFII mRNA and protein levels were significantly downregulated (Fig. 4G,H; supplementary material Fig. S2). These expression data indicate that COUP-TFII expression is diminished in BRG1-deficient endothelial cells both in vivo and in vitro.
Fig. 4.
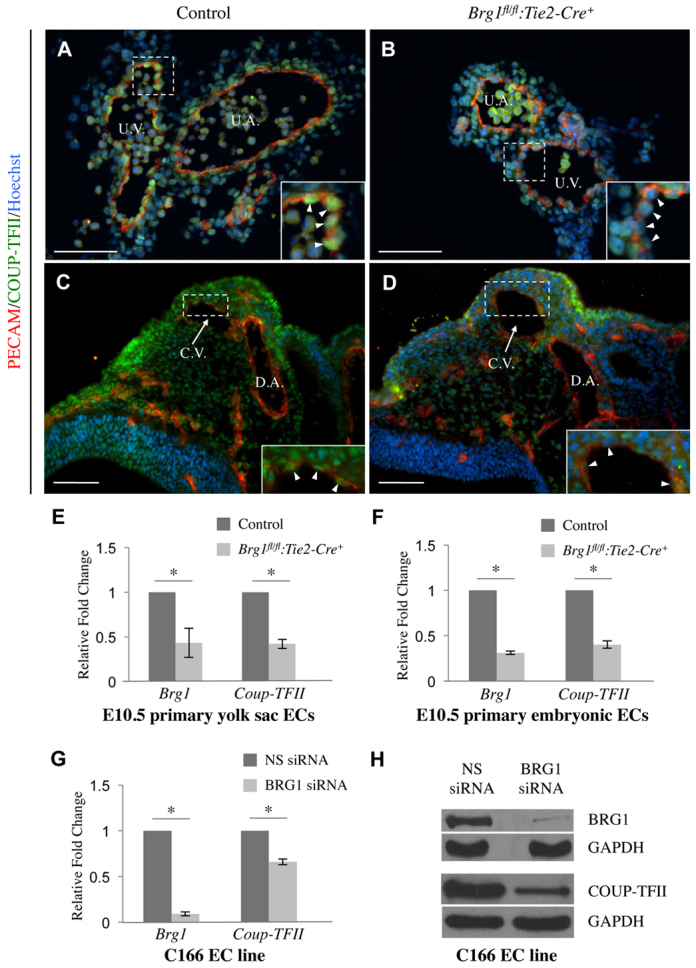
COUP-TFII expression is downregulated in Brg1-deficient endothelial cells. (A-D) Cryosections of littermate control and Brg1fl/fl:Tie2-Cre+ tissues were immunostained for the endothelial cell marker PECAM1 (red), COUP-TFII (green) and the nuclear marker Hoechst (blue). (A,B) Cross-sectioned E10.5 umbilical vessels were immunostained, and although COUP-TFII was expressed in umbilical vein (U.V.) endothelial cells in the control section (A), it was significantly diminished in Brg1fl/fl:Tie2-Cre+ venous endothelial cells (B). U.A., umbilical artery.(C,D) E9.75 embryos were cross-sectioned and immunostained. COUP-TFII was expressed in endothelial cells of the cardinal vein (C.V.) in the control section (C) but was downregulated in Brg1fl/fl:Tie2-Cre+ C.V. endothelial cells (D). D.A., dorsal aorta. For A-D, insets show magnified views of the boxed regions and arrowheads indicate individual endothelial cells. Scale bars: 100 μm. (E,F) Primary endothelial cells (ECs) were isolated from E10.5 control and Brg1fl/fl:Tie2-Cre+ tissues, RNA was purified and cDNA was synthesized. Samples from individual littermate control and Brg1fl/fl:Tie2-Cre+ yolk sacs (E) or embryos (F) were processed for qPCR analysis of Brg1 and COUP-TFII expression. Data from three independent experiments were combined and are presented as relative fold change over the expression levels in control cells±s.em. Significant differences were calculated using a two-tailed Student’s t-test (*P<0.05). (G,H) C166 endothelial cells were transfected with nonspecific (NS) or BRG1-specific siRNA for 24 hours. (G) RNA was isolated, cDNA was synthesized and qPCR for Brg1 or COUP-TFII was performed. Data from three independent experiments were combined and are presented as relative fold change over the expression levels in NS siRNA-treated cells±s.e.m. Significant differences were calculated using a two-tailed Student’s t-test (*P<0.05).(H) Protein samples were subjected to western blot analysis with antibodies that recognize BRG1, COUP-TFII or GAPDH.
COUP-TFII is a BRG1 target gene
To test whether BRG1 directly regulates COUP-TFII expression, we assessed BRG1 binding to the murine COUP-TFII promoter in endothelial cells. We used a comparative sequence alignment program (www.DCODE.org) to identify conserved regions of the COUP-TFII promoter in various species (Fig. 5A). We then designed chromatin immunoprecipitation (ChIP) primers towards two highly conserved promoter regions (-4.7 kb and -1.2 kb) and one nonconserved promoter region (-2.5 kb) located upstream of the COUP-TFII transcription start site (TSS). We also designed ChIP primers immediately upstream of the TSS at -0.3 kb. Our ChIP experiments in C166 endothelial cells showed BRG1 bound the -0.3 kb region immediately upstream of the COUP-TFII TSS as well as the two highly conserved regions at -1.2 kb and -4.7 kb (Fig. 5B). We also assessed BRG1 binding to the COUP-TFII promoter in human umbilical vein endothelial cells (HUVECs) and in human aortic endothelial cells. Unlike the C166 cell line, which we found expressed a mixture of arterial and venous markers, these primary cells differentially expressed COUP-TFII and other venous and arterial markers (data not shown). We found BRG1 did not bind to the -4.7 kb COUP-TFII promoter region in HUVECs or aortic endothelial cells, but it bound to the -1.2 kb region in both human cell types (supplementary material Fig. S3). Furthermore, BRG1 significantly bound the -0.3 kb promoter region in HUVECs but not in aortic endothelial cells. Although the difference in BRG1 binding to this region between the two cell types was small, our findings may indicate the -0.3 kb region plays an important role in differentiating COUP-TFII expression in venous and arterial endothelial cells. Altogether, our results demonstrate that BRG1 associates with COUP-TFII regulatory regions in human and murine endothelial cells, and indicate that COUP-TFII is a direct BRG1 target gene.
Fig. 5.
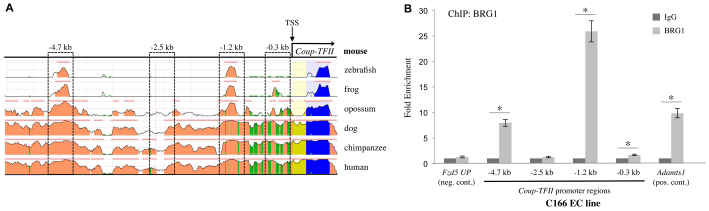
BRG1 binds to the COUP-TFII promoter in endothelial cells. (A) Alignment of the murine COUP-TFII promoter region with sequences from zebrafish, frog, opossum, dog, chimpanzee and human genomes from the NCBI DCODE website (http://www.dcode.org). Peak heights indicate degree of sequence homology; pink bars above peaks denote evolutionarily conserved regions; yellow represents the COUP-TFII 5′ untranslated region; blue indicates COUP-TFII exon 1. Boxed regions were selected for further analysis of BRG1 binding. Numbers above boxed regions denote approximate distances upstream of the COUP-TFII transcription start site (TSS). (B) Chromatin immunoprecipitation (ChIP) assays were performed on C166 endothelial cells using antibodies against BRG1 or isotype-matched non-specific IgG as a negative control. DNA was isolated and amplified by qPCR to determine whether BRG1 bound to various COUP-TFII promoter regions. Significant BRG1 binding was detected at the -0.3 kb, the -1.2 kb and the -4.7 kb promoter regions. A region upstream of the Fzd5 promoter (Fzd5 UP) was used as a negative control BRG1-binding region, and the Adamts1 promoter served as a positive control BRG1-binding region, as previously described (Griffin et al., 2011). Data from four independent experiments were combined and are presented as fold enrichment over the level of ChIP with negative control IgG antibodies±s.e.m. Significant differences were calculated using a two-tailed Student’s t-test (*P<0.05).
BRG1 mediates chromatin remodeling at the COUP-TFII promoter
In order to determine the functional consequence of BRG1 binding to the COUP-TFII promoter, we knocked down BRG1 in C166 endothelial cells and performed ChIP with an antibody to total histone H3 (H3). H3 binding correlates with nucleosome density and indicates whether chromatin is arranged in a closed or open conformation. In the absence of BRG1, H3 was enriched at the -0.3 kb and the -1.2 kb regions of the COUP-TFII promoter, indicating chromatin was highly compacted in those regions (Fig. 6A). Conversely, H3 occupancy was decreased at the -4.7 kb promoter region upon BRG1 knockdown, implying that chromatin was decompacted in that region (Fig. 6A). These results suggest BRG1 differentially remodels chromatin in various regions of the COUP-TFII promoter. To determine whether BRG1 impacts the ability of transcriptional machinery to bind the COUP-TFII promoter, we next assessed the ability of RNA polymerase II (RNAPolII) to ChIP to the COUP-TFII promoter in the absence of BRG1. We found RNAPolII binding near the COUP-TFII TSS was significantly decreased in BRG1 knockdown endothelial cells (Fig. 6B).
Fig. 6.
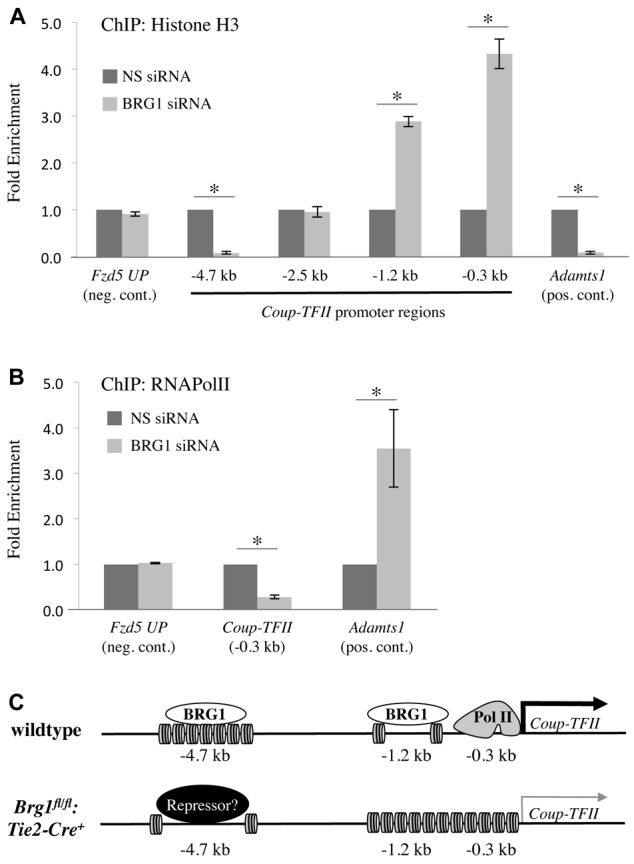
BRG1 remodels chromatin at the COUP-TFII promoter and influences accessibility of transcriptional machinery. (A,B) C166 endothelial cells were transfected with nonspecific (NS) or BRG1-specific siRNA for 24 hours prior to processing for ChIP assays. (A) ChIP with an antibody against total histone H3 was used to determine nucleosome density at various regions of the COUP-TFII promoter. Nucleosome density was significantly decreased at the -4.7 kb region of the COUP-TFII promoter but was significantly increased at the -1.2 kb and -0.3 kb promoter regions following BRG1 knockdown. (B) ChIP with an antibody against RNA polymerase II (RNAPolII) indicated its ability to bind the -0.3 kb region of the COUP-TFII promoter was significantly decreased following BRG1 knockdown. For A and B, a region upstream of the Fzd5 promoter (Fzd5 UP) was used as a negative control region, and the Adamts1 promoter served as a positive control region for BRG1-induced changes in nucleosome density or RNAPolII binding, respectively. Data from three independent experiments were combined and are presented as fold enrichment over the levels of ChIP with the H3 or RNAPolII antibodies in NS siRNA transfected cells±s.e.m. Significant differences were calculated using a two-tailed Student’s t test (*P<0.05). (C) Model of how BRG1 epigenetically promotes COUP-TFII expression. In wild-type endothelial cells, BRG1 binds the -4.7 kb region of the COUP-TFII promoter, where it mediates chromatin compaction. BRG1 also binds the -1.2 kb and -0.3 kb regions of the promoter, where it mediates chromatin decondensation, thereby allowing binding of RNAPolII close to the COUP-TFII transcription start site. These events promote the expression of COUP-TFII in wild-type cells. By contrast, in Brg1fl/fl:Tie2-Cre+ endothelial cells, which lack BRG1, the -4.7 kb COUP-TFII promoter region undergoes chromatin decondensation, potentially allowing for binding of a transcriptional repressor protein. Likewise, the -1.2 kb and -0.3 kb regions of the promoter undergo chromatin compaction, thereby inhibiting efficient RNAPolII binding and diminishing COUP-TFII expression.
We also assessed enrichment of lysine 9 acetylation on histone H3 (H3K9Ac) within the COUP-TFII promoter in the presence and absence of BRG1. This covalent epigenetic mark is typically associated with chromatin decompaction and transcriptional activation when found close to a transcription start site (Nishida et al., 2006). ChIP analysis revealed significantly diminished H3K9Ac enrichment at the -0.3 kb and -1.2 kb regions of the COUP-TFII promoter upon BRG1 knockdown in C166 endothelial cells (supplementary material Fig. S4). This finding is even more striking when the increased nucleosome density in these regions following BRG1 knockdown is taken into account (Fig. 6A). It is also consistent with the decreased RNAPolII binding we see near the COUP-TFII TSS after BRG1 knockdown (Fig. 6B). Interestingly, H3K9Ac is enriched at the -4.7 kb region of the COUP-TFII promoter after BRG1 knockdown (supplementary material Fig. S4). Considering this enrichment is seen in conjunction with chromatin decompaction (Fig. 6A), H3K9 acetylation per nucleosome is strikingly high. Although BRG1 has no histone acetytransferase or deacetylase activities on its own, it can associate with or recruit co-regulatory complexes containing these functions (Fry and Peterson, 2001; Trotter and Archer, 2008). Therefore, H3K9 acetylation and deacetylation at the sites we examined within the COUP-TFII promoter may occur simultaneously with or secondarily to chromatin-remodeling mediated by BRG1.
Altogether, our data show BRG1 binds and remodels the COUP-TFII promoter at three sites, impacting the ability of transcriptional machinery to access the TSS (Fig. 6C). In the presence of BRG1, the -1.2 kb and -0.3 kb sites are decompacted, allowing RNAPolII to bind the promoter and activate transcription of COUP-TFII. Conversely, BRG1 appears to facilitate the compaction of the -4.7 kb COUP-TFII promoter site. We speculate that BRG1 may act at this region to prevent a repressor protein from binding and inhibiting COUP-TFII expression in venous endothelial cells.
Wnt signaling does not contribute to extra-embryonic venous specification
The highly conserved -4.7 kb COUP-TFII promoter region we found bound by BRG1 in C166 endothelial cells (Fig. 5B) has previously been shown to be bound by the central Wnt signaling mediator β-catenin in preadipocytes (Okamura et al., 2009). Because BRG1 promotes Wnt signaling and directly co-activates certain Wnt target genes in yolk sac vasculature (Griffin et al., 2011), we questioned whether BRG1 and β-catenin co-activate COUP-TFII expression in extra-embryonic veins. Using a Wnt signaling reporter transgenic mouse line (BAT-gal) (Maretto et al., 2003), we found yolk sac vitelline veins and umbilical veins had more reporter activity than vitelline and umbilical arteries (supplementary material Fig. S5A-D). This unexpected finding suggests that Wnt signaling plays an important role in extra-embryonic veins. However, when we crossed vascular β-catenin loss-of-function mice (Catnbfl/fl:Tie2-Cre+) onto the arterial Efnb2LacZ reporter line, Catnbfl/fl:Tie2-Cre+ vitelline veins did not show the same aberrant reporter activity as seen in Brg1fl/fl:Tie2-Cre+ yolk sac veins (compare supplementary material Fig. S5F and Fig. 2B). Furthermore, cross-sections of E10.5 control and Catnbfl/fl:Tie2-Cre+ umbilical veins showed comparable levels of COUP-TFII expression upon immunostaining (supplementary material Fig. S5G,H). Finally, qPCR analysis revealed COUP-TFII mRNA levels were not significantly changed in β-catenin knockdown C166 endothelial cells compared with control cells (supplementary material Fig. S5I). Our combined results indicate Wnt signaling is not crucial for venous specification or COUP-TFII expression in extra-embryonic veins, so the role of elevated Wnt signaling in these vessels remains unknown.
Notch pathway genes are upregulated and Ephb4 is downregulated in Brg1 mutant endothelial cells
COUP-TFII inhibits multiple components of the Notch signaling pathway to promote venous specification in endothelial cells (You et al., 2005; Chen et al., 2012). Nrp1 and Foxc1, two upstream mediators of Notch signaling, and Hey2, a downstream effector of Notch signaling, are directly targeted by COUP-TFII in human umbilical vein endothelial cells (HUVECs) (Chen et al., 2012). In order to determine whether these and other components of the Notch signaling pathway are impacted by Brg1 deletion, we performed qPCR on primary endothelial cells isolated from E10.5 control and Brg1fl/fl:Tie2-Cre+ embryos. As predicted, we saw significant upregulation of the Notch pathway genes and arterial markers Hey1, Dll4, Hey2 and Foxc1 (Fig. 7A). We did not see transcriptional changes in the upstream Notch signaling mediator and COUP-TFII target gene Nrp1, but this finding is consistent with published findings from COUP-TFII knockdown HUVECs (Chen et al., 2012). Therefore, deletion of Brg1 results in upregulation of multiple arterial Notch signaling pathway genes, as would be predicted in endothelial cells with downregulated COUP-TFII.
Fig. 7.

BRG1 impacts expression of genes downstream of COUP-TFII signaling. (A) Primary endothelial cells (ECs) were isolated from E10.5 control and Brg1fl/fl:Tie2-Cre+ embryos, RNA was purified, and cDNA was synthesized. Expression levels of Brg1, the arterial markers (red) Nrp1, Hey1, Dll4, Hey2 and Foxc1, and the venous markers (blue) COUP-TFII, Ephb4 and Nrp2 were measured by qPCR. Data from three independent experiments were combined and are presented as relative fold change over the normalized expression level of each gene in control cells (dotted line) ±s.e.m. Significant differences were calculated using a two-tailed Student’s t-test (*P<0.05). (B) C166 cells were transfected with increasing amounts (0.02 ng, 0.2 ng, and 2 ng) of empty vector or comparable amounts of a BRG1 expression plasmid for 24 hours. RNA was isolated, cDNA was synthesized and qPCR for Brg1, COUP-TFII and Ephb4 was performed. Data from three independent experiments were combined and are presented as relative fold change over the normalized expression level of each gene in cells transfected with corresponding amounts of empty vector (dotted line). Bars represent ±s.e.m.; significant differences were calculated using a two-tailed Student’s t-test (*P<0.05). (C) C166 endothelial cells were transfected with nonspecific (NS) siRNA, BRG1-specific siRNA or BRG1 siRNA plus a COUP-TFII expression plasmid for 24 hours. RNA was isolated, cDNA was synthesized and qPCR for Hey2 was performed. Bars represent ±s.e.m. from three independent experiments; significant differences were calculated using a two-tailed Student’s t-test (*P<0.05). (D) Model of how BRG1 impacts venous specification. In arterial endothelial cells (ECs), Notch signaling promotes expression of arterial markers such as Ephrin B2. In venous ECs, BRG1 epigenetically promotes expression of COUP-TFII, presumably in cooperation with an unknown venous-specific co-regulatory protein or transcription factor. COUP-TFII directly inhibits Nrp1 and Foxc1, two upstream mediators of the Notch signaling pathway. COUP-TFII also directly inhibits the downstream Notch effector Hey2 (Chen et al., 2012). As a result of COUP-TFII-mediated Notch pathway inhibition, arterial marker expression is suppressed and the venous marker EPHB4 is expressed.
As arterial-venous malformations (AVMs) are associated with misregulated vascular Notch signaling (Lawson et al., 2001; Duarte et al., 2004; Krebs et al., 2004; Carlson et al., 2005; Kim et al., 2008; Krebs et al., 2010), we also examined Brg1fl/fl:Tie2-Cre+ mutants for abnormal arterial-venous fusion events. We found no evidence of AVMs in serial sections of Brg1fl/fl:Tie2-Cre+ mutants. However, Coup-TFIIfl/fl:Tie2-Cre+ mutants likewise show no evidence of arterial-venous fusions (You et al., 2005), so the lack of AVMs in our Brg1fl/fl:Tie2-Cre+ mutants is not unexpected.
We also analyzed transcriptional changes in the venous marker Ephb4. Ephb4 was significantly downregulated in Brg1fl/fl:Tie2-Cre+ endothelial cells (Fig. 7A) and in BRG1 knockdown C166 cells (supplementary material Fig. S6A). EPHB4 protein levels were likewise diminished in Brg1fl/fl:Tie2-Cre+ cardinal vein endothelial cells (supplementary material Fig. S6B,C). These results are reminiscent of the decreased Ephb4 transcripts seen in COUP-TFII knockdown HUVECs and the decreased EPHB4 expression seen in COUP-TFIIfl/fl:Tie2-Cre+ embryonic veins (You et al., 2005; Chen et al., 2012). We also analyzed expression of the venous marker Nrp2, which is a direct COUP-TFII target gene in developing lymphatic vessels (Lin et al., 2010), but we saw no evidence of its downregulation in E10.5 Brg1fl/fl:Tie2-Cre+ endothelial cells (Fig. 7A). Nevertheless, the downregulation of Ephb4 seen in Brg1-deficient endothelial cells suggested that BRG1 promotes venous specification at midgestation.
To further substantiate our evidence that BRG1 promotes venous specification, we overexpressed BRG1 in C166 endothelial cells and assessed transcription of the venous markers COUP-TFII and Ephb4. Both genes were upregulated with increasing expression of exogenous BRG1 (Fig. 7B). Together with our evidence from loss-of-function experiments showing that COUP-TFII and Ephb4 expression are downregulated in Brg1-deficient endothelial cells, these gain-of-function experiments support our conclusion that BRG1 promotes venous specification in vascular endothelium.
The most downstream effector of Notch signaling that is directly inhibited by COUP-TFII is the Notch target gene and transcription factor Hey2 (Chen et al., 2012). HEY2 may play a key role in venous specification as it mediates inhibition of Ephb4 expression in developing zebrafish veins (Zhong et al., 2001). In addition to seeing Hey2 transcription upregulated in primary E10.5 Brg1fl/fl:Tie2-Cre+ endothelial cells (Fig. 7A), we also saw Hey2 significantly upregulated in BRG1 knockdown C166 endothelial cells (Fig. 7C). This upregulation of Hey2 was presumably secondary to decreased COUP-TFII expression as exogenous COUP-TFII significantly reduced the aberrant Hey2 expression seen in BRG1 knockdown cells (Fig. 7C). Our findings corroborate the current model of how COUP-TFII inhibits Notch signaling to promote venous specification and add new insight into epigenetic mechanisms for regulating COUP-TFII expression in veins (Fig. 7D).
DISCUSSION
COUP-TFII promotes venous specification by suppressing Notch signaling in endothelial cells (You et al., 2005), but the mechanism by which it does so has been unclear. Recent work by Chen et al. shows COUP-TFII directly inhibits transcription of two upstream mediators of Notch signaling: the transcription factor Foxc1 and the VEGF co-receptor Nrp1 (Chen et al., 2012). FOXC1 promotes expression of the Notch ligand Dll4 (Hayashi and Kume, 2008), and NRP1 mediates VEGF signaling along with VEGFR2 to promote Notch signaling (Lawson et al., 2002; Swift and Weinstein, 2009). Therefore COUP-TFII-mediated downregulation of Foxc1 and Nrp1 suppresses Notch signaling in veins. In addition, COUP-TFII directly inhibits the Notch target gene Hey2 in venous endothelial cells (Chen et al., 2012). Several pieces of evidence indicate HEY2 plays a crucial role in arterial specification. First, when Hey2 and the paralogous gene Hey1 are simultaneously targeted for deletion in mice, the arterial marker Efnb2 is significantly reduced in embryonic arteries (Fischer et al., 2004). Likewise, Nrp1 is reduced in Hey1/Hey2-deficient arteries, indicating that HEY2 may feed back to regulate upstream mediators of Notch signaling (Fischer et al., 2004). Furthermore, HEY2 activates an artery-specific gene expression program when ectopically expressed in veins (Chi et al., 2003). Altogether, COUP-TFII-mediated inhibition of Hey2, Foxc1 and Nrp1 profoundly impacts arterial/venous specification by downregulating Notch signaling in endothelial cells. Despite its important influence on vessel identity, however, nothing is known about what lies upstream of COUP-TFII and regulates its expression in veins. In this study, we present evidence that the chromatin-remodeling enzyme BRG1 directly promotes expression of COUP-TFII in venous endothelial cells by remodeling the COUP-TFII promoter to make it accessible to transcriptional machinery.
As BRG1 is not a transcription factor, it cannot be the only regulator of COUP-TFII expression in veins. Indeed, loss of BRG1 did not eliminate COUP-TFII expression completely from primary or cultured endothelial cells. Moreover, we found BRG1 expressed in both arteries and veins in vivo, whereas COUP-TFII expression is restricted to veins (You et al., 2005). Both of these points indicate that a venous-specific transcription factor or combination of transcription factors co-regulates COUP-TFII expression with the help of BRG1. We initially thought Wnt signaling transcription factors were ideal venous-specific candidates for promoting COUP-TFII expression in extra-embryonic veins. This hypothesis was based on our observation that Wnt signaling reporter activity occurred preferentially in yolk sac and umbilical veins rather than arteries. It was also based on a published report that Wnt signaling promotes COUP-TFII expression in preadipocytes (Okamura et al., 2009). Further support for this hypothesis came from our finding that BRG1 co-activates expression of certain Wnt target genes in extra-embryonic vasculature (Griffin et al., 2011). However, we found no evidence that the central Wnt signaling component β-catenin is required for extra-embryonic COUP-TFII expression or venous specification. Instead, Wnt signaling appears to play a more crucial role in promoting arterial specification at midgestation (Corada et al., 2010). Therefore, the role of the prominent Wnt signaling we detected in extra-embryonic veins remains unknown, and the venous-specific transcription factors that work with BRG1 to promote COUP-TFII expression still require identification. The BRG1-binding sites that we identified within the COUP-TFII promoter could serve as starting points in experimental searches for venous-specific transcription factors that regulate COUP-TFII expression. Alternatively, the BRG1-binding sites may serve as guideposts for identifying arterial-specific inhibitors of COUP-TFII, as we found BRG1 binding to be mostly similar in human arterial and venous endothelial cells.
BRG1-containing SWI/SNF complexes exercise temporally and spatially specific control over target genes during developmental processes (Ho and Crabtree, 2010). Therefore, one outstanding issue is whether BRG1 differentially impacts the timing and location of COUP-TFII expression in developing vasculature. This research article demonstrates that BRG1 promotes venous COUP-TFII expression during early stages of vascular development, but it is not clear whether BRG1 is required for COUP-TFII maintenance on more mature veins, as Brg1fl/fl:Tie2-Cre+ embryos die from anemia by E11.0 (Griffin et al., 2008). To address this issue, vascular Brg1 excision must be induced at later stages of embryonic development. Later induction of Brg1 excision will also help determine whether BRG1 plays crucial roles in lymphatic specification and tumor angiogenesis - two processes for which COUP-TFII expression and maintenance are required (Lin et al., 2010; Qin et al., 2010; Srinivasan et al., 2010).
In conclusion, our data indicate that BRG1 promotes venous specification through COUP-TFII induction during vascular development. This study delineates a novel role for chromatin remodeling activity in the regulation of blood vessel identity and broadens our understanding of how epigenetic processes influence vascular development.
Supplementary Material
Acknowledgments
We thank Mr Vijay Muthukumar for technical assistance and mouse colony maintenance, and Griffin lab members for critiquing this manuscript. We also thank Dr Hendra Setiadi for HUVECs and Dr Sophia Tsai for the COUP-TFII expression plasmid, and for sharing pre-published results with us.
Footnotes
Funding
This work was supported by grants from the National Institutes of Health to C.T.G. [R00HL087621, P20GM103441 and R01HL111178]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.087379/-/DC1
References
- Adams R. H., Wilkinson G. A., Weiss C., Diella F., Gale N. W., Deutsch U., Risau W., Klein R. (1999). Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 13, 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brault V., Moore R., Kutsch S., Ishibashi M., Rowitch D. H., McMahon A. P., Sommer L., Boussadia O., Kemler R. (2001). Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128, 1253–1264 [DOI] [PubMed] [Google Scholar]
- Carlson T. R., Yan Y., Wu X., Lam M. T., Tang G. L., Beverly L. J., Messina L. M., Capobianco A. J., Werb Z., Wang R. (2005). Endothelial expression of constitutively active Notch4 elicits reversible arteriovenous malformations in adult mice. Proc. Natl. Acad. Sci. USA 102, 9884–9889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Qin J., Cheng C. M., Tsai M. J., Tsai S. Y. (2012). COUP-TFII is a major regulator of cell cycle and Notch signaling pathways. Mol. Endocrinol. 26, 1268–1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi J. T., Chang H. Y., Haraldsen G., Jahnsen F. L., Troyanskaya O. G., Chang D. S., Wang Z., Rockson S. G., van de Rijn M., Botstein D., et al. (2003). Endothelial cell diversity revealed by global expression profiling. Proc. Natl. Acad. Sci. USA 100, 10623–10628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corada M., Nyqvist D., Orsenigo F., Caprini A., Giampietro C., Taketo M. M., Iruela-Arispe M. L., Adams R. H., Dejana E. (2010). The Wnt/beta-catenin pathway modulates vascular remodeling and specification by upregulating Dll4/Notch signaling. Dev. Cell 18, 938–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C. D., Griffin C. T. (2012). The chromatin-remodeling enzymes BRG1 and CHD4 antagonistically regulate vascular Wnt signaling. Mol. Cell. Biol. 32, 1312–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C. D., Davis R. B., Ingram K. G., Griffin C. T. (2012). Chromatin-remodeling complex specificity and embryonic vascular development. Cell. Mol. Life Sci. 69, 3921–3931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte A., Hirashima M., Benedito R., Trindade A., Diniz P., Bekman E., Costa L., Henrique D., Rossant J. (2004). Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev. 18, 2474–2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A., Schumacher N., Maier M., Sendtner M., Gessler M. (2004). The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes Dev. 18, 901–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry C. J., Peterson C. L. (2001). Chromatin remodeling enzymes: who’s on first? Curr. Biol. 11, R185–R197 [DOI] [PubMed] [Google Scholar]
- Garcia-Cardeña G., Comander J., Anderson K. R., Blackman B. R., Gimbrone M. A., Jr (2001). Biomechanical activation of vascular endothelium as a determinant of its functional phenotype. Proc. Natl. Acad. Sci. USA 98, 4478–4485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebuhr T. C., Kovalev G. I., Bultman S., Godfrey V., Su L., Magnuson T. (2003). The role of Brg1, a catalytic subunit of mammalian chromatin-remodeling complexes, in T cell development. J. Exp. Med. 198, 1937–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerety S. S., Wang H. U., Chen Z. F., Anderson D. J. (1999). Symmetrical mutant phenotypes of the receptor EphB4 and its specific transmembrane ligand ephrin-B2 in cardiovascular development. Mol. Cell 4, 403–414 [DOI] [PubMed] [Google Scholar]
- Griffin C. T., Brennan J., Magnuson T. (2008). The chromatin-remodeling enzyme BRG1 plays an essential role in primitive erythropoiesis and vascular development. Development 135, 493–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin C. T., Curtis C. D., Davis R. B., Muthukumar V., Magnuson T. (2011). The chromatin-remodeling enzyme BRG1 modulates vascular Wnt signaling at two levels. Proc. Natl. Acad. Sci. USA 108, 2282–2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves D. C., Crabtree G. R. (2011). ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 21, 396–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H., Kume T. (2008). Foxc transcription factors directly regulate Dll4 and Hey2 expression by interacting with the VEGF-Notch signaling pathways in endothelial cells. PLoS ONE 3, e2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog Y., Kalcheim C., Kahane N., Reshef R., Neufeld G. (2001). Differential expression of neuropilin-1 and neuropilin-2 in arteries and veins. Mech. Dev. 109, 115–119 [DOI] [PubMed] [Google Scholar]
- Ho L., Crabtree G. R. (2010). Chromatin remodelling during development. Nature 463, 474–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones E. A., Yuan L., Breant C., Watts R. J., Eichmann A. (2008). Separating genetic and hemodynamic defects in neuropilin 1 knockout embryos. Development 135, 2479–2488 [DOI] [PubMed] [Google Scholar]
- Kim Y. H., Hu H., Guevara-Gallardo S., Lam M. T., Fong S. Y., Wang R. A. (2008). Artery and vein size is balanced by Notch and ephrin B2/EphB4 during angiogenesis. Development 135, 3755–3764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisanuki Y. Y., Hammer R. E., Miyazaki J., Williams S. C., Richardson J. A., Yanagisawa M. (2001). Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 230, 230–242 [DOI] [PubMed] [Google Scholar]
- Kokubo H., Miyagawa-Tomita S., Nakazawa M., Saga Y., Johnson R. L. (2005). Mouse hesr1 and hesr2 genes are redundantly required to mediate Notch signaling in the developing cardiovascular system. Dev. Biol. 278, 301–309 [DOI] [PubMed] [Google Scholar]
- Koo B. K., Lim H. S., Song R., Yoon M. J., Yoon K. J., Moon J. S., Kim Y. W., Kwon M. C., Yoo K. W., Kong M. P., et al. (2005). Mind bomb 1 is essential for generating functional Notch ligands to activate Notch. Development 132, 3459–3470 [DOI] [PubMed] [Google Scholar]
- Krebs L. T., Shutter J. R., Tanigaki K., Honjo T., Stark K. L., Gridley T. (2004). Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev. 18, 2469–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs L. T., Starling C., Chervonsky A. V., Gridley T. (2010). Notch1 activation in mice causes arteriovenous malformations phenocopied by ephrinB2 and EphB4 mutants. Genesis 48, 146–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson N. D., Scheer N., Pham V. N., Kim C. H., Chitnis A. B., Campos-Ortega J. A., Weinstein B. M. (2001). Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development 128, 3675–3683 [DOI] [PubMed] [Google Scholar]
- Lawson N. D., Vogel A. M., Weinstein B. M. (2002). sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev. Cell 3, 127–136 [DOI] [PubMed] [Google Scholar]
- le Noble F., Moyon D., Pardanaud L., Yuan L., Djonov V., Matthijsen R., Bréant C., Fleury V., Eichmann A. (2004). Flow regulates arterial-venous differentiation in the chick embryo yolk sac. Development 131, 361–375 [DOI] [PubMed] [Google Scholar]
- Lin F. J., Chen X., Qin J., Hong Y. K., Tsai M. J., Tsai S. Y. (2010). Direct transcriptional regulation of neuropilin-2 by COUP-TFII modulates multiple steps in murine lymphatic vessel development. J. Clin. Invest. 120, 1694–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F. J., Qin J., Tang K., Tsai S. Y., Tsai M. J. (2011). Coup d’Etat: an orphan takes control. Endocr. Rev. 32, 404–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maretto S., Cordenonsi M., Dupont S., Braghetta P., Broccoli V., Hassan A. B., Volpin D., Bressan G. M., Piccolo S. (2003). Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc. Natl. Acad. Sci. USA 100, 3299–3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida H., Suzuki T., Kondo S., Miura H., Fujimura Y., Hayashizaki Y. (2006). Histone H3 acetylated at lysine 9 in promoter is associated with low nucleosome density in the vicinity of transcription start site in human cell. Chromosome Res. 14, 203–211 [DOI] [PubMed] [Google Scholar]
- Okamura M., Kudo H., Wakabayashi K., Tanaka T., Nonaka A., Uchida A., Tsutsumi S., Sakakibara I., Naito M., Osborne T. F., et al. (2009). COUP-TFII acts downstream of Wnt/beta-catenin signal to silence PPARgamma gene expression and repress adipogenesis. Proc. Natl. Acad. Sci. USA 106, 5819–5824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J., Chen X., Xie X., Tsai M. J., Tsai S. Y. (2010). COUP-TFII regulates tumor growth and metastasis by modulating tumor angiogenesis. Proc. Natl. Acad. Sci. USA 107, 3687–3692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R. S., Geng X., Yang Y., Wang Y., Mukatira S., Studer M., Porto M. P., Lagutin O., Oliver G. (2010). The nuclear hormone receptor Coup-TFII is required for the initiation and early maintenance of Prox1 expression in lymphatic endothelial cells. Genes Dev. 24, 696–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift M. R., Weinstein B. M. (2009). Arterial-venous specification during development. Circ. Res. 104, 576–588 [DOI] [PubMed] [Google Scholar]
- Thurston G., Yancopoulos G. D. (2001). Gridlock in the blood. Nature 414, 163–164 [DOI] [PubMed] [Google Scholar]
- Trindade A., Kumar S. R., Scehnet J. S., Lopes-da-Costa L., Becker J., Jiang W., Liu R., Gill P. S., Duarte A. (2008). Overexpression of delta-like 4 induces arterialization and attenuates vessel formation in developing mouse embryos. Blood 112, 1720–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotter K. W., Archer T. K. (2008). The BRG1 transcriptional coregulator. Nucl. Recept. Signal. 6, e004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. J., Greer P., Auerbach R. (1996). Isolation and propagation of yolk-sac-derived endothelial cells from a hypervascular transgenic mouse expressing a gain-of-function fps/fes proto-oncogene. In Vitro Cell. Dev. Biol. Anim. 32, 292–299 [DOI] [PubMed] [Google Scholar]
- Wang H. U., Chen Z. F., Anderson D. J. (1998). Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 93, 741–753 [DOI] [PubMed] [Google Scholar]
- Yao L., Pan J., Setiadi H., Patel K. D., McEver R. P. (1996). Interleukin 4 or oncostatin M induces a prolonged increase in P-selectin mRNA and protein in human endothelial cells. J. Exp. Med. 184, 81–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You L. R., Lin F. J., Lee C. T., DeMayo F. J., Tsai M. J., Tsai S. Y. (2005). Suppression of Notch signalling by the COUP-TFII transcription factor regulates vein identity. Nature 435, 98–104 [DOI] [PubMed] [Google Scholar]
- Zhong T. P., Rosenberg M., Mohideen M. A., Weinstein B., Fishman M. C. (2000). gridlock, an HLH gene required for assembly of the aorta in zebrafish. Science 287, 1820–1824 [DOI] [PubMed] [Google Scholar]
- Zhong T. P., Childs S., Leu J. P., Fishman M. C. (2001). Gridlock signalling pathway fashions the first embryonic artery. Nature 414, 216–220 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.