

Abstract
Background and Objectives
GLPG0259 is a small-molecule inhibitor of mitogen-activated protein kinase-activated protein kinase 5 (MAPKAPK5), a kinase enzyme that plays a role in important inflammatory pathways. The main objectives of the phase I clinical studies in early development were to characterize the pharmacokinetics, tolerability, and safety of GLPG0259 in healthy subjects, including the development of a solid dosage form (free-base pellets and fumarate salt capsules) and the potential for interaction of GLPG0259 with methotrexate.
Subjects and Methods
Four phase I studies were initiated. Study 1 was a randomized, double-blind, placebo-controlled study to evaluate the safety, tolerability, and pharmacokinetics of single ascending doses (1.5–150 mg) and multiple oral doses (20 and 50 mg once daily) of GLPG0259 in healthy male subjects (n = 34). Study 2 was a randomized, double-blind, placebo-controlled study to evaluate the safety, tolerability, and pharmacokinetics of oral multiple ascending doses of GLPG0259 (25–75 mg once daily) given for 14 days to healthy male subjects, and to get preliminary information on the potential pharmacokinetic interaction between GLPG0259 and methotrexate (n = 24). Studies 3 and 4 were open-label, randomized, crossover studies to compare the oral bioavailability of two solid dosage forms of GLPG0259 (a capsule) relative to an oral solution after a 100 mg or 50 mg single dose and to evaluate the effect of food on these formulations (n = 12 for each study).
Main Outcome Measures
The non-compartmental pharmacokinetic parameters for plasma concentrations of GLPG0259 were determined, and a population pharmacokinetic model of GLPG0259 was developed to support the planning of the number and timing of the sparse samples to be taken per patient in the phase II study. Safety and tolerability data are also summarized.
Results
The absorption of GLPG0259 was slow, with a decrease in the absorption rate with increasing dose, and there was decreased elimination, with an apparent terminal elimination half-life of 26.0 hours. On the basis of statistical analysis of variance, the exposure to GLPG0259 increased in proportion to the dose over a 30–150 mg single-dose range and a 25–75 mg repeated-dose range. Between- and within-subject variability in GLPG0259 pharmacokinetics was low/moderate (coefficient of variation [CV] 16–30%). After once-daily repeated dosing, steady-state plasma concentrations were reached at between 5 and 8 dosing days, which is consistent with the long apparent elimination half-life of GLPG0259. Food increased the bioavailability of GLPG0259 given in a solid dosage form. Co-administration of GLPG0259 with a single dose of methotrexate 7.5 mg did not result in any change in the pharmacokinetic profiles of either GLPG0259 or methotrexate.
Conclusion
In summary, the investigation of safety/tolerability and pharmacokinetics in the early development phase showed that single and repeated doses of GLPG0259 were safe and well tolerated. The most common adverse event reported was mild gastrointestinal discomfort. The pharmacokinetics characterized in healthy male subjects showed no major obstacles and supports a once-daily oral regimen in patients.
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune inflammatory and joint degenerative disease, which affects almost 1% of the adult population worldwide, with onset classically occurring between the ages of 30 and 50 years, and a higher prevalence in women. The disease is characterized by pain, stiffness, and restricted mobility due to persistent symmetrical inflammation of the synovial membranes of multiple joints, which ultimately results in irreversible damage of the joint cartilage and bone.[1–3]
Development of the disease involves an inflammatory response of the synovial membrane, accompanied by infiltration of a variety of immune cells, which leads to the build-up and maintenance of a cytokine network. One of the cytokines central to this network is tumor necrosis factor (TNF), as is clearly demonstrated by the clinical success of TNF blockers in treating RA. TNF and other proinflammatory cytokines contribute to cartilage and bone erosion by inducing release of degradative enzymes, such as matrix metalloproteinases (MMPs), and stimulating the release of receptor-activated NFκB-ligand (RANKL), which triggers differentiation of hematopoeitic cells into bone-resorbing osteoclasts. When left untreated, the disease leads to significant disability associated with high economic costs.
In recent years, the therapeutic management of patients with RA has undergone major evolution. Up to 10 years ago, therapeutic approaches relied on synthetic disease-modifying anti-rheumatic drugs (DMARDs) such as methotrexate and sulphasalazine, which had only partial clinical benefit and were associated with significant toxicity. A considerable advance in the effective treatment of RA came from the introduction of the biologic therapeutics that neutralize cytokines or their receptors (TNFα and interleukin [IL]-6) or that inhibit cellular activation (B-cell or T-cell activation).[4,5]
However, because of the high production costs, inconvenience of parenteral administration, increased risk of infections, and potential immunogenicity of biologics, there is still a need for less expensive and orally administered drugs.[4] Hence, the development of small-molecule inhibitors targeting disease-relevant signal transduction pathways is being pursued by various companies.[6]
GLPG0259 is a small-molecule inhibitor of mitogen-activated protein kinase–activated protein kinase 5 (MAPKAPK5), a kinase enzyme that plays a role in important inflammatory pathways. Inhibition of MAPKAPK5 with GLPG0259 represents a novel mechanism of action in the treatment of RA. MAPKAPK5 belongs to a family of mitogen-activated protein kinases that play an important role in several cellular processes, including inflammation, proliferation, and differentiation. MAPKAPK5 is involved in a transduction pathway in RA patients that ultimately leads to secretion of catabolic enzymes such as MMP1, which cause damage to the bone and cartilage in these patients. GLPG0259 is a potent inhibitor of MAPKAPK5, and in vitro it reduces the release of several mediators of inflammation and bone degradation, such as MMP1, MMP13, TNF, and IL6. After oral administration in mice, GLPG0259 reduces paw inflammation as well as bone destruction in the mouse collagen-induced arthritis (CIA) model of human RA at a dose of 1 mg/kg and higher. Even in mice with late-stage CIA disease, GLPG0259 reduces inflammation and bone destruction.
The main objectives of the phase I clinical studies in early development were to characterize the pharmacokinetics, tolerability, and safety of GLPG0259 in healthy subjects, including the development of a solid dosage form and the potential for interaction of GLPG0259 with methotrexate (the anchor drug in RA patients).
However, an exploratory phase II study in a small number of RA patients with an insufficient response to methotrexate showed no significant clinical benefit of GLPG0259 compared with placebo, and the development of GLPG0259 was discontinued (Westhovens R et al., unpublished data).[7]
Subjects and Methods
All studies were conducted in accordance with accepted standards for the protection of subject safety and welfare, and the principles of the Declaration of Helsinki and its amendments, and were in compliance with Good Clinical Practice. Protocols and informed consents were approved by the Ziekenhuis Netwerk Antwerpen (ZNA) Institutional Review Board (Antwerp, Belgium). All healthy participants gave written informed consent prior to study initiation.
Study Designs
Study 1: First-in-Human, Single Ascending and Multiple Oral Doses
This was a randomized, double-blind, placebo-controlled, single-center study to evaluate the safety, tolerability, and pharmacokinetics of single ascending and multiple oral doses of GLPG0259 in healthy subjects. Eligible subjects (aged 18–50 years, body mass index [BMI] 18–30 kg/m2) were in good health with no clinically significant deviation from normal in terms of medical history, physical examinations, electrocardiograms (ECGs), or clinical laboratory determinations. Subjects were excluded from the study if they had medical history of abnormal platelet function or a history of a current immunosuppressive condition.
The study was divided into two parts:
Part 1 (n = 16 subjects): Single escalating dose intake of GLPG0259 or matching placebo (1.5–150 mg) was studied in two cohorts of eight subjects (active or placebo in a 3 : 1 ratio) in an alternating panel design. There was a minimum 7-day washout period between treatments for each subject within a cohort and at least 3 days for each dose increment between cohorts.
Part 2 (n = 18 subjects): Multiple dose intakes (20 mg and 50 mg once daily) of GLPG0259 or a matching placebo over 5 days were studied in two cohorts of nine subjects (active or placebo in a 2 : 1 ratio). There was a minimum period of 7 days between the two cohorts.
Treatment allocation was determined by a computer-generated randomization schedule. Subjects were admitted to the clinical unit on the evening prior to dosing (day -1) and were confined until 24 hours after the last dose.
In both parts, GLPG0259 free-base solution (1–10 mg/mL in 40% [w/v] hydroxypropyl-ß–cyclodextrin, pH 3) or a matching placebo was given using a graduated syringe. A volume of 200 mL of water was given to each subject immediately at the time of dosing. Treatments were administered after a standard breakfast (i.e. four slices of whole-wheat bread, one slice of salami, one slice of cheddar cheese, one tablespoon of butter and jam, totaling 590 kilocalories) except during the last dosing period of part 1, where the treatment was administered after an overnight fast to assess the food effect on GLPG0259 bioavailability. Drinks were standardized to at least 1000 mL of mineral water per day.
Blood samples for pharmacokinetics were collected at regular intervals over 24 hours (part 1: 1.5–15 mg; part 2: 20 mg and 50 mg on day 1), 4 days (part 1: 30–150 mg), or 7 days postdose (part 2: 20 mg and 50 mg on day 5). Blood was collected in tubes containing lithium heparinate as an anticoagulant in order to obtain plasma for analysis of the concentrations of GLPG0259. Within 30 minutes after blood collection, the plasma was separated in a refrigerated centrifuge (4–8°C) for 10 minutes at approximately 1500 g, transferred into two polypropylene tubes with at least 500 μL of plasma per tube, and stored at -20°C until analysis. Three urine fractions were collected in part 2 on days 1 and 5 over a 24-hour period to determine the amount of GLPG0259 excreted in urine. After homogenization and recording of the total weight, two 10 mL samples for the GLPG0259 assay were stored at or below -20°C until analysis.
Study 2: Multiple Ascending Oral Doses and Methotrexate Drug-Drug Interaction
This was a phase I, randomized, double-blind, placebo-controlled, single-center study to evaluate the safety, tolerability, and pharmacokinetics of oral multiple ascending doses of GLPG0259 given for 14 days to healthy subjects (n = 24), and to get preliminary information on the potential pharmacokinetic interaction between GLPG0259 and methotrexate. The criteria for subject eligibility were the same as those listed for study 1.
A total of 24 healthy male subjects were randomized into three cohorts of eight subjects dosed orally once daily with either placebo or GLPG0259 25 mg, 50 mg, or 75 mg (active or placebo in a 3 : 1 ratio). In addition, the subjects from the 50 mg cohort received one single dose of methotrexate 7.5 mg on two occasions: 7 days prior to dosing with GLPG0259 (on day -7) and on day 14 at the same time as the last GLPG0259 dose.
GLPG0259 free base (10 mg/mL in 40% [w/v] hydroxypropyl-ß–cyclodextrin, pH 3) or a matching placebo was administered once daily for 14 days, using a syringe, as for study 1. Subjects in the 50 mg dose group were additionally administered an oral dose of methotrexate 7.5 mg (3 tablets of Ledertrexate® 2.5 mg; Wyeth-Pfizer) on two occasions. A dose of 4 mg of folic acid (Folavit®; Kela Pharma NV) was administered 24 hours after each methotrexate administration as a preventive measure for methotrexate toxicity. Folic acid was administered after all safety and pharmacokinetic assessments had been done.
Blood samples for pharmacokinetics were collected at regular intervals over 24 hours (on days 1 and 13 [in the 50 mg cohort only]) or over 7 days after the last dose on day 14 (i.e. up to day 21) to assess plasma concentrations of GLPG0259. Blood sample handling was similar to that described for study 1. For methotrexate (in the 50 mg cohort only), blood samples were collected at regular intervals over 24 hours (on day -7 and day 14) in tubes containing lithium heparinate, in order to obtain plasma, and were stored at -20°C until analysis.
Study 3: Oral Relative Bioavailability and the Food Effect
This was a phase I, open-label, randomized, three-period, three-treatment crossover study to compare the oral bioavailability of a solid dosage form of GLPG0259 (a capsule) relative to an oral solution, and to evaluate the effect of food on oral bioavailability of GLPG0259 formulated as a capsule in healthy subjects (n = 12). The criteria for subject eligibility were the same as those listed for study 1.
The treatments consisted of an oral dose of a 50 mg GLPG0259 free-base solution given after an overnight fast (treatment A), a GLPG0259 fumarate capsule (equivalent to 50 mg free base) given after an overnight fast (treatment B), and a GLPG0259 fumarate capsule (equivalent to 50 mg free base) given 30 minutes after the start of a high-fat, high-calorie breakfast (treatment C). Each subject was administered treatments A, B, and C in one of the two treatment sequences (i.e. ABC or ACB) determined by a computer-generated randomization schedule. There was at least a 7-day washout period between treatments for each subject. Subjects were admitted to the clinical unit on the evening prior to dosing (day -1) and were confined until 24 hours after the last dose.
For treatment A, GLPG0259 free base was administered as 5 mL of 10 mg/mL in 40% (w/v) hydroxypropyl-ß–cyclodextrin (pH 3), using a syringe. A volume of 235 mL of water was given to each subject immediately at the time of dosing. Capsules to be administered for treatments B and C were filled with 50 mg of GLPG0259 as a fumarate salt. One capsule was given with 240 mL of water alone (treatment B) or after a high-fat, high-calorie breakfast as described in the FDA Guidance for Industry[8] (i.e. two eggs fried in butter, two slices of bacon, two slices of toast with butter, 113 g of hash brown potatoes, and 240 mL of whole milk, totaling 800–1000 kilocalories). The subjects took the 50 mg capsule with 240 mL of water, within 10 minutes after the high-fat, high-calorie breakfast. The breakfast had to start 30 minutes prior to administration of the study drug, and the subjects had to eat their breakfast within 20 minutes.
Blood samples for pharmacokinetics were collected at regular intervals over 96 hours to assess plasma concentrations of GLPG0259. Blood sample handling was similar to that described for study 1.
Study 4: Oral Relative Bioavailability of Two Solid Dosage Forms
This was a phase I, randomized, open label, two-period, two-treatment crossover study to compare the oral bioavailability of two solid oral formulations of GLPG0259 after single-dose intake in healthy subjects (n = 12). The criteria for subject eligibility were the same as those listed for study 1.
The two treatments consisted of an oral dose of two fumarate capsules containing GLPG0259 (equivalent to 25 mg free base) given exactly 30 minutes after the start of a high-fat, high-calorie breakfast (treatment A) and a single free-base pellet capsule containing GLPG0259 50 mg given exactly 30 minutes after the start of a high-fat, high-calorie breakfast (treatment B). Each subject was administered treatments A and B in one of the two treatment sequences (i.e. AB or BA) determined by a computer-generated randomization schedule, with at least a 10-day washout period between treatments. Subjects were admitted to the clinical unit on the evening prior to dosing (day -1) and were confined until 24 hours after the last dose.
Capsules administered in fed conditions were taken within 10 minutes after the high-fat, high-calorie breakfast, as in study 3.
Blood samples for pharmacokinetics were collected at regular intervals over 96 hours to assess plasma concentrations of GLPG0259. Blood sample handling was similar to that described for study 1.
Safety Assessments
In all four studies, general safety was evaluated by the incidence of adverse events (AEs) through non-leading questioning, clinical laboratory parameters (hematology, biochemistry, and urinalysis), vital signs, 12-lead ECGs, and physical examinations.
Bioanalytic and Pharmacokinetic Methods
GLPG0259
Plasma GLPG0259 concentrations were determined using a validated liquid-chromatography–mass spectrometry/mass spectrometry (LC–MS/MS) assay. In brief, the internal standard (deuterated GLPG0259; 20 μL at 0.25 μg/mL) was added to plasma samples and then processed by liquid–liquid extraction. The evaporated and reconstituted samples were injected into a Sciex API 4000™ LC–MS/MS equipped with a short high-pressure liquid chromatography (HPLC) column. GLPG0259 was detected with multiple reaction monitoring. Quantification was performed using peak area ratios and standard curves (with 1/x2 linear least-squares regression) prepared from calibration standards. The lower limit of quantification was 0.200 ng/mL. The between- and within-run precision for quality controls, expressed as coefficients of variation (CVs), were no greater than 13.9% and 7.50%, respectively, with deviations from nominal concentrations of no more than 12.0%.
A method adapted from the plasma bioanalytic method was used to determine the concentrations of GLPG0259 in urine. The internal standard (deuterated GLPG0259; 20 μL at 0.5 μg/mL) was added to 20 μL of the urine sample. The corresponding solution was diluted 50-fold and injected directly into a Sciex API 4000™ LC–MS/MS. The lower limit of quantification was 2.00 ng/mL. The within-run precision for quality controls, expressed as the CV, was no greater than 6.7%, with deviations from nominal concentrations of no more than 6.5%.
Plasma GLPG0259 concentrations were analyzed by a non-compartmental method. The maximum plasma drug concentration (Cmax) and time to reach Cmax (tmax) values were observed directly from the data. The terminal elimination rate constant (λz) was determined by log-linear regression analysis of the elimination phase. The apparent terminal elimination half-life (t1/2,λz), calculated as t1/2,λz = Ln2/λz, was reported only if more than three datapoints were used for linear regression to determine λz with an adjusted r2 value of ≥0.900. Area under the plasma concentration–time curve (AUC) values over the collection interval (AUCt), over the dosing interval (AUCτ), or extrapolated to infinity (AUC∞) were determined using standard non-compartmental methods (WinNonLin® version 5.2 software; Pharsight Corporation, Mountain View, CA, USA). The relative bioavailability (Frel) was calculated as the ratio between the AUCs for the test formulations (fumarate capsules or free-base pellet capsules) and the AUCs for the reference formulations (solution or fumarate capsules) from studies 3 and 4. After multiple dosing, the accumulation of GLPG0259 was estimated as the ratio between the steady-state AUCτ and the day 1 AUCτ (Rac(AUC)). The following urine parameters were determined after multiple dosing for 5 days (study 1 part 2): the amount of GLPG0259 excreted unchanged in urine (Ae24h), expressed as a percentage of the dose, and renal clearance (CLR) over 24 hours (CLR24h).
Methotrexate
Plasma methotrexate concentrations were determined using a validated LC–MS/MS assay. In brief, the internal standard (deuterated GLPG0259; 200 μL at 25 ng/mL) was added to plasma samples and then processed by liquid–liquid extraction. The evaporated and reconstituted samples were injected into a Sciex API 4000™ LC–MS/MS equipped with a short HPLC column. Methotrexate was detected with multiple reaction monitoring. Quantification was performed using peak area ratios and standard curves (with 1/x2 linear least-squares regression) prepared from calibration standards. The lower limit of quantification was 5.00 ng/mL. The between- and within-run precision for quality controls, expressed as CVs, were no greater than 7.40% and 8.16%, respectively, with deviations from nominal concentrations of no more than 8.0%.
The plasma methotrexate concentrations were analyzed by a non-compartmental method, and the following parameters were assessed: Cmax, tmax, t1/2,λz, AUCt, and AUC∞.
Statistical Analyses
All statistical analyses were conducted using SAS® version 9.1 software (SAS Institute Inc., Cary, NC, USA).
For the pharmacokinetic analyses of the four clinical studies, the descriptive statistics analysis included arithmetic means and CVs for Cmax, AUC, t1/2,λz, Ae24h, and CLR24h; the medians and ranges for tmax; and the geometric means and CVs for Rac(AUC) and Frel.
Clinical safety was addressed by assessing AEs, physical examinations, laboratory assessments, ECGs, and vital sign results in a descriptive manner. Descriptive statistics and shift tables (according to normal ranges) were calculated for each parameter at every timepoint and in each treatment group. A treatment-emergent AE analysis was performed.
The following inferential statistics were performed for each study, with a statistical significance level of p < 0.0500.
Study 1
Dose proportionality was tested on dose-normalized and natural log–transformed GLPG0259 parameters (Cmax normalized to a 1 mg dose [Cmax/dose] and AUC from 0 to 24 hours [AUC24h] normalized to a 1 mg dose [AUC24h/dose]) after single fed dosing by means of mixed-effects analysis of variance (ANOVA) with the cohort and dose as fixed effects and the subject (nested within the cohort) as a random effect. In the case of a significant dose effect being observed on the parameters listed above, comparison between doses was performed using Tukey’s test. The tmax, being a discrete variable, was analyzed using a non–parametric Kruskal-Wallis test to assess the dose proportionality.
For part 2, a mixed-effects ANOVA was performed on natural log–transformed GLPG0259 parameters (Cmax/dose, AUC24h/dose, t1/2,λz, Ae24h, and CLR24h) with the day, dose, and day-by-dose interaction as fixed effects and the subject as a random effect. Dose proportionality for Rac(AUC) was evaluated from the adapted mixed-effects ANOVA of AUC24h/dose. A Wilcoxon–Mann-Whitney non-parametric test was used to assess the dose proportionality of tmax. The time to reach steady state was assessed by visual inspection of the trough plasma drug concentrations as well as by means of a mixed-effects ANOVA on Ln-transformed GLPG0259 trough plasma drug concentrations. Comparison between days was performed using Tukey’s test.
The food effect was assessed using geometric mean ratios of the observed pharmacokinetic parameters (Cmax, AUC24h, AUC∞, and t1/2,λz) for GLPG0259, with and without food, and the corresponding 90% confidence intervals (CIs) for the ratios. Calculation of 90% CIs for the geometric mean ratios was based on a mixed-effects model for the natural log–transformed parameters, with the dietary condition as a fixed effect and the subject as a random effect. A Wilcoxon–Mann-Whitney non-parametric test was used to assess the food effect on tmax.
Study 2
Dose proportionality of GLPG0259 pharmacokinetics and steady-state assessment were tested using the same statistical model as described for study 1 part 2. The effect of GLPG0259 on methotrexate pharmacokinetics (day 14 versus day -7) and the effect of methotrexate on GLPG0259 pharmacokinetics (day 14 versus day 13) were separately assessed on natural log–transformed parameters (Cmax, tmax, AUC, and t1/2,λz), using a mixed-effects ANOVA model with the day as a fixed effect and the subject as a random effect. The geometric mean ratio (i.e. the point estimate) of these pharmacokinetic parameters between days 14 and 13 for GLPG0259 and between days 14 and -7 for methotrexate was estimated from this model, using the least-squares mean (LSM) together with the 90% CI.
Studies 3 and 4
For both studies, the comparison between treatments was assessed on Ln-transformed parameters (Cmax, AUC24h, AUC∞, and t1/2,λz) by means of a mixed-effects ANOVA. The point estimate was calculated as the geometric mean of the individual ratios of each parameter for the test/reference treatments and expressed as a percentage. The 90% CI of the point estimates was calculated using the mean square error of the ANOVA. As tmax is a discrete variable dependent on selected blood sampling times, the same comparisons were assessed using a non-parametric test. The 90% non-parametric CIs for the treatment differences were calculated.
Population Pharmacokinetic Model
A population pharmacokinetic model was developed with data from the three first phase I studies (at the time of performing the population pharmacokinetic analysis, study 4 had not been performed yet), which included 54 subjects who received the active treatment within the dose range of 1.5–150 mg on at least one occasion (n = 6 at 1.5, 5, and 15 mg; n = 18 at 20–30 mg; n = 24 at 50 mg; n = 12 at 60–75 mg and 100 mg; n = 6 at 150 mg) as fumarate salt capsules or free-base solution given in either the fasted or fed state. The model that was developed was then used to support the planning of the number and timing of the sparse samples to be taken per patient in the 3-month phase II study. An exploratory graphical analysis of the pharmacokinetics of GLPG0259 was performed. The graphical analysis consisted of plotting and comparing individual profiles and the smoothes of dose-normalized profiles. Dose linearity was evaluated by comparing the dose-normalized profiles. The exploratory graphical analysis plots were also scrutinized for food and formulation effects. All analyses were performed in accordance with appropriate guidelines.[9,10] The population pharmacokinetic analyses were performed using NONMEM® version 7.1.0 software.[11] The NONMEM® model fitting used the first-order conditional estimation (FOCE) method with the interaction option throughout. S-Plus version 6.2 software was used for exploratory graphical analysis. R software (version 2.12.2)[12] was used for evaluation of goodness of fit and model evaluation. The program WinPOPT (version 1.2.1)[13] was used to aid selection of the timing and number of samples to be taken per patient in phase II.
Results
Safety and Tolerability
With the exception of a single subject who discontinued study 1 because of a nephrolithiasis while on placebo (reported as a serious AE [SAE]), all subjects completed the studies. The four studies showed a consistent pattern of AEs. Nausea, abdominal discomfort, and loose stools were the most frequently reported AEs, showing a dose-related pattern of incidence and severity from a dose of 50 mg upward. The feeding status or type of formulation had no influence on these AEs. All other AEs were typical phase I environment events, such as somnolence, fatigue, headache, oropharyngeal pain, and nasopharyngitis. No clinically relevant trends or changes were observed in the median laboratory and urinalysis values over time. A single case of a mild alanine aminotransferase increase was observed in a subject at the 75 mg dose in the second study. Across the four studies, no clinically relevant trends or changes were observed in the median vital sign values and ECG parameters over time. No treatment-emergent abnormalities related to vital signs or ECG parameters were observed in more than one subject during the trials. None of the abnormalities related to vital signs or ECG parameters were considered clinically relevant by the investigators. After multiple dosing, the maximum tolerated dose was established as being 50 mg once daily.
GLPG0259 Single-Dose Pharmacokinetics (Study 1)
GLPG0259 plasma concentration–time data are plotted in figures 1 and 2 (linear and semi-logarithmic plots), and the pharmacokinetic parameters are listed in table I. At the three lowest doses (up to 15 mg), λz could not be reliably estimated in most of the subjects, because of insufficient datapoints to characterize the terminal elimination phase. In addition, for some subjects at the highest doses (≥30 mg), the AUC∞ was poorly estimated, with an extrapolated AUC from 24 hours to infinity that represented more than 20% of the total AUC. Consequently, the t1/2,λz and AUC∞ of these subjects were not included in the summary statistics, and no inferential statistical analysis was performed on these two parameters. After a single oral administration to healthy, fed subjects, GLPG0259 was absorbed slowly, with the median tmax increasing with the dose from 2 to 7 hours (table I). The terminal plasma elimination phase of GLPG0259 was parallel for doses ≥30 mg and displayed a monophasic profile (figure 1).
Fig. 1.
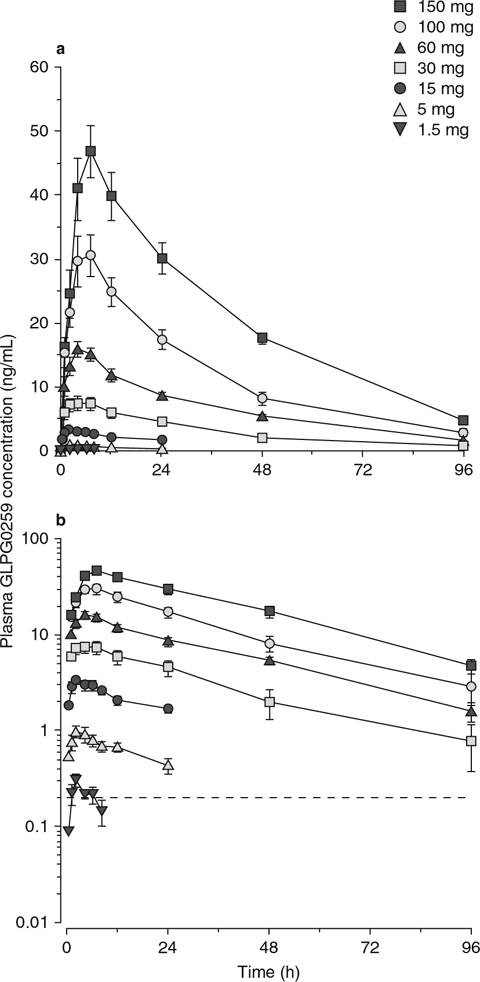
Mean (± standard error of the mean) plasma GLPG0259 concentrations after single oral doses in fed healthy subjects (n = 6 per dose group): (a) linear and (b) semi-logarithmic plots.
Table I.

GLPG0259 pharmacokinetic parameters after a single oral dose in fed healthy subjects (n = 6 per dose group)
After single dosing, Cmax and AUC24h increased proportionally within the 15–100 mg and 30–150 mg dose ranges (table I). A significant dose effect on tmax was observed, with a higher median value observed at the two highest doses. Although no statistical analysis was performed on t1/2,λz, no noticeable difference in this parameter was observed, with a mean value of about 26.0 hours (range 25.5–26.4 hours).
GLPG0259 Repeated-Dose Pharmacokinetics (Studies 1 and 2)
GLPG0259 plasma concentration–time data are plotted in figure 2, and the pharmacokinetic parameters are listed in table II. As was already evident from the single-dose pharmacokinetics, GLPG0259 was absorbed slowly, with a trend toward an increase in tmax with increased dosing (table II).
Fig. 2.

Mean (± standard error of the mean) plasma GLPG0259 concentrations after once-daily repeated oral dosing in fed healthy subjects: (a) dosing for 5 days (n = 6 per dose group); (b) dosing for 14 days (n = 6 per dose group).
Table II.

GLPG0259 pharmacokinetic parameters after once-daily repeated oral dosing in fed healthy subjects (n = 6 per dose group)
The steady-state GLPG0259 plasma concentration was reached at between 4 and 8 dosing days (figure 2, table III). After the last dose, plasma elimination of GLPG0259 over time displayed a monophasic profile, with a t1/2,λz of about 39 hours (range 35.0–41.6 hours). An approximate 2.5-fold increase in AUC24h and Cmax of GLPG0259, similar for all doses, was observed after once-daily dosing, which was consistent with the long GLPG0259 t1/2,λz. After repeated administration, GLPG0259 did not deviate from dose proportionality, with AUC24h and Cmax increasing in proportion to the dose within the 20–75 mg dose range. Overall, the between-subject variability in AUC24h and Cmax at steady state was low/moderate (between-subject CV range 16–30%) as was the within-subject variability, which was derived from the square root of the mean square error of the ANOVA (the CVs of AUC and Cmax ranged between 9.8% and 20%; data not shown).
Table III.

Trough plasma GLPG0259 concentrations after once-daily repeated oral dosing in fed healthy subjects (n = 6 per dose group)
Excretion of unchanged GLPG0259 in urine was rapid, with about 64–88% excreted within the first 12 hours (data not shown). The Ae24h of GLPG0259 represented 4.99% and 10.4% of the dose administered after single and multiple dosing, respectively, of 50 mg of GLPG0259 for 5 days (table II). The increase in the amount of GLPG0259 excreted in urine between the first and last doses mirrored the accumulation of GLPG0259 observed in plasma. As a consequence, the CLR24h remained constant between the first and last doses. At the 20 mg dose, the increase in Ae24h between the first and last doses (from 3.47% to 4.75%) was smaller than the increase in exposure (from 116 to 261 ng · h/mL), resulting in a slight decrease in CLR with time (from 5.95 to 3.74 L/h). However, these differences were not statistically different and could have been due to high variability in individual Ae24h values (range 0.685–12.0%; CV 81.4%) compared with AUC24h on day 5 (range 197–351 ng · h/mL; CV 19.3%).
Drug-Drug Interaction with Methotrexate (Study 2)
GLPG0259 and methotrexate plasma concentration–time data are plotted in figure 3, and GLPG0259 and methotrexate pharmacokinetic parameters with summary statistical analyses are presented in table IV. Regarding GLPG0259, co-administration of methotrexate 7.5 mg did not significantly alter the rate and extent of absorption of GLPG0259, with point estimates for Cmax and AUC24h of 102.67% and 102.11%, respectively. Although the t1/2,λz of GLPG0259 could be estimated on one occasion only, there was no modification of the elimination, as shown by the superimposable elimination phases with or without methotrexate in figure 3. It must be noted that even if the study was not powered to analyze the influence of methotrexate on GLPG0259 pharmacokinetics, using the 90% CI approach, the intervals were narrow and their boundaries fell within the 80–125% bioequivalence range for both Cmax and AUC24h (table IV). These results are explained by the low/moderate within-subject variability in GLPG0259 pharmacokinetics (<20%) and suggest that a sample size of 12 subjects would be sufficient to show bioequivalence between two treatments.
Fig. 3.

Mean (± standard error of the mean) plasma concentrations of (a) GLPG0259 and (b) methotrexate after administration of each drug alone or in combination to fed healthy subjects (n = 6).
Table IV.

Summary statistics for GLPG0259 and methotrexate pharmacokinetic parameters (n = 6)
The plasma pharmacokinetic parameters of methotrexate observed in this study were in agreement with those reported previously for the methotrexate 7.5 mg dose.[14,15] When methotrexate was co-administered with GLPG0259 50 mg, the rate of absorption of methotrexate was slightly but not statistically significantly decreased, with a point estimate for Cmax of 89.63% (figure 3, table IV). The extent of absorption (AUC∞) and the elimination (t1/2,λz) of methotrexate were not affected by GLPG0259, and their point estimates were 118.22% and 110.64%, respectively.
Bioavailability and Food Interaction Studies (Studies 1, 3, and 4)
As shown in figure 4a, food did not have an impact on the rate and extent of absorption of GLPG0259 given as 100 mg of free-base oral solution, with a Cmax of 31.8 ng/mL (versus 31.0 ng/mL in the fasted state) and an AUC24h of 562 ng · h/mL (versus 572 ng · h/mL in the fasted state) [table V], and corresponding point estimates of 89.67% (90% CI 74.71, 107.61) and 100.42% (90% CI 83.46, 120.83), respectively (table VI).
Fig. 4.
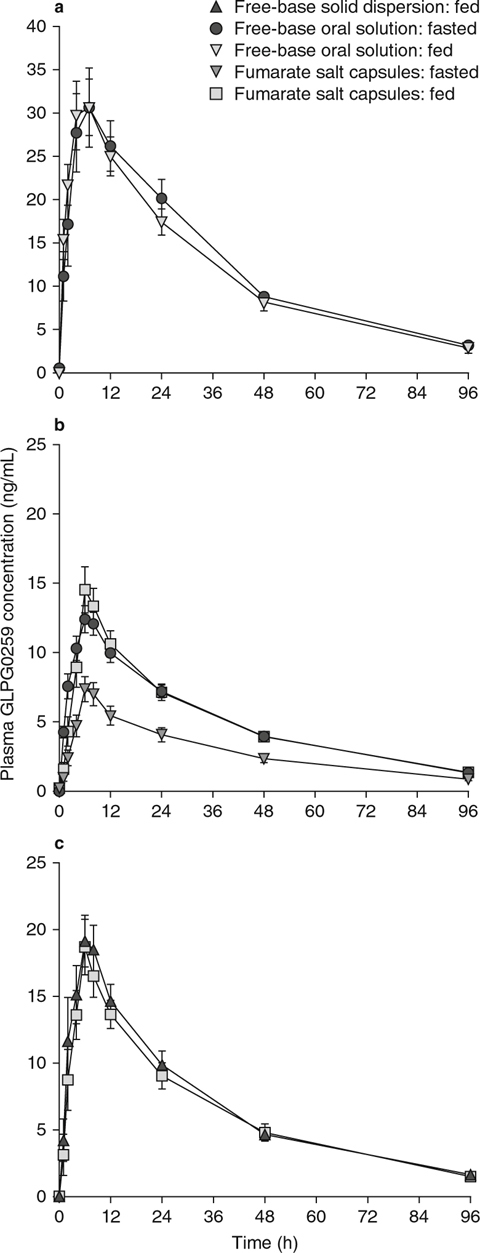
GLPG0259 pharmacokinetic profiles after a single oral dose of GLPG0259 given to healthy subjects as (a) free-base oral solution in the fasted and fed states; (b) free-base oral solution in the fasted state and fumarate salt capsules in the fasted and fed states; or (c) fumarate salt capsules in the fed state and free-base solid dispersion capsules in the fed state.
Table V.

GLPG0259 pharmacokinetic parameters after a single oral dose of GLPG0259 given as various oral formulations to fasted or fed healthy subjects (n = 6 or 12 per formulation)
Table VI.
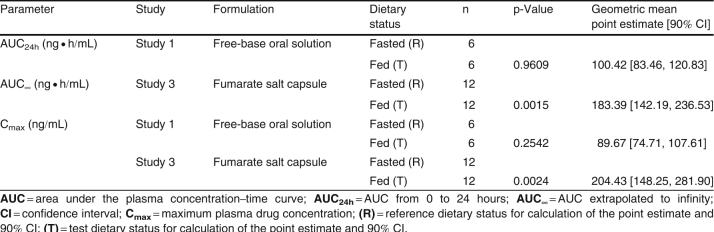
Table VI. Statistical analysis of the food effect on GLPG0259 pharmacokinetic parameters
Compared with GLPG0259 free-base oral solution, the bioavailability (both Cmax and AUC∞) of a single 50 mg dose of GLPG0259 given as the fumarate salt in capsule form in the fasted state was decreased by about 45% (table VII). No change in the absorption rate (tmax 6 versus 7 hours) or t1/2,λz (31.6 versus 29.6 hours) was noted. This decrease in bioavailability was prevented by dosing the GLPG0259 fumarate salt capsule in the fed state (figure 4b). In such conditions, the solid dosage form led to bioavailability comparable to that obtained with the GLPG0259 free-base oral solution administered in fasted conditions, as shown by a Cmax of 15.2 ng/mL (versus 12.8 ng/mL) and an AUC∞ of 542 ng · h/mL (versus 536 ng · h/mL) [table V].
Table VII.
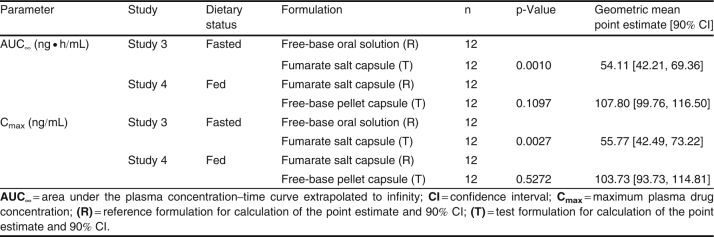
Table VII. Statistical analysis of the formulation effect on GLPG0259 pharmacokinetic parameters
Finally, a-head-to-head comparison of two solid dosage forms was investigated after a single 50 mg dose was given in the fed state as capsules of GLPG0259 fumarate salt and GLPG0259 free-base solid dispersion as coated pellets. The two formulations compared well, as shown by Cmax values of 20.4 ng/mL versus 19.8 ng/mL and AUC∞ values of 713 ng · h/mL versus 670 ng · h/mL for the free-base solid dispersion and fumarate salt, respectively (table V), with corresponding point estimates of 103.73% (90% CI 93.73, 114.81) and 107.80% (90% CI 99.76, 116.50), respectively (table VI).
Even if these three studies were not powered to compare formulations, using the 90% CI approach, the interval boundaries for both Cmax and AUC were close to or even fell within (study 4) the 80–125% bioequivalence range.
Population Pharmacokinetics of GLPG0259
The exploratory graphical analysis from study 1 (a single ascending dose) revealed that the elimination of GLPG0259 was independent of the dose, but that the dose-normalized profiles were not superimposable within the entire dose range (1.5–150 mg), that tmax occurred later at higher doses, and that there appeared to be no influence of food on the absorption of the solution formulation. After multiple doses, steady-state GLPG0259 plasma concentrations were reached after 4–5 days. The dose non-linearity observed after single dose administration was not apparent after multiple doses where a smaller dose range of 25–75 mg was given (data not shown). The graphical analysis also showed that the Frel for GLPG0259 was lower for the capsule formulation than for the solution formulation, but that food increased the bioavailability of the capsule to the same level as that of the solution formulation. These findings are in good agreement with the conclusions drawn from traditional pharmacokinetic analysis of the data from these three studies.
Structural Model Development
Initially, one- and two-compartment disposition models with simple first-order absorption were compared. Inter-individual variability (IIV) was included on all parameters, and a proportional residual error model was used. The two-compartment model was superior. Next, food and formulation effects were included on Frel, and the absorption sub-model was expanded to a sequential zero- then first-order absorption process for each of the solution and capsule formulations. Ultimately, only the duration of the zero-order input process (D1) differed between the two formulations, followed by the same first-order absorption-rate constant (ka). The statistical model was refined in the next step; IIV was included initially on all structural model parameters but was not able to be estimated on inter-compartmental clearance (Q) and so was removed. Next, inter-occasion variability (IOV) was introduced on Frel, ka, and D1. The introduction of IOV on Frel and D1 resulted in the IIV values for these two parameters being extremely small and considered negligible, and so IIV was removed. The proportional residual error model used at the start of the analysis was found to be adequate and was retained throughout model development.
Population Pharmacokinetic Model of GLPG0259
The final population pharmacokinetic model was a two-compartment disposition model with sequential zero- then first-order disposition. The exploratory analysis had clearly shown that dose was a potentially important covariate. The final model contained an influence of dose on the parameters ka and Frel. Dose was included as a covariate on Frel as a power model; Frel increased with increasing dose (figure 5b). Dose was also included as a covariate on ka as a maximum effect (Emax) model; ka decreased with increasing dose up to 50 mg and was then reasonably constant (figure 5a). During the evaluation of dose as a covariate, the parameterization used to describe ka was altered to be equal to λz plus a constant (flip-flop pharmacokinetics). As a result, t1/2,λz could be calculated correctly.
Fig. 5.
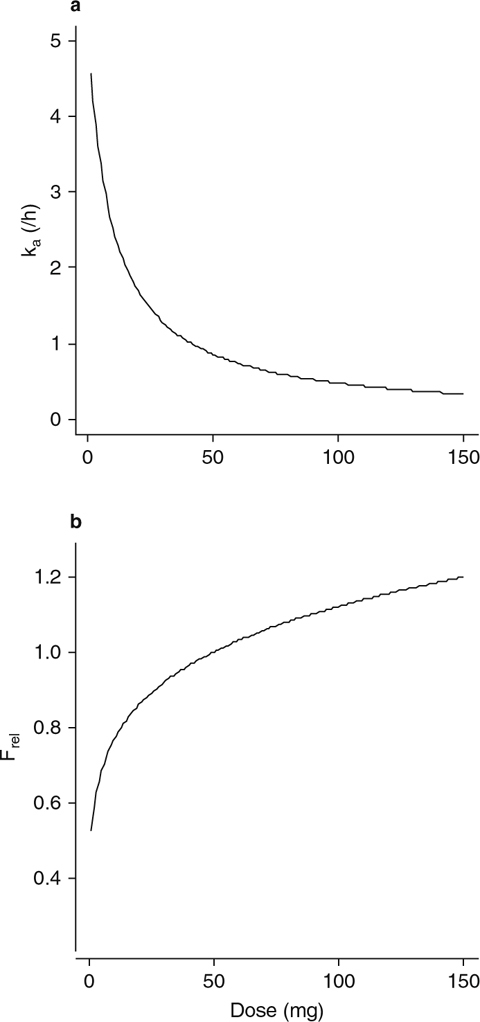
Influence of dose on (a) the first-order absorption rate constant and (b) relative bioavailability in the final population pharmacokinetic model. F rel = relative bioavailability; k a = first-order absorption rate constant.
Formulation had an effect on D1, which was estimated to be 0.317 hours for the solution formulation and 2.66 hours for the capsule formulation. Formulation also had an effect on Frel, which was estimated to be 0.489 of the Frel for the capsule formulation. The presence of food (a high-fat breakfast) was also found to influence Frel; Frel was 1.89 times greater in the presence of food with the capsule formulation only. Consequently, food was included as a covariate on Frel for the capsule formulation only, and formulation was included as a covariate on Frel and D1. IIV terms were included on the apparent total body clearance (CL/F), apparent volumes of distribution in the central and peripheral compartments (V1/F and V2/F, respectively), and ka. IOV was included on Frel, D1, and ka. A proportional error model was used to describe the residual variability. The parameter estimates for the final population pharmacokinetic model are presented in table VIII.
Table VIII.

GLPG0259 parameter estimates for the final population pharmacokinetic model
The residual variability for the final model (15.0%) was low and showed that the final population pharmacokinetic model described the vast majority of the variability in the data. The value of CL/F estimated for GLPG0259 was 79.3 L/h and was estimated with high precision (relative standard error [SE] 4.0%). The estimate of V1/F was 3030 L and was also precise (relative SE 4.4%). The values for CL/F and V1/F could be used to obtain the t1/2,λz for GLPG0259, which was calculated to be 26.5 hours. In general, all of the parameters associated with the disposition of GLPG0259 were estimated precisely (IIV around 20%). Parameters associated with absorption were less precisely estimated (IIV and IOV ranged between 20% and 75%), indicating that the majority of the overall variability in the pharmacokinetics of GLPG0259 was due to absorption. The value of ka at a dose of 50 mg was calculated to be 0.88/hour.
The goodness-of-fit plots for the final population pharmacokinetic model of GLPG0259 are shown in figures 6 and 7.
Fig. 6.
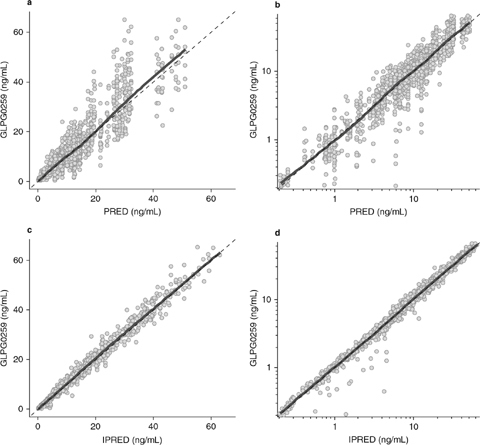
Goodness-of-fit plots: observed data are plotted on the y-axes, and population predictions [graphs (a) and (b)] and individual model predictions [graphs (c) and (d)] are plotted on the x-axes. Graphs () and (c) are on a linear scale, and graphs (b) and (d) are on a logarithmic scale. The dashed datalines are identity lines, and the thick solid datalines are smoothes through the data. The smooth lines lie very close to the identity lines, for both the population and individual predictions, indicating that the structural model describes the data well. IPRED = individual predictions; PRED = population predictions.
Fig. 7.

Goodness-of-fit plots: (a) conditional weighted residuals versus population predictions; (b) absolute individual weighted residuals versus individual predictions; (c) conditional weighted residuals versus time after dose; (d) conditional weighted residuals versus continuous time. The dashed datalines are zero lines, and the thick solid datalines are smoothes through the data. The lack of trends in graphs (), (c), and (d) again indicates that the structural model describes the data well. The lack of a trend in the smooth line in graph (b) shows that the proportional error model is appropriate for describing the residual error. CWRESI = conditional weighted residuals; IPRED = individual predictions; ∣IWRES∣ = absolute individual weighted residuals; PRED = population predictions.
Model qualification of the final model, using a visual predictive check (VPC) and a numerical predictive check (NPC), showed that the model was a good description of the data (figure 8).
Fig. 8.
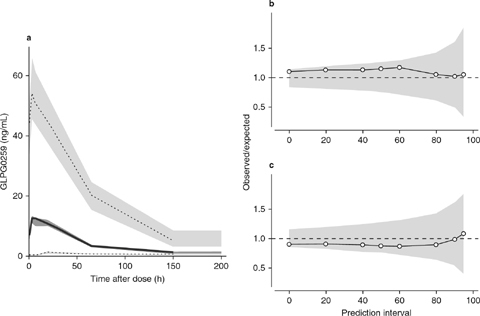
(a) Visual predictive check; (b) numerical predictive check (upper prediction interval limit); and (c) numerical predictive check (lower prediction interval limit). In graph (), the thick solid dataline shows the median of the observed data, and the dark gray shading shows the model-predicted 95% confidence interval around the median. The dotted datalines are the limits between which 95% of the observed data are found, and the light gray shading shows the model-predicted 95% confidence intervals around those limits. In graphs (b) and (c), the thin solid datalines and white datapoints show the ratios between the actual and expected numbers of points for (b) the upper prediction interval and (c) the lower prediction interval indicated on the x-axes, and the light gray shading shows the uncertainty of the model around the ratio of 1. The dashed datalines are identity lines, with no difference between the actual and expected numbers.
Sample time optimization was performed using the WinPOPT library two-compartment model with first-order absorption. This is a simpler model than the final population pharmacokinetic model, adjusted to reflect the structure of the library model prior to performing the sample time optimization. The absorption process was simplified from the sequential zero- then first-order process to a first-order process only, and the IOV terms for D1, Frel, and ka were also removed. The actual parameter values used for the sample time optimization are presented in table IX. The simplified model retained the influence of dose on ka, thus the value for ka (0.403/hour) is that calculated for a 50 mg dose. The results of sample time optimization are shown in table X.
Table IX.

GLPG0259 parameter estimates used for sample time optimization
Table X.

GLPG0259 parameter estimates used for sample time optimization
The gold-standard design (six samples per subject after both the 7th and 84th doses) criterion value was set at 100%. Further, the imprecision in the estimated CL/F value under this design was only 4.2%, indicating that the design was able to estimate CL/F well.
The poor design (a single sample per subject after each of the 7th, 14th, 28th, 56th, and 84th doses, at 2 hours postdose) gave a criterion value that was 0.026% of that for the gold-standard design, and CL/F was estimated extremely imprecisely.
Design no. 4, where a single sample was taken per subject but at different times per visit and always in the afternoon (thus at 5, 6, 7, 8, and 9 hours postdose across the visits) gave rise to a criterion ratio of 4.1%, and CL/F was estimated with 64.4% imprecision. Thus design no. 4 was not very good but was a considerable improvement over the poor design. Design no. 5 was similar to design no. 4, but samples were allowed to be taken both in the morning and in the afternoon, and the criterion ratio improved to 8.0% and CL/F was estimated with 22.1% imprecision. As can be seen in table IX, various designs were tested, but the greatest improvement came when the spread of the timing of the samples over the dosing interval was as wide as possible across the visits (design no. 8), and the criterion ratio was 25.8% and CL/F was estimated with 6.2% imprecision. Allowing more than one sample to be taken on one of the visits (design nos. 11 and 12) did not improve the criterion ratio or improve the precision with which CL/F was estimated, probably because a design with five samples per subjects was already adequate as a sparse sample design.
Discussion
After single and daily repeated administration, GLPG0259 was slowly absorbed and eliminated. On the basis of a statistical ANOVA, the exposure to GLPG0259 increased in proportion to the dose over a 30–150 mg single-dose range and a 25–75 mg repeated-dose range.
In the population pharmacokinetic model developed with data from the three first phase I studies, the Frel for GLPG0259 increased with increasing dose, while the ka decreased with increasing dose up to 50 mg and was then reasonably constant. Conversely to the conclusion drawn from the ANOVA on dose-normalized parameters, these changes in Frel and ka detected during the development of the population pharmacokinetic model would be a sign of non–dose-proportional pharmacokinetics. It is not unusual to observe deviation from dose proportionality within a dose range as wide as 1.5–150 mg. In addition, a population approach is much more sensitive than standard statistical analysis for finding and characterizing dose non-linearity.[16] More data would be needed, especially at higher dose levels, to refine the model and the relation of ka and Frel to the dose to draw definitive conclusions on the dose linearity of GLPG0259 pharmacokinetics. The most frequently reported AEs following repeated administration with GLPG0259 were related to gastrointestinal disorders (loose stools, nausea, abdominal pain, or discomfort). These events, reported only at doses of 50 mg and higher, could be explained by the residence time of GLPG0259 in the gastrointestinal tract. Indeed in a whole-body autoradiography with [14C]-radiolabeled compound administered in a mouse model (3 mg/kg [14C]-GLPG0259), a huge amount of radioactivity was localized 4 and 8 hours postdose in the small and large intestine contents, as well as in the gallbladder, suggesting slow and incomplete absorption and/or intestinal secretion directly or via the bile (data not shown). Apart from gastrointestinal disorders, no systemic AEs were reported after repeated dosing with GLPG0259. Thus an increase in Frel with increasing dose should not be of concern as long as systemic exposure in humans remains below the ‘no observed adverse effect level’ (NOAEL) exposures in animal species.
Methotrexate is an antifolate used with varying degrees of success in the treatment of cancer and autoimmune diseases.[17] Low-dose pulse methotrexate has emerged as the anchor drug in patients with RA because of its favorable risk-benefit profile.[18] Methotrexate is mainly eliminated by the kidney as intact drug, regardless of the route of administration. Glomerular filtration is the predominant pathway, with an additional active secretory process via organic anion transporters (OATs). Active biliary secretion also plays a role in methotrexate elimination, with variable amounts of methotrexate available for enterohepatic recirculation. Many drugs currently used in RA are known to interact with methotrexate pharmacokinetics: chloroquine reduces intestinal absorption; non-steroidal anti-inflammatory drugs can lead to a decrease in renal blood flow and glomerular filtration, and can compete with drug transporters for active renal tubular secretion; and calcium folinate has been shown to shorten the mean residence time of methotrexate in the kidney and liver.[15] GLPG0259 was eliminated by metabolism as well as renal excretion. Total body clearance of GLPG0259, predicted using allometric scaling of intravenous data from several animal species corrected for their maximum lifespan, as described by Mahmood,[19] was moderate, with a value of 54 L/h (data not shown). CLR determined in healthy subjects accounts for about 9 L/h of the total body clearance. As reported previously, the presence of radioactivity in the gallbladder after [14C]-GLPG0259 administration in a mouse model may suggest the elimination of GLPG0259 or metabolites via bile secretion and a possibility for re-absorption and enterohepatic recirculation. As GLPG0259 was intended to be developed for use as a monotherapy or in combination with drugs such as methotrexate, and taking into account the common routes of elimination of both methotrexate and GLPG0259, it was of interest to get preliminary information on the potential for drug-drug interaction between these two compounds at an early stage in drug development. Although this analysis was performed on a small subset of subjects (n = 6), no modification of the absorption or the elimination of methotrexate was noted after a daily dose of GLPG0259 50 mg. The t1/2,λz values for methotrexate estimated with and without GLPG0259 were about 3.4 and 3.1 hours, respectively. The range of boundary values for t1/2,λz reported in the literature is quite large (6–69 hours).[17] This variability may be partly related to differences in blood sampling between studies. The terminal log-linear phase cannot be determined accurately if the sampling interval is too short and/or too few blood samples are collected after 12 hours postdose.[15,17] Concerning GLPG0259, concomitant dosing with methotrexate had no impact on its bioavailability (Cmax and AUC24h).
Although the GLPG0259 free-base oral solution showed good bioavailability, this formulation is not easy to handle in long-term trials. Consequently, development of an oral solid dosage form was envisaged. GLPG0259 free base is poorly soluble in aqueous media, and its solubility decreases with increasing pH (<0.01 mg/mL at pH 7). Two approaches were developed in parallel to overcome this low solubility and to improve compound bioavailability after dosing in a solid dosage form. The first approach was a salt screening, which resulted in the selection of the fumarate salt for further formulation development work. The water solubility of the GLPG0259 fumarate salt, as compared with that of the free base, was increased to 1.9–2.7 mg/mL. The impact of the improvement in solubility was confirmed in a comparative bioavailability study in fasted dogs. In that study, GLPG0259 fumarate salt (suspension in 20% [w/v] hydroxypropyl-ß–cyclodextrin, pH 3, or as crystalline powder in capsule form) resulted in plasma exposure similar to that of GLPG0259 free base in suspension in 20% acidified hydroxypropyl-ß–cyclodextrin, but 4-fold higher plasma exposure than that of GLPG0259 free-base crystalline powder in capsule form (data not shown). In humans, administration of GLPG0259 fumarate salt as a crystalline powder in capsule form leads to 50% lower bioavailability than that of GLPG0259 free base given as a solution in 40% (w/v) hydroxypropyl-ß–cyclodextrin, pH 3 (study 3). The lower performance of the fumarate capsule in humans than in dogs is explained by the higher percentage of hydroxypropyl-ß–cyclodextrin (40% versus 20%) in the liquid formulation, which enhances GLPG0259 free-base solubility. Concomitant food intake with the solid dosage form prevents this decrease in bioavailability by increasing the solubility further. The second approach was the improvement of GLPG0259 solubility by physical modifications of the drug substance – in particular, the development of solid dispersion formulations with GLPG0259 free base in an amorphous form homogenously dispersed in a polymer matrix. The free-base solid dispersion as a powder or pellets filled into capsules was tested in fasted dogs, and both solid dispersion formulations showed GLPG0259 plasma exposure similar to that of the fumarate salt as a crystalline powder in capsule form. Similar results were obtained in humans (study 4).
In the Biopharmaceutical Classification System, drugs are classified according to measurements of solubility and permeability.[20] Regarding GLPG0259, it is a poorly soluble compound, with solubility that decreases with increased pH. The absorption of GLPG0259 was not measured in vivo in humans (there are no data after intravenous dosing), but its permeability assessed using the well established in vitro system, based on the human adenocarcinoma cell line Caco-2, was good, with an apparent permeability coefficient (Papp) of 12.4 10-6 cm/s and limited efflux (Papp B2A/Papp A2B = 2). Thus on the basis of these data, GLPG0259 could be classified as a class 2 compound, with in vivo drug dissolution then being the rate-limiting step for its absorption. Salt selection or solid dispersion development allows this issue to be overcome and increases the solubility and dissolution rate of GLPG0259, leading to an improvement in the bioavailability of the oral solid dosage forms to be used in future clinical trials.
Conclusion
In summary, the investigation of safety/tolerability and pharmacokinetics in the early development phase showed that single and repeated doses of GLPG0259 were safe and well tolerated. The most common AE reported was mild gastrointestinal discomfort. The pharmacokinetics characterized in healthy male subjects showed no major obstacles and supports a once-daily oral regimen in patients.
Acknowledgments
The authors would like to acknowledge Drs. E. Vets, L. Gheyle, and W. Haazen from SGS Life Science Services Clinical Pharmacology Unit (Antwerp, Belgium) for conducting these studies, and Mr. Romuald Sable from SGS Life Sciences Services (Wavre, Belgium) for plasma sample analysis.
This work was supported by a grant from the Flemish Government (IWT-Vlaanderen/Institute for the Promotion of Innovation through Science and Technology in Flanders; grant no. IWT070374). All authors are employee of Galapagos SASU or Galapagos NV and own stock or stock options in the company.
References
- 1.Smolen J.S., Steiner G. Therapeutic strategies for rheumatoid arthritis. Nat Rev Drug Discov. 2003;2:473–88. doi: 10.1038/nrd1109. [DOI] [PubMed] [Google Scholar]
- 2.Smolen J.S., Aletaha D., Koeller M., et al. New therapies for treatment of rheumatoid arthritis. Lancet. 2007;370:1861–74. doi: 10.1016/S0140-6736(07)60784-3. [DOI] [PubMed] [Google Scholar]
- 3.Firestein G.S. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–61. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 4.Van Vollenhoven R. Treatment of rheumatoid arthritis: state of the art 2009. Nat Rev Rheumatology. 2009;5:531–41. doi: 10.1038/nrrheum.2009.182. [DOI] [PubMed] [Google Scholar]
- 5.McInnes I., O’Dell J.R. State-of-the-art: rheumatoid arthritis. Ann Rheum Dis. 2010;69:1898–906. doi: 10.1136/ard.2010.134684. [DOI] [PubMed] [Google Scholar]
- 6.Yazici Y., Regens A.L. Promising new treatments for rheumatoid arthritis: the kinase inhibitors. Bull NYU Hosp Jt Dis. 2011;69:233–7. [PubMed] [Google Scholar]
- 7.Westhovens R., De Keyser F., Rekalov D., et al. A twelve-week exploratory phase II trial of GLPG0259 versus placebo in patients with active rheumatoid arthritis and inadequate response to methotrexate [abstract no. 2237] Arthritis Rheum. 2011;63(10):2237. [Google Scholar]
- 8.Center for Drug Evaluation and Research [CDER], Food and Drug Administration, US Department of Health and Human Services. Guidance for industry: food-effect bioavailability and fed bioequivalence studies. Rockville (MD): CDER: 2002 Dec [online]. Available from URL: http://www.fda.gov/downloads/regulatoryinformation/guidances/ucm126833.pdf [Accessed 2012 Jul 31]
- 9.Center for Drug Evaluation and Research [CDER], Food and Drug Administration, US Department of Health and Human Services. Guidance for industry: population pharmacokinetics. Rockville (MD): CDER: 1999 Feb [online]. Available from URL: http://www.fda.gov/downloads/ScienceResearch/SpecialTopics/WomensHealthResearch/UCM133184.pdf [Accessed 2012 Jul 31]
- 10.Committee for Medicinal Products for Human Use, European Medicines Agency [EMEA]. Guideline on reporting the results of population pharmacokinetic analyses. London: EMEA, 2007 Jun 21 [online]. Available from URL: http://www.emea.europa.eu/pdfs/human/ewp/18599006enfin.pdf [Accessed 2012 Jul 31]
- 11.Beal SL, Sheiner LB, Boeckmann AJ, et al., editors. NONMEM users guides. Ellicott City (MD): Icon Development Solutions, 1989–2009
- 12.R Development Core Team. The R project for statistical computing [online]. Available from URL: http://www.r-project.org/ [Accessed 2012 Jul 31]
- 13.WinPOPT Development Team. WinPOPT [online]. Available from URL: http://www.winpopt.com/index.htm [Accessed 2012 Jul 31]
- 14.Kremer J.M., Hamilton R.A. The effects of nonsteroidal antiinflammatory drugs on methotrexate (MTX) pharmacokinetics: impairment of renal clearance of MTX at weekly maintenance doses but not at 7.5 mg. J Rheumatol. 1995;22:2072–7. [PubMed] [Google Scholar]
- 15.Bannwarth B., Péhourcq F., Schaeverbeke T., et al. Clinical pharmacokinetics of low-dose methotrexate in rheumatoid patients. Clin Pharmacokinet. 1996;30:194–210. doi: 10.2165/00003088-199630030-00002. [DOI] [PubMed] [Google Scholar]
- 16.Jonsson E.N., Wade J.R., Karlsson M.O. Nonlinearity detection: advantages of nonlinear mixed-effects modeling. AAPS PharmSci. 2000;2:E32. doi: 10.1208/ps020332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen D.D., Azarnoff D.L. Clinical pharmacokinetics of methotrexate. Clin Pharmacokinet. 1978;3:1–13. doi: 10.2165/00003088-197803010-00001. [DOI] [PubMed] [Google Scholar]
- 18.Smolen J.S., Landewé R., Breedveld F.C., et al. Eular recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs. Ann Rheum Dis. 2010;69:964–75. doi: 10.1136/ard.2009.126532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mahmood I. Application of allometric principles for the predictions of pharmacokinetics in human and veterinary drug development. Adv Drug Deliv Rev. 2007;59:1177–92. doi: 10.1016/j.addr.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 20.Amidon G.L., Lennernas H., Shah V.P., et al. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–20. doi: 10.1023/A:1016212804288. [DOI] [PubMed] [Google Scholar]