

Highlights
► We hypothesize that neurodegeneration in anti-NGF is due to NGF/proNGF imbalance. ► We propose that the proNGF co- receptor sortilin contributes to the neurodegeneration. ► We analyzed anti-NGF mice crossed to sortilin knockout mice. ► Sortilin loss partially protected AD10 anti-NGF mice from neurodegeneration.
Keywords: proNGF, Sortilin, Alzheimer’s disease, Amyloid β, Tau, Choline acetyltransferase
Abstract
Sortilin is a member of the family of vacuolar protein sorting 10 protein domain receptors which has emerged as a co-receptor in cell death and neurodegeneration processes mediated by proneurotrophins.
Here we tested the possibility that sortilin deficiency interferes with behavioral and neuropathological endpoints in a chronic Nerve Growth factor (NGF)-deprivation model of Alzheimer’s disease (AD), the AD10 anti-NGF mouse. AD10 mice show cholinergic deficit, increased APP processing and tau hyper-phosphorylation, resulting in behavioral deficits in learning and memory paradigms assessed by novel object recognition and Morris water maze tests. Sort1−/− mice were crossed with AD10 anti-NGF mice and the neurodegenerative phenotype was studied. We found that the loss of sortilin partially protected AD10 anti-NGF mice from neurodegeneration. A protective effect was observed on non-spatial memory as assessed by novel object recognition, and histopathologically at the level of Aβ and BFCNs, while the phosphotau increase was unaltered by knocking out sortilin. We suggest that sortilin might be involved in different aspects of neurodegeneration in a complex way, supporting the view that sortilin functions in the CNS are broader than being a co-receptor in proneurotrophin and neurotrophin signaling.
1. Introduction
The neurotrophin NGF is synthesized from a precursor, proNGF, which displays biological activities distinct from those of mature NGF [1]. ProNGF induces neurodegeneration and cell death under acute and chronic situations of the nervous system upon binding to p75NTR and sortilin receptors [2].
We extensively characterized a mouse model [3,4] in which neurodegeneration is achieved by the expression of a recombinant antibody which selectively blocks the activity of mature NGF [5]. We hypothesized that an imbalance of the NGF to proNGF ratio would account for the neurodegeneration observed in the anti-NGF transgenic mice [6,7] and provided supporting evidence by crossing anti-NGF to p75NTR−/− mice [6,7].
In this study, using a similar approach, we explored the role of the pro-neurotrophin co-receptor sortilin in the progression of neurodegeneration in AD10 anti-NGF mice, a line of transgenic mice in which anti-NGF antibodies are obligatorily expressed in lymphocytes and therefore are initially only found in serum and later, after disruption of the blood brain barrier, also in the brain [8]. AD10 mice develop a central neurodegeneration characterized by cholinergic deficit, tau hyperphosphorylation, amyloid β (Aβ) accumulation derived from the altered processing of endogenous mouse APP, and behavioral deficits [8]. AD10 anti-NGF mice were crossed with Sort1−/− mice and the neurodegenerative phenotype was studied. We found that visual working memory and cholinergic deficit in the medial septum are fully rescued in aged mice, while β amyloid and hyperphosphorylated tau are mildly, or not at all, affected by sortilin deficiency.
2. Materials and methods
2.1. Animals
Sortilin-deficient mice were engineered by targeted gene deletion of 126 bp from exon 14 and 303 bp of the subsequent intron of sortilin [9]. Homozygous Sort1−/− mice were crossed with homozygous AD10 anti-NGF mice [8]. Heterozygous AD10×Sort1 +/− mice from the first crossing were interbred and sex and age matched wild type, AD10, Sort1−/− and AD10×Sort1−/− mice were used for analysis. Genotype was determined by standard PCR, using the oligonucleotides described in Supplementary methods. In total, 117 mice were analyzed in this study across three different age groups: 3 months (WT = 9; AD10 n = 15; Sort1−/− n = 10; AD10×Sort1−/− n = 14), 6 months (WT = 6; AD10 n = 11; Sort1−/− n = 6; AD10×Sort1−/− n = 12) and 12 months (WT = 9; AD10 n = 8; Sort1−/− n = 6; AD10×Sort1−/− n = 11).
2.2. Behavioral tests
Object recognition test was performed over three consecutive days as described [10]. A detailed description of the Morris water maze test has been provided in Supplementary methods.
2.3. Tissue collection and immunohistochemistry
After behavioral analysis, mice were anaesthetized with an excess of 2,2,2-tribromethanol (400 mg/kg) and intracardially perfused with a 4% solution of paraformaldehyde in PBS. Brains were processed for immunohistochemical analysis as described [3,11]. Primary antibodies concentration is provided in Supplementary methods.
2.4. Stereology
Stereological analysis on ChAT-immunoreactive neurons, number of Aβ clusters of dystrophic neurites, AT8-immunoreactive neurons in the hippocampus and lateral entorhinal cortex was performed as described [8,12].
2.5. Statistical analysis
Statistical analysis was performed using the SigmaSTAT program version 3.5 (Systat Software Inc., San Jose, CA). The alpha was set at 0.05 and a normality and equal variance test were first performed. One-way ANOVA or Kruskal Wallis ANOVA was used for multiple comparisons, followed by Bonferroni or Holm-Sidak post hoc tests.
3. Results
To determine the contribution of sortilin on the progressive neurodegeneration induced by anti-NGF antibodies, homozygous Sort1−/− mice were bred to homozygous AD10 anti-NGF mice and newborns were intercrossed. Littermates with the following genotypes were analyzed: wild type (WT), homozygous sortilin deficient animals (Sort1−/−), AD10 mice and AD10 mice homozygous for Sort1 null alleles (AD10×Sort1−/−). Mice were examined at 3, 6, and 12 months of age, corresponding to incipient, intermediate and full-blown neurodegeneration in AD10 mice, respectively [8].
3.1. Sortilin loss protects AD10 mice from late non-spatial memory but not from spatial memory deficits
Non-spatial memory was analyzed by the object recognition test. Mice from all genotypes spent the same time in exploring the two identical objects to which they were exposed during the sample phase of the test at all ages examined (Supplementary Fig. 1A–C, respectively at 3, 6 and 12 months of age). In the test phase, at 3 months of age, mice from all genotypes explored more the new object (Supplementary Fig. 1D, P < 0.05). At 6 months of age, AD10 displayed the expected memory deficit [8], as they explored equally the new and the familiar object (Fig 1A), while AD10×Sort1−/− mice, similarly to WT, and Sort1−/− mice, continued to explore more the new object (Fig. 1A, P < 0.05). Surprisingly, at 12 months of age the non-spatial memory deficit appeared also in Sort1−/− mice (Fig. 1B), while AD10×Sort1−/− mice continued to explore more the new object than the old one (Fig. 1B, P < 0.05). Next, we explored the possibility that loss of sortilin may also affect spatial memory deficits in wild type and AD10 mice, using the Morris water maze test. Mice from the different genotypes swam with the same speed at all tested ages, with the exception of 6 month-old Sort1−/− mice which swam faster than the other groups of mice (Supplementary Fig. 2A, C and E). At 3 and 6 months of age, mice from all genotypes learned to recognize the location of the platform (Supplementary Fig. 2B,D), while at 12 months of age, AD10 mice showed, as previously documented [8], a deficit in learning where the platform was (Fig. 1C, P < 0.05). This deficit was partially rescued in AD10×Sort1−/− mice (Fig. 1C). At all ages, AD10 mice showed a spatial memory deficit during the probe phase (Fig. 1D–F). Sort1−/− mice did not show spatial memory deficits at 3 months of age (Fig. 1D), while an impairment was observed in older mice (Fig. 1E and F, respectively at 6 and 12 months of age). Sortilin deficiency was insufficient to protect AD10 mice from spatial memory deficit, since, at 3 and 12 months of age, AD10×Sort1−/− showed an impaired memory of platform location (Fig. 1D and F), while 6 months old AD10×Sort1−/− mice showed no spatial memory deficit (Fig. 1E). We conclude that sortilin loss, per se, determines memory deficits (Supplementary Table 1), while, in the context of NGF deprivation, it fully protects from non-spatial memory deficits and, in a more limited and time restricted way, from spatial memory deficits.
Fig. 1.

Effects of sortilin deficiency on memory deficits. (A) At 6 months of age AD10 transgenic mice show a deficit in non-spatial working memory which is absent in Sort1−/− and AD10×Sort1−/− mice, (B) At 12 months of age both AD10 and Sort1−/− mice showed a memory deficit which was prevented in AD10×Sort1−/− mice, (C) During the acquisition phase of the Morris water maze test performed at 12 months of age, AD10 mice did not learn as well as WT mice or Sort1−/− mice while AD10×Sort1−/− showed an intermediate acquisition pattern and (D–F) During the probe phase, only WT mice were able to remember the target quadrant at all ages, while AD10 mice showed always a spatial memory impairment. The deficit appeared also in 6 (E) and 12 (F) month-old Sort1−/− mice while the crossing of AD10 to Sort1−/− mice only temporally protected from spatial memory deficit at 6 months of age (E). In A, B bars are representative of mean ± SEM. ∗P < 0.05 new versus old object. In C–F Bars and points are representative of mean ± SEM. In C, ∗P < 0.05 versus WT mice. In D–F, ∗P < 0.05 time in target quadrant versus adjacent and opposite quadrants.
3.2. Genetic inactivation of Sortilin prevents the increase of Aβ immunoreactive dystrophic neurites in aged AD10 mice
To examine the influence of sortilin gene inactivation on the amyloid pathology in AD10 mice, we evaluated the number of clusters of dystrophic neurites immunoreactive for Aβ/APP, which appear in the 6 months-old AD10 hippocampus [8]. The number of Aβ/APP clusters was not significantly different in 3 months old AD10, Sort1−/− or AD10×Sort1−/− mice, with respect to WT (Fig. 2A). 6 months old AD10 mice showed the expected increase in the number of Aβ/APP clusters compared to WT (Fig. 2A, P < 0.05 and [8]), Sort1−/− have the same number of Aβ/APP clusters as WT mice, while AD10×Sort1−/− mice showed an equivalent number of Aβ/APP clusters to AD10 mice (Fig. 2A). However, at 12 months of age, sortilin loss in AD10 mice resulted in a twofold decrease in the number of Aβ/APP clusters compared to AD10 mice (Fig. 2A, B and D). Surprisingly, at 12 months, Sort1−/− mice showed an equivalent number of Aβ/APP immunoreactive dystrophic neurites to age-matched AD10 mice (Fig. 2A–C). Thus, inactivation of sortilin in AD10 mice delays the amyloidogenic process, determining a marked protection at 12 months of age. However, at this age, sortilin loss per se, appears to be amyloidogenic.
Fig. 2.
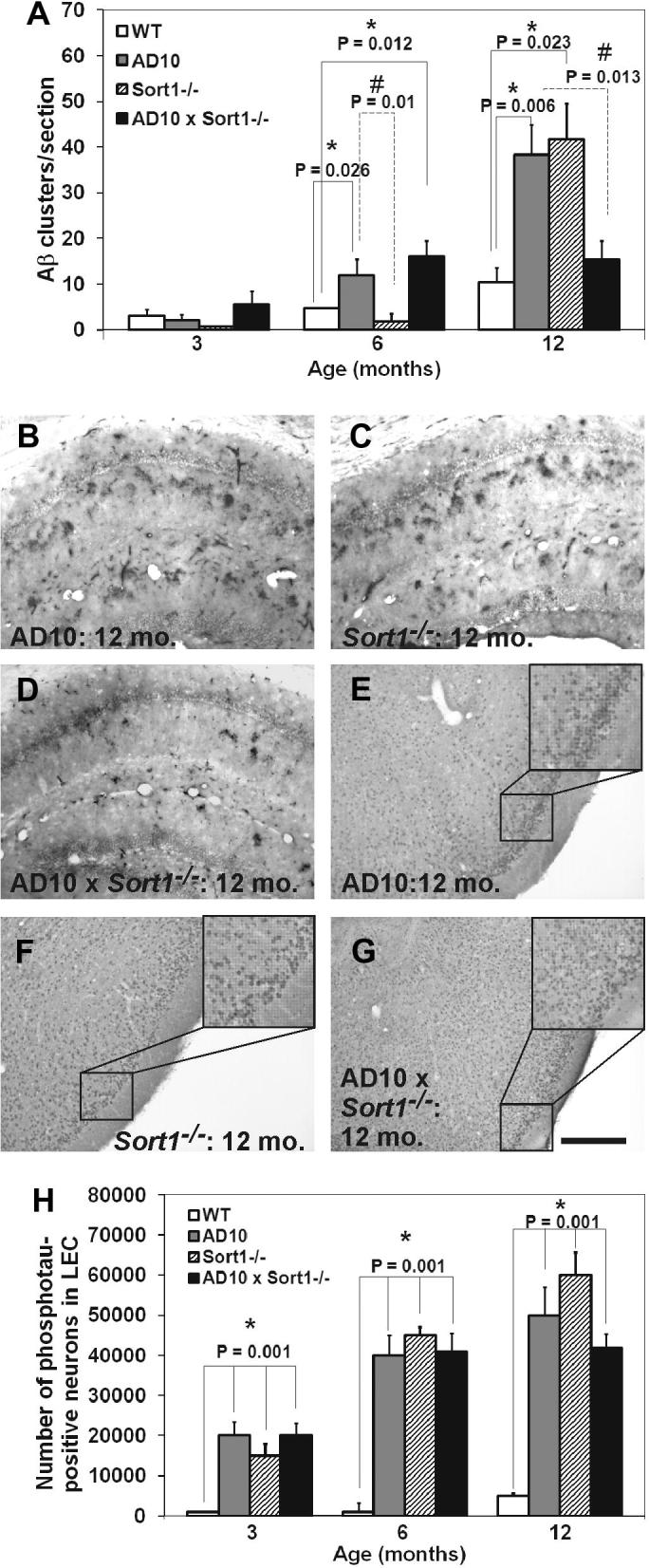
Measurements of APP/Aβ clusters of dystrophic neurites in the hippocampus and of phosphorylated tau in lateral entorhinal cortex. (A) Quantification of the number of clusters at 3, 6 and 12 months of age. (B–D) Qualitative images of APP/Aβ amyloid clusters in (B) AD10, (C) Sort1−/− and (D) AD10×Sort1−/− hippocampus at 12 months of age. (E–G) Qualitative images of phosphotau-imunoreactive neurons in the lateral entorhinal cortex from (E) AD10 (E) Sort1−/− and (G) AD10×Sort1−/− mice at 12 months of age. (H) Quantification of the number of phospho-immunoreactive neurons at 3, 6 and 12 months of age in the LEC. All transgenic mice show an increase of phospho-tau immunoreactive neurons from 3 months of age with respect to WT mice. Bars are representative of mean ± SEM. ∗P < 0.05 versus WT mice. #P < 0.05 versus AD10 and Sort1−/− mice. Scale bar = 250 μm.
3.3. Loss of sortilin does not prevent mislocalization and increase of phosphorylated tau
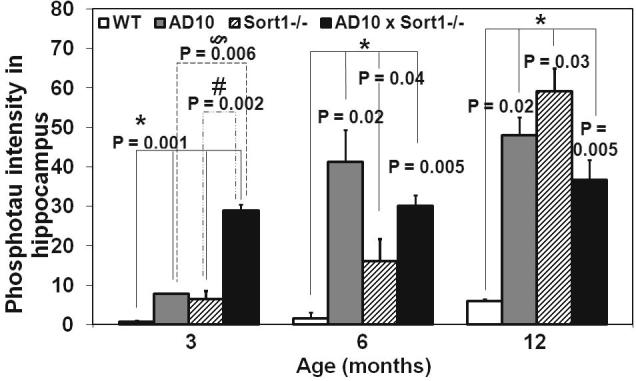
The effects of sortilin loss on phosphorylated tau was analyzed by immunohistochemistry with the phospho-tau specific antibody mAb AT8. In 3 months old AD10 mice, AT8 labels neurons in the lateral entorhinal cortex (LEC) (Fig. 2H, P < 0.05 versus WT mice) and, to a lesser extent, in the hippocampus (Supplementary Fig. 3), with a prominent somatodendritic localization (not shown). At the same age, Sort1−/− and AD10×Sort1−/− mice showed an equivalent number of AT8-immunoreactive neurons in LEC (Fig. 2H), while, in the hippocampus, sortilin deficiency in AD10 mice determined a threefold increase in the number of AT8-immunoreactive neurons (Supplementary Fig. 3). With age, the number of AT8-immunoreactive hippocampal neurons increases in AD10 and, with a delayed time-course, in Sort1−/− mice (Fig. 2E, F and H, P < 0.05 versus WT mice; Supplementary Fig. 3). Crossing of AD10 to Sort1−/− mice did not block the increase of AT8-immunoreactive neurons in LEC and hippocampus, at all ages (Fig. 2A, G and H, P < 0.05; Supplementary Fig. 3). Thus, Sort1−/− mice show an increased expression of phospho-tau, and loss of sortilin in the AD10 background does not prevent the increase in somatodendritic phosphorylated tau in different brain regions of old mice.
3.4. Sortilin deficiency partially rescues cholinergic deficit in AD10 mice
The effects of sortilin deficiency on the expression of choline acetyltranferase (ChAT), in neurons of the nucleus basalis of Meynert (NBM) and of the medial septum/diagonal band of Broca (MS/DBH), were studied by ChAT immunohistochemistry. In the NBM of AD10, Sort1−/− and AD10×Sort1−/− mice, a significant decrease in the number of ChAT-immunoreactive neurons was first observed at 6 months of age (Fig. 3A and C; P < 0.05 versus WT mice). This decrease persisted in 12-month old AD10 mice (Fig. 3A and C, P < 0.05 versus WT mice) but was absent in age-matched Sort1−/− mice (Fig. 3A and C) and partially rescued in AD10×Sort1−/− mice (Fig. 3A and C). In the MS/DBH of AD10, as well as of Sort1−/− and AD10×Sort1−/− mice, cholinergic deficits appear earlier than in NBM, already at 3 months (Fig. 3B, P < 0.05 versus WT mice). 6 months old AD10 and AD10×Sort1−/− mice showed a further decrease in the number of MS/DBH cholinergic neurons (Fig. 3B, P < 0.05 versus WT mice), while Sort1−/− mice show normal levels (Fig. 3B). Twelve months old AD10 mice still showed a decreased number of ChAT-immunoreactive neurons (Fig. 3B and D, P < 0.05 versus WT mice, that was partially rescued in AD10×Sort1−/− mice and normal absent in Sort1−/− (Fig. 3B and D). We concluded that sortilin loss partially rescues AD10 mice from cholinergic deficit in the BF nucleus at late stages of neurodegeneration.
Fig. 3.

Measurements of the number of cholinergic neurons in the NBM and MS/DBH. (A, B) Quantification of the number of ChAT-immunoreactive neurons at 3, 6 and 12 months of age in (A) NBM and (B) MS/DBH, (C) qualitative images of ChAT-immunoreactive neurons in NBM from WT, AD10, Sort1−/− and AD10×Sort1−/− mice at 12 months of age, (D) qualitative images of ChAT-imunoreactive neurons in MS/DBH from WT, AD10, Sort1−/− and AD10×Sort1−/− mice at 12 months of age, (E) quantification of the number of TrkA-immunoreactive neurons at 12 months of age in MS/DBH and (F) Qualitative images of TrkA-immunoreactive neurons in MS/DBH from WT, AD10, Sort1−/− and AD10×Sort1−/− mice at 12 months of age. Bars are representative of mean ± SEM. ∗P < 0.05 versus WT mice. #P < 0.05 versus AD10 mice. Scale bar in B = 250 μm. Scale bars in C, F = 250 μm.
3.5. Sortilin deficiency restores TrkA expression in the MS/DBH of AD10 mice
The expression of the NGF receptor TrkA is essential for neurotrophic actions of NGF on cholinergic neurons [13]. TrkA immunohistochemistry showed that the number of TrkA-immunoreactive neurons was strongly reduced in AD10 mice with respect to WT mice (Fig. 3E and F; P < 0.05), whereas this number was reverted in AD10×Sort1−/− mice (Fig. 3E and F; P < 0.05). Thus, we concluded that sortilin might influence the expression of TrkA on cholinergic neurons, and, in turn, it might restore the correct neurotrophic activity of NGF.
4. Discussion
Sortilin is an important regulator of neuronal survival and function [14], controlling the release of proneurotrophic factors and acting as a as co-receptor for them, together with p75NTR, [15–17]. A role for proNGF as an inducer of neurodegeneration was proposed [6] on the basis of the neurodegeneration observed in a line of anti-NGF mice [4,17], in which the selective neutralization of mature NGF with respect to proNGF leads to an NGF/proNGF imbalance.
The characterization of sortilin deficient mice has increased the knowledge of its functions at peripheral and central level. Sortilin loss abolishes apoptosis in mouse retina during development [9], protects lesioned corticospinal neurons against cell death [9], aggravates Trk receptor phenotypes present in p75NTR deficient mice [18], and increases embryonic lethality and sympathetic neuropathy in TrkA−/− heterozygous mice [18]. Only few reports studied Sort1−/− mice at the level of the Central Nervous System, with the exception of the effects of sortilin deficiency on corticospinal lesions [9] We found that Sort1−/− mice show an early decrease of the number of ChAT-immunoreactive neurons in the BF and an increased expression of phosphorylated tau and Aβ in cortical regions and in the hippocampus (Supplementary Table 1). These findings match with the overall observed decrease in memory functions, such as non-spatial and spatial memory in Sort1−/− mice (Supplementary Table 1). Thus, our first conclusion is that sortilin might play a neuroprotective role in the BF, hippocampus and cortical regions. Consistently, sortilin mRNA is decreased in AD11 anti-NGF mice [19]. The increase of Aβ in aged Sort1−/− mice could be explained by the broad actions of sortilin, acting not only as a receptor for proNGF but also for neurotensin [20] and progranulin [21], both of which are suggested to play a neuroprotective role [22,23]. Neurotensin has been found to be decreased in several brain areas of human AD brains [24–26], and neurotensin analogues rescue memory deficits [27]. Both null and missense progranulin mutations have been observed in AD patients [28,29]. Thu, loss of sortilin would mimick a decreased signaling of neurotensin and/or progranulin, explaining the observed neurodegenerative processes in Sort1−/− mice.
The second conclusion is that loss of sortilin does not completely protect AD10 anti-NGF mice from neurodegeneration. A mild protective effect can be observed at the level of Aβ and BFCNs, in the latter case probably linked to the re-expression of TrkA on these cells, while the increase of phosphotau in AD10 mice is not affected at all by knocking out sortilin. The partial rescue of Aβ in AD10×Sort1−/− mice might appear in contradiction with the increase found in Sort1−/− mice. We interpret this protective role as evidence for the prevalence of proNGF signaling in the Aβ formation in AD10 mice, in line with what we observed crossing AD10 to p75NTR−/− [7]. This reinforces the view that proNGF signaling through p75NTR and sortilin, in a context of neurotrophic imbalance or deficit as in AD10 mice, is pro-amyloidogenic. The lack of effects on phosphorylated tau, in presence of a rescue of Aβ might appear in contradiction with the serial “amyloid hypothesis”, whereby elevation of Aβ precedes and drives other AD features, including hyperphosphorylation of tau [30] and favour a dual pathway model, whereby Aβ and tau would be downstream to a common upstream driver for neurodegeneration [31]. Our results suggest that sortilin may be part of the recently postulated upstream driver, represented by NGF/proNGF signaling [32].
More generally, we conclude that in AD10 mice, the role of sortilin as a mediator of proNGF actions in different brain areas is not as essential as that of p75NTR [7], possibly due to the pleiotropic actions of sortilin in the CNS or to the fact that sortilin activity might be substituted, in a subset of cell populations, by other members of the VPS10P receptor family [14,33]. Indeed, we must also keep into account our unexpected finding on the Sort1−/−, phenotype, showing that, in the CNS, sortilin can be neuroprotective. This conclusion should be put in the context of studies showing that sortilin, besides being a partaker to cell death (including BFCNs) induced by all the different proneurotrophins, is required for the cleavage of APP to Aβ by BACE1 and for the toxic actions of Aβ oligomers in cortical neurons [34]. On the other hand, it has been recently reported that in neurons lacking sortilin the production of sAPPα [35] and the catabolism of the amyloid peptide by APOE are decreased [36]. However, most of these experiments were performed in cell cultures, thus extrapolating sortilin from the complexity of its role in the brain. Thus, sortilin has been recently found to be required for the axonal transport of Trk neurotrophin receptors, leading to an enhancement of neurotrophic signaling [18,34]. Thus, the expectation of selectively knocking-out proNGF signaling by removing sortilin might be too simplistic, given the complexities of sortilin functions in vivo, which are more multifaceted than expected. Further studies in different transgenic models of AD are required to dissect further the relationship and significance of the different pathways in which this important receptor is involved.
Acknowledgments
The authors thank Prof. Luciano Domenici (University of L’Aquila, Italy) for support and critical discussion. This work was funded by EU FP6 MEMORIES Project (no 037831) to A.C. and A.N. The Lundbeck Foundation to A.N., and by Fondazione Roma and Telethon (grant no. GGP05234) to A.C.
Appendix A. Supplementary data
Supplementary Figure 1.

Supplementary Figure 2.

Supplementary Figure 3.
Effects of Sortilin deficiency on memory deficits: object recognition test.
Effects of sortilin deficiency on memory deficits: Morris water maze test.
Measurements of phosphorylated tau in hippocampus as determined by immunostaining with mAb AT8.
References
- 1.Lee R., Kermani P., Teng K.K., Hempstead B.L. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945–1948. doi: 10.1126/science.1065057. [DOI] [PubMed] [Google Scholar]
- 2.Skeldal S., Matusica D., Nykjaer A., Coulson E.J. Proteolytic processing of the p75 neurotrophin receptor: A prerequisite for signalling?: Neuronal life, growth and death signalling are crucially regulated by intra-membrane proteolysis and trafficking of p75(NTR) Bioessays. 2011;33:614–625. doi: 10.1002/bies.201100036. [DOI] [PubMed] [Google Scholar]
- 3.Capsoni S., Ugolini G., Comparini A., Ruberti F., Berardi N., Cattaneo A. Alzheimer-like neurodegeneration in aged antinerve growth factor transgenic mice. Proc. Natl. Acad. Sci. USA. 2000;97:6826–6831. doi: 10.1073/pnas.97.12.6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruberti F., Capsoni S., Comparini A., Di Daniel E., Franzot J., Gonfloni S., Rossi G., Berardi N., Cattaneo A. Phenotypic knockout of nerve growth factor in adult transgenic mice reveals severe deficits in basal forebrain cholinergic neurons, cell death in the spleen, and skeletal muscle dystrophy. J. Neurosci. 2000;20:2589–2601. doi: 10.1523/JNEUROSCI.20-07-02589.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cattaneo A., Capsoni S., Paoletti F. A new generation of non invasive NGF-based therapies for Alzheimer’s disease. In: Martinez A, editor. Emerging drugs Targets for Alzheimer’s disease. RCS Publishing; 2009. [Google Scholar]
- 6.Capsoni S., Cattaneo A. On the molecular basis linking nerve growth factor (NGF) to Alzheimer’s disease. Cell. Mol. Neurobiol. 2006;26:619–633. doi: 10.1007/s10571-006-9112-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Capsoni S., Tiveron C., Vignone D., Amato G., Cattaneo A. Dissecting the involvement of tropomyosin-related kinase A and p75 neurotrophin receptor signaling in NGF deficit-induced neurodegeneration. Proc. Natl. Acad. Sci. USA. 2010;107:12299–12304. doi: 10.1073/pnas.1007181107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Capsoni S., Tiveron C., Amato G., Vignone D., Cattaneo A. Peripheral neutralization of nerve growth factor induces immunosympathectomy and central neurodegeneration in transgenic mice. J. Alzheimers Dis. 2010;20:527–546. doi: 10.3233/JAD-2010-091357. [DOI] [PubMed] [Google Scholar]
- 9.Jansen P., Giehl K., Nyengaard J.R., Teng K., Lioubinski O., Sjoegaard S.S., Breiderhoff T., Gotthardt M., Lin F., Eilers A., Petersen C.M., Lewin G.R., Hempstead B.L., Willnow T.E., Nykjaer A. Roles for the pro-neurotrophin receptor sortilin in neuronal development, aging and brain injury. Nat. Neurosci. 2007;10:1449–1457. doi: 10.1038/nn2000. [DOI] [PubMed] [Google Scholar]
- 10.Capsoni S., Marinelli S., Ceci M., Vignone D., Amato G., Malerba F., Paoletti F., Meli G., Viegi A., Pavone F., Cattaneo A. Intranasal “painless” human nerve growth factor slows amyloid neurodegeneration and prevents memory deficits in App X PS1 mice. PLoS One. 2012;7:e37555. doi: 10.1371/journal.pone.0037555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Capsoni S., Giannotta S., Cattaneo A. Beta-amyloid plaques in a model for sporadic Alzheimer’s disease based on transgenic anti-nerve growth factor antibodies. Mol. Cell. Neurosci. 2002;21:15–28. doi: 10.1006/mcne.2002.1163. [DOI] [PubMed] [Google Scholar]
- 12.Capsoni S., Covaceuszach S., Ugolini G., Spirito F., Vignone D., Stefanini B., Amato G., Cattaneo A. Delivery of NGF to the brain: Intranasal versus ocular administration in Anti-NGF transgenic mice. J. Alzheimers Dis. 2009;16:371–388. doi: 10.3233/JAD-2009-0953. [DOI] [PubMed] [Google Scholar]
- 13.Mobley W.C., Rutkowski J.L., Tennekoon G.I., Gemski J., Buchanan K., Johnston M.V. Nerve growth factor increases choline acetyltransferase activity in developing basal forebrain neurons. Brain Res. 1986;387:53–62. doi: 10.1016/0169-328x(86)90020-3. [DOI] [PubMed] [Google Scholar]
- 14.Nykjaer A., Willnow T.E. Sortilin: a receptor to regulate neuronal viability and function. Trends Neurosci. 2012;35:261–270. doi: 10.1016/j.tins.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 15.Gliemann J., Hermey G., Nykjaer A., Petersen C.M., Jacobsen C., Andreasen P.A. The mosaic receptor sorLA/LR11 binds components of the plasminogen-activating system and platelet-derived growth factor-BB similarly to LRP1 (low-density lipoprotein receptor-related protein), but mediates slow internalization of bound ligand. Biochem. J. 2004;381:203–212. doi: 10.1042/BJ20040149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teng H.K., Teng K.K., Lee R., Wright S., Tevar S., Almeida R.D., Kermani P., Torkin R., Chen Z.Y., Lee F.S., Kraemer R.T., Nykjaer A., Hempstead B.L. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J. Neurosci. 2005;25:5455–5463. doi: 10.1523/JNEUROSCI.5123-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tauris J., Gustafsen C., Christensen E.I., Jansen P., Nykjaer A., Nyengaard J.R., Teng K.K., Schwarz E., Ovesen T., Madsen P., Petersen C.M. Proneurotrophin-3 may induce Sortilin-dependent death in inner ear neurons. Eur. J. Neurosci. 2011;33:622–631. doi: 10.1111/j.1460-9568.2010.07556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vaegter C.B., Jansen P., Fjorback A.W., Glerup S., Skeldal S., Kjolby M., Richner M., Erdmann B., Nyengaard J.R., Tessarollo L., Lewin G.R., Willnow T.E., Chao M.V., Nykjaer A. Sortilin associates with trk receptors to enhance anterograde transport and neurotrophin signaling. Nat. Neurosci. 2011;14:54–61. doi: 10.1038/nn.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.D’Onofrio M., Arisi I., Brandi R., Di Mambro A., Felsani A., Capsoni S., Cattaneo A. Early inflammation and immune response mRNAs in the brain of AD11 anti-NGF mice. Neurobiol. Aging. 2011;32:1007–1022. doi: 10.1016/j.neurobiolaging.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 20.Mazella J., Zsurger N., Navarro V., Chabry J., Kaghad M., Caput D., Ferrara P., Vita N., Gully D., Maffrand J.P., Vincent J.P. The 100-kDa neurotensin receptor is gp95/sortilin, a non-G-protein-coupled receptor. J. Biol. Chem. 1998;273:26273–26276. doi: 10.1074/jbc.273.41.26273. [DOI] [PubMed] [Google Scholar]
- 21.Hu F., Padukkavidana T., Vaegter C.B., Brady O.A., Zheng Y., Mackenzie I.R., Feldman H.H., Nykjaer A., Strittmatter S.M. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68:654–667. doi: 10.1016/j.neuron.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giordano V., Peluso G., Iannuccelli M., Benatti P., Nicolai R., Calvani M. Systemic and brain metabolic dysfunction as a new paradigm for approaching Alzheimer’s dementia. Neurochem. Res. 2007;32:555–567. doi: 10.1007/s11064-006-9125-8. [DOI] [PubMed] [Google Scholar]
- 23.Pickford F., Marcus J., Camargo L.M., Xiao Q., Graham D., Mo J.R., Burkhardt M., Kulkarni V., Crispino J., Hering H., Hutton M. Progranulin is a chemoattractant for microglia and stimulates their endocytic activity. Am. J. Pathol. 2011;178:284–295. doi: 10.1016/j.ajpath.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrier I.N., Cross A.J., Johnson J.A., Roberts G.W., Crow T.J., Corsellis J.A., Lee Y.C., O’Shaughnessy D., Adrian T.E., McGregor G.P. Neuropeptides in Alzheimer type dementia. J. Neurol. Sci. 1983;62:159–170. doi: 10.1016/0022-510x(83)90196-x. [DOI] [PubMed] [Google Scholar]
- 25.Benzing W.C., Mufson E.J., Jennes L., Armstrong D.M. Reduction of neurotensin immunoreactivity in the amygdala in Alzheimer’s disease. Brain Res. 1990;537:298–302. doi: 10.1016/0006-8993(90)90372-i. [DOI] [PubMed] [Google Scholar]
- 26.Nemeroff C.B., Kizer J.S., Reynolds G.P., Bissette G. Neuropeptides in Alzheimer’s disease: A postmortem study. Regul. Pept. 1989;25:123–130. doi: 10.1016/0167-0115(89)90254-1. [DOI] [PubMed] [Google Scholar]
- 27.Azmi N., Norman C., Spicer C.H., Bennett G.W. Effects of a neurotensin analogue (PD149163) and antagonist (SR142948A) on the scopolamine-induced deficits in a novel object discrimination task. Behav. Pharmacol. 2006;17:357–362. doi: 10.1097/01.fbp.0000224382.63744.20. [DOI] [PubMed] [Google Scholar]
- 28.Brouwers N., Sleegers K., Engelborghs S., Maurer-Stroh S., Gijselinck I., van der Zee J., Pickut B.A., Van den Broeck M., Mattheijssens M., Peeters K., Schymkowitz J., Rousseau F., Martin J.J., Cruts M., De Deyn P.P., Van Broeckhoven C. Genetic variability in progranulin contributes to risk for clinically diagnosed Alzheimer disease. Neurology. 2008;71:656–664. doi: 10.1212/01.wnl.0000319688.89790.7a. [DOI] [PubMed] [Google Scholar]
- 29.Cortini F., Fenoglio C., Guidi I., Venturelli E., Pomati S., Marcone A., Scalabrini D., Villa C., Clerici F., Dalla Valle E., Mariani C., Cappa S., Bresolin N., Scarpini E., Galimberti D. Novel exon 1 progranulin gene variant in Alzheimer’s disease. Eur. J. Neurol. 2008;15:1111–1117. doi: 10.1111/j.1468-1331.2008.02266.x. [DOI] [PubMed] [Google Scholar]
- 30.Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 31.Small S.A., Duff K. Linking abeta and tau in late-onset Alzheimer’s disease: a dual pathway hypothesis. Neuron. 2008;60:534–542. doi: 10.1016/j.neuron.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Capsoni S., Brandi R., Arisi I., D’Onofrio M., Cattaneo A. A dual mechanism linking NGF/proNGF imbalance and early inflammation to Alzheimer’s disease neurodegeneration in the AD11 anti-NGF mouse model. CNS Neurol. Disord. Drug Targets. 2011;10:635–647. doi: 10.2174/187152711796235032. [DOI] [PubMed] [Google Scholar]
- 33.Pallesen L.T., Vaegter C.B. Sortilin and SorLA regulate neuronal sorting of trophic and dementia-linked proteins. Mol. Neurobiol. 2012;45:379–387. doi: 10.1007/s12035-012-8236-2. [DOI] [PubMed] [Google Scholar]
- 34.Fortress A.M., Buhusi M., Helke K.L., Granholm A.C. Cholinergic degeneration and alterations in the TrkA and p75NTR balance as a result of pro-NGF injection into aged rats. J. Aging Res. 2011;2011:460543. doi: 10.4061/2011/460543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gustafsen C., Glerup S., Pallesen L.T., Olsen D., Andersen O.M., Nykjær A., Madsen P., Petersen C.M. Sortilin and SorLA display distinct roles in processing and trafficking of amyloid precursor protein. J. Neurosci. 2013;33:64–71. doi: 10.1523/JNEUROSCI.2371-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carlo A.S., Gustafsen C., Mastrobuoni G., Nielsen M.S., Burgert T., Hartl D., Rohe M., Nykjaer A., Herz J., Heeren J., Kempa S., Petersen C.M., Willnow T.E. The pro-neurotrophin receptor sortilin is a major neuronal APOE receptor for catabolism of amyloid-peptide in the brain. J. Neurosci. 2013;33:358–370. doi: 10.1523/JNEUROSCI.2425-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effects of Sortilin deficiency on memory deficits: object recognition test.
Effects of sortilin deficiency on memory deficits: Morris water maze test.
Measurements of phosphorylated tau in hippocampus as determined by immunostaining with mAb AT8.