

Abstract
Background
Elucidating the genomic basis of adaptation and speciation is a major challenge in natural systems with large quantities of environmental and phenotypic data, mostly because of the scarcity of genomic resources for non-model organisms. The Atlantic molly (Poecilia mexicana, Poeciliidae) is a small livebearing fish that has been extensively studied for evolutionary ecology research, particularly because this species has repeatedly colonized extreme environments in the form of caves and toxic hydrogen sulfide containing springs. In such extreme environments, populations show strong patterns of adaptive trait divergence and the emergence of reproductive isolation. Here, we used RNA-sequencing to assemble and annotate the first transcriptome of P. mexicana to facilitate ecological genomics studies in the future and aid the identification of genes underlying adaptation and speciation in the system.
Description
We provide the first annotated reference transcriptome of P. mexicana. Our transcriptome shows high congruence with other published fish transcriptomes, including that of the guppy, medaka, zebrafish, and stickleback. Transcriptome annotation uncovered the presence of candidate genes relevant in the study of adaptation to extreme environments. We describe general and oxidative stress response genes as well as genes involved in pathways induced by hypoxia or involved in sulfide metabolism. To facilitate future comparative analyses, we also conducted quantitative comparisons between P. mexicana from different river drainages. 106,524 single nucleotide polymorphisms were detected in our dataset, including potential markers that are putatively fixed across drainages. Furthermore, specimens from different drainages exhibited some consistent differences in gene regulation.
Conclusions
Our study provides a valuable genomic resource to study the molecular underpinnings of adaptation to extreme environments in replicated sulfide spring and cave environments. In addition, this study adds to the increasing number of genomic resources in the family Poeciliidae, which are widely used in comparative analyses of behavior, ecology, evolution, and medical genetics.
Keywords: De novo assembly, Ecological speciation, Expression analysis, Poeciliidae, RNA sequencing, Transcriptome
Background
A fundamental challenge in evolutionary biology is the mechanistic integration from genomic variation to fitness of organisms in their natural environment. Linking genomes to fitness is requisite to understand adaptation, speciation, and the interactions between the two processes [1,2]. In the past, different model systems have been used to either understand the genomic basis of phenotypes, or the fitness effects of phenotypic variation in response to different environmental conditions in nature, but in only few systems have we a thorough understanding about the ecological context in which phenotypic traits evolve and the genomic basis of respective traits. Notable exceptions include heavy metal tolerance in Arabidopsis[3], eco-morphological differentiation in lake trout [4], whitefish [5,6], and marine snails [7], the reduction of armor in freshwater sticklebacks [8,9], and changes in fur coloration in beach mice [10].
Elucidating the genomic basis of adaptation and speciation in systems with in- depth knowledge of ecological sources of selection and phenotypic trait variation has been hindered by the lack of genomic resources, which are often only available for model organisms. However, next generation sequencing (NGS) techniques provide a promising tool in this endeavor [11-13]. While whole genome sequencing on replicated sets of individuals is still expensive, focusing on transcribed portions of the genome (the transcriptome) has become increasingly popular. Such transcriptomic studies focus on sequencing cDNA libraries constructed from mRNA isolated from specific tissues (RNA-seq [14,15]), and in combination with barcoding technologies [16,17], can provide large amounts of sequence and expression level data for comparative transcriptomic studies.
We provide a first characterization of the transcriptome of the Atlantic molly, Poecilia mexicana (Poeciliidae). This livebearing fish species is widely distributed in freshwater environments along the Atlantic versant from northeastern Mexico into lower Central America [18,19]. Poecilia mexicana has been used as a study organism in animal behavior and behavioral ecology [20-22], predator–prey interactions [23,24], sensory ecology [25-27], and life history evolution [28,29]. Furthermore, the species is the maternal ancestor of a unisexual hybrid species, the Amazon molly (P. formosa), and is thus frequently investigated to address questions about the evolution and maintenance of sexual reproduction [30-32].
Most importantly, P. mexicana is an emerging model system to study adaptation to extreme environments and ecological speciation, as the species has colonized both hydrogen sulfide-rich and cave habitats in southern Mexico [33]. Hydrogen sulfide (H2S) is a potent toxicant lethal for most metazoans even in micro-molar amounts by inhibiting cellular respiration [34,35]. The absence of light in caves inhibits the use of visual senses, and cave-dwellers must cope with perpetual darkness, especially if they evolved from diurnal surface-dwelling forms like in poeciliids [36,37]. Extreme habitats harbor distinct ecotypes of P. mexicana that have evolved convergently across independently colonized sulfidic springs and caves [38,39]. Compared to conspecifics in adjacent non-sulfidic surface habitats, which harbor the ancestral populations, extremophile populations diverged in eye size, body shape, and gill morphology [38,40], physiology [38], life history strategies [41,42], and behavior [43,44]. Despite the lack of physical barriers between extreme and adjacent normal habitats, gene flow across habitat types is eminently low [45], and reproductive isolation is at least partially mediated by natural and sexual selection against immigrants [46-48].
Despite the well-characterized selective environments and phenotypic variation in this system, there are currently no genomic resources to start addressing questions about the genomic changes underlying trait divergence. Such resources are particularly required to test whether fixed positively selected mutations and changes in gene expression patterns show similar patterns of convergence across replicated environmental gradients, or whether unique genomic changes in each evolutionary replicate essentially precipitated similar phenotypic effects. To address such questions in the future, and to start building genomic resources for other study areas using P. mexicana (and close relatives such as the sailfin molly P. latipinna, the amazon molly P. formosa, and the guppy P. reticulata), we used RNA sequencing to obtain a first transcriptome of this species. We particularly focused on gill tissues, since many physiological processes involved in the maintenance of homeostasis take place here [49,50], and used six individuals from ancestral, non-sulfidic populations to facilitate de novo transcriptome assembly in absence of a reference genome. Our key objectives were to (1) create a database of the gill transcriptome in P. mexicana for future comparative studies, (2) to annotate transcripts based on functional annotations in reference databases, (3) to compare the P. mexicana transcriptome to other published fish transcriptomes, including the guppy (Poecilia reticulata), medaka (Oryzias latipes), threespined stickleback (Gasterosteus aculeatus), and zebrafish (Danio rerio), and (4) to identify potential candidate genes of interest in the study of extremophile poeciliids.
Construction and content
Sample collection methods
Gill samples for transcriptomic profiling were obtained from fish collected in their natural environment. Three adult females were collected in both the Arroyo Rosita (Río Pichucalco drainage) and Arroyo Bonita (Río Tacotalpa drainage). Both river drainages originate in the mountains of the Sierra Madre de Chiapas of Southern Mexico and flow into the wide floodplains of northern Tabasco, where they ultimately join the Río Grijalva (see Additional file 1: Figure S1 for a map). Collection locations were intermediate gradient, non-sulfidic streams with similar structure and water chemistry (see [38] for detailed environmental data). Locations were chosen to represent the most eastern (Tacotalpa) and most western (Pichucalco) drainages inhabited by sulfide spring fishes in Southern Mexico [38]. Analyses were restricted to specimens from habitats with similar environmental conditions (i.e., non-sulfidic surface streams) to facilitate de novo assembly.
Fish were caught with a seine (2 × 6 m). Immediately after capture, fish were sacrificed, measured and weighed, and gill tissue was extracted from both sides of the body using previously sterilized scissors and forceps. Tissues were preserved in 2 ml of RNAlater (Ambion, Inc.) and stored on ice during transport to the laboratory. Experiments were approved by the Institutional Animal Care and Use Committee of Oklahoma State University (ACUP AS10-15).
RNA isolation and RNAseq library construction
RNA was isolated from gills by pulverizing 50–100 mg of tissue frozen in liquid nitrogen in individual tubes with a Covaris Cryoprep at setting 3. RNA was then extracted with Qiagen’s RNeasy Plus mini kit. PolyA+ mRNA was prepared from 50 μg total RNA using Invitrogen’s Dynabeads mRNA purification kit. RNA was bound and eluted twice to Dynabeads to minimize ribosomal RNA contamination. mRNA was fragmented to an average size of 400 nt using NEB’s mRNA Fragmentation Module by incubation at 94°C for 4 minutes. Fragmented mRNA was purified using Agencourt RNAClean XP beads and eluted in 12 μl ddH2O. First strand cDNA was synthesized in a 20 μl reaction using Invitrogen’s Double-stranded cDNA kit, primed with 1 μl of a mix of random hexamers:oligo dT primers (2 μg:1 μg), and incubated with Superscript II at 45°C for one hour. The first strand cDNA reaction was used directly in NEBNext mRNA Second Strand Synthesis kit.
After second strand cDNA synthesis, the reaction was purified with Agencourt Ampure XP beads and eluted in 25 μl water. Double stranded cDNA was used as input for Illumina sequencing library preparation with end-repair using the NEBNext end-repair kit, A-tailing with Taq polymerase, ligation with Truseq barcoded adapters, and amplification with Kapa Library Amplification Readymix. All steps were cleaned up with Ampure XP beads. RNAseq libraries were quantified on an Agilent 2100 Bioanalyzer High Sensitivity DNA chip and pooled based on nM concentration. Libraries were sequenced on an Illumina HiSeq 2000 with paired-end 101 bp reads.
De novo assembly
Reads were sorted by barcode and trimmed to 87 basepairs (5 bases were trimmed from the beginning of each read and 11 bases from the end of the read due to lower sequence quality at the beginning and end). All reads with remaining ambiguous bases were removed and only paired reads were used for the analysis, which resulted in removal of less than 5% of reads. Data from the six individuals was concatenated for de novo assembly using Trinity [51], with the default settings. To remove possible spurious transcripts and very lowly expressed transcripts, a modified version of RSEM (RNA-Seq by Expectation Maximization [52]) available with the Trinity package was applied, and transcripts with an FPKM (fragments per kilobase of exon per million fragments mapped) less than 0.1 were removed. To remove isoforms and paralogs, we conducted a reciprocal blast to our own dataset. For sequences with >97% similarity, we only retained the longest sequence for further analysis. Finally, we tested for predicted open reading frames (ORFs) using OrfPredictor [53]. Only sequences with a predicted ORF were retained for subsequent analyses. Six transcripts were validated using Invitrogen SuperScript One-Step reverse-transcriptase-polymerase chain reaction (RT-PCR) with primers designed for each transcript (Additional file 2: Table S1). Nested internal primers were used in a second PCR step with Kapa Library Amplification Readymix polymerase (Kapa Biosystems), the product of which was Sanger sequenced for validation.
Comparison to other transcriptomes
We compared the reduced dataset to the NCBI Unigene records (ftp://ftp.ncbi.nih.gov/repository/UniGene/) for medaka (Oryzias latipes; 21,803 transcripts), three-spined stickleback (Gasterosteus aculeatus; 18,681 transcripts), and zebrafish (Danio rerio; 52,653 transcripts). All datasets were downloaded on 11/1/2011. We also compared our data set to the guppy (Poecilia reticulata; 71,138 transcripts) transcriptome [54], which was obtained from http://www.bio.fsu.edu/kahughes/Databases.html. Reciprocal similarity searches were conducted using tblastx with an E-value threshold of 0.001.
Transcriptome annotation
To annotate transcripts, we first conducted a blast search of all unique contigs with a predicted ORF against the SwissProt database (http://ca.expasy.org/sprot/; blastx, critical E-value = 0.001; database accessed 11/04/2011) using Blast2GO [55-57]. Any sequences that did not have a match in SwissProt were subsequently blasted against the NCBI non-redundant (NR) protein database (blastx, critical E-value = 0.001; database accessed 12/03/2011). This procedure was employed because the SwissProt database provides more informative functional annotations, but the NR database is larger and has the possibility to annotate sequences not available in SwissProt. For each sequence, we retained the top blast hit for subsequent analysis. Lastly, contigs with no match in either database were translated and searched against the Pfam-A and Pfam-B protein families databases [58] with an E-value cut-off of 0.01, and against the Rfam database for non-coding RNA families [59].
Sequences with a match in either the SwissProt or NR database were subsequently annotated with Gene Ontology (GO) IDs [60] as implemented in Blast2GO. GO IDs describe gene product characteristics and are hierarchically organized in terms of biological processes, molecular functions, and cellular components. Due to the hierarchical organization, GO annotations can be simplified to a smaller set of high-level GO terms (GO slims). We obtained GO slims through Blast2GO with the generic slim developed by the GO Consortium (http://www.geneontology.org/GO.slims.shtml).
To compare transcriptome annotation in P. mexicana to previously published annotations of P. reticulata[54], the reduced P. reticulata dataset (http://www.bio.fsu.edu/kahughes/Databases.html) was re-annotated with the same procedure as outlined above for P. mexicana to reduce effects of differential methodologies and database access dates. Differential representation of records in each GO slim term was then compared between the two species by counting the number of sequences associated with each GO slim category. We tested for differences in representation for each GO slim category with a Chi-square test.
Finally, we searched the P. mexicana transcriptome for candidate genes of interest in future comparative studies. We particularly focused on genes related to environmental stress and living in extreme environments. To that end, we searched our annotation database for gene products known to be involved in general stress and oxidative stress responses. Since P. mexicana has also colonized several springs with high concentrations of hydrogen sulfide and severe hypoxia, we also searched for gene products related to sulfide detoxification and metabolism as well as hypoxia induced responses.
SNP discovery
To facilitate future research on genomic variation in P. mexicana, we developed a database of single nucleotide polymorphisms (SNPs). Putative SNPs were identified by mapping trimmed RNAseq reads to the reference transcriptome using Burrows-Wheeler Aligner (BWA [61]). We then applied the Genome Analysis Toolkit (GATK [62]) to the mapped reads for PCR duplicate removal, base quality score recalibration, and indel realignment. SNP and INDEL discovery as well as genotyping was performed across all 6 samples using standard hard filtering parameters [63]. The resulting SNPs were annotated as synonymous or non-synonymous using an in-house script based on the open reading frame predicted for each transcript. We also quantified the number of fixed alternate alleles between P. mexicana individuals from the two drainages.
Differential expression analysis
We tested whether there are differences in gene expression patterns in the three biological replicates of P. mexicana from the two different drainages. Trimmed reads were mapped to the reference transcriptome using Bowtie [64] and RSEM [52] within the Trinity package suite [51]. Mapped read counts were highly correlated between individuals (among and within drainages; Pearson correlation: r ≥ 0.91 for log-transformed data and r ≥ 0.99 for un-transformed data in all cases). We then used the edgeR package of Bioconductor [65-67] to identify genes that are differentially expressed between the drainages. We used the common dispersion estimated from a negative binomial implemented in edgeR. To reduce bias in our analyses due to low or high expression in single individuals, we filtered lowly expressed transcripts and those that were only expressed in a small number of samples by selectively retaining transcripts with at least one count per million in at least 3 samples. Of the 53,245 original transcripts, 21,480 meet these criteria. Sequences that were differentially expressed across drainages were annotated based on the procedure described above.
Utility
Transcriptome assembly
Sequencing gill tissue transcriptomes in six females of P. mexicana yielded over 70 million reads (Table 1), which represented – on average – 23.7-fold coverage of the transcriptome for each individual; taking the transcriptome size of 49.5 Mb, the sum of the number of bases in our assembled transcriptome, as a reference. We assembled reads, using Trinity, into 80,111 contigs. We excluded 14,982 contigs with an FPKM less than 0.1. After a reciprocal blast search within the P. mexicana transcriptome, we maintained the longest of those contigs presenting more than 97% similarity to each other. This filtering procedure allowed us to discard 11,884 smaller, redundant contigs and to retain 53,245 contigs representing putatively unique loci. Over 99% of these loci exhibited a predicted open reading frame (ORF), and only 331 sequences were removed from further analyses because they did not have an ORF. Data files with sequence information were deposited in the Sequence Read Archive on Genbank (Study Accession ID: SRP014728).
Table 1.
Sequencing and assembly statistics for Illumina sequencing used for the assembly of the P. mexicana transcriptome
| Average number of reads (± SD) | 11,698,857 (2,363,343) |
|---|---|
| Total number of reads |
70,193,146 |
| Average number of base pairs (± SD) |
1,181,584,624 (238,697,721) |
| Total number of base pairs |
7,089,507,746 |
| Average coverage (± SD) |
23.8 (4.4) |
| Total number of assembled contigs |
80,111 |
| Total number of unique loci |
53,245 |
| Total number of unique loci with predicted ORF |
52,914 |
| Mean contig length (basepairs) (± SD) |
932 (1004) |
| Maximum contig length | 15,623 |
Data presented represents reads from N = 6 bar coded individuals.
The frequency distribution of contig lengths is depicted in Figure 1. As expected, the number of mapped reads per contig was significantly correlated with contig length (Pearson correlation on log-transformed values: r = 0.87, P < 0.001). The average (± SE) level of gene expression controlled for contig length was 19.4 (± 1.2) FPKM, ranging from a minimum of 0.02 to a maximum of 33,072.50.
Figure 1.
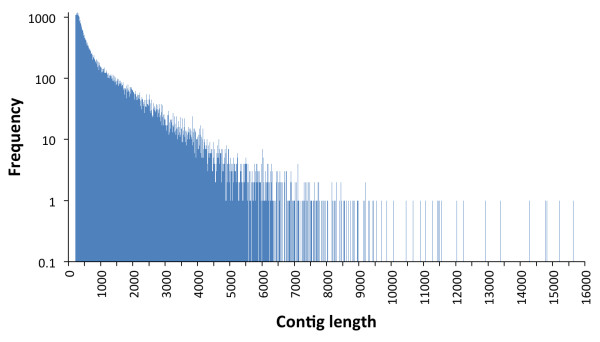
Frequency distribution of assembled contigs by size.
Six predicted transcripts were selected for RT-PCR and sequencing validation (Additional file 2: Table S1). All six predicted transcripts were validated via direct sequencing.
Comparison to other fish transcriptomes
We compared the P. mexicana transcriptome to data available from four other freshwater fishes (guppy, medaka, stickleback, and zebrafish; Table 2) using reciprocal blast searches. Mapping unique P. mexicana contigs to the transcriptomes of these species resulted in blast matching of more than 50% (51-74%) in each species examined. In contrast, matching was lower (29-55%) when mapping contigs from those species to the P. mexicana dataset. The difference in the percent mapping between the species partially reflects the difference in the sizes of the databases and the phylogenetic divergence among species under consideration (Additional file 3: Figure S2).
Table 2.
Results of reciprocal blast searches of the P. mexicana transcriptome to the database of the guppy (Poecilia reticulata) [54], and Uniprot databases of medaka (Oryzias latipes), zebrafish (Danio rerio), and stickleback (Gasterosteus aculeatus) [68]
| |
P. mexicana to focal species |
Focal species to P. mexicana |
||||
|---|---|---|---|---|---|---|
| Unique P. mexicana transcripts | Unique hits in focal species | % coverage in focal species | Unique focal species transcripts | Unique hits in P. mexicana | % coverage in P. mexicana | |
| Guppy |
29,671 |
46,634 |
65.6 |
46,953 |
29,337 |
55.2 |
| Medaka |
19,447 |
15,337 |
70.3 |
14,958 |
18,277 |
34.4 |
| Zebrafish |
25,013 |
26,871 |
51.0 |
26,870 |
24,370 |
45.8 |
| Stickleback | 18,037 | 13,807 | 73.9 | 13,554 | 17,024 | 28.8 |
The table lists the number of unique transcripts that map to the reference species’ transcriptome, the number of unique transcripts that received hits in the reference species, and the percent coverage in the references species database.
Annotation
The P. mexicana transcriptome was annotated by blast searches against the SwissProt and the NCBI non-redundant (NR) protein databases. Overall, 26,317 contigs (49.7%) had matches in the SwissProt database. These represented 17,814 unique records. Of the remaining sequences, 3,475 (6.6%) had a match to 2,497 unique records in the NR database. Of the unmatched contigs, 766 had matches in the Pfam database and 31 had matches in the Rfam database, indicating at least part of unmatched contigs represent real transcript.
The 29,792 sequences with a match in the SwissProt or NR databases were further annotated with Gene Ontology (GO) terms based on the Uniprot database, which yielded results for 22,184 (74.5%) of sequences. Of these, 13,002 sequences were annotated with a biological process GO term, 13,623 with a molecular function, and 14,430 with a cellular component. The relative frequency of level 2 GO terms is visualized in Figure 2. We also compared the representation of records in each generic slim between P. mexicana and P. reticulata. While representation was qualitatively very similar between the two species, 20 out of 30 level 5 GO categories exhibited significant differences across species even when accounting for the effects for multiple testing (Chi2 ≥ 13.545, P ≤ 0.0002, α’ = 0.001).
Figure 2.

Blast2GO assignment for 17,286 P. mexicana and 9,721 P. reticulata sequences. Significant differences in the frequency of different categories are highlighted with an asterisk. The numbers next to each colored slice of the pie chart represent the number of genes in the respective category.
By mining our annotation database, we found a diverse set of candidate genes for future genomic studies related to life in extreme environments (Table 3). The candidate list includes genes involved in general and oxidative stress responses. Particularly, we found multiple gene products associated with different functions from the heat shock protein family and could detect several components of antioxidant systems. Due to the severe hypoxia and high concentrations of hydrogen sulfide in some P. mexicana habitats, we also surveyed our data for genes previously associated with responses to hypoxia and sulfide detoxification. Most notably, we recorded a hypoxia specific transcription factor and several components involved in regulating aerobic and anaerobic metabolism. Furthermore, our database contains records for sulfide:quinone oxidoreductase, rhodanese, and several other key proteins involved in the metabolic processing of toxic hydrogen sulfide.
Table 3.
Candidate genes with annotations from the SwissProt (SP) database that are involved in environmental stress tolerance, hypoxia, and sulfide metabolism
| Category | Gene description | Contig ID | Accession number | Percent coverage | E-value | Number of contigs |
|---|---|---|---|---|---|---|
| General stress responses | ||||||
| Stress-activated protein kinase signaling pathways [69] |
Stress-activated protein kinase (JNK3) |
comp49081_c0_seq1 |
P53779 |
97 |
2.71E-63 |
1 |
| Stress-activated protein kinase kinase (SAPKK4) |
comp30986_c0_seq1 |
O14733 |
96 |
2.43E-153 |
1 |
|
| Highly inducible heat shock proteins [70,71] |
Hsp 70 |
comp24087_c0_seq1 |
P27541 |
92 |
3.65E-103 |
1 |
| comp34171_c0_seq1 |
Q91233 |
98 |
1.48E-25 |
2 |
||
| Hsp A2 |
comp24673_c0_seq1 |
P54652 |
88 |
1.47E-54 |
1 |
|
| Hsp A4 |
comp12212_c0_seq1 |
Q61316 |
91 |
3.38E-158 |
1 |
|
| comp7589_c0_seq1 |
P34932 |
79 |
<1.00E-180 |
2 |
||
| Hsp 90 |
comp48547_c0_seq1 |
P07900 |
77 |
7.45E-61 |
2 |
|
| comp5586_c0_seq1 |
Q4R4P1 |
76 |
<1.00E-180 |
1 |
||
| comp32336_c0_seq1 |
O61998 |
77 |
8.80E-33 |
3 |
||
| Constitutive heat shock proteins [70,71] |
Hsp83 |
comp28290_c0_seq1 |
P12861 |
95 |
1.42E-43 |
3 |
| Hsc20 |
comp17434_c0_seq1 |
Q8K3A0 |
64 |
3.26E-60 |
1 |
|
| Hsc70 |
comp29081_c0_seq1 |
Q9U639 |
80 |
3.69E-88 |
1 |
|
| Hsc71 |
comp7828_c0_seq1 |
P08108 |
98 |
<1.00E-180 |
4 |
|
| Small heat shock proteins [70,71] |
Hsp β8 |
comp23233_c0_seq1 |
Q9UJY1 |
67 |
6.80E-66 |
1 |
| Oxidative stress responses | ||||||
| Antioxidant systems [72,73] |
Catalase |
comp3837_c0_seq1 |
Q9PT92 |
93 |
<1.00E-180 |
1 |
| (Cu & Zn) superoxide dismutase |
comp21322_c0_seq1 |
Q751L8 |
61 |
2.29E-30 |
1 |
|
| comp496_c0_seq1 |
O73872 |
87 |
3.87E-88 |
2 |
||
| comp17555_c0_seq1 |
P82205 |
64 |
2.05E-33 |
1 |
||
| comp28438_c0_seq1 |
Q9C0N4 |
46 |
2.83E-09 |
1 |
||
| (Mn) superoxide dismutase |
comp29573_c0_seq1 |
P41978 |
57 |
6.78E-51 |
1 |
|
| comp1897_c0_seq1 |
P07895 |
86 |
5.13E-131 |
1 |
||
| Glutathione peroxidases |
comp37533_c0_seq1 |
Q4AEH3 |
61 |
6.22E-27 |
2 |
|
| comp18221_c0_seq1 |
Q4RSM6 |
90 |
5.39E-121 |
1 |
||
| comp559_c0_seq1 |
Q4AEI2 |
80 |
9.44E-87 |
1 |
||
| comp841_c0_seq1 |
P00435 |
81 |
8.33E-86 |
1 |
||
| Thioredoxin and glutathione reductase |
comp2236_c0_seq1 |
Q86VQ6 |
85 |
<1.00E-180 |
1 |
|
| Thioredoxin |
comp26993_c0_seq1 |
Q9BDJ3 |
72 |
4.09E-16 |
1 |
|
| comp572_c0_seq1 |
Q9DGI3 |
90 |
7.86E-20 |
1 |
||
| Methallothioneins [73] |
Metal-responsive element-binding transcription factor 2 |
comp17857_c0_seq1 |
Q02395 |
70 |
3.28E-164 |
1 |
| Hypoxia induced responses | ||||||
| Transcription factors [74] |
Hypoxia-inducible factor 1α |
comp436_c1_seq1 |
Q9YIB9 |
76 |
1.65E-110 |
1 |
| comp2126_c0_seq1 |
Q98SW2 |
78 |
<1.00E-180 |
3 |
||
| Oxygen transport [74] |
Erythropoietin |
comp50800_c0_seq1 |
Q5IGQ0 |
90 |
7.05E-47 |
1 |
| Hemoglobin β chain |
comp12875_c0_seq1 |
P84652 |
87 |
1.01E-78 |
1 |
|
| Myoglobin |
comp390_c1_seq1 |
Q9DGJ1 |
91 |
5.58E-72 |
1 |
|
| Aerobic/anaerobic metabolism [74] |
Malate dehydrogenase |
comp11493_c0_seq1 |
P11708 |
83 |
3.41E-37 |
1 |
| Succinate dehydrogenase |
comp898_c0_seq1 |
Q7ZVF3 |
94 |
<1.00E-180 |
2 |
|
| Citrate synthase |
comp44107_c0_seq1 |
Q91V92 |
77 |
6.21E-39 |
1 |
|
| Phosphoglycerate mutase |
comp703_c0_seq1 |
P18669 |
94 |
6.82E-163 |
1 |
|
| Phosphoglycerate kinase |
comp21645_c0_seq1 |
Q60HD8 |
76 |
2.87E-163 |
1 |
|
| α-enolase |
comp324_c0_seq1 |
Q9PVK2 |
96 |
<1.00E-180 |
1 |
|
| Lactate dehydrogenase (A chain, B chain, C chain) |
comp4088_c0_seq1 |
Q92055 |
99 |
<1.00E-180 |
1 |
|
| comp192_c0_seq1 |
P20373 |
98 |
<1.00E-180 |
1 |
||
| comp17481_c0_seq1 |
Q06176 |
97 |
7.12E-128 |
2 |
||
| Glycogen phosphorylase |
comp28770_c0_seq1 |
Q9XTL9 |
79 |
2.43E-178 |
3 |
|
| Metabolic rate suppression [74] |
α-tropomyosin |
comp1635_c0_seq2 |
P84335 |
99 |
4.56E-56 |
2 |
| comp1488_c0_seq1 |
P13105 |
96 |
1.61E-37 |
1 |
||
| Myosin heavy chain |
comp37734_c0_seq1 |
Q63357 |
87 |
2.22E-41 |
1 |
|
| Insulin-like growth factor binding protein 1 |
comp12811_c0_seq1 |
P24591 |
55 |
5.49E-43 |
1 |
|
| Sulfide detoxification | ||||||
| Sulfide metabolism and toxicity [75-77] |
Sulfide:quinone oxidoreductase |
comp1919_c0_seq1 |
Q9Y6N5 |
85 |
<1.00E-180 |
1 |
| Sulfite oxidase |
comp25579_c0_seq1 |
P07850 |
78 |
2.59E-93 |
2 |
|
| Sulfur dioxygenase (ETHE1) |
comp1681_c0_seq1 |
Q9DCM0 |
78 |
3.54E-104 |
2 |
|
| Thiosulfate sulfurtransferase (Rhodanese) |
comp2624_c0_seq1 |
Q8NFU3 |
62 |
2.19E-28 |
2 |
|
| comp12736_c0_seq1 |
Q3U269 |
71 |
1.22E-176 |
1 |
||
| Mercaptopyruvate sulfurtransferase |
comp1051_c1_seq7 |
P97532 |
74 |
1.81E-15 |
1 |
|
| Cytochrome c oxidase complex (complex III subunit 6, subunit 3) |
comp513_c0_seq3 |
P07919 |
84 |
1.75E-18 |
1 |
|
| comp4_c0_seq1 | Q96133 | 93 | 3.16E-141 | 1 | ||
For each candidate genes, we reported the database and accession number, percent similarity and E-value, as well as the total number of contigs that matched to a specific database record.
SNP discovery
We identified SNPs by mapping RNAseq reads back to the reference transcriptome using BWA and calling SNPs using the GATK pipeline [61,62]. Sites were limited to those for which data for all six individuals were available, and 106,524 sites had confident genotype calls across all six individuals. 18,703 transcripts had at least 1 SNP with a median of 4 SNPs and mean of 5.7 SNPs per transcript. In this set, there are 41,777 synonymous and 21,706 non-synonymous mutations in coding sequences. 43,228 SNPs were found in untranslated regions. The transcript with the largest number of SNPs (74 SNPs) was comp1948_c0_seq1 (bromodomain containing 2 protein; 3,679 nt long).
Of the 106,524 SNPs, 1,566 sites are fixed for alternate alleles between the two drainages; 280 of these represented non-synonymous differences in 250 contigs. Sites with SNP calls in all individuals were combined into an excel spreadsheet for accessibility to researchers in the field. This database is available under: http://www.sulfide-life.info/mtobler/databases.
Differential expression across drainages
We compared expression levels in biological replicates across the two drainages investigated to identify loci that are differentially expressed in P. mexicana from the two drainages. 21,480 transcripts were analyzed for differential expression. 382 transcripts (representing less than 2% of analyzed transcripts) showed evidence for differential expression (with a P-value ≤ 0.01) between the two drainages. 229 transcripts (59.9% of differentially expressed transcripts) were up-regulated in the Pichucalco compared to Tacotalpa drainage, and 153 genes were up-regulated in Tacotalpa compared to Pichucalco drainage (Figure 3).
Figure 3.
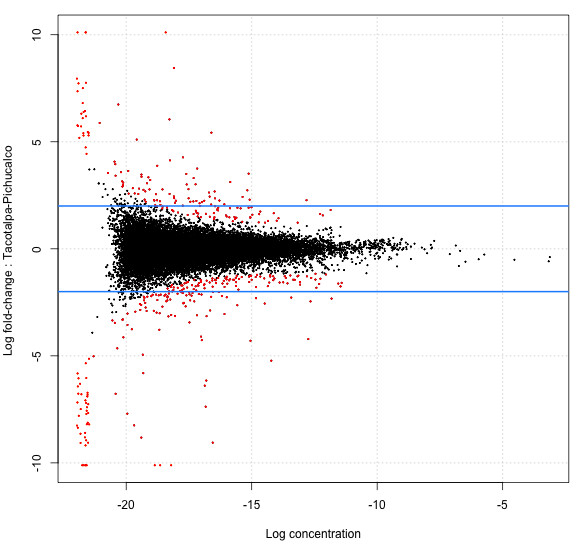
Plot of the log-fold change between the two drainages versus the log-concentration for each transcript. The most differentially expressed transcripts (P ≤ 0.01) are colored in red. The blue lines are a log-fold change of 2, indicating a fold change of 4.
Of the 382 differentially expressed transcripts, 251 had hits either in the SwissProt or NR databases in our annotation database; 172 of them were also annotated with GO IDs. Overall, 87 sequences were annotated with a molecular function term (these mostly related to binding and catalytic activity), 82 sequences with biological process term (mostly relating to metabolic processes, cellular processes, and biological regulation), and 83 sequences with a cellular component term (mostly cell and organelle; see Additional file 4: Figure S3 for details).
Discussion
This study used RNA sequencing and de novo assembly to characterize and annotate the transcriptome of Poecilia mexicana, a livebearing fish used in various evolutionary ecology studies. We identified over 53,000 putatively unique loci, which mapped to over 20,300 unique sequences in the SwissProt and NR databases. Reciprocal blast searches to other published fish transcriptomes indicated that P. mexicana transcripts detected in our study provided high concordance (between 51 and 74%) with the guppy, medaka, stickleback, and zebrafish transcriptomes. We also validated six of the predicted transcripts using RT-PCR and sequencing. This provides strong evidence that the contigs we assembled in absence of a reference genome largely represent real transcripts and not assembly error.
To further investigate the congruence of our draft P. mexicana transcriptome to that of the most closely related species available, the congeneric guppy, we annotated both transcriptomes with gene ontology (GO) IDs for quantitative comparison. While the GO ID representation uncovered for P. mexicana qualitatively matched that of other fish transcriptomes [e.g., [54,78], we found significant deviations in the majority of GO ID representations between P. mexicana and the guppy. These differences in GO ID representations across the two closely related species likely stem from differences in methodologies between the studies, specifically the tissue choice and sample preparation. Our approach captured a high proportion of the guppy transcriptome (66% of transcripts identified by blast; only 55% of transcripts were matched in the reciprocal comparison). This is surprising because we focused solely on the gills of females, while the guppy transcriptome was based on a broader sample of tissues (brain and body) from both sexes [54]. This suggests that many loci are transcribed at sufficient levels in gill tissue for transcriptome assembly. Based on comparative analyses with other fish transcriptomes, we achieved a similar (or higher) degree of representation with our approach; we attribute this to our focus on a single sex, a single tissue, and a single ecotype primarily to mitigate potential problems in de novo assembly in absence of a reference genome. For example, reciprocal blast searches to model organism transcriptomes consistently revealed higher congruence for P. mexicana (this study) than in the guppy [54]. In summary, our approach resulted in high coverage and high congruence with other available fish transcriptomes, which renders the P. mexicana transcriptome a useful resource for developing genomic tools in future evolutionary ecology studies.
Among the most peculiar aspects of P. mexicana’s biology is its tendency to invade extreme environments in the form of caves and toxic, hydrogen sulfide-rich springs. Populations in extreme environments are characterized by convergent patterns of adaptive trait divergence and ongoing ecological speciation [38,40]. The genomic tools developed here will allow for an increased focus on elucidating the genetic basis of evolution in extreme environments. Annotation of transcripts based on matches to major databases revealed the presence of a diversity of candidate genes relevant to dealing with physiochemical stressors, which will be instrumental for hypothesis testing in upcoming comparative studies between ecotypes. These candidate genes pertain to general stress responses, such as heat shock proteins and oxidative stress responses, that could provide insights about the mechanisms underlying selection against immigrants across ecologically divergent habitat types [46-48]. Candidate genes also include more system-specific pathways pertaining to sulfide metabolism and hypoxia-induced responses. Sulfide spring ecotypes are perpetually exposed to high concentrations of hydrogen sulfide and low oxygen concentrations [79]; hence, analyzing structural changes in candidate proteins and changes in gene regulation across ecotypes residing in sulfidic and non-sulfidic environments should be a high priority.
Sulfide springs have been independently colonized by P. mexicana in at least three different drainages [38], and future analyses of ecotype differentiation between sulfidic and non-sulfidic habitats also have to anticipate potential drainage specific effects. We identified over 1,500 putatively fixed alleles between the two drainages, 280 of which were characterized as non-synonymous mutations in coding regions. Our sample size of three individuals per drainage does not have adequate power to call fixed differences with certainty, nor can we draw any conclusions about the adaptive value of different alleles. However, the present analysis corroborates some previously detected geographic structuring across drainages [38] and provides a set of markers for future studies.
Besides allelic variation across drainages, we were also able to document significant variation in gene regulation. We uncovered some noise in the expression patterns quantified, with single individuals having highly up- or down-regulated gene expression for certain loci. Individual expression outliers could be driven by a variety of factors in the sampling procedure; e.g., age, reproductive state, history of parasitization, and other potentially relevant factors, which were not quantified for individual specimens, could all affect transcription of particular genes. Nonetheless, the expression analysis allowed us to identify over 350 transcripts that were consistently up- or downregulated between the drainages. These differences in gene regulation may be due to genetic divergence among populations of different drainages, or they may reflect plastic differences in gene expression in response to large-scale environmental factors. Clearly, uncovering the proximate and ultimate mechanisms underlying differential gene regulation need to be explored in future studies.
Conclusion
The newly sequenced, assembled, and annotated transcriptome of P. mexicana provides a valuable genomic resource to study the molecular underpinnings of adaptation to extreme environment in replicated sulfide spring and cave environments. This dataset also contributes to the growing number of genomic resources available for species of the family Poeciliidae [e.g., [54,80-86], which are broadly studied as model organisms for behavior, ecology, evolution, and medical genetics [87]. Together, these newly developed genomic tools provide valuable resource for ecological genomics projects, since we can build upon an extensive collection of data on phenotypic variation and the evolutionary forces shaping this variation across populations and species in this family (see [87,88] for broad overviews).
Availability and requirements
Data files with raw sequence information were deposited in the Sequence Read Archive on Genbank (Study Accession ID: SRA056996). The transcriptome, annotation summaries, and SNP data is publicly available at http://www.sulfide-life.info/mtobler/databases.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JLK, MP, and MT conceived of the study and participated in design and implementation of the study. LRA and MT conducted fieldwork. MCY and JLK conducted RNA-seq sample and library preparation. JLK, CNP, MT performed all computational and statistical analysis, and JLK and MT prepared the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Map of the study locations with the three major towns in the area for orientation. Site (1) represents Arroyo Rosita in the Río Pichucalco drainage; site (2) Arroyo Bonita in the Río Tacotalpa drainage. The insert depicts the location of the study area (black square) in Mexico.
Transcripts for RT-PCR validation and corresponding primers.
Phylogenetic relationships between species used in the comparative transcriptome analysis after Li et al. [89].
{kind=link}
Blast2GO assignment for 172 annotated sequences that were differentially expressed between P. mexicana from the Tacotalpa and Pichucalco river drainages. The numbers next to each colored slice of the pie chart represent the number of genes in the respective category.
Contributor Information
Joanna L Kelley, Email: joanna.l.kelley@gmail.com.
Courtney N Passow, Email: courtney.passow@okstate.edu.
Martin Plath, Email: mplath@bio.uni-frankfurt.de.
Lenin Arias Rodriguez, Email: leninariasrodriguez@hotmail.com.
Muh-Ching Yee, Email: yee@stanford.edu.
Michael Tobler, Email: michi.tobler@okstate.edu.
Acknowledgments
We thank the local community in Teapa and Tapijulapa for their hospitality and access to the study sites. We thank Z. Culumber, M. Palacios, and R. Riley for assistance in the field, O. E. Cornejo, R. McCoy, B. Haas, and K. K. Dhillon for technical advice, and C. D. Bustamante for financial support. Permits were kindly provided by the Mexican government (DGOPA.00093.120110.-0018). This study was funded by a grant from the National Science Foundation (NSF) to MT and JLK (IOS-1121832), and National Institute of Health National Research Service Award postdoctoral fellowship to JLK (GM087069).
References
- Wolf JBW, Lindell J, Backström N. Speciation genetics: current status and evolving approaches. Philosophical Transactions of the Royal Society B-Biological Sciences. 2010;365:1717–1733. doi: 10.1098/rstb.2010.0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schluter D. Evidence for ecological speciation and its alternative. Science. 2009;323:737–741. doi: 10.1126/science.1160006. [DOI] [PubMed] [Google Scholar]
- Turner TL, Bourne EC, Von Wettberg EJ, Hu TT, Nuzhdin SV. Population resequencing reveals local adaptation of Arabidopsis lyrata to serpentine soils. Nat Genet. 2010;42:260–263. doi: 10.1038/ng.515. [DOI] [PubMed] [Google Scholar]
- Goetz F, Rosauer D, Sitar S, Goetz G, Simchick C, Roberts S, Johnson R, Murphy C, Bronte CR, MacKenzie S. A genetic basis for the phenotypic differentiation between siscowet and lean lake trout (Salvelinus namaycush) Mol Ecol. 2010;19:176–196. doi: 10.1111/j.1365-294X.2009.04481.x. [DOI] [PubMed] [Google Scholar]
- Renaut S, Nolte AW, Bernatchez L. Mining transcriptome sequences towards identifying single nucleotide polymorphisms in lake whitefish pairs (Coregonus spp., Salmonidae) Mol Ecol. 2010;19:115–131. doi: 10.1111/j.1365-294X.2009.04477.x. [DOI] [PubMed] [Google Scholar]
- Jeukens J, Bittner D, Knudsen R, Bernatchez L. Candidate genes and adaptive radiation: insights from transcriptional adaptation to the limnetic niche among coregonine fishes (Coregonus spp., Salmonidae) Mol Biol Evol. 2009;26:155–166. doi: 10.1093/molbev/msn235. [DOI] [PubMed] [Google Scholar]
- Galindo J, Grahame JW, Butlin RK. An EST-based genome scan using 454 sequencing in the marine snail Littorina saxatilis. J Evol Biol. 2010;23:2004–2016. doi: 10.1111/j.1420-9101.2010.02071.x. [DOI] [PubMed] [Google Scholar]
- Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, Cresko WA. Population genomics of parallel adaptation in threespined stickleback using sequenced RAD tags. PLoS Genet. 2010;6:e1000862. doi: 10.1371/journal.pgen.1000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett RDH, Rogers SM, Schluter D. Natural selection on a major armor gene in threespine stickleback. Science. 2008;322:255–257. doi: 10.1126/science.1159978. [DOI] [PubMed] [Google Scholar]
- Hoekstra H, Hirschmann RJ, Bundey RA, Insel PA, Crossland JP. A single amino acid mutation contributes to adaptive beach mouse color pattern. Science. 2006;313:101–104. doi: 10.1126/science.1126121. [DOI] [PubMed] [Google Scholar]
- Gilad Y, Pritchard JK, Thornton K. Characterizing natural variation using next-generation sequencing technologies. Trends Ecol Evol. 2009;25:463–471. doi: 10.1016/j.tig.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luikart G, England PR, Tallmon D, Jordan S, Taberlet P. The power and promise of population genomics: from genotyping to genome typing. Nat Rev Genet. 2003;4:981–994. doi: 10.1038/nrg1226. [DOI] [PubMed] [Google Scholar]
- Stinchcombe JR, Hoekstra HE. Combining population genomics and quantitative genetics: finding genes underlying ecologically important traits. Heredity. 2008;100:158–170. doi: 10.1038/sj.hdy.6800937. [DOI] [PubMed] [Google Scholar]
- Marguerat S, Bähler J. RNA-seq: from technology to biology. Cell Mol Life Sci. 2010;67:569–579. doi: 10.1007/s00018-009-0180-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Gabriel S. Combinatorics and next-generation sequencing. Nat Biotechnol. 2009;27:826–827. doi: 10.1038/nbt0909-826. [DOI] [PubMed] [Google Scholar]
- Smith AM, Heisler LE, St. Onge RP, Farias-Hesson E, Wallace IM, Bodeau J, Harris AN, Perry KM, Giaever G, Pourmand N. et al. Highly-multiplexed barcode sequencing: an efficient method for parallel analysis of pooled samples. Nucleic Acids Res. 2010;38:e142. doi: 10.1093/nar/gkq368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RR, Minckley W, Norris S. Freshwater fishes of Mexico. Chicago: University of Chicago Press; 2005. [Google Scholar]
- Menzel BW, Darnell RM. Systematics of Poecilia mexicana (Pisces, Poeciliidae) in Northern Mexico. Copeia. 1973;1973:225–237. doi: 10.2307/1442961. [DOI] [Google Scholar]
- Tobler M, Schlupp I, Plath M. Costly interactions between the sexes: combined feects of male sexual harassment and female choice? Behav Ecol. 2011;4:723–729. [Google Scholar]
- Marler CA, Ryan MJ. Origin and maintenance of a female mating preference. Evolution. 1997;51:1244–1248. doi: 10.2307/2411053. [DOI] [PubMed] [Google Scholar]
- Plath M, Parzefall J, Körner K, Schlupp I. Sexual selection in darkness? Female mating preferences in surface- and cave-dwelling Atlantic mollies, Poecilia mexicana (Poeciliidae, Teleostei) Behav Ecol Sociobiol. 2004;55:596–601. doi: 10.1007/s00265-003-0750-9. [DOI] [Google Scholar]
- Tobler M, Schlupp I, Plath M. Predation of a cave fish (Poecilia mexicana, Poeciliidae) by a giant water-bug (Belostoma, Belostomatidae) in a Mexican sulfur cave. Ecol Entomol. 2007;32:492–495. doi: 10.1111/j.1365-2311.2007.00892.x. [DOI] [Google Scholar]
- Plath M, Riesch R, Culumber ZW, Streit B, Tobler CM. Giant waterbug (Belostoma sp.) predation on a cavefish (Poecilia mexicana): effects of female body size and gestational state. Evol Ecol Res. 2011;13:133–144. [Google Scholar]
- Körner KE, Schlupp I, Plath M, Loew ER. Spectral sensitivity of mollies: comparing surface- and cave-dwelling Atlantic mollies, Poecilia mexicana. J Fish Biol. 2006;69:54–65. doi: 10.1111/j.1095-8649.2006.01056.x. [DOI] [Google Scholar]
- Tobler M, Coleman SW, Perkins BD, Rosenthal GG. Reduced opsin gene expression in a cave-dwelling fish. Biol Lett. 2010;6:98–101. doi: 10.1098/rsbl.2009.0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parzefall J. A review of morphological and behavioural changes in the cave molly, Poecilia mexicana, from Tabasco, Mexico. Environ Biol Fishes. 2001;62:263–275. doi: 10.1023/A:1011899817764. [DOI] [Google Scholar]
- Riesch R, Plath M, Schlupp I. Toxic hydrogen sulfide and dark caves: life-history adaptations in a livebearing fish (Poecilia mexicana, Poeciliidae) Ecology. 2010;91:1494–1505. doi: 10.1890/09-1008.1. [DOI] [PubMed] [Google Scholar]
- Riesch R, Tobler M, Plath M, Schlupp I. Offspring number in a livebearing fish (Poecilia mexicana, Poeciliidae): reduced fecundity and reduced plasticity in a population of cave mollies. Environ Biol Fishes. 2009;84:89–94. doi: 10.1007/s10641-008-9392-0. [DOI] [Google Scholar]
- Heubel KU, Hornhardt K, Ollmann T, Parzefall J, Ryan MJ, Schlupp I. Geographic variation in female mate-copying in the species complex of a unisexula fish, Poecilia formosa. Behaviour. 2008;145:1041–1064. doi: 10.1163/156853908784474533. [DOI] [Google Scholar]
- Schartl M, Wilde B, Schlupp I, Parzefall J. Evolutionary origin of a parthenoform, the Amazon molly Poecilia formosa, on the basis of a molecular genealogy. Evolution. 1995;49:827–835. doi: 10.2307/2410406. [DOI] [PubMed] [Google Scholar]
- Schlupp I. The evolutionary ecology of gynogenesis. Annual Rev Ecology, Evolution and Syst. 2005;36:399–417. doi: 10.1146/annurev.ecolsys.36.102003.152629. [DOI] [Google Scholar]
- Tobler M, Plath M. In: Ecology and evolution of poeciliid fishes. Evans J, Pilastro A, Schlupp I, editor. Chicago: University of Chicago Press; 2011. Living in extreme habitats; pp. 120–127. [Google Scholar]
- Bagarinao T. Sulfide as an environmental factor and toxicant: tolerance and adaptations in aquatic organisms. Aquat Toxicol. 1992;24:21–62. doi: 10.1016/0166-445X(92)90015-F. [DOI] [Google Scholar]
- Grieshaber MK, Völkel S. Animal adaptations for tolerance and exploitation of poisonous sulfide. Annu Rev Physiol. 1998;60:33–53. doi: 10.1146/annurev.physiol.60.1.33. [DOI] [PubMed] [Google Scholar]
- Howarth FG. High-stress subterranean habitats and evolutionary change in cave-inhabiting arthropods. Am Nat. 1993;142:S65–S77. doi: 10.1086/285523. [DOI] [PubMed] [Google Scholar]
- Langecker TG. In: Ecosystems of the world 30: Subterranean Ecosystems. Wilkens H, Culver DC, Humphreys WF, editor. Amsterdam: Elsevier Science; 2000. The effect of continuous darkness on cave ecology and cavernicolous evolution; pp. 135–157. [Google Scholar]
- Tobler M, Palacios M, Chapman LJ, Mitrofanov I, Bierbach D, Plath M, Arias-Rodriguez L, de Leon FJ G, Mateos M. Evolution in extreme environments: replicated phenotypic differentiation in livebearing fish inhabiting sulfidic springs. Evolution. 2011;65:2213–2228. doi: 10.1111/j.1558-5646.2011.01298.x. [DOI] [PubMed] [Google Scholar]
- Tobler M, Riesch R, de Leon FJ G, Schlupp I, Plath M. A new and morphologically distinct cavernicolous population of Poecilia mexicana (Poeciliidae, Teleostei) Environ Biol Fishes. 2008;82:101–108. doi: 10.1007/s10641-007-9258-x. [DOI] [Google Scholar]
- Tobler M, DeWitt TJ, Schlupp I, de Leon FJ G, Herrmann R, Feulner P, Tiedemann R, Plath M. Toxic hydrogen sulfide and dark caves: Phenotypic and genetic divergence across two abiotic environmental gradients in Poecilia mexicana. Evolution. 2008;62:2643–2649. doi: 10.1111/j.1558-5646.2008.00466.x. [DOI] [PubMed] [Google Scholar]
- Riesch R, Plath M, de Leon Garcia FJ, Schlupp I. Convergent life-history shifts: toxic environments result in big babies in two clades of poeciliids. Naturwissenschaften. 2010;97:133–141. doi: 10.1007/s00114-009-0613-y. [DOI] [PubMed] [Google Scholar]
- Riesch R, Schlupp I, Langerhans RB, Plath M. Shared and unique patterns of embryo development in extremophile poeciliids. PLoS One. 2011;6:e27377. doi: 10.1371/journal.pone.0027377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath M, Schlupp I. Parallel evolution leads to reduced shoaling behavior in two cave dwelling populations of Atlantic mollies (Poecilia mexicana, Poeciliidae, Teleostei) Environ Biol Fishes. 2008;82:289–297. doi: 10.1007/s10641-007-9291-9. [DOI] [Google Scholar]
- Riesch R, Duwe V, Herrmann N, Padur L, Ramm A, Scharnweber K, Schulte M, Schulz-Mirbach T, Ziege M, Plath M. Variation along the shy-bold continuum in extremophile fishes (Poecilia mexicana, Poecilia sulphuraria) Behav Ecol Sociobiol. 2009;63:1515–1526. doi: 10.1007/s00265-009-0780-z. [DOI] [Google Scholar]
- Plath M, Hermann C, Schröder R, Riesch R, Tobler M, de Leon FJ G, Schlupp I, Tiedemann R. Locally adapted fish populations maintain small-scale genetic differentiation despite perturbation by a catastrophic flood event. BMC Evol Biol. 2010;10:256. doi: 10.1186/1471-2148-10-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobler M. Does a predatory insect contribute to the divergence between cave- and surface adapted fish populations? Biol Lett. 2009;5:506–509. doi: 10.1098/rsbl.2009.0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobler M, Riesch R, Tobler CM, Schulz-Mirbach T, Plath M. Natural and sexual selection against immigrants maintains differentiation among micro-allopatric populations. J Evol Biol. 2009;22:2298–2304. doi: 10.1111/j.1420-9101.2009.01844.x. [DOI] [PubMed] [Google Scholar]
- Plath M, Riesch R, Oranth A, Dzienko J, Karau N, Schiessl A, Stadler S, Wigh A, Zimmer C, Arias-Rodriguez L. et al. Complementary effects of natural and sexual selection against immigrants maintains differentiation between locally adapted fish. Naturwissenschaften. 2010;97:769–774. doi: 10.1007/s00114-010-0691-x. [DOI] [PubMed] [Google Scholar]
- Evans DH, Claiborne JB, editor. The physiology of fishes. 3. Boca Raton: Taylor & Francis; 2006. [Google Scholar]
- Evans TG, Hammill E, Kaukinen K, Schulze AD, Patterson DA, English KK, Curtis JMR, Miller KM. Transcriptomics of envrionmental acclimatization and survival in wild adult Pacific sockeye salmon (Oncorhynchus nerka) during spawing migration. Mol Ecol. 2011;20:4472–4489. doi: 10.1111/j.1365-294X.2011.05276.x. [DOI] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng QD. et al. Full-length transcriptome assemby from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–U130. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min XJ, Butler G, Storm R, Tsang A. OrfPredictor: predicting protein-coding regions in EST-derived sequences. Nucleic Acids Res. 2005;33:W677–W680. doi: 10.1093/nar/gki394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser BA, Weadick CJ, Janowitz I, Rodd FH, Hughes KA. Sequencing and characterization of the guppy (Poecilia reticulata) transcriptome. BMC Genomics. 2011;12:202. doi: 10.1186/1471-2164-12-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Goetz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Goetz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ, Robles M, Talon M, Dopazo J, Conesa A. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36:3420–3435. doi: 10.1093/nar/gkn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Goetz S. Blast2GO: a comprehensive suite for functional analysis in plant genomics. Int J Plant Genomics. 2008;2008:1–13. doi: 10.1155/2008/619832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J. et al. The Pfam protein families database. Nucleic Acids Res. 2012;40:D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner PP, Daub J, Tate J, Moore BL, Osuch HI, Griffiths-Jones S, Finn RD, Nawrocki EP, Kolbe DL, Eddy SR. et al. Rfam: wikipedia, clans and the "decimal" release. Nucleic Acids Res. 2011;39:141–145. doi: 10.1093/nar/gkq1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology Consortium. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–D261. doi: 10.1093/nar/gkh036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pristo M, Banks E, Poplin R, Garimella K, Maguire J, Hartl C, Philippakis A, del Angel G, Rivas MA, Hanna M. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, Smyth GK. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics. 2007;23:2881–2887. doi: 10.1093/bioinformatics/btm453. [DOI] [PubMed] [Google Scholar]
- Robinson MD, Smyth GK. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics. 2008;9:321–332. doi: 10.1093/biostatistics/kxm030. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium. Reorganizing the protein space at the Universal Protein Source (UniProt) Nucleic Acids Res. 2012;40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posas F, Nebrada AR, editor. Stress-activated protein kinases. Berlin: Springer; 2008. [Google Scholar]
- Feder ME, Hofmann GE. Heat-shock proteins, molecular chaperones, and the stress response: Evolutionary and ecological physiology. Annual Rev Physiology. 1999;61:243–282. doi: 10.1146/annurev.physiol.61.1.243. [DOI] [PubMed] [Google Scholar]
- Korsloot A, Van Gestel CAM, Van Straalen NM. Envrionmental stress and cellular response in arthropods. Boca Raton: CRC Press; 2004. [Google Scholar]
- Kohen R, Nyska A. Oxidation of biological systems: oxidative stress phenomena, antioxidants, redox reactions, and methords for their quantification. Toxicol Pathol. 2002;30:620–650. doi: 10.1080/01926230290166724. [DOI] [PubMed] [Google Scholar]
- Van Straalen NM, Roelofs D. Ecological genomics. Oxford: Oxford University Press; 2006. [Google Scholar]
- Richards JG. In: Fish physiology: hypoxia. 27. Richards JG, Farrell AP, Brauner CJ, editor. New York: Academic; 2009. Metabolic and molecular responses of fish to hypoxia; pp. 443–485. [Google Scholar]
- Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- Lagoutte E, Mimoun S, Andriamihaja M, Chaumontet C, Blachier F, Bouillaud F. Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochimica Et Biophysica Acta. 2010;1797:1500–1511. doi: 10.1016/j.bbabio.2010.04.004. [DOI] [PubMed] [Google Scholar]
- Ip YK, Kuah SSL, Chew SF. Strategies adopted by the mudskipper Boleophthalmus boddaerti to survive sulfide exposure in normoxia or hypoxia. Physiol Biochem Zool. 2004;77:824–837. doi: 10.1086/423746. [DOI] [PubMed] [Google Scholar]
- Elmer KR, Fan S, Gunter HM, Jones JC, Boekhoff S, Kuraku S, Meyer A. Rapid evolution and selection inferred from the transcriptomes of sympatric crater lake cichlids. Mol Ecol. 2010;19(Suppl. 1):197–211. doi: 10.1111/j.1365-294X.2009.04488.x. [DOI] [PubMed] [Google Scholar]
- Tobler M, Schlupp I, Heubel K, Riesch R, de Leon Garcia FJ, Giere O, Plath M. Life on the edge: Hydrogen sulfide and the fish communities of a Mexican cave and surrounding waters. Extremophiles. 2006;10:577–585. doi: 10.1007/s00792-006-0531-2. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Wang Y, Wang S, Liu J, Warren W, Mitreva M, Walter RB. Transcriptome analysis of female and male Xiphophorus maculatus Jp 163 A. PLoS One. 2011;6:e18379. doi: 10.1371/journal.pone.0018379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boswell MG, Wells MC, Kirk LM, Ju Z, Zhang Z, Booth RE, Walter RB. Comparison of gene expression responses to hypoxia in viviparous (Xiphophorus) and oviparous (Oryzias) fishes using a medaka microarray. Comp Biochem Physiol C. 2009;149:258–265. doi: 10.1016/j.cbpc.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Shen YJ, Catchen J, Garcia T, Amores A, Beldorth I, Wagner J, Zhang ZP JP, Warren W, Schartl M. et al. Identification of transcriptome SNPs between Xiphophorus lines and species for assessing allele specific gene expression within F-1 interspecies hybrids. Comp Biochem Physiol C. 2012;155:102–108. doi: 10.1016/j.cbpc.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter RB, Rains JD, Russell JE, Guerra TM, Daniels C, Johnston DA, Kumar J, Wheeler A, Kelnar K, Khanolkar VA. et al. A microsatellite genetic linkage map for Xiphophorus. Genetics. 2004;168:363–372. doi: 10.1534/genetics.103.019349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi N, Hoffmann M, Willing E-M, Lanz C, Weigel D, Dreyer C. Genetic linkage map of the guppy, Poecilia reticulata, and quantitative trait loci analysis of male size and colour variation. Proc R Soc B. 2009;276:2195–2208. doi: 10.1098/rspb.2008.1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyer C, Hoffmann M, Lanz C, Willing E-M, Riester M, Warthmann N, Sprecher A, Tripathi N, Henz SR, Weigel D. ESTs and EST-linked polymorphisms for genetic mapping and phylogenetic reconstruction in the guppy, Poecilia reticulata. BMC Genomics. 2007;8:269. doi: 10.1186/1471-2164-8-269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willing E-M, Bentzen P, Van Oosterhout C, Hoffmann M, Cable J, Breden F, Weigel D, Dreyer C. Genome-wide nucleotide polymorphisms reveal population history and adaptive divergence in wild guppies. Mol Ecol. 2010;19:968–984. doi: 10.1111/j.1365-294X.2010.04528.x. [DOI] [PubMed] [Google Scholar]
- Evans JP, Pilastro A, Schlupp I, editor. Ecology and evolution of poeciliid fishes. Chicago: The University of Chicago Press; 2011. [Google Scholar]
- Meffe GK, Snelson FF, editor. Ecology and evolution of lifebearing fishes (Poeciliidae) New Jersey: Prentice Hall; 1989. [Google Scholar]
- Li C, Orti G, Zhang G, Lu G. A practical approach to phylogenomics: the phylogeny of ray-finned fish (Actinopterygii) as a case study. BMC Evol Biol. 2007;7:44. doi: 10.1186/1471-2148-7-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Map of the study locations with the three major towns in the area for orientation. Site (1) represents Arroyo Rosita in the Río Pichucalco drainage; site (2) Arroyo Bonita in the Río Tacotalpa drainage. The insert depicts the location of the study area (black square) in Mexico.
Transcripts for RT-PCR validation and corresponding primers.
Phylogenetic relationships between species used in the comparative transcriptome analysis after Li et al. [89].
Blast2GO assignment for 172 annotated sequences that were differentially expressed between P. mexicana from the Tacotalpa and Pichucalco river drainages. The numbers next to each colored slice of the pie chart represent the number of genes in the respective category.