

Abstract
Objective: The aim of this study was to evaluate the pharmacokinetic profile of lesogaberan in healthy subjects after single oral and intravenous administration of 14C-labeled lesogaberan and non-14C-labeled lesogaberan.
Study Design: This was an open-label, single-center, randomized, two-way crossover, phase I study.
Participants: Ten healthy male subjects took part in the study.
Intervention: Volunteers were randomized to receive a single dose of either orally dosed (100 mg) or intravenously infused (20 mg) non-14C-labeled lesogaberan, and then orally (100 mg) or intravenously (20 mg) administered 14C-labeled lesogaberan in a crossover design. Treatment periods were separated by a washout period of at least 7 days.
Main Outcome Measures Analyses of the rate and route of excretion, dose recovery, area under the plasma concentration versus time curve (AUC), AUC to the last quantifiable concentration, maximal plasma concentration (Cmax), time to Cmax, the apparent elimination half-life, bioavailability, total clearance, renal clearance, fraction of the bioavailable dose excreted unchanged in the urine, cumulative amount of drug excreted unchanged in urine, and the apparent volume of distribution at steady state of lesogaberan.
Results: Lesogaberan was rapidly and extensively absorbed from the gastrointestinal tract and Cmax was achieved within 1–2 hours of oral dosing. The terminal half-life of lesogaberan was between 11 and 13 hours. Renal clearance accounted for approximately 22% of total body clearance. Based on the recovery of administered radioactivity, approximately 84% of the dose was excreted into the urine either as the parent compound or as water-soluble metabolite(s). There were no safety concerns raised during the study.
Conclusion: Orally administered lesogaberan is rapidly absorbed with high bioavailability and the majority of the dose is excreted by the kidneys either as the parent compound or as metabolites. The major elimination pathway for lesogaberan in man is metabolism.
Introduction
Pharmacologic agents such as GABAB receptor agonists (e.g. baclofen) have been shown to inhibit transient lower esophageal sphincter relaxations (TLESRs), a major cause of reflux events in both healthy subjects and patients with gastroesophageal reflux disease (GERD).[1,2] As such, GABAB agonists offer a rational therapeutic add-on treatment to proton pump inhibitors (PPIs) for patients with GERD who have a partial response to PPIs, which is the current standard of care for GERD.[3] Lesogaberan (AZD3355) is a primarily peripherally restricted GABAB receptor agonist[4] that has previously been shown to inhibit the frequency of TLESRs in both healthy subjects[5] and patients with GERD.[6] In the latter pharmacodynamic study in GERD patients,[6] for example, lesogaberan at an oral dose of 65 mg twice daily reduced the mean number of reflux events by approximately 35% versus placebo. A subsequent phase II study in GERD patients attests to the potential clinical efficacy of lesogaberan at this dose.[7] The aim of the present study, therefore, was to further investigate the pharmacokinetic profile of lesogaberan, including the rate and route of excretion of total radioactivity following oral and intravenous dosing with 14C-labeled and non-14C-labeled lesogaberan. A secondary objective of the study was to assess the safety and tolerability of lesogaberan in terms of adverse events, blood pressure, pulse rate, electrocardiography, physical examination, and laboratory safety testing.
Subjects and Methods
This was a phase I, open-label, randomized, two-way crossover study (study code: D9120C00017) to evaluate the pharmacokinetic profile of lesogaberan after oral and intravenous administration in healthy subjects. The study was conducted at a single center in the UK, in accordance with ethical principles and standards described in the Declaration of Helsinki and the International Conference on Harmonisation (ICH)/Good Clinical Practice (GCP) guidelines, and was approved by an independent ethics committee. Written informed consent was obtained from all subjects prior to the commencement of the study.
Study Population
Healthy male subjects aged 35–55 years with a bodyweight of 65–100 kg, a body mass index of 19–30 kg/m2, and clinically normal physical findings and laboratory values were eligible for inclusion. In order to ensure safety, subject exclusion criteria included (i) significant clinical illness occurring in the 2 weeks prior to the study; (ii) history of mental, cardiac, renal, hepatic, neurologic, or significant gastrointestinal disease; (iii) a history of, or ongoing hypersensitivities; (iv) a history of gallstones; or (v) any condition/concomitant medication that could potentially modify the pharmacokinetics of the study drug.
Study Design and Treatments
The study design is presented in figure 1. At the pre-entry screening visit, subjects were provided with information about the study and informed consent was collected. Following the pre-study screening visit, the study consisted of two treatment periods separated by a washout of at least 7 days.
Fig. 1.
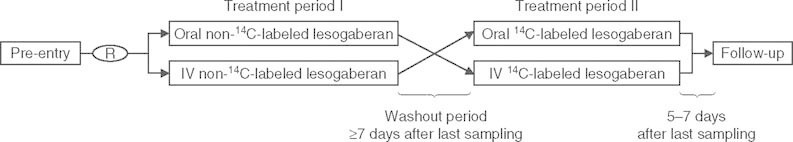
Study design. = intravenous; R = randomization.
In the first treatment period (72 hours in-house stay), subjects were randomized (50 : 50 ratio) to receive either lesogaberan dosed orally (100 mg) or intravenously (20 mg, 75 minute infusion of a 2 mg/mL solution). The solution for injection was used for both oral and intravenous dosing and administered to fasted subjects (overnight fast). The oral dose was administered with approximately 200 mL of water. Standardized meals and snacks were provided during the stay at the study center.
On the second dosing occasion (168 hours in-house stay), all subjects received 14C-labeled lesogaberan in a crossover design under conditions similar to the first treatment period (i.e. subjects who previously received orally administered lesogaberan received intravenously administered lesogaberan and vice versa). Subjects only received one dose of 14C-labeled lesogaberan during the study to minimize exposure risk and to remain within the guidance of WHO category I study limits of radioactivity. Standardized meals and snacks were provided during the stay at the study center, as for the first treatment period.
A post-study follow-up visit was scheduled 5–7 days after discharge from the second treatment period or early discontinuation.
Sample Collection
Blood samples (2 mL) for the determination of plasma lesogaberan concentrations were collected during both treatment periods after oral (sampling times: 0, 0.25, 0.5, 0.75, 1, 1.25, 1.5, 2, 3, 4, 6, 8, 12, 18, 24, 36, 48, 60, and 72 hours post-dose) and intravenous (sampling times: 0, 0.08, 0.25, 0.5, 0.75, 1, 1.25, 1.33, 1.5, 1.75, 2, 2.5, 3, 4, 6, 8, 12, 18, 24, 30, 36, 48, 60, and 72 hours post-dose) administration. Additional blood samples for the determination of total radioactivity (sampling times: 96, 120, 144, and 168 hours post-dose) were taken during the second treatment period after both oral and intravenous administration.
Urine samples for the determination of urine lesogaberan concentrations were collected from pre-dose until 72 hours post-dose during both treatment periods in the following time intervals: pre-dose, 0–4, 4–8, 8–12, 12–24, 24–48, and 48–72 hours post-dose. Additional samples were collected during the second treatment period in 24-hour time intervals from 72 to 168 hours post-dose. Urine was sampled for 216 hours post-dose for the determination of total radioactivity.
Fecal samples for the determination of total radioactivity were collected from pre-dose to 216 hours after dose intake.
Further study procedures during both treatment periods included assessments of electrocardiography, blood pressure, and pulse at pre-dose, 1 hour, and 24 hours post-dose, safety laboratory tests at pre-dose and 24 hours post-dose, and daily recording of adverse events.
Information regarding the metabolic profile, quantification of metabolites, and metabolite identification of lesogaberan in excreta and plasma samples will be presented in a subsequent publication.
Analytical Methods
Plasma and urine samples for analysis of lesogaberan concentrations were stored and shipped at less than -20°C. Samples were analyzed by an accredited laboratory (PRA International-Bioanalytical Laboratory B.V., Assen, the Netherlands) by liquid chromatography and mass spectrometric detection (lower limits of quantification were 0.03 μmol/L [plasma] and 1.00 μmol/L [urine]) within a timeframe and protocol consistent with previously established stability and analytical data. The total radioactivity in plasma, urine, and feces was analyzed at and reported by Covance Laboratories Limited (Harrogate, UK) according to a previously validated protocol.
The lower limit of quantification (LLOQ) of total radioactivity in the plasma samples was based upon the radioactive concentration of the background value (determined from pre-dose plasma samples), the weight of the sample taken, and the specific radioactivity of the dose formulation. For excreta samples, the LLOQ was taken as twice the background disintegrations per minute value of the corresponding pre-dose sample type.
Pharmacokinetic and Statistical Methods
The sample size was based on previous pharmacokinetic studies of this nature and no formal sample-size calculations were performed. Descriptive statistics were calculated for all pharmacokinetic and safety variables using SAS® (version 8.2).
The following pharmacokinetic variables were estimated for lesogaberan by non-compartmental methods using WinNonlin Enterprise version 4.1 (Pharsight Corporation, Mountain View, CA, USA): total area under the plasma concentration versus time curve (AUC); AUC from time zero until the last quantifiable concentration (AUCt); observed maximum plasma concentration (Cmax); the time to reach Cmax (tmax); terminal plasma half-life (t1/2); oral bioavailability (F); total body clearance (CL); renal clearance (CLR); fraction of dose excreted unchanged into the urine following intravenous administration; cumulative amount of the drug excreted unchanged in the urine (following intravenous administration only); and the apparent volume of distribution at steady-state (Vss). The amounts of total radioactivity in the urine and feces (expressed as a percentage of the dose) were also calculated.
Estimates and 95% confidence intervals (CIs) of the true geometric means were calculated for AUC, AUCt, Cmax, t1/2, F, CL, CLR and Vss (arithmetic mean was calculated for tmax). For the cumulative amount of the drug excreted unchanged in the urine and the fraction of the dose excreted unchanged into the urine, estimates and CIs of the true geometric means, or estimates and CIs of the true means were calculated depending on the observed frequency distribution.
Results
A total of ten subjects were enrolled in the study and randomized to treatment. All subjects completed the study and were included in both the pharmacokinetic and safety analyses. A summary of subject baseline characteristics is presented in table I.
Table I.

Subject baseline characteristics (n = 10)
The estimated geometric mean pharmacokinetic variables for orally and intravenously administered lesogaberan for the whole study population (n = 10) are presented in table II. The oral bioavailability of lesogaberan was high (F = 0.67). Following intravenous administration, Vss and CL of lesogaberan were 208.7 L and 22.9 L/hour, respectively. CLR accounted for 5.2 L/hour of total clearance; a total of 31.2 μmol, or 22.6% of the dose, was excreted unchanged into the urine following intravenous administration. CLR of lesogaberan following oral and intravenous dosing was similar, as was the amount excreted unchanged into the urine following adjustments for dose and bioavailability.
Table II.
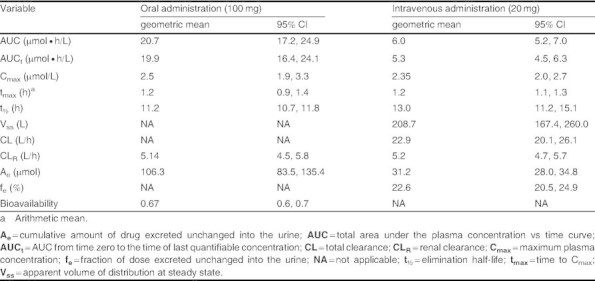
Estimated geometric means and 95% CIs for the pharmacokinetic variables of lesogaberan (n = 10)
The pharmacokinetic variables calculated from 14C-labeled lesogaberan data (table III) were similar to those for non-14C-labeled lesogaberan. The pharmacokinetic profiles of orally and intravenously dosed 14C-labeled lesogaberan are presented in figure 2.
Table III.

Estimated geometric means and ranges for the pharmacokinetic variables of 14C-labeled lesogaberan dosed orally (n = 5) and intravenously (n = 5)
Fig 2.
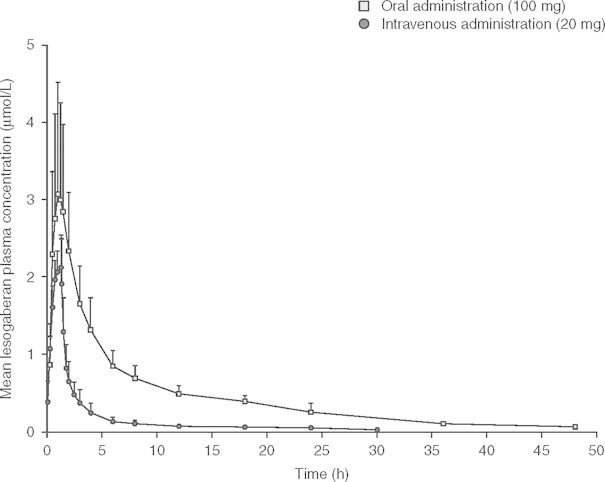
Mean (± SD) plasma concentration versus time profiles of orally (100 mg) and intravenously (20 mg) administered 14C-labeled lesogaberan (n = 5).
The mean (± SD) recovery of total radioactivity, expressed as a percentage of the total dose, from the urine and feces over a 216-hour sampling period following oral dosing of lesogaberan was 84.2 ± 5.6% and 9.0 ± 1.5%, respectively. The corresponding values for intravenously administered lesogaberan were 83.9 ± 2.3% and 7.1 ± 1.3%, respectively. The renal elimination and fecal excretion of radioactivity were almost complete after 216 hours and 168 hours, respectively. Plasma concentrations for total radioactivity were higher than those of lesogaberan throughout the majority of the sampling period, and thus indicate the presence of circulating metabolite(s).
In total, nine of ten subjects reported 22 adverse events during active treatment (table IV). All of the adverse events were transient and mild to moderate in intensity. The most frequently reported adverse events that were experienced by more than one subject in each treatment group were paresthesia (short-lasting cutaneous sensations: oral administration, n = 6; intravenous administration, n = 8) and headache (oral administration, n = 2; intravenous administration, n = 2). There were no serious adverse events, adverse events leading to study discontinuation, or clinically relevant changes in vital signs, ECG, or physical findings throughout the study.
Table IV.

Number of subjects with adverse events after oral and intravenous administration of 14C-labeled and non-14C-labeled lesogaberan (n = 10)a
Discussion
This is the first study to report on the pharmacokinetic profile, including routes of excretion, of orally and intravenously administered lesogaberan in humans. An oral dose of 100 mg was chosen for study in accordance with the predicted dose range because the therapeutic dose has not yet been established. Results show that lesogaberan was readily absorbed from the gastrointestinal tract following oral administration, with a terminal plasma half-life of 11.2 hours. The estimates of bioavailability, Vss, CL, and the fraction of the dose of lesogaberan excreted unchanged into the urine were in-line with the results of previous early-stage clinical studies.[8] A relatively large Vss (approximately 200 L), coupled with no binding to plasma proteins,[9] indicates that lesogaberan demonstrates extensive distribution in the tissues. Overall, the main conclusion of the present study is that the major route of elimination (clearance of parent compound in this case) is metabolism.
While further studies are planned to determine the metabolic pathway and metabolites of lesogaberan, it is apparent that the kidney represents the major route of clearance of drug-related material because 84% of administered radioactivity was excreted into the urine either as the parent compound or as water-soluble metabolite(s). CLR of lesogaberan following intravenous administration, however, only accounted for approximately 22% of CL. Plasma concentrations of total radioactivity were also higher than lesogaberan plasma concentrations during a significant proportion of the sampling period. Together, such findings indicate that metabolism is a presumed major route of elimination, followed by renal clearance of drug-related material, and further studies are planned to investigate the metabolic disposition of lesogaberan in terms of specific metabolic pathways. Further research may also be warranted to determine the effect of renal and hepatic impairment on the disposition of lesogaberan because this will be relevant for clinical use.
Previous studies in humans were used to determine the duration of the biologic sampling periods to ensure that virtually no radioactive lesogaberan remained to be excreted after the sampling was completed.[8] The oral dose (100 mg) was selected on the basis that this dose level is expected to be within the range of therapeutic doses.[7] The intravenous dose and infusion rate were set to yield a similar maximal plasma concentration but lower total systemic exposure, compared with the oral dose.
Both the oral and intravenous doses of lesogaberan were well tolerated, and neither dosing regimens raised any safety concerns. This finding, albeit limited by the small size of the study, is in-line with those of previous studies that reported that lesogaberan is well tolerated in oral doses of up to 500 mg.[8]
Conclusion
In this study in healthy subjects, lesogaberan administered orally was rapidly absorbed with high bioavailability, extensively metabolized (based on clearance of the parent compound), and the majority of the dose was excreted by the kidneys either as the parent compound or as water-soluble metabolites.
Acknowledgements
The authors acknowledge Deborah Sandall (Principle Investigator) and the research team at AstraZeneca (Macclesfield, UK and Mölndal, Sweden) for conducting the study; PRA International-Bioanalytical Laboratory B.V. (Assen, the Netherlands) and Covance Laboratories Limited (Harrogate, UK) for the analyses of biologic samples; and Andrew Stead and Simon Lancaster, from inScience Communications, a Wolters Kluwer business, who provided medical writing support funded by AstraZeneca.
The study was funded by AstraZeneca R & D, Mölndal, Sweden, the manufacturer of lesogaberan. All authors are present employees of AstraZeneca.
References
- 1.Lidums I, Lehmann A, Checklin H, et al. Control of transient lower esophageal sphincter relaxations and reflux by the GABAB agonist baclofen in normal subjects. Gastroenterology. 2000;118(1):7–13. doi: 10.1016/S0016-5085(00)70408-2. [DOI] [PubMed] [Google Scholar]
- 2.van Herwaarden MA, Samsom M, Rydholm H, et al. The effect of baclofen on gastro-oesophageal reflux, lower oesophageal sphincter function and reflux symptoms in patients with reflux disease. Aliment Pharmacol Ther. 2002;16(9):1655–62. doi: 10.1046/j.1365-2036.2002.01325.x. [DOI] [PubMed] [Google Scholar]
- 3.Lehmann A, Jensen JM, Boeckxstaens GE. GABAB receptor agonism as a novel therapeutic modality in the treatment of gastroesophageal reflux disease. Adv Pharmacol. 2010;58:287–313. doi: 10.1016/S1054-3589(10)58012-8. [DOI] [PubMed] [Google Scholar]
- 4.Lehmann A, Antonsson M, Holmberg AA, et al. (R)-(3-amino-2-fluoropropyl) phosphinic acid (AZD3355), a novel GABAB receptor agonist, inhibits transient lower esophageal sphincter relaxation through a peripheral mode of action. J Pharmacol Exp Ther. 2009;331(2):504–12. doi: 10.1124/jpet.109.153593. [DOI] [PubMed] [Google Scholar]
- 5.Boeckxstaens GE, Rydholm H, Lei A, et al. Effect of lesogaberan, a novel GABAB-receptor agonist, on transient lower oesophageal sphincter relaxations in male subjects. Aliment Pharmacol Ther. 2010;31(11):1208–17. doi: 10.1111/j.1365-2036.2010.04283.x. [DOI] [PubMed] [Google Scholar]
- 6.Boeckxstaens GE, Beaumont H, Mertens V, et al. Effects of lesogaberan on reflux and lower esophageal sphincter function in patients with gastroesophageal reflux disease. Gastroenterology. 2010;139(2):409–17. doi: 10.1053/j.gastro.2010.04.051. [DOI] [PubMed] [Google Scholar]
- 7.Boeckxstaens GE, Beaumont H, Hatlebakk JG, et al. A novel reflux inhibitor lesogaberan (AZD3355) as add-on treatment in patients with GORD with persistent reflux symptoms despite proton pump inhibitor therapy: a randomised placebo-controlled trial. Gut. In press [DOI] [PubMed]
- 8.Data on file . AstraZeneca. 2007. [Google Scholar]
- 9.Data on file . AstraZeneca. 2001. [Google Scholar]