

Abstract
Introduction
Chronic obstructive pulmonary disease (COPD) is a heterogeneous disease with pulmonary and extra-pulmonary manifestations. Although COPD is a complex disease, diagnosis and staging are still based on simple spirometry measurements. Different COPD phenotypes exist based on clinical, physiological, immunological and radiological observations. Cigarette smoking is the most important risk factor for COPD, but only 15–20% of smokers develop the disease, suggesting a genetic predisposition. Unfortunately, little is known about the pathogenesis of COPD, and even less on the very first steps that are associated with an aberrant response to smoke exposure. This study aims to investigate the underlying local and systemic inflammation of different clinical COPD phenotypes, and acute effects of cigarette smoke exposure in individuals susceptible and non-susceptible for the development of COPD. Furthermore, we will investigate mechanisms associated with corticosteroid insensitivity. Our study will provide valuable information regarding the pathogenetic mechanisms underlying the natural course of COPD.
Methods and analysis
This cross-sectional study will include young and old individuals susceptible or non-susceptible to develop COPD. At a young age (18–40 years) 60 ‘party smokers’ will be included who are called susceptible or non-susceptible based on COPD prevalence in smoking family members. In addition, 30 healthy smokers (age 40–75 years) and 110 COPD patients will be included. Measurements will include questionnaires, pulmonary function, low-dose CT scanning of the lung, body composition, 6 min walking distance and biomarkers in peripheral blood, sputum, urine, exhaled breath condensate, epithelial lining fluid, bronchial brushes and biopsies. Non-biased approaches such as proteomics will be performed in blood and epithelial lining fluid.
Ethics and dissemination
This multicentre study was approved by the medical ethical committees of UMC Groningen and Utrecht, the Netherlands. The study findings will be presented at conferences and will be reported in peer-reviewed journals.
Trial registration
ClinicalTrials.gov, NCT00807469 (study 1) and NCT00850863 (study 2).
Keywords: COPD, Inflammation, Susceptibility, Corticosteroid insensitivity, Smoking
Article summary.
Article focus
This article describes the study protocol of a cross-sectional study investigating the underlying local and systemic inflammation of different clinical Chronic obstructive pulmonary disease (COPD) phenotypes, and acute effects of cigarette smoke exposure in individuals susceptible and non-susceptible for the development of COPD.
Key messages
Young (18–40) and older (40–75) individuals who are susceptible and non-susceptible to develop COPD are included.
All groups are extensively phenotyped by clinical, physiological, immunological and radiographical characterisation. Furthermore, effects of acute smoking are studied.
Strengths and limitations of the study
Extensive characterisation of a well-defined study population, providing valuable information regarding the pathogenic mechanisms underlying the natural cause of COPD.
Recruitment of a population with high and low familiar risk to develop COPD.
Background
Chronic obstructive pulmonary disease (COPD) is a major cause of morbidity and mortality worldwide.1 The disease is characterised by persistent and progressive expiratory airflow obstruction (postbronchodilator FEV1/FVC<0.70) and its severity is based on FEV1%predicted.1 Cigarette smoking is the most important risk factor for COPD in the western world, but only 15–20% of young smokers will eventually develop the disease, suggesting a genetic predisposition. So far, the genetic background of these susceptible smokers has not been elucidated.2 Unravelling the underlying pathogenetic mechanisms of COPD is difficult because it takes 20–30 years of smoking before susceptible smokers develop established COPD. Also, there is the problem that COPD has many clinical expressions, and we recently have started to learn how to phenotype this heterogeneous disease. Finally, there are many other risk factors that may modulate the complex interaction between the genetic background and smoking, like in utero events, microbial infections, dietary factors, physical inactivity and pharmacological treatment.
It is well accepted that the spirometry measurements (FEV1 and FVC) are largely insufficient to diagnose and classify COPD.1 With the increased recognition of various clinical expressions of COPD, consensus is growing that COPD represents a spectrum of overlapping diseases with important extra-pulmonary consequences. Phenotypes of COPD may be classified according to four domains: clinical, physiological, immunological and radiographical.3
Clinical distinctions are generally based on dyspnoea scores, frequency of exacerbations, body mass, muscle wasting, corticosteroid responsiveness, depression/anxiety, comorbidity and healthy status.4
Physiological distinctions may be based on the degree of airflow limitation, decline in lung function, bronchodilator responsiveness, airway hyper-responsiveness, CO diffusion capacity, hyperinflation, body-plethysmography, bio-impedance and exercise tolerance.
Immunological features comprise the type and severity of local and systemic immunological processes in the lung and systemic compartment. In blood leucocytes, cytokines and mediators may affect the functionality of extra-pulmonary tissues and organs, leading to COPD-associated comorbid conditions.
Radiographic distinctions may be based on the presence of various forms and severity of emphysema, thickened large airways and small airways abnormality on high-resolution CT scans.
Although systemic inflammation and multiorgan pathology have been put forward as important features of COPD, surprisingly little is known about the underlying pathogenesis. Most COPD studies in this field included small numbers of individuals, focused on more severe stages of COPD, characterised subjects clinically on the basis of a few arbitrary pulmonary measurements, did not take into account the genetic background and paid limited attention to different aspects of systemic inflammation. In addition, most studies assumed that assessment of cytokines by multiplex assays (eg, Luminex) is sufficient to accurately describe the systemic inflammatory response. Unfortunately, many caveats are present that preclude a complete insight in this response, for example:
Not all cytokines implicated in COPD are known;
Little effort is taken to measure anti-inflammatory cytokines (the balance between proinflammatory and anti-inflammatory signals will probably determine the extent and type of inflammation);
Different proinflammatory cytokines can act as heterologous antagonists (inhibit the effects of other cytokines);
The kinetics of cytokines are very dynamic and no consensus is present regarding an optimal single time point for blood collection.
In the present study we set out to characterise systemic inflammation by an alternative approach. Innate immune cells will be used as integrators of proinflammatory and anti-inflammatory signals. We hypothesise that subtle changes in the phenotype of granulocytes and monocytes are caused by an ‘inflammatory imprinting’ of these cells.
Cigarette smoking is the main risk factor for developing COPD. Repetitive acute effects of cigarette smoke exposure may accumulate and after many years lead to irreversible lung damage. To understand the changes in the lung due to chronic smoking we believe that it is important to first investigate the exact immunological responses to an acute smoke exposure event, particularly in ‘naive’ lungs that are not yet affected by chronic smoke exposure. The acute (<24 h) effects of smoking in humans, animals and cell cultures have been extensively reviewed some years ago by van der Vaart et al.5 If we integrate all the available data on acute smoking we are able to construct a hypothetical time frame for acute effects of smoking (figure 1). One of the very first insults on the bronchial system is by oxidants present in cigarette smoke. After local depletion of antioxidants, the first oxidative stress products can be measured within 1 h. These products will disappear within 6 h. There is a surprisingly fast influx of inflammatory cells; even faster than the synthesis of some pro-inflammatory cytokines (tumour necrosis factor (TNF) α, interleukin (IL)-1β, IL-8). The exact time period at which the proteinase/antiproteinase balance is affected is unknown; however, protein degradation is measurable within 6 h after smoking. Unfortunately, until now, only a few studies have investigated the acute effects of cigarette smoking in humans.5 These studies included only small numbers of individuals, characterised subjects mainly on basis of pulmonary measurements, paid no attention to the genetic background and paid limited attention to different aspects of pulmonary inflammation.
Figure 1.
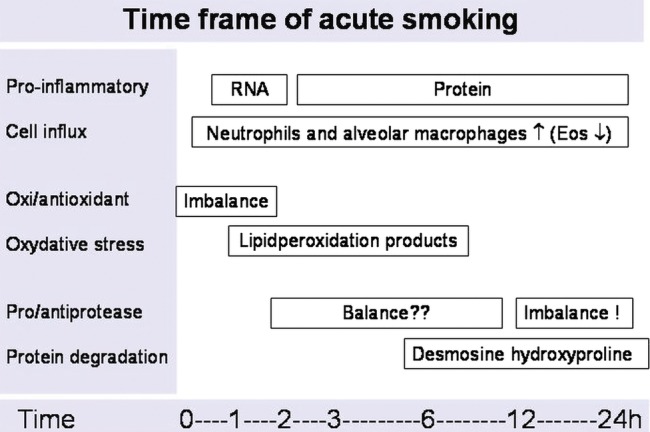
Acute smoking effects in the lung.
Corticosteroids provide little therapeutic benefit in a relatively large group of COPD patients, despite their broad anti-inflammatory effects. Our goal is to identify common markers in peripheral blood monocytes, skin and lung epithelial cells that might contribute to corticosteroid insensitivity. Recently, the GLUCOLD study demonstrated beneficial effects on airway wall inflammation and decline in lung function yet with large interindividual differences.6 In vitro studies have shown that the ability of dexamethasone to suppress cytokine release (eg, IL-8) from alveolar macrophages are impaired in COPD patients as compared with healthy smokers.7 Furthermore, alveolar macrophages from healthy smokers are more resistant to corticosteroids than macrophages from non-smokers.8 This relative steroid insensitivity may, in part, be explained by a suppressive effect of cigarette smoke-induced oxidative stress. This suppression may particularly play a role in the airway epithelium, where cells are in first contact with cigarette smoke and form an important source of mediators involved in the induction of neutrophilic airway inflammation (eg, the chemoattractant IL-8). It may well be that corticosteroid insensitivity is gradually acquired by smoking in COPD, and one might hypothesise that smokers who develop COPD are more prone to have signs of corticosteroid insensitivity.
General hypotheses
There is a clear need to understand all the factors that contribute to the development of COPD and its different phenotypes better. This study focuses on the pathogenesis and clinical expression of smoking-induced COPD, studied both in the pulmonary and the systemic compartments. The following general hypothesis is put forward by our consortium (figure 2):
Figure 2.

General hypothesis about the role of systemic inflammation.
COPD is a multi-organ disease situated in both the lung and extra-pulmonary organs and tissues. Dysfunction of the latter tissues is exemplified by muscle atrophy, impaired muscle oxidative capacity, osteoporosis, atherosclerosis and heart failure. A low-grade systemic inflammation plays a pivotal role in the induction and perpetuation of this multi-organ disease. Smoking and persistent production of inflammatory mediators from the lung are inducers of systemic inflammation. Other risk factors such as diet deficiencies, sedentary life style and frequent infections contribute independently to further amplification of systemic inflammation. In more advanced COPD the extra-pulmonary pathology starts to contribute to disease severity and a vicious circle of persistent difficulty to treat inflammation. Consequently, local and systemic inflammation should be reduced in all stages of disease by reversing negative lifestyle factors and applying successful anti-inflammatory treatment modalities. In more advanced stages multimodal interventions additionally should improve impaired tissue functions. An important contributing problem is the relative corticosteroid insensitivity of both lung and peripheral tissue responses in COPD.
Aims of the study
- To assess systemic and local inflammation at baseline in:
- Young healthy individuals with low number of pack years smoking who have a high and low familial risk to develop COPD;
- Older individuals with higher number of pack years who either have normal lung function or COPD.
- We hypothesise that young susceptible individuals and COPD patients demonstrate a higher degree and different type of local and systemic inflammation at baseline.
To study systemic and local inflammation after acute smoke exposure in the above groups. We hypothesise that young susceptible individuals and COPD patients demonstrate a higher and aberrant local and systemic inflammatory response to cigarette smoke.
To compare in bronchial epithelial cells and peripheral blood mononuclear cells (PBMCs) corticosteroid responsiveness in vitro between susceptible and non-susceptible individuals. To study in these cells the effects of cigarette smoking and to elucidate underlying mechanisms of corticosteroid unresponsiveness.
To determine whether the type and severity of the systemic inflammatory response contributes to the clinical outcome of COPD. We hypothesise that the type and severity of systemic inflammation have profound effects on the clinical picture of COPD.
To investigate the relationship between downstream genetic effects (transcriptome and proteome) and specific COPD phenotypes in peripheral blood and lung tissue (induced sputum, bronchial biopsies and epithelial lining fluid).
Methods
Study population
In total, 200 old and young individuals who are susceptible or not susceptible to develop COPD will be recruited (table 1). At old age (>40 years), 30 healthy smokers (>20 pack years) and 110 COPD patients (>10 pack years) will be enrolled in the study. At a young age, 60 ‘party smokers’ with a normal lung function will be included with a high or low prevalence of COPD in smoking family members (see table 1). Party smoking was defined by irregularly smoking and/or able to quit smoking for at least 2 days. Exclusion criteria are: α-1-antitrypsin deficiency, acute pulmonary infections (like tuberculosis, pneumonia, flu and tracheo-bronchitis), prior history of significant inflammatory lung disease other than COPD (sarcoidosis, pulmonary fibrosis, silicosis, etc), active infections (such as hepatitis A–C, cystitis, gastroenteritis, etc), treatment with antibiotics or corticosteroids within 8 weeks, taking part in another study and recent diagnosis of cancer. Medication such as non-steroidal anti-inflammatory drugs and immunosuppressive agents which could affect the results of the study will be excluded, as well as substance abuse. Comorbidities that might lead to study-related (serious) adverse events will be excluded on the basis of an arbitrary selection of conditions listed in the ACE-27 comorbidity scale.9
Table 1.
Study population
| Disease | No | Age (years) | Smoking status | Pack years | FEV1/VC (%) | FEV1 (% predicted) |
|---|---|---|---|---|---|---|
| Non-susceptible | ||||||
| Healthy (A) | 30 | 18–40 | Party smoking | 0–10 | >70 | >85 |
| Healthy (B) | 30 | 40–75 | Ex or current | >20 | >70 | >85 |
| Susceptible | ||||||
| Healthy (C) | 30 | 18–40 | Party smoking | 0–10 | >70 | >85 |
| COPD | ||||||
| Stage I (D1) | 30 | 40–75 | Ex or current | >10 | ≤70 | >80 |
| Stage II (D2) | 30 | 40–75 | >10 | ≤70 | 50–80 | |
| Stage III (D3) | 30 | 40–75 | >10 | ≤70 | 30–50 | |
| Stage IV (D4)* | 20 | 40–75 | >10 | ≤70 | <30 | |
| <53 | <30% | |||||
Susceptibility in young individuals is based on family history. Not susceptible means that none of the smoking family members who are at least 40 years of age have COPD. Susceptible means that the prevalence of COPD in smoking family members older than 40 years is high: 2 of 2, 2 of 3 or 3 of 3, 3 of 4 or 4 of 4. Party smoking was defined by irregularly smoking and/or able to quit smoking for at least 2 days. α-1-antitrypsin deficiency is excluded.
*Patients with a FEV1 30–50% predicted in combination with chronic respiratory failure also have stage IV.
COPD, chronic obstructive pulmonary disease.
Study design
This study is a bi-centre cross-sectional study that takes place at University Medical Centers Utrecht (UMC Utrecht) and Groningen (UMC Groningen). Participating subjects will undergo extensive clinical characterisation (table 2). Local and systemic inflammation will be investigated in several ways. Special attention will be paid to acute smoking and corticosteroid insensitivity in selected subgroups.
Table 2.
Measurements
| Measurements | Group |
| Clinical | |
| Demographics | All |
| Physical examination | All |
| Peripheral blood (routine measurements) | All |
| Presence of metabolic syndrome | All |
| ECG | B, D1-4 |
| BODE index | B, D1-4 |
| Fagerstrom Smoking Questionnaire | All |
| St George's Respiratory Questionnaire (SGRQ) | D1-4 |
| Clinical COPD (chronic obstructive pulmonary disease) Questionnaire (CCQ) | D1-4 |
| SQUASH | All |
| Urine (microproteins) | All |
| AGE (Advanced Glycation Endproducts)-reader | All |
| Skin blanching test | All |
| Physiological | |
| Flow volume+reversibility | All |
| Body plethysmography | All |
| CO diffusion | All |
| Methacholine challenge test | A, B, C, D1-3 |
| Bioelectrical impedance | All |
| Six minute walking distance | B, D1-4 |
| Immunological | |
| Sputum induction (only baseline) | A, B, C, D2 |
| Peripheral blood (systemic inflammation) | All |
| Peripheral blood 4× (acute smoking) | A, B, C, D2 |
| Exhaled breath condensate 3× (acute smoking) | A, B, C, D2 |
| Exhaled CO 5× (acute smoking) | A, B, C, D2 |
| Bronchial biopsy 2× (acute smoking) | A, B, C, D2 |
| Epithelial lining fluid 2× (acute smoking) | A, B, C, D2 |
| Epithelial brushes 2× (acute smoking) | A, B, C, D2 |
| Radiographical | |
| Low dose HRCT-scan lung | All |
HRCT, high resolution computed tomography.
Clinical outcomes
Demographic variables include: age, sex, smoking habits, education, profession, other exposures, height and weight. Risk factors of the metabolic syndrome will be determined including blood pressure, waist hip circumference, lipid profile and fasting glucose (table 2). Questionnaires will be the Clinical COPD Questionnaire (CCQ), the St George's Respiratory Questionnaire (SGRQ), the Dutch Fagerstrom test for nicotine dependence and the Short QUestionnaire to ASsess Health-enhancing physical activity (SQUASH).10–12 Exacerbation frequency will be recorded in COPD patients. The BODE-index will be calculated on the basis of FEV1, 6 min walking distance (6MWD), body mass index (BMI) and MRC-dyspnoea score.4 In urine (micro) protein concentration will be assessed. Corticosteroid sensitivity will be measured by cutaneous vasoconstrictor response to topical budesonide using the skin blanching test.13 Budesonide dissolved in 95% ethanol will be applied to the skin using eight different concentrations (0–1000 μg/ml). Blanching will be scored with a 7-point scale: 0–3 (increasing with steps of 0.5; 0=no blanching and 3=intense blanching). Cumulative oxidative stress will be measured in the skin using the non-invasive advanced glycation endproducts (AGE) reader (DiagnOptics, Groningen, The Netherlands).14
Physiological outcomes
Spirometry will be performed according to international guidelines (ERS 2005).15 We will assess FEV1, FEV1/FVC, IVC, FEF50, FEF75, reversibility to salbutamol, TLC, FRC (body box) and CO diffusion. Methacholine challenge tests are performed according to international guidelines (ERS 2005), using serial doubling concentrations of methacholine-bromide (0.03–38.4 mg/ml) with the 2 min tidal breathing method at 5 min intervals. The 6MWD will be determined according to the American Thoracic Society published guidelines of 2002.16 Individuals should walk at their own pace, can stop if necessary and are allowed to use oxygen. Body composition will be estimated using single frequency (50 kHz) bioelectrical impedance (Biostat 500), and fat-free mass will be calculated with the disease-specific equation of Schols et al.17
Immunological outcomes
Lung inflammation
Sputum will be induced and processed according to a validated and standardised technique,18 with some modifications. Differential cell counts of eosinophils, neutrophils, lymphocytes, macrophages and epithelial and squamous cells will be performed on May Grünwald Giemsa (MGG)-stained cytospins by a qualified cytopathologist.
Exhaled breath condensate will be collected using the EcoScreen (Jaeger, Hoechberg, Germany). Hydrogen peroxide, pH, 8-isoprostane, nitrite, nitrate, 4-hydroxy-2-nonenal and malondialdehyde will be measured.
Bronchoscopy will be performed using established guidelines,19–21 and six bronchial biopsies will be taken from subsegmental carinae in the right or left lower lobe. Epithelial morphology, epithelial proliferation and basement membrane thickness will be measured.22 Submucosal density of inflammatory cells (AA1, EG2, CD68, CD3, CD4, CD4CD25, CD8, mast cells and neutrophils) will be quantitated in a semiautomated way.22 Expression of E-cadherin, vascular endothelial growth factor, intercellular adhesion molecule, vascular cell adhesion molecule, E-selectin, P-selectin, AGEs and receptor for advanced glycation end-products will be measured.
Epithelial lining fluid will be sampled by advancing three microsample probes (BC-401C, Olympus, Tokyo, Japan) in the lumen of the left main bronchus.23 24 Cytokines will be measured by Luminex (Linco, Nuclilab BV, Ede, The Netherlands). Ninety per cent of the epithelial lining fluid (ELF) will be used for proteomic analysis. Briefly, each trypsin-digested sample will be labelled (iTRAQ Reagent 8-plex, ABSciex, Foster City, California, USA) according to the manufacturer's protocol. The individually labelled digests will be combined into a single-sample mixture and subjected to strong cation-exchange chromatography (AKTA Purifier, GE Healthcare Biosciences AB, Uppsala, Sweden). The resulting peptide-containing fractions will be separated by reversed-phase chromatography (Ultimate 3000 nanoflow liquid chromatography system, Dionex, Amsterdam, The Netherlands). Fractions of 12 s will be spotted on matrix-assisted laser-desorption ionisation (MALDI) targets (Probot, Dionex, Amsterdam, The Netherlands) and mass spectrometric analysis will be carried out on a 4800 Proteomics Analyzer MALDI TOF/TOF instrument (Applied Biosystems, Foster City, California, USA) controlled by the 4000 Series Explorer V.3.5 software. Proteins will be identified using Protein Pilot software V.2.0 (Applied Biosystems).
Bronchial epithelial cells will be harvested from the right or left main bronchus by brushing as described elsewhere.25 Brushed epithelial cells will be cultured to enable corticosteroid sensitivity experiments. In these experiments, cultured bronchial epithelial cells will be incubated in vitro with steroids and the effects on chemokine production (IL-8, growth regulated oncogene-alpha (GRO-α) and regulated and normal T cell expressed and secreted (RANTES)) and MMP/TIMP expression (mRNA) will be established. In addition, in PBMC the following parameters will be studied: (1) plasma levels of chemokines/inflammatory cytokines, (2) in vitro effects of steroids on TNF-α, IL-1β, IL-10, TGF-β, signalling pathways (western/electrophoretic mobility shift assay), toll-like receptors and CD14 expression as well as genes with a glucocorticoid response element (GRE) in their promoter, for example, β2-adrenergic receptor, mapkinase phosphatase-1 (MAPKP-1, FoxP3 (ELISA/RTPCR).
Systemic inflammation
Systemic inflammation will be measured in peripheral blood using several methods to study systemic activation of innate immune cells at four different levels:
Expression of established and new markers on innate immune cells associated with preactivation.26 27 The established markers include proteins that are upregulated on the cell surface upon activation of neutrophils in vitro, and can be measured by flow cytometry: CD11b (Mac-1), CD18 (integrin β2 chain), CD66b (CAECAM-8) and CD63 (LAMP-3). New markers directed against active integrins and Fc-receptors have been shown to be useful in detecting more subtle activation such as induced by cytokines: active Mac-1 (CD11b/clone CBRM1/5 28), active β1-integrin chain (CD29/ clone N2929) and active FcγRII (CD32/clones A1730). These latter markers will be used to detect subtle priming signals affecting the function of leucocytes in peripheral blood.
Determination of the sensitivity of innate immune cells for stimuli. One of the first changes which can be observed in response to inflammatory stimuli in vivo is a change in sensitivity for innate immune stimuli such as fMet-Leu-Phe (fMLF). Little activation is associated with an enhanced responsiveness, whereas pronounced systemic activation is associated with decreased responsiveness for fMLF.31 Therefore, the responsiveness of leucocytes for fMLF will be measured as a read-out for systemic inflammatory signals in vivo.
Genomic and proteomic analysis of innate immune cells in vivo.32 Total mRNA and proteins are collected from leucocytes and will be analysed by unsupervised genomic and proteomics techniques. Proteomics will be carried out by 2D-difference gel electrophoresis (2D-DIGE).33
Multiplex analysis of the presence of proinflammatory and anti-inflammatory cytokines in plasma/serum. Serum samples will be analysed for the presence of multiple cytokines and chemokines by luminex technology.34
Systemic inflammation will also be measured in peripheral blood using PBMCs:
Expression of intracellular and cell-surface markers of adaptive immune cells (Th1, Th2, Th17, Treg, B and natural killer cells) will be measured by flow cytometry.
Lung and systemic inflammation after acute smoking
Young and old subjects who are susceptible or not susceptible to develop COPD will smoke three cigarettes in 1 h. Exhaled CO, blood samples and urine, exhaled breath condensate, bronchial biopsies, epithelial lining fluid and epithelial brushes will be collected at baseline and after smoking according to the scheme in table 3. Exhaled CO will be measured at baseline to check if individuals did not smoke recently, and after smoking to check if individuals inhaled cigarette smoke sufficiently. A first bronchoscopy will be performed after 24 h. Bronchial biopsies, epithelial brushes and microprobe sampling of epithelial lining fluid will be collected. Six weeks after the acute smoking procedure a second bronchoscopy will be performed as a baseline measurement, obtaining the same specimen.
Table 3.
Acute smoke model
| Baseline | Smoking three cigarettes | 5 min | 2 h | 24 h | 6 weeks | |
|---|---|---|---|---|---|---|
| Exhaled CO | x | x | x | x | x | |
| Blood | x | x | ||||
| Exhaled breath condensate | x | x | x | |||
| Urine | x | x | x | |||
| Bronchial Biopsies | x | x | ||||
| Epithelial brush | x | x | ||||
| Microsampling probe (ELF) | x | x |
Samples collected at baseline and during the acute smoking procedure including sputum supernatant, serum, plasma, DNA and RNA of blood, urine, exhaled breath condensate, epithelial lining fluid, epithelial brushes and bronchial biopsies will be stored for further analyses.
Radiological outcomes
All subjects will undergo a low-dose CT scan at full inspiration and expiration. Exposure settings will be 30 mAs at 90 kVp for patients weighing less then 50 kg, 30 mAs at 120 kVp for patients weighing between 50 and 80 kg and 30 mAs at 140 kVp for those weighing more than 80 kg without dose modulation. During expiration the exposure settings will be 20 mAs at 90 kVp (body mass <80 kg) or 20 mAs at 120 kVp (body mass >80 kg). Emphysematous lung changes will be quantitated using automated software on low-dose CT scanning images developed in UMC Utrecht.
Sample size calculation
We concluded that the limited data in the literature do not allow us to calculate a reliable sample size according to a formal power-analysis. In general, 20–30 subjects per group are needed in studies to detect a significant proinflammatory or anti-inflammatory effect in sputum, BAL or bronchial biopsies. Looking to the available acute smoking studies in the literature this seems sufficient to detect an effect at least in exhaled breath condensate.
Statistical analyses
Demographic variables such as age, sex, smoking habits, education, work, other exposures, height and weight will be expressed as means (SD) or medians (IQR) as appropriate for continuous variables, and number (percentages) for dichotomous variables, according to group. Exacerbation frequency will be described (with percentage) per groups. Spirometry data (FEV1, FEV1/FVC, IVC, FEF50, FEF75, reversibility to salbutamol, TLCO TLC, FRC (body box), CO diffusion and methacholine challenge tests), and data indicative of systemic inflammation will be described similarly.
Comparisons between groups with regard to all the aforementioned variables will be tested using χ2 tests in case of comparison of proportions, and parametric (like the unpaired t test) or non-parametric tests (like the M-W-U test/Wilcoxon rank sum) as appropriate according to the distribution of the residuals. To test changes within groups over time at various visits, additionally paired variants of the before-mentioned tests will be used as appropriate (eg, the paired-t test and the Wilcoxon signed rank test).
Linear or logistic regression will be used to further analyse differences between groups in the aforementioned outcome variables taking confounding factors into account. Techniques like linear mixed effects models will be used to estimate changes in variables over time.
Ethics and dissemination
The two studies are registered at clinicaltrial.gov (identifier study 1: NCT00807469 and identifier study 2: NCT 00850863). These two studies have been judged by the medical ethical committee of UMC Groningen and additionally study 2 has been minimally judged by the medical ethical committee of UMC Utrecht. The study findings will be presented at conferences and will be reported in peer-reviewed journals.
Discussion
There is a large backlog in the recognition of different phenotypes of COPD and their underlying immunopathological processes. This importantly hinders the appropriate diagnosis, treatment and prognosis of this disabilitating disease. Currently, lung function (FEV1 and FEV1/FVC) is still the standard for the diagnosis and classification of COPD.1 However, there is general consensus that FEV1 poorly correlates with important patient-centred outcomes such as quality of life, symptoms and exercise capacity.35 Celli et al4 showed an association between FEV1 and mortality when FEV1 was combined with MRC-dyspnoea score, 6MWD and BMI. The so-called BODE index was put forward as a composite measure to characterise COPD in a more realistic way. In the last decade different approaches have been put forward to characterise COPD leading to at least 16 different phenotypes.36 Although clinically relevant in terms of presentation, triggers and treatment response these phenotypes do not necessarily give insight into the underlying disease processes of COPD. In this perspective the term intermediate phenotype or endotype has been put forward to describe a subtype of a disease which is defined by a distinct functional or pathophysiological mechanism.37 Together with genetic and environmental factors intermediate phenotypes may explain the clinical presentation of a heterogeneous disease like COPD. Accordingly, the present study will phenotype the induction and progression of COPD and associate this with underlying pathophysiological mechanisms in a biased as well as non-biased way. As smoking is the most important environmental risk factor for COPD we will use an acute smoking model to evaluate differences in smoking-induced acute mechanisms differentially expressed between individuals with a high and low risk for development of COPD.
Recently, a large prospective cohort study (ECLIPSE) was initiated to study the natural course of COPD in order to gain more insight into the underlying pathogenetic mechanisms.38 The ECLIPSE study is a 3-year observational study including current and ex-smoking COPD patients and healthy controls with and without a smoking history. Indeed, the ECLIPSE study confirmed that the clinical manifestations of COPD are highly variable and that the degree of airflow limitation does not capture the heterogeneity of the disease.39 In particular, the rate of change in FEV1 among patients with COPD was highly variable, with increased rates of decline among current smokers, patients with bronchodilator reversibility and with emphysema.40 Several new susceptibility genes have been identified in the ECLIPSE study,41 42 as well as potentially useful biomarkers.43–45 However, in contrast to our study, ECLIPSE does not include young subjects and is, therefore, not able to investigate the susceptibility for COPD at a young age. ECLIPSE investigates aspects of systemic inflammation (C reactive protein, TNF-α, IL-6, IL-8, serum surfactant protein D (SP-D)), but does not investigate the activation state of circulating neutrophils and lymphocytes, nor does it perform unbiased proteomic analyses of epithelial lining fluid and peripheral blood neutrophils. Therefore, our study will complement ECLIPSE data by focusing on the pathogenesis of local and systemic inflammation by using unique approaches to link genomic and inflammatory phenotypes in all stages of COPD from preclinical to advanced disease.
The present study has already been started and recruitment is still ongoing. The study population has been described in ClinicalTrial.gov (NCT00807469 and NCT 00850863) and was divided into nine groups. Initially, we planned to distinguish susceptible individuals into a ‘susceptible’ and ‘very susceptible’ group. The group of ‘old’ very susceptible individuals should include early-onset COPD (FEV1/FVC<70%, FEV1<40% predicted, age<53 years) and COPD with low number of pack years (FEV1/FVC<70%, FEV1<80% predicted, pack years <5). The group of ‘young’ very susceptible individuals should have included young individuals with family members with early-onset COPD or COPD with low smoke exposure. Despite an intensive search among lung transplantation (LTx) candidates/recipients and their family members we were not able to recruit this group in sufficiently high numbers. Therefore, we decided to combine the susceptible and very susceptible groups.
COPD is often accompanied by different comorbidities, especially cardiovascular conditions, which also affect the prognosis of the disease as well as quality of life and cost of COPD.46 47 Consequently, we do not exclude subjects with cardiovascular comorbidity conditions unless the condition was acute or too severe. We use the selected grades 1–3 comorbidity list in ACE-279 to exclude patients with comorbidities within grade 2 or 3 in all organ systems except the respiratory system. We also exclude subjects with systemic inflammatory diseases such as rheumatoid arthritis, because we might investigate systemic inflammation related to other systemic inflammatory diseases.
In conclusion, this study will provide valuable information regarding the pathogenetic mechanisms underlying the development of COPD, which in the future will help us to develop new targets for the management of different phenotypes of COPD.
Supplementary Material
Footnotes
Contributors: NHTtH, J-WL, DSP, LK, RB, AJMvO, WT and HMB participated in the design and supervision of the study. ATLTL and SJMH were involved in the patient-related investigations and contributed equally to the manuscript. LF performed the proteomic analyses of epithelial lining fluid. RFH investigated in vitro corticosteroid sensitivity of epithelial cells. All the authors read and approved the final manuscript.
Funding: This research was carried out in the framework of the Top Institute Pharma project T1-108 ‘Acute and chronic inflammatory responses-COPD and smoking’, with partners University Medical Center Groningen (UMCG), University of Groningen (RUG), GRIAC Research Institute Groningen, University Medical Center Utrecht (UMCU), Nycomed BV, GlaxoSmithKline and Foundation TI Pharma. The corticosteroid sensitivity part of study 1 was also carried out in the framework of the Top Institute Pharma project T1-201 ‘COPD, transition of systemic inflammation into multiorgan pathology’, with partners University Medical Center Groningen, University Medical Center Utrecht, University Medical Center Maastricht, Nycomed BV, GlaxoSmithKline, Danone, AstraZeneca and Foundation TI Pharma.
Competing interests: WT has received payment for lectures from Chiesi, GSK and Roche diagnostics. DSP has received an unrestricted educational grant for research from AstraZeneca and Chiesi; fees for consultancies by DSP were given to the University of Groningen by AZ, Boehringer Ingelheim, Chiesi, GSK, Nycomed and TEVA; NHTtH received grants from GlaxoSmithKline, Boehringer Ingelheim, Nycomed and Chiesi. ATLTL, SJMH, LF, RB, RFH, IH, AJMvO, HMB, J-WL and LK have no competing interests to declare.
Ethics approval: This multicenter study was approved by the medical ethical committees of UMC Groningen and Utrecht, the Netherlands.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Global initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease, 2010. http://www.goldcopd.org/uploads/users/files/GOLDReport_April112011.pdf (accessed date 2011)
- 2.Joos L, Pare PD, Sandford AJ. Genetic risk factors of chronic obstructive pulmonary disease. Swiss Med Wkly 2002;132:27–37 [DOI] [PubMed] [Google Scholar]
- 3.Friedlander AL, Lynch D, Dyar LA, et al. Phenotypes of chronic obstructive pulmonary disease. COPD 2007;4:355–84 [DOI] [PubMed] [Google Scholar]
- 4.Celli BR, Cote CG, Marin JM, et al. The body-mass index, airflow obstruction, dyspnea, and exercise capacity index in chronic obstructive pulmonary disease. N Engl J Med 2004;350:1005–12 [DOI] [PubMed] [Google Scholar]
- 5.van der Vaart H, Postma DS, Timens W, et al. Acute effects of cigarette smoke on inflammation and oxidative stress: a review. Thorax 2004;59:713–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lapperre TS, Snoeck-Stroband JB, Gosman MM, et al. Effect of fluticasone with and without salmeterol on pulmonary outcomes in chronic obstructive pulmonary disease: a randomized trial. Ann Intern Med 2009;151:517–27 [DOI] [PubMed] [Google Scholar]
- 7.Culpitt SV, Rogers DF, Shah P, et al. Impaired inhibition by dexamethasone of cytokine release by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2003;167:24–31 [DOI] [PubMed] [Google Scholar]
- 8.Russell RE, Culpitt SV, DeMatos C, et al. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 2002;26:602–9 [DOI] [PubMed] [Google Scholar]
- 9.Paleri V, Wight RG. Applicability of the adult comorbidity evaluation—27 and the Charlson indexes to assess comorbidity by notes extraction in a cohort of United Kingdom patients with head and neck cancer: a retrospective study. J Laryngol Otol 2002;116:200–5 [DOI] [PubMed] [Google Scholar]
- 10.Vink JM, Willemsen G, Beem AL, et al. The Fagerstrom test for nicotine dependence in a Dutch sample of daily smokers and ex-smokers. Addict Behav 2005;30:575–9 [DOI] [PubMed] [Google Scholar]
- 11.Wendel-Vos GC, Schuit AJ, Saris WH, et al. Reproducibility and relative validity of the short questionnaire to assess health-enhancing physical activity. J Clin Epidemiol 2003;56:1163–9 [DOI] [PubMed] [Google Scholar]
- 12.Jones PW, Quirk FH, Baveystock CM. The St George's Respiratory Questionnaire. Respir Med 1991;85(Suppl B):25–31; discussion 33–7 [DOI] [PubMed] [Google Scholar]
- 13.Livingston E, Chaudhuri R, McMahon AD, et al. Systemic sensitivity to corticosteroids in smokers with asthma. Eur Respir J 2007;29:64–71 [DOI] [PubMed] [Google Scholar]
- 14.Meerwaldt R, Graaff R, Oomen PH, et al. Simple non-invasive assessment of advanced glycation endproduct accumulation. Diabetologia 2004;47:1324–30 [DOI] [PubMed] [Google Scholar]
- 15.Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J 2005;26:319–38 [DOI] [PubMed] [Google Scholar]
- 16.ATS Committee on Proficiency Standards for Clinical Pulmonary Function Laboratories ATS statement: guidelines for the six-minute walk test. Am J Respir Crit Care Med 2002;166:111–17 [DOI] [PubMed] [Google Scholar]
- 17.Baarends EM, van Marken Lichtenbelt WD, Wouters EF, et al. Body-water compartments measured by bio-electrical impedance spectroscopy in patients with chronic obstructive pulmonary disease. Clin Nutr 1998;17:15–22 [DOI] [PubMed] [Google Scholar]
- 18.Snoeck-Stroband JB, Lapperre TS, Gosman MM, et al. Chronic bronchitis sub-phenotype within COPD: inflammation in sputum and biopsies. Eur Respir J 2008;31:70–7 [DOI] [PubMed] [Google Scholar]
- 19.Workshop summary and guidelines: investigative use of bronchoscopy, lavage, and bronchial biopsies in asthma and other airway diseases. J Allergy Clin Immunol 1991;88:808–14 [DOI] [PubMed] [Google Scholar]
- 20.Hattotuwa K, Gamble EA, O'Shaughnessy T, et al. Safety of bronchoscopy, biopsy, and BAL in research patients with COPD. Chest 2002;122:1909–12 [DOI] [PubMed] [Google Scholar]
- 21.British Thoracic Society Bronchoscopy Guidelines Committee, a Subcommittee of Standards of Care Committee of British Thoracic Society . British Thoracic Society guidelines on diagnostic flexible bronchoscopy. Thorax 2001;56(Suppl 1):i1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Broekema M, ten Hacken NH, Volbeda F, et al. Airway epithelial changes in smokers but not in ex-smokers with asthma. Am J Respir Crit Care Med 2009;180:1170–8 [DOI] [PubMed] [Google Scholar]
- 23.Komaki Y, Sugiura H, Koarai A, et al. Cytokine-mediated xanthine oxidase upregulation in chronic obstructive pulmonary disease's airways. Pulm Pharmacol Ther 2005;18:297–302 [DOI] [PubMed] [Google Scholar]
- 24.Ishizaka A, Watanabe M, Yamashita T, et al. New bronchoscopic microsample probe to measure the biochemical constituents in epithelial lining fluid of patients with acute respiratory distress syndrome. Crit Care Med 2001;29:896–8 [DOI] [PubMed] [Google Scholar]
- 25.Kelsen SG, Mardini IA, Zhou S, et al. A technique to harvest viable tracheobronchial epithelial cells from living human donors. Am J Respir Cell Mol Biol 1992;7:66–72 [DOI] [PubMed] [Google Scholar]
- 26.Luijk B, Lindemans CA, Kanters D, et al. Gradual increase in priming of human eosinophils during extravasation from peripheral blood to the airways in response to allergen challenge. J Allergy Clin Immunol 2005;115:997–1003 [DOI] [PubMed] [Google Scholar]
- 27.Koenderman L, Kanters D, Maesen B, et al. Monitoring of neutrophil priming in whole blood by antibodies isolated from a synthetic phage antibody library. J Leukoc Biol 2000;68:58–64 [PubMed] [Google Scholar]
- 28.Diamond MS, Springer TA. A subpopulation of Mac-1 (CD11b/CD18) molecules mediates neutrophil adhesion to ICAM-1 and fibrinogen. J Cell Biol 1993;120:545–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johansson MW, Kelly EA, Busse WW, et al. Up-regulation and activation of eosinophil integrins in blood and airway after segmental lung antigen challenge. J Immunol 2008;180:7622–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanters D, ten Hove W, Luijk B, et al. Expression of activated Fc gamma RII discriminates between multiple granulocyte-priming phenotypes in peripheral blood of allergic asthmatic subjects. J Allergy Clin Immunol 2007;120:1073–81 [DOI] [PubMed] [Google Scholar]
- 31.Pillay J, Hietbrink F, Koenderman L, et al. The systemic inflammatory response induced by trauma is reflected by multiple phenotypes of blood neutrophils. Injury 2007;38:1365–72 [DOI] [PubMed] [Google Scholar]
- 32.Oudijk EJ, Lo Tam Loi AT, Langereis JD, et al. Functional antagonism by GM-CSF on TNF-alpha-induced CD83 expression in human neutrophils. Mol Immunol 2008;46:91–6 [DOI] [PubMed] [Google Scholar]
- 33.Langereis JD, Franciosi L, Ulfman LH, et al. GM-CSF and TNFalpha modulate protein expression of human neutrophils visualized by fluorescence two-dimensional difference gel electrophoresis. Cytokine 2011;56:422–9 [DOI] [PubMed] [Google Scholar]
- 34.de Jager W, te Velthuis H, Prakken BJ, et al. Simultaneous detection of 15 human cytokines in a single sample of stimulated peripheral blood mononuclear cells. Clin Diagn Lab Immunol 2003;10:133–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huijsmans RJ, de Haan A, ten Hacken NN, et al. The clinical utility of the GOLD classification of COPD disease severity in pulmonary rehabilitation. Respir Med 2008;102:162–71 [DOI] [PubMed] [Google Scholar]
- 36.O'Neil SE, Lundback B, Lotvall J. Proteomics in asthma and COPD phenotypes and endotypes for biomarker discovery and improved understanding of disease entities. J Proteomics 2011;75:192–201 [DOI] [PubMed] [Google Scholar]
- 37.Anderson GP. Endotyping asthma: new insights into key pathogenic mechanisms in a complex, heterogeneous disease. Lancet 2008;372:1107–19 [DOI] [PubMed] [Google Scholar]
- 38.Vestbo J, Anderson W, Coxson HO, et al. Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points (ECLIPSE). Eur Respir J 2008;31:869–73 [DOI] [PubMed] [Google Scholar]
- 39.Agusti A, Calverley PM, Celli B, et al. Characterisation of COPD heterogeneity in the ECLIPSE cohort. Respir Res 2010;11:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vestbo J, Edwards LD, Scanlon PD, et al. Changes in forced expiratory volume in 1 s over time in COPD. N Engl J Med 2011;365:1184–92 [DOI] [PubMed] [Google Scholar]
- 41.Qiu W, Cho MH, Riley JH, et al. Genetics of sputum gene expression in chronic obstructive pulmonary disease. PLoS One 2011;6:e24395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castaldi PJ, Cho MH, Litonjua AA, et al. The Association of Genome-Wide Significant Spirometric Loci with Chronic Obstructive Pulmonary Disease Susceptibility. Am J Respir Cell Mol Biol 2011;45:1147–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lomas DA, Silverman EK, Edwards LD, et al. Serum surfactant protein D is steroid sensitive and associated with exacerbations of COPD. Eur Respir J 2009;34:95–102 [DOI] [PubMed] [Google Scholar]
- 44.Sin DD, Miller BE, Duvoix A, et al. Serum PARC/CCL-18 concentrations and health outcomes in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2011;183:1187–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pillai SG, Kong X, Edwards LD, et al. Loci identified by genome-wide association studies influence different disease-related phenotypes in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2010;182:1498–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sin DD, Anthonisen NR, Soriano JB, et al. Mortality in COPD: role of comorbidities. Eur Respir J 2006;28:1245–57 [DOI] [PubMed] [Google Scholar]
- 47.Anthonisen NR, Skeans MA, Wise RA, et al. The effects of a smoking cessation intervention on 14.5-year mortality: a randomized clinical trial. Ann Intern Med 2005;142:233–9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.