

Abstract
Hearing loss (HL) is a congenital disease with a high prevalence, and patients with hearing loss need early diagnosis for treatment and prevention. The GJB2, MT-RNR1, and SLC26A4 genes have been reported as common causative genes of hearing loss in the Korean population and some mutations of these genes are the most common mutations associated with hearing loss. Accordingly, we developed a method for the simultaneous detection of seven mutations (c.235delC of GJB2, c.439A>G, c.919-2A>G, c.1149+3A>G, c.1229C>T, c.2168A>G of SLC26A4, and m.1555A>G of the MT-RNR1 gene) using multiplex SNaPshot minisequencing to enable rapid diagnosis of hereditary hearing loss. This method was confirmed in patients with hearing loss and used for genetic diagnosis of controls with normal hearing and neonates. We found that 4.06% of individuals with normal hearing and 4.32% of neonates were heterozygous carriers. In addition, we detected that an individual is heterozygous for two different mutations of GJB2 and SLC26A4 gene, respectively and one normal hearing showing the heteroplasmy of m.1555A>G. These genotypes corresponded to those determined by direct sequencing. Overall, we successfully developed a robust and cost-effective diagnosis method that detects common causative mutations of hearing loss in the Korean population. This method will be possible to detect up to 40% causative mutations associated with prelingual HL in the Korean population and serve as a useful genetic technique for diagnosis of hearing loss for patients, carriers, neonates, and fetuses.
Introduction
Congenital hearing loss (HL) is a common sensory impairment that occurs in approximately one in 1000 neonates, and more than 50% of cases are hereditary [1]. The occurrence of HL in early childhood could have serious effects on language acquisition, while later onset of severe hearing defect compromises sociality [2]. Currently, the best provision for hereditary HL is prevention of outbreak through early diagnosis and continuous management. Therefore, a precise rapid genetic diagnosis may be the most effective support for treatment and prevention of HL.
More than over 150 loci have been mapped in HL, and 57 genes of them have been shown to cause nonsyndromic HL [3]. Among these, mutations of the GJB2 gene (OMIM 121011) are the most common cause of hereditary HL in many Caucasian populations [4]. One of them, mutation c.35delG, accounts for up to 70% of the pathologic alleles in European and Caucasian populations [5]. Unlike Caucasian populations, GJB2 mutations seem to account for a lower percentage of hereditary HL in East Asian populations; however, approximately 43% of the GJB2 hearing loss are caused by c.235delC mutation in Koreans [6]–[8]. Moreover, mutations of the SLC26A4 gene (OMIM 605646), which cause both Pendred syndrome and nonsyndromic HL with enlarged vestibular aqueduct, appear to be common in Asian populations [9]. Especially, five mutations of this gene account for up to 90% of all mutant alleles [8], [10]. Mitochondrial DNA mutations have also been reported in both nonsyndromic and syndromic HL. Among these mutations, only the m.1555A>G mutation of the MT-RNR1 gene (OMIM 561000) has been identified repeatedly in Koreans with HL by our previous studies [11], [12]. Based on the prevalence of mutations from the previous studies, we selected 7 mutations which show the high frequencies in Korean hearing loss population and designed the method to detect effectively mutant alleles through rapid screening: c.235delC of GJB2, c.919-2A>G and c.2168A>G of SLC26A4 and m.1555A>G of the MT-RNR1 gene. All of the 7 mutations selected in this study had been reported that mutations cause abnormal function. The c.235delC mutation of GJB2 gene caused a loss of targeting activity to the cell membrane and severe deterioration of gap junction activity [13]. The m.1555A>G in MT-RNR1 gene made the human mitochondrial ribosome more bacteria-like and alter binding sites for aminoglycosides [14]. The p.M147V and p.H723R mutations of SLC26A4 gene caused loss of Cl−/HCO3 − and Cl−/I− exchange activity [15]–[17]. The p.T410M mutation in SLC26A4 gene caused loss of iodide efflux [17], [18]. The c.919-2A>G and c.1149+3A>G mutations of SLC26A4 gene resulted in premature termination of translation on gene expression, respectively [10], [19]. Accordingly, these mutations have been screened by priority during genetic testing of HL. Because all of these mutations are recessive in their effect except the mitochondrial mutation (m.1555A>G), analysis of individual genotype for these mutations can provide crucial genetic information for inheritance of autosomal recessive hearing loss.
In this study, we developed a rapid and low cost multiplex genetic diagnosis of common mutations in Koreans using SNaPshot minisequencing for patients with HL and heterozygous carriers.
Materials and Methods
Subjects and DNA preparation
To set up the SNaPshot minisequencing methods, five patients with mutations in GJB2, MT-RNR1, and SLC26A4 and one normal hearing individual without any mutations were used as positive and normal controls, respectively. One hundred ninety-seven unrelated Koreans with normal hearing from Kyungpook National University Hospital were used as controls aged 20 and 64 years. In addition, 139 Korean neonates of unrelated individuals who were born in Kyungpook National University Hospital were recruited for this study with the consent of their parents. Written informed consent was obtained from all participants, and this study was approved by the local ethics committee.
The genomic DNA of normal controls and neonates was extracted from peripheral blood and buccal cells using a FlexiGene DNA kit (Qiagen, Hilden, Germany) or a Gentra Puregene Buccal Cell kit (Qiagen, Hilden, Germany) following the manufacturer's instructions.
PCR multiplex amplification
Fourteen primers were used for amplification of the mutation region, and amplification was conducted in a reaction mixture with a final volume of 25 μl that contained 20 ng of DNA template, 0.2 mM deoxynucleotide, 1 X multiplex polymerase chain reaction (PCR) primer mixture that consisted of seven pairs of primers, 1 X h-Taq reaction buffer, and 0.5 U of h-Taq DNA Polymerase (Solgent, Daejeon, Korea) (Table 1). The PCR conditions were as follows: initial denaturation at 95°C for 15 min, followed by 40 cycles of denaturation at 94°C for 20 sec, annealing at 57°C for 20 sec, and extension at 72°C for 40 sec and then final extension at 72°C for 5 min. Five microliters of the PCR products were separated and visualized following electrophoresis on a 2% agarose gel. Shrimp alkaline phosphatase (SAP) (USB, Cleveland, OH, USA) and exonuclease I (USB, Cleveland, OH, USA) were used for post-PCR purification of the examined PCR products.
Table 1. Multiplex PCR primer sequences.
| Gene | Exon | PCR primers (5′→3′) | Concentration* (pmol/μl) | Product size (bp) | Nucleotide change | Protein change | |
| GJB2 | 2 | F | TCTTTTCCAGAGCAAACCGC | 0.2 | 416 | c.235delC | p.L79CfsX3 |
| R | GATGCGGACCTTCTGGGTTT | 0.2 | |||||
| MT-RNR1 | - | F | CGTCACCCTCCTCAAGTATACTTC | 0.04 | 137 | m.1555A>G | - |
| R | GCTTTGTGTTAAGCTACACTCTGG | 0.04 | |||||
| SLC26A4 | 5 | F | TTTTTAAACCCTATGCAGACACA | 0.5 | 175 | c.439A>G | p.M147V |
| R | TTAATACAGTTCCATTGCTGCTG | 0.5 | |||||
| SLC26A4 | 7 | F | CAAAATCCCAGTCCCTATTCCTA | 0.4 | 363 | c.919-2A>G | - |
| R | GGTTGTTTCTTCCAGATCACACAC | 0.4 | |||||
| SLC26A4 | 9 | F | GCTTGTTCTCGGAGATGCTG | 0.15 | 301 | c.1149+3A>G | - |
| R | AGTGATGCAGTGTGTCTATTCC | 0.15 | |||||
| SLC26A4 | 10 | F | GGATCGTTGTCATCCAGTCTC | 0.5 | 488 | c.1229C>T | p.T410M |
| R | TTACCAGGCCATCTGTCTCC | 0.5 | |||||
| SLC26A4 | 19 | F | CCTGGGCAATAGAATGAGACTC | 0.15 | 227 | c.2168A>G | p.H723R |
| R | AAATGGAACCTTGACCCTCTTG | 0.15 | |||||
Final concentration in the reaction mixture.
SNaPshot minisequencing reaction
The single base extension (SBE) reaction was performed in a reaction mixture with a final volume of 15 μl that contained 2.45 μl of purified multiplex PCR product, 1 X extension primer mixture (Table 2), and 3 μl of SNaPshot Multiplex Ready Reaction Mix (Applied Biosystems, Foster City, CA, USA). The reaction mixture was subjected to 30 SBE cycles of denaturation at 96°C for 10 sec, primer annealing at 50°C for 5 sec, and primer extension at 60°C for 30 sec. SAP (USB, Cleveland, OH, USA) was used for post-SBE purification of the SBE reaction products.
Table 2. Extension primer sequences.
| Gene | Mutation | Extension primers (5′→3′) | Concentration* (pmol/μl) | Primer size (bp) | WT allele (peak color) | MT allele (peak color) | |
| GJB2 | c.235delC | F | ACGATCACTACTTCCCCATCTCCCACATCCGGCTATGGGCC | 4 | 41 | C (Black) | T (Red) |
| MT-RNR1 | m.1555A>G | F | TTAACTAAAACCCCTACGCATTTATATAGAGGAG | 0.07 | 34 | A (Green) | G (Blue) |
| SLC26A4 | c.439A>G | F | TTAATAACTGATTAATTGTTAGAGACTTTTTTTCCCCAGGA CCTTTTCCAGTGGTGAGTTTA | 0.05 | 62 | A (Green) | G (Blue) |
| SLC26A4 | c.919-2A>G | F | AAGTTCAGCATTATTTGGTTGACAAACAAGGAATTATTAAA ACCAATGGAGTTTTTAACATCTTTTGTTTTATTTC | 0.03 | 76 | A (Green) | G (Blue) |
| SLC26A4 | c.1149+3A>G | R | TATGTTTTTTTCCTGTTTCCAGCCCTATAAAACCAGTTCAGCA AAAGGGCACCCA | 0.07 | 55 | A (Red) | G (Black) |
| SLC26A4 | c.1229C>T | F | CCTTTGGGATCAGCAACATCTTCTCAGGATTCTTCTCTTGTTTT GTGGCCACCACTGCTCTTTCCCGCA | 6.67 | 69 | C (Black) | T (Red) |
| SLC26A4 | c.2168A>G | F | GGTTCTTTGACGACAACATTAGAAAGGACACATTCTTTTTGACG GTCC | 0.07 | 48 | A (Green) | G (Blue) |
Final concentration in the reaction mixture.
For electrophoresis, 2 μl of purified multiplex SBE reaction products were mixed with 0.3 μl of GeneScan 120 LIZ Size Standard (Applied Biosystems, Foster City, CA, USA) and 7.7 μl of Hi-Di Formamide (Applied Biosystems, Foster City, CA, USA) and denatured at 95°C for 2 min. The fluorescently labeled fragments were resolved by capillary electrophoresis on an ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The resulting data were analyzed with the GeneMapper v3.7 (Applied Biosystems, Foster City, CA, USA) software.
Sanger sequencing
Specific DNA fragments containing each mutation were amplified by PCR to confirm the genotype. PCR was conducted using a reaction mixture composed of 0.2 mM deoxynucleotide, 10 pmol of each forward and reverse primer, and 0.25 U of h-Taq DNA Polymerase (Solgent, Daejeon, Korea) in a final volume of 25 μl. The PCR conditions were as follows: initial denaturation at 95°C for 15 min, followed by 35 cycles of denaturation at 94°C for 20 sec, annealing at 55°C, depending on the primers for 40 sec, and extension at 72°C for 50 sec followed by final extension at 72°C for 5 min. The quality of the PCR products was examined by electrophoresis on 2% agarose gels. SAP (USB, Cleveland, OH, USA) and exonuclease I (USB, Cleveland, OH, USA) were used for purification of the examined PCR products. The sequences of the purified PCR products were obtained by direct sequencing using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA). Extended products were purified by ethanol precipitation. A 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) was used to resolve the products and the data were analyzed using the Chromas Lite v2.01 (Technelysium Pty Ltd., Tewantin, QLD, Australia) software.
Results
Development of the multiplex amplification and multiplex SNaPshot minisequencing
The seven genomic segments containing each mutation were amplified in a single multiplex PCR reaction, after which the amplified DNA fragments were separated and visualized on agarose gel (Figure 1). We developed a genetic diagnostic technique consisting of SNaPshot multiplex minisequencing of seven mutations in three genes. Five positive controls for the mutant allele of the seven mutations and one normal control with wild-type alleles were analyzed and correctly genotyped in all reactions (Figure 2). Normal controls were genotyped to wild-type in every mutant region (Figure 2, first panel). Positive controls were genotyped mutant types in each mutant region (Figure 2, second to sixth panels). Relative differences in peak heights between each allele were observed because fluorescence emission can be directly influenced by interaction between other fluorophores. In addition, the results of SNaPshot minisequencing were compared with those of Sanger sequencing of control samples, and these results were matched exactly.
Figure 1. The products of multiplex amplification.
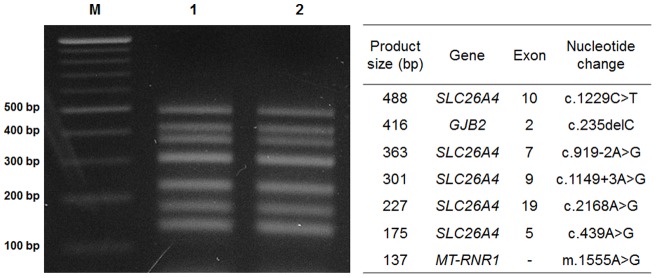
Agarose gel electrophoresis pattern of digested PCR products stained with ethidium bromide shows the multiplex amplification products of wild type (lane 1), mutant type (lane 2), 100 bp ladder marker (lane M), and corresponding size (bp) for each band of multiplex PCR products (table).
Figure 2. Electropherograms of Genescan Analysis of the SNaPshot multiplex minisequencing.

On the top, a control DNA sample with the position and nucleotide of the wild type allele indicated. Following, electropherograms for the most frequent hearing loss mutations in homozygote, compound heterozygote forms on GJB2, SLC26A4 genes, and homoplasmy in the MT-RNR1 gene.
SNaPshot minisequencing in the normal hearing controls and neonates
To investigate the frequency of these common mutations in the Korean normal hearing population, the genotypes of 197 normal hearing controls and 139 neonates were analyzed for the seven mutations using SNaPshot multiplex minisequencing. Analysis revealed a 20-year-old female with normal hearing that had heteroplasmy of m.1555A>G in the MT-RNR1 gene, which is the most common cause of aminoglycoside-induced HL. In addition, a 35-year-old female with normal hearing was found to have two different heterozygous alleles (c.235delC in GJB2 and c.2168A>G in SLC26A4 gene). Moreover, eight normal hearing controls and six neonates were diagnosed as carriers of each mutation. In the normal hearing controls, c.235delC of the GJB2 gene and c.919-2A>G and c.2168A>G of the SLC26A4 gene mutations were detected in one, two and four heterozygotes, respectively (Table 3, left panel). In the neonates, three, two and one individuals were found to be heterozygous carriers of the c.235delC GJB2 gene mutation, c.919-2A>G and c.2168A>G SLC26A4 mutation, respectively (Table 3, right panel). These genotypes diagnosed by the SNaPshot multiplex minisequencing matched those determined by direct sequencing.
Table 3. Frequencies of seven hearing loss mutations in normal controls and neonates.
| Gene | Mutation | Normal hearing controls | Neonates | ||
| No. of alleles (n = 394) | Allele frequency (%) | No. of alleles (n = 278) | Allele frequency (%) | ||
| GJB2 | c.235delC | 2 | 0.51 | 3 | 1.08 |
| MT-RNR1 | m.1555A>G heteroplasmy | 1 | 0.51 | 0 | 0 |
| SLC26A4 | c.439A>G | 0 | 0 | 0 | 0 |
| SLC26A4 | c.919-2A>G | 2 | 0.51 | 2 | 0.72 |
| SLC26A4 | c.1149+3A>G | 0 | 0 | 0 | 0 |
| SLC26A4 | c.1229C>T | 0 | 0 | 0 | 0 |
| SLC26A4 | c.2168A>G | 5 | 1.27 | 1 | 0.36 |
Discussion
The SNaPshot minisequencing technique, which was developed at first by Smith et al. [20], has been utilized in various branches of biology, including population genetics and phylogenetics, with a high level of accuracy and effectiveness. Several studies have applied this technique for haplogroup classification of human mitochondrial DNA, which provides crucial information for phylogenetic analysis and forensic identification of individuals [21], [22]. Simultaneous identification of several different species using the multiplex SNaPshot reaction has also been attempted in many studies, and their strong possibility of industrial application has been authenticated [23], [24].
Recently, SNaPshot minisequencing has been used for effective detection of numerous pathogenic mutations that cause human hereditary disorders such as CYP21, PIK3CA, CYP3A5, MDR1, and BRCA1/2 genes [25]–[30]. Especially, it is greatly effective at genetic diagnosis of heterogeneous diseases that have a number of causative genes, as well as large single gene diseases because this technique allows simultaneous detection of several variations in a single reaction. Most heterogeneous diseases have several major common causative mutations generated from different genes. Therefore, SNaPshot multiplex minisequencing of selected common mutations provides temporal and economic efficiency rather than direct sequencing of all genes.
Based on previous genetic studies of HL in the Korean population, we selected the seven most common mutations that lead to hereditary HL [8]–[12], [31]. Multiplex genotyping of these seven mutations was successfully performed using SNaPshot minisequencing, which produced accurate data and a detection rate of the mutations that was found to account for up to 40% of the causative mutant alleles associated with prelingual HL in Koreans [8]. These results suggest that this strategy is suitable for use as a precise and rapid genetic diagnosis tool.
To verify the applicability of this technique as a genetic diagnosis tool and a tool for building a population genetic database of type and frequency of mutations in the Korean population, we analyzed genotypes of normal hearing adults and neonates. As a result, we detected heterozygous carriers successfully. Specifically, we found that 4.06% of individuals with normal hearing and 4.32% of neonates were heterozygous carriers. In addition, we identified one carrier having two different heterozygous alleles, and one normal hearing having possibility to cause the HL. The frequencies of two mutations (GJB2 c.235delC and c.2168A>G of the SLC26A4 gene) presented that the allele frequency of c.235delC mutation (0.52%) in normal hearing controls by other studies were very similar to the result of this study (0.51%) [10], [31]–[33], and the c.2168A>G mutation presented higher frequency than the other SLC26A4 mutations in this study and the previous studies in Koreans [9], [10], [31]. The c.439A>G, c.1149+3A>G, and c.1229C>T mutations of SLC26A4 were not detected in this study. Likewise, results of this study show similar results of previous studies. These findings suggest that the developed strategy can be highly effective for identification of carriers of mutations associated with HL in the Korean population. Moreover, these results indicate the potential for mitigation and prevention of HL via genetic counseling before the HL begins to possible detection of carriers having each heterozygous allele in different genes and individuals with normal hearing having genes with the potential to cause disease.
This method enables the detection of seven mutations associated with HL at once and costs less than $12 per sample from blood or buccal swab to genotype analysis (based on $1 = 1133.90 KRW), making this method rapid and cost-effective when compared with other diagnostic tools using PCR-RFLP or real-time quantitative PCR [34]–[37]. Bardien et al. elucidated that an estimated cost comparison between the two methods was less than $16 per sample for the SNaPshot method versus less than $30 per sample for the PCR-RFLP method (based on $1 = 10.39 ZAR) [37]. These results verified economical effectiveness of the SNaPshot method compared with other methods. Additionally, the analysis is quick and easy because the results of SNaPshot minisequencing show different colored peaks that reflect single bases of mutations required for analysis. Furthermore, the method can be easily performed in any location equipped with a thermal cycler and DNA sequencer. Because each mutation is simultaneously but independently amplified using specific primers, it is simple and easy to add more mutations by inserting the primers for multiplex PCR and SBE reaction. Thus, if primers are inserted for SBE reaction of other mutations identified in Korean HL, this method will be applicable to identification of genetic diagnosis of more individuals. The present science technology is very advanced, so it is possible to screen large-scale sequences in the short-term using next generation sequencing (NGS). However, the method is still somewhat expensive; therefore, known mutations should be filtered prior to NGS to identify the causative mutations. Nevertheless, our method will be fast and efficient as a method of first-pass screening. To date, this technique has been found to be useful for detection of mutations in fetal DNA from maternal plasma [38], [39], as well as both blood DNA from adults and small amounts of DNA from buccal swabs of neonates. Therefore, the developed method will make diagnosis of hereditary HL in fetuses possible as well.
Because hereditary HL is heterogeneous disorder and the major causative gene in Korean population has not been identified, it is almost impossible that all patients with genetic HL are diagnosed by single standardized diagnosis platform, even if it is highly effective techniques to detect a number of mutations. Nevertheless, three genes (GJB2, SLC26A4, and MT-RNR1) are the most common causative genes in Korean population, and their 7 mutations selected in this study account for up to 70% of hearing loss caused by these 3 genes [8]. It obviously suggests that detection of these 7 frequent mutations has the highest diagnosis rate than any other mutations. For this reasons, primary mutation screening of these 3 genes are essentially being required in all genetic studies of hearing loss. It means that analysis of these 7 major mutations using SNaPshot minisequencing tool is very useful for researches of hereditary hearing loss.
In conclusion, we successfully developed a rapid, accurate, robust, and cost-effective genetic tool for diagnosis of HL using SNaPshot minisequencing. It is possible to detect up to 40% causative mutations associated with prelingual HL in the Korean population using this method, and this technique is applicable to other fields including genetic diagnosis of fetuses or first-pass screening prior to NGS screening.
Funding Statement
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology (2011-0028066) and by a grant from the Korea Health Technology Research & Development Project, Ministry of Health & Welfare, Republic of Korea. (A111345). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Marazita ML, Ploughman LM, Rawlings B, Remington E, Arnos KS, et al. (1993) Genetic epidemiological studies of early-onset deafness in the U.S. school-age population. Am J Med Genet 46: 486–491. [DOI] [PubMed] [Google Scholar]
- 2. Petit C, Levilliers J, Hardelin JP (2001) Molecular genetics of hearing loss. Annu Rev Genet 35: 589–646. [DOI] [PubMed] [Google Scholar]
- 3. Lin X, Tang W, Ahmad S, Lu J, Colby CC, et al. (2012) Applications of targeted gene capture and next-generation sequencing technologies in studies of human deafness and other genetic disabilities. Hear Res 288: 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cohn ES, Kelley PM (1999) Clinical phenotype and mutations in connexin 26 (DFNB1/GJB2), the most common cause of childhood hearing loss. Am J Med Genet 89: 130–136. [PubMed] [Google Scholar]
- 5. Gasparini P, Rabionet R, Barbujani G, Melchionda S, Petersen M, et al. (2000) High carrier frequency of the 35delG deafness mutation in European populations. Genetic Analysis Consortium of GJB2 35delG. Eur J Hum Genet 8: 19–23. [DOI] [PubMed] [Google Scholar]
- 6. Lee KY, Choi SY, Bae JW, Kim S, Chung KW, et al. (2008) Molecular analysis of the GJB2, GJB6 and SLC26A4 genes in Korean deafness patients. Int J Pediatr Otorhinolaryngol 72: 1301–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yan D, Park HJ, Ouyang XM, Pandya A, Doi K, et al. (2003) Evidence of a founder effect for the 235delC mutation of GJB2 (connexin 26) in east Asians. Hum Genet 114: 44–50. [DOI] [PubMed] [Google Scholar]
- 8. Shin J-W, Lee S-C, Lee H-K, Park H-J (2012) Genetic Screening of GJB2 and SLC26A4 in Korean Cochlear Implantees: Experience of Soree Ear Clinic. Clin Exp Otorhinolar 5: S10–S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Park HJ, Shaukat S, Liu XZ, Hahn SH, Naz S, et al. (2003) Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet 40: 242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Park HJ, Lee SJ, Jin HS, Lee JO, Go SH, et al. (2005) Genetic basis of hearing loss associated with enlarged vestibular aqueducts in Koreans. Clin Genet 67: 160–165. [DOI] [PubMed] [Google Scholar]
- 11. Bae JW, Lee KY, Choi SY, Lee SH, Park HJ, et al. (2008) Molecular analysis of mitochondrial gene mutations in Korean patients with nonsyndromic hearing loss. Int J Mol Med 22: 175–180. [PubMed] [Google Scholar]
- 12. Bae JW, Kim DB, Choi JY, Park HJ, Lee JD, et al. (2012) Molecular and Clinical Characterization of the Variable Phenotype in Korean Families with Hearing Loss Associated with the Mitochondrial A1555G Mutation. PLoS One 7: e42463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Choung YH, Moon SK, Park HJ (2002) Functional study of GJB2 in hereditary hearing loss. Laryngoscope 112: 1667–1671. [DOI] [PubMed] [Google Scholar]
- 14. Guan MX (2011) Mitochondrial 12S rRNA mutations associated with aminoglycoside ototoxicity. Mitochondrion 11: 237–245. [DOI] [PubMed] [Google Scholar]
- 15. Yoon JS, Park HJ, Yoo SY, Namkung W, Jo MJ, et al. (2008) Heterogeneity in the processing defect of SLC26A4 mutants. J Med Genet 45: 411–419. [DOI] [PubMed] [Google Scholar]
- 16. Ishihara K, Okuyama S, Kumano S, Iida K, Hamana H, et al. (2010) Salicylate restores transport function and anion exchanger activity of missense pendrin mutations. Hear Res 270: 110–118. [DOI] [PubMed] [Google Scholar]
- 17. Dossena S, Nofziger C, Tamma G, Bernardinelli E, Vanoni S, et al. (2011) Molecular and functional characterization of human pendrin and its allelic variants. Cell Physiol Biochem 28: 451–466. [DOI] [PubMed] [Google Scholar]
- 18. Taylor JP, Metcalfe RA, Watson PF, Weetman AP, Trembath RC (2002) Mutations of the PDS gene, encoding pendrin, are associated with protein mislocalization and loss of iodide efflux: implications for thyroid dysfunction in Pendred syndrome. J Clin Endocrinol Metab 87: 1778–1784. [DOI] [PubMed] [Google Scholar]
- 19. Yang JJ, Tsai CC, Hsu HM, Shiao JY, Su CC, et al. (2005) Hearing loss associated with enlarged vestibular aqueduct and Mondini dysplasia is caused by splice-site mutation in the PDS gene. Hear Res 199: 22–30. [DOI] [PubMed] [Google Scholar]
- 20. Smith WM, Van Orsouw NJ, Fox EA, Kolodner RD, Vijg J, et al. (1998) Accurate, high-throughput “snapshot” detection of hMLH1 mutations by two-dimensional DNA electrophoresis. Genet Test 2: 43–53. [DOI] [PubMed] [Google Scholar]
- 21. Paneto GG, Kohnemann S, Martins JA, Cicarelli RM, Pfeiffer H (2011) A single multiplex PCR and SNaPshot minisequencing reaction of 42 SNPs to classify admixture populations into mitochondrial DNA haplogroups. Mitochondrion 11: 296–302. [DOI] [PubMed] [Google Scholar]
- 22. Grignani P, Peloso G, Achilli A, Turchi C, Tagliabracci A, et al. (2006) Subtyping mtDNA haplogroup H by SNaPshot minisequencing and its application in forensic individual identification. Int J Legal Med 120: 151–156. [DOI] [PubMed] [Google Scholar]
- 23. La Neve F, Civera T, Mucci N, Bottero MT (2008) Authentication of meat from game and domestic species by SNaPshot minisequencing analysis. Meat Sci 80: 216–224. [DOI] [PubMed] [Google Scholar]
- 24. Huang CH, Chang MT, Huang MC, Lee FL (2011) Application of the SNaPshot minisequencing assay to species identification in the Lactobacillus casei group. Mol Cell Probes 25: 153–157. [DOI] [PubMed] [Google Scholar]
- 25. Fiorentino F, Magli MC, Podini D, Ferraretti AP, Nuccitelli A, et al. (2003) The minisequencing method: an alternative strategy for preimplantation genetic diagnosis of single gene disorders. Mol Hum Reprod 9: 399–410. [DOI] [PubMed] [Google Scholar]
- 26. Speiser PW, White PC (2003) Congenital adrenal hyperplasia. N Engl J Med 349: 776–788. [DOI] [PubMed] [Google Scholar]
- 27. Li SY, Wang W, Li JM, Wang Z, Wen RY, et al. (2011) PIK3CA mutation is an independent indicator of malignant phenotype and prognosis in breast cancer. Zhonghua Zhong Liu Za Zhi 33: 605–608. [PubMed] [Google Scholar]
- 28. Dudarewicz M, Baranska M, Rychlik-Sych M, Trzcinski R, Dziki A, et al. (2012) C3435T polymorphism of the ABCB1/MDR1 gene encoding P-glycoprotein in patients with inflammatory bowel disease in a Polish population. Pharmacol Rep 64: 343–350. [DOI] [PubMed] [Google Scholar]
- 29. Garcia-Roca P, Medeiros M, Reyes H, Rodriguez-Espino BA, Alberu J, et al. (2012) CYP3A5 polymorphism in Mexican renal transplant recipients and its association with tacrolimus dosing. Arch Med Res 43: 283–287. [DOI] [PubMed] [Google Scholar]
- 30. Kirchhoff T, Gaudet MM, Antoniou AC, McGuffog L, Humphreys MK, et al. (2012) Breast cancer risk and 6q22.33: combined results from Breast Cancer Association Consortium and Consortium of Investigators on Modifiers of BRCA1/2. PLoS One 7: e35706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Song MJ, Lee ST, Lee MK, Ji Y, Kim JW, et al. (2011) Estimation of carrier frequencies of six autosomal-recessive Mendelian disorders in the Korean population. J Hum Genet 57: 139–144. [DOI] [PubMed] [Google Scholar]
- 32. Han SH, Park HJ, Kang EJ, Ryu JS, Lee A, et al. (2008) Carrier frequency of GJB2 (connexin-26) mutations causing inherited deafness in the Korean population. J Hum Genet 53: 1022–1028. [DOI] [PubMed] [Google Scholar]
- 33. Park HJ, Hahn SH, Chun YM, Park K, Kim HN (2000) Connexin26 mutations associated with nonsyndromic hearing loss. Laryngoscope 110: 1535–1538. [DOI] [PubMed] [Google Scholar]
- 34. Zhao J, Wu LQ, Feng Y, Hu H, Pan Q, et al. (2009) [Rapid detection of the hot spot gene mutations in Chinese patients with nonsyndromic hearing loss by polymerase chain reaction-restrictive fragment length polymorphism]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 26: 518–520. [DOI] [PubMed] [Google Scholar]
- 35. Kato T, Nishigaki Y, Noguchi Y, Ueno H, Hosoya H, et al. (2010) Extensive and rapid screening for major mitochondrial DNA point mutations in patients with hereditary hearing loss. J Hum Genet 55: 147–154. [DOI] [PubMed] [Google Scholar]
- 36. Kokotas H, Grigoriadou M, Hatzaki A, Antoniadi T, Giannoulia-Karantana A, et al. (2010) Easy, rapid, and cost-effective methods for identifying carriers of recurrent GJB2 mutations causing nonsyndromic hearing impairment in the Greek population. Genet Test Mol Biomarkers 14: 189–192. [DOI] [PubMed] [Google Scholar]
- 37. Bardien S, Human H, Harris T, Hefke G, Veikondis R, et al. (2009) A rapid method for detection of five known mutations associated with aminoglycoside-induced deafness. BMC Med Genet 10: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lo YM, Corbetta N, Chamberlain PF, Rai V, Sargent IL, et al. (1997) Presence of fetal DNA in maternal plasma and serum. Lancet 350: 485–487. [DOI] [PubMed] [Google Scholar]
- 39. Poon LL, Leung TN, Lau TK, Chow KC, Lo YM (2002) Differential DNA methylation between fetus and mother as a strategy for detecting fetal DNA in maternal plasma. Clin Chem 48: 35–41. [PubMed] [Google Scholar]