

Abstract
Purpose
Tumors with oncogenic dependencies on the HER family of receptor tyrosine kinases (RTKs) often respond well to targeted inhibition. Our previous work suggested that many cell lines derived from squamous cell carcinomas of the head and neck (SCCHNs) depend on autocrine signaling driven by HER2/3 dimerization and high-level co-expression of HRG. Additionally, results from a Phase I trial of MEHD7495A, a dual-action antibody that blocks ligand binding to EGFR and HER3, suggest that high-level HRG expression was associated with clinical response in SCCHN patients. Here we explore the hypothesis that high-level HRG expression defines a subpopulation of SCCHNs with activated HER3.
Experimental Design
qRT-PCR expression profiling was performed on >750 tumors of diverse origin, including >150 therapy-naïve, primary, and recurrent SCCHNs. Activated HER3, defined by immunoprecipitation of phospho-HER3, was compared to HRG expression in SCCHN samples. Paracrine versus autocrine expression was evaluated using RNA-in situ hybridization.
Results
SCCHN tumors express the highest levels of HRG compared to a diverse collection of other tumor types. We show that high HRG expression is associated with activated HER3, whereas low HRG expression is associated with low HER3 activation in SCCHN tumors. Furthermore, HRG expression is higher in recurrent SCCHN compared to patient-matched therapy naïve specimens.
Conclusions
HRG expression levels define a biologically distinct subset of SCCHN patients. We propose that high-level expression of HRG is associated with constitutive activation of HER3 in SCCHN and thus defines an actionable biomarker for interventions targeting HER3.
Introduction
Approximately 52,140 new cases of squamous cell carcinoma of the head and neck (SCCHN) were diagnosed and 11,460 people are estimated to have died from this disease in the United States last year [1]. Curative interventions for SCCHN include surgery, radiation, and combined radio-chemotherapy. The overall 5-year relative survival rate for primary SCCHN is approximately 60%. However, the 5-year relative survival rate is only 35% for patients diagnosed with metastatic disease [2]. The poor outcomes in patients with advanced SCCHN clearly indicate the need for more effective therapies in this population [3].
Signaling through the epidermal growth factor receptor (EGFR) pathway is a major driver of SCCHN [4]. EGFR is overexpressed in up to 90% of all SCCHN [5], [6]. EGFR inhibition with cetuximab has proven to be a successful therapeutic strategy, albeit with somewhat limited long-term clinical benefits due to intrinsic or acquired resistance [7]. Tyrosine kinase inhibitors (TKIs), such as erlotinib, gefitinib, and lapatinib, that target EGFR and or HER2 have been investigated in clinical studies of SCCHN but have not demonstrated a survival advantage in randomized trials [8], [9], [10].
Recently we showed that HER3 signaling also plays an important role in SCCHN [11], [12]. These preclinical studies suggested that constitutive, high-level activation of HER3 signaling can occur in the absence of direct genetic activation of HER3 (e.g. mutation or amplification) or its ligand (mutation or amplification of Heregulin (HRG)). In addition, this activation did not occur as a result of mutation or amplification of the HER3 co-receptors EGFR or HER2. Rather, we demonstrated that chronic HER3 signaling was driven by high-level and potentially autocrine expression of the alpha and beta forms of Neuregulin 1 (NRG1), collectively known as (HRG) [13], [14] in cell lines derived from SCCHN. Moreover, in a recent phase I trial of an antibody that blocks ligand binding to EGFR and HER3, the two patients who exhibited partial responses had SCCHN tumors that expressed high levels of HRG, suggesting a potential causal relationship between high HRG expression and activity of MEHD7495A [12], [15]. Thus high expression of HRG could define a population of tumors that may have an oncogenic dependency on ligand-activated signaling via HER3. To further explore the hypothesis that high-level HRG expression defines a sub-population of SCCHN that may be sensitive to agents targeting HER3 and to identify other potential target indications, we evaluated the expression of HRG in large cohorts of patient samples from multiple solid tumor indications and surveyed the activation status of HER3 in SCCHN tumor samples.
Materials and Methods
Ethics Statement
First-line NSCLC samples
All patient samples were obtained as part of a completed Phase II clinical trial and is listed at http://clinicaltrials.gov/ct2/show/NCT00480831. All samples were collected and analyzed with written informed consent. The protocol for this trial was reviewed by an external review board (The Copernicus Group IRB; http://www.cgirb.com/) prior submission to the regulatory agencies as well as by the review boards associated with each site.
Second/third line NSCLC samples
All patient samples were obtained as part of a completed Phase II clinical trial and is listed at http://clinicaltrials.gov/ct2/show/NCT00854308. All samples were collected and analyzed with written informed consent. The protocol for this trial was reviewed by an external review board (The Copernicus Group IRB; http://www.cgirb.com/) prior submission to the regulatory agencies as well as by the review boards associated with each site.
First-line CRC samples
All patient samples were obtained as part of a completed Phase II clinical trial and is listed at http://clinicaltrial.gov/ct2/show/NCT00636610. All samples were collected and analyzed with written informed consent. The protocol for this trial was reviewed by an external review board (The Copernicus Group IRB; http://www.cgirb.com/) prior submission to the regulatory agencies as well as by the review boards associated with each site.
Tissue Specimens
A total of 754 tumor specimens were used in this study: 127 SCCHN (All stages, primary and recurrent), 117 surgically resected NSCLC (Stage I–IV), 102 NSCLCs from patients with untreated metastatic disease (Stage III–IV), 82 NSCLCs from patients who went on to fail front-line standard-of-care but who ultimately received 2L therapy (Stage III–IV), 29 metastatic platinum refractory ovarian cancers, 149 therapy-naïve metastatic colorectal cancers, 44 primary and metastatic melanomas, and 29 samples from patients with triple-negative breast cancer (stage I–IV). A separate cohort of 19 fresh frozen SCCHNs were used to correlate phosphor-HER3 levels by IP-western with HRG transcript. In addition, a separate series of matched samples from a cohort of 28 patients (biopsies from initial diagnosis and at time of first recurrence) with recurrent SCCHN was obtained. Tables summarizing the available pathologic and demographic variables of these patients and their tumors may be found in the supplementary information. All samples except 1st Line NSCLC, 2/3L NSCLC, and 1st Line metastatic CRC (referenced under the Ethics Statement) were obtained from commercial sources following IRB approval (http://www.libertyirb.com/) and appropriate confirmation of written informed consent.
Cell Lines and HRG Induction
Cell lines were obtained from commercial sources. All cell lines were genotyped and tested for mycoplasma prior to experimentation. Cell lines were incubated in RPMI1640 (Sigma) with 10% FBS (Hyclone) at 37 degrees Celsius in a humidified atmosphere with 5% CO2. For the HRG induction experiments, cells were plated in 10 cm dishes at 50% confluence and allowed to adhere to the plates overnight. Cells were then washed with serum-free medium and then incubated for 24 hours in RPMI1640 supplemented with 0.5% serum. The samples were treated with 10 µg/ml MEHD7495A or vehicle at 22 hours and then treated with 3 nm of HRG (Genentech protein synthesis core facility). After 10 minutes, the cells were washed and then lysed as described below. Western analysis was performed using standard protocols. The following antibodies were purchased from Cell Signaling Technologies: pHER3 (Y1289; #4791), AKT (#9272), pAKT (Ser473; #9271), pERK (T202/Y204; #9101), ERK (#9102). The total HER3 (sc-285) and HSP90 (sc-7947) antibodies were purchased from Santa Cruz Biotechnologies.
Immunoprecipitations and Immunoblotting
For immunoprecipitation of tumor tissue, tumor content was verified by H&E requiring a minimum of 50% tumor (most samples had >75% tumor tissue). Tumors were minced on dry ice, then homogenized in chilled lysis buffer (BioWorld, 22040045-2) supplemented with phosphatase and protease inhibitors (Sigma, P5726-5ML, Roche, 13146100). 25 µl anti-HER3 antibody (Santa Cruz, sc-285-G) and 15 µl Dynabeads (Invitrogen, 100.07D) were added to 1–2 mg soluble protein per immunoprecipitation. Samples were then allowed to rotate overnight at 4°C. Beads were then separated using a magnet, and washed three times in lysis buffer. Protein was eluted in sample buffer (Invitrogen, NP0007 and NP0009) by boiling at 95°C for 5 minutes. Western blotting was performed using standard protocols. 15 µl of eluted protein was loaded per sample and immunodetection was performed using pHER3 (Y1289) (Cell Signaling Technology; #4791) or pan p-Tyr (#9441, Cell Signaling Technology). Total HER3 was evaluated in the tumor lysates prior to immunoprecipitation with sc-285-G.
Tissue Processing for Nucleic Acids
For RNA extraction tissue sections were submerged in 300 µl RLT buffer (Qiagen) and homogenized using a gentleMACS Octo Dissociator (Miltenyi Biotec). The samples were then split in half for DNA prep (DNAeasy, Qiagen) and RNA prep (TriZol, Invitrogen), as per the manufacturer’s instructions.
Fluidigm Expression Analysis
Gene expression analysis was performed on the cell lines and formalin-fixed paraffin embedded tumor samples using the BioMark 96×96 gene expression platform (Fluidigm). For the tumors, 2 µl of total RNA was reverse-transcribed to cDNA and pre-amplified in a single reaction using Superscript III/Platinum Taq (Invitrogen) and Pre-amplification reaction mix (Invitrogen). The pre-amplification reaction was performed at a final dilution of 0.05x original Taqman assay concentration (Applied Biosystems). The thermocycling conditions were as follows: 1 cycle of 50°C for 15 min, 1 cycle of 70°C for 2 min, then 14 cycles of 95°C for 15 sec and 60°C for 4 min.
Pre-amplified cDNA was diluted 1.94-fold and then amplified using Taqman Universal PCR MasterMix (Applied Biosystems) on the BioMark BMK-M-96.96 platform (Fluidigm) according to the manufacturer’s instructions. All samples were assayed in triplicate. Two custom-designed assays targeting the reference genes, AL-1377271 and VPS-33B, were included in the expression panel. Primer/probe sequences are available upon request. A mean of the Ct values for the two reference genes was calculated for each sample, and expression levels of HRG and HER3 were determined using the delta Ct (dCt) method as follows: Mean Ct (Target Gene) - Mean Ct (Reference Genes).
Analysis of Distribution of HRG Expression and Cut-off Determination
The bimodal distribution of log10 (HRG) in HNSCC motivates the fitting of the mixture of two normal distributions, one corresponding to the lack of over-expression and the other to over-expression. Let xi denote the log10 HRG expression for the ith sample (i = 1,…,n). The likelihood for the mixture model is:
where 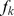 is the normal density function of the kth component, and (
is the normal density function of the kth component, and (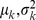 ) denote the corresponding mean and variance parameters. Maximum likelihood estimates of model parameters were obtained via the EM algorithm [16]. Briefly, the E-step computes the conditional probability that the ith sample belongs to the kth component of the mixture given the current parameter estimates. The M-step computes the mixing proportions, means and variances given the current probabilities. The process is then iterated to converge. Posterior probability of component membership was computed for each sample, and the cutoff was selected at the value where the posterior probabilities for the two components were equal. Model fitting was performed using the R package mixtools [17].
) denote the corresponding mean and variance parameters. Maximum likelihood estimates of model parameters were obtained via the EM algorithm [16]. Briefly, the E-step computes the conditional probability that the ith sample belongs to the kth component of the mixture given the current parameter estimates. The M-step computes the mixing proportions, means and variances given the current probabilities. The process is then iterated to converge. Posterior probability of component membership was computed for each sample, and the cutoff was selected at the value where the posterior probabilities for the two components were equal. Model fitting was performed using the R package mixtools [17].
Dual Color Chromogenic RNA-ISH
Dual color RNA in situ hybridization was performed by Advanced Cell Diagnostics (Fremont, CA). The HRG probe set has 31 pairs of oligos (62 total) covering nt 1082–3001 of transcript NM_013964. The HER3 probe set is essentially a pool of two probe sets which together cover all of the transcript variants: 20 pairs of oligos (40 total) covering nt 1962–2945 of NM_001982. 14 pairs of oligos (28 total) covering nt 108–899 of NM_001005915. Probes designed against cyclophilin B (PPIB; positive) and the bacterial gene dihydrodipicolinate reductase (DapB; negative) were used as controls. Images were scanned by a Hamamatsu Nanozoomer Digital Slide scanner, running Nanozoomer software, with a 40× objective and 8-bit camera [2], [15]. The MatLab (MathWorks, Natick, MA) scoring algorithm consists of the following steps: A region of interest with a minimum of 75% tumor cells was manually defined by a pathologist for each section, then a haematoxylin mask was created to identify nuclei, followed by a blue mask (HRG) and a red mask (HER3) was applied. Individual “cells” were defined by the haematoxylin mask in order to unambiguously separate cells whereupon blue or red dot counts were tabulated for each cell.
Scanned images were also analyzed using Definiens Developer (Munich, AG), using the RGB (red, green and blue) spectra. The same region of interest was used for analysis as in the MATLAB method. The region was subdivided into tiled regions of approximately 300 um height and width and analyzed at full resolution. Both color intensity (balance of red, green, and blue intensity values) and object size were used as criteria to distinguish between cell nuclei, HRG, and HER3, from background. Heavily overlapping nuclei, which were impossible to spectrally separate were excluded from analysis, in order to avoid bias.
Both methods were used to derive each of the following groups of cells: HER3+/HRG+, HER3+/HRG−, HER3−/HRG+, and HER3−/HRG−. Autocrine signal was defined as (HER3+/HRG+)/((HER3+/HRG+)+(HER3+/HRG−)+(HER3−/HRG+)).
Results
To identify potential target indications and patient populations that might be sensitive to HER3 inhibition, we profiled the expression of HRG and HER3 in more than 750 individual tumor specimens using a high throughput multiplex qRTPCR system [18]. These tumor specimens included a large cohort of both primary and metastatic squamous and non-squamous NSCLCs derived from patients eligible for first and second line chemotherapy. In addition, we analyzed samples from patients with metastatic colorectal carcinoma (CRC), therapy-naïve and recurrent SCCHN, surgically resected melanoma, surgically resected triple-negative breast (TNB) cancer, and surgically resected ovarian cancers including those from patients with primary and acquired platinum-refractory disease (Pl/R Ova) (Table S1).
SCCHNs expressed the highest median levels of HRG compared to all other tumor types examined (Figure 1A; Figure S1A). In addition, a significant subset (approx. 40%) of these SCCHNs expressed higher levels of HRG than any other tumor type (Mann-Whitney test p<0.0001; Figure S1A). Furthermore, HRG expression exhibited a bimodal distribution in SCCHN when plotted on a log10 scale (Figure 1B, Figure S1A). A two-component Gaussian mixture distribution was used to estimate the inflection point between high and low expression, which was found to be near 1.5 on arbitrary linear scale. The misclassification rates, based on the fitted models, were 9.2% and 6.6% for the high expression and low expression populations, respectively.
Figure 1. Analysis of HRG expression in the indicated epithelial tumors.

A) qRTPCR of HRG in the indicated tumor types shows that SCCHNs have the highest median expression of HRG. NSCLC = non-small cell lung cancer; TI–III = pathological stage I–III; CRC = metastatic colorectal carcinoma; Pl/R Ova = platinum refractory ovarian cancers; SCCHN = squamous cell carcinoma of the head and neck; TNB = triple negative breast cancer. B) Statistical analysis of HRG expression in SCCHNs. The two mixture distributions and their joined distribution are depicted in red, green and black lines above. The dotted line indicates the inflection point between the high and low components of expression of HRG in SCCHN, which is set at 0.3689 on the logarithm scale. The sensitivity of identifying HRG overexpressed population using a cutoff of 0.3689 in the logarithm scale (∼1.45 on the linear scale) is 90.8% and the attendant specificity is 93.4%. C) IP-westerns for pHER3 (Y1289), pTyr, and total HER3 in 19 fresh frozen SCCHNs. MCF7 is a negative control; MCF7+ HRG and PCI6A are positive controls. The controls for the pTyr blot were run separately whereas the controls for the pHER3 were run concurrently. D) qRTPCR for HRG in 18/19 SCCHNs (overlap with the IP-western). Black lines below the x-axis indicate the tumors with detectable pHER3 by IP-western.
We have previously demonstrated that in some SCCHN cell lines high-level co-expression of HRG and HER3 leads to constitutive HER3 signaling, which can be blocked by the HER3 arm of MEHD7495A [11]. We have also shown that MEHD7495A blocks ligand dependent activation of both HER3 and EGFR in vivo [12]. To confirm and extend our previous findings, we serum-starved several cell lines for 24 hours and then induced them using HRG with or without pre-incubation with MEHD7495A. As expected, HRG induced pHER3 as well as downstream activation of AKT in most cell lines (Figure S1C). MEHD7495A blocked this activation in all lines except EBC-1, which is known to harbor an amplification of c-MET.
To determine if high HRG expression was associated with activated HER3 in SCCHN tumors, we performed qRT-PCR for HRG and immunoprecipitation (IP) of total HER3 followed by Western blot for pHER3 and p-Tyrosine in fresh frozen tumor specimens from patients with therapy naïve SCHNN (Figure 1C & D; Table S1). We selected pHER3 as a read-out for this association because it is the most proximal and experimentally feasible (in human tumors) measure of ligand-activation of HER3 and thus the most relevant endpoint to the mechanism of action for MEHD7495A. All tumors with high HRG expression (at or near the low-point within the bimodal distribution) were positive for pHER3 by IP-Western; conversely, 5/7 tumors with the lowest expression of HRG were negative for pHER3 (two-tailed sign test, P = 0.0386).
To further characterize SCCHN patient populations with tumors that show high HRG expression we obtained tumor samples from patients with surgically resectable SCCHN as well as tumor samples from patients with recurrent disease. HRG expression was higher in the recurrent setting compared to primary resectable disease (Figure 2A). These findings could not be explained by differences in site of origin, HPV status, or any other available clinicopathological information (data not shown).
Figure 2. Expression of HRG in primary vs. recurrent SCCHN. A).
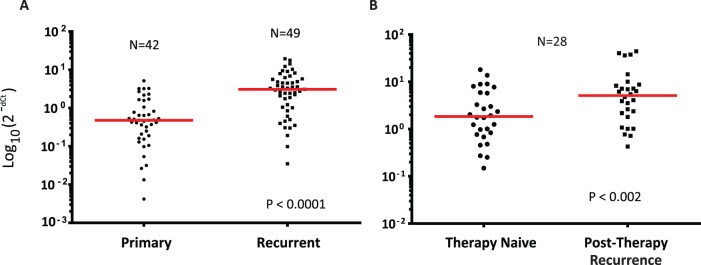
qRTPCR analysis comparing the levels of HRG expression in unmatched primary and recurrent SCCHN specimens. B) qRTPCR analysis comparing the levels of HRG expression in matched primary and recurrent SCCHN. In both cases HRG levels appear to be higher in the recurrent disease setting compared to primary resected SCCHN.
The finding that HRG expression may differ in primary versus recurrent disease could suggest that HRG expression increases as a consequence of prior therapy, or because high HRG expression is a prognostic factor associated with an increased likelihood of recurrence in patients with SCCHN. To explore this question further, we obtained a series of patient matched primary tumors and relapse specimens and compared HRG expression in both cohorts by qRTPCR. As shown in Figure 2B (Table 1 & 2), HRG expression is significantly higher in the recurrent setting compared to the primary disease in matched patient specimens (Wilcoxon signed rank test; P = 0.002).
Table 1. Detailed evaluation of HRG and HER3 expression in matched therapy naïve and post-chemotherapy SCCHNs.
| Primary• | NRG1 | HER3 | % Autocrine | Recurrance† | NRG1 | HER3 | % Autocrine | ||||
| MatLab | Tumor Cells | Tumor Area | MatLab | Tumor Cells | Tumor Area | ||||||
| RxN 1 | 0.77 | 2.06 | 8.43% | 38.0% | 19.2% | PC 1 | 2.38 | 5.70 | 10.1% | 36.8% | 27.5% |
| RxN 2 | 13.71 | 3.36 | 4.41% | 21.3% | 10.3% | PC 2 | 4.09 | 5.83 | 4.3% | 17.9% | 8.3% |
| RxN 3 | 1.77 | 4.88 | 7.62% | 27.6% | 13.6% | PC 3 | 7.18 | 6.15 | 15.0% | 36.9% | 24.8% |
| RxN 4 | 8.99 | 0.62 | #N/A | #N/A | #N/A | PC 4 | 36.13 | 2.02 | 10.0% | 34.6% | 24.0% |
| RxN 5 | 2.03 | 11.68 | #N/A | #N/A | #N/A | PC 5 | 3.53 | 12.44 | 12.7% | 40.4% | 25.0% |
| RxN 6 | 0.46 | 6.58 | 3.21% | 41.9% | 29.7% | PC 6 | 0.77 | 6.89 | 2.1% | 42.9% | 27.9% |
| RxN 7 | 3.28 | 3.07 | #N/A | #N/A | #N/A | PC 7 | 6.32 | 5.49 | 5.5% | 30.0% | 15.7% |
| RxN 8 | 1.92 | 4.23 | 23.96% | 47.6% | 39.1% | PC 8 | 3.90 | 5.50 | #N/A | #N/A | #N/A |
| RxN 9 | 1.74 | 1.46 | 17.36% | 40.3% | 31.5% | PC 9 | 40.27 | 6.63 | 6.9% | 37.5% | 24.7% |
| RxN 10 | 1.01 | 3.09 | 5.51% | 35.7% | 23.3% | PC 10 | 1.80 | 3.95 | #N/A | #N/A | #N/A |
| RxN 11 | 5.91 | 5.10 | 7.97% | 31.8% | 20.9% | PC 11 | 6.93 | 9.09 | 2.9% | 23.9% | 12.7% |
| RxN 12 | 7.71 | 4.35 | 9.86% | 31.3% | 18.9% | PC 12 | 7.14 | 7.66 | 1.8% | 10.9% | 3.5% |
| RxN 13 | 0.84 | 6.16 | 14.92% | 54.9% | 45.9% | PC 13 | 5.44 | 5.69 | 14.0% | 32.3% | 18.7% |
| RxN 14 | 1.25 | 0.34 | 5.36% | 26.7% | 17.2% | PC 14 | 2.17 | 2.40 | #N/A | #N/A | #N/A |
| RxN 15 | 8.88 | 2.14 | 0.29% | 7.8% | 3.7% | PC 15 | 14.43 | 3.13 | 6.6% | 26.5% | 14.5% |
| RxN 16 | 18.10 | 7.72 | 27.30% | 58.6% | 38.5% | PC 16 | 6.19 | 2.91 | 1.9% | 13.0% | 5.9% |
| RxN 17 | 0.98 | 7.12 | 16.57% | 46.0% | 37.6% | PC 17 | 1.08 | 8.24 | 11.0% | 37.1% | 27.1% |
| RxN 18 | 0.25 | 1.33 | 9.63% | 53.9% | 39.3% | PC 18 | 0.72 | 4.41 | 8.0% | 67.1% | 56.5% |
| RxN 19 | 0.48 | 2.53 | #N/A | #N/A | #N/A | PC 19 | 1.02 | 2.81 | 17.8% | 62.4% | 48.5% |
| RxN 20 | 5.75 | 3.90 | #N/A | #N/A | #N/A | PC 20 | 44.21 | 3.28 | 14.7% | 51.3% | 43.5% |
| RxN 21 | 0.15 | 2.32 | 7.75% | 35.6% | 22.7% | PC 21 | 4.76 | 5.35 | 13.9% | 47.3% | 37.0% |
| RxN 22 | 0.68 | 0.03 | 1.45% | 8.9% | 3.0% | PC 22 | 0.43 | 0.07 | 0.4% | 16.5% | 6.4% |
| RxN 23 | 2.68 | 2.16 | 18.50% | 60.2% | 48.3% | PC 23 | 1.01 | 8.17 | 4.5% | 43.0% | 30.0% |
| RxN 24 | 3.01 | 1.34 | 1.40% | 41.0% | 28.2% | PC 24 | 8.25 | 2.03 | 22.1% | 56.6% | 44.0% |
| RxN 25 | 0.27 | 2.40 | 0.30% | 29.9% | 21.1% | PC 25 | 2.62 | 1.90 | #N/A | #N/A | #N/A |
| RxN 26 | 2.35 | 11.53 | 18.58% | 57.9% | 49.8% | PC 26 | 37.51 | 2.39 | 4.4% | 31.5% | 20.9% |
| RxN 27 | 8.01 | 7.47 | 25.61% | 51.2% | 39.7% | PC 27 | 10.06 | 5.41 | 48.3% | 71.3% | 61.7% |
| RxN 28 | 1.24 | 4.95 | 8.93% | 26.9% | 16.9% | PC 28 | 8.72 | 1.71 | 11.3% | 30.0% | 21.1% |
RxN corresponds to therapy naïve patients;
PC corresponds to Post-Chemo patients.
% Autocrine cells were based on tumor cell content only; qRTPCR was performed on macrodissected specimens as outlined in the methods section.
Table 2. Summary of the Relationship Between HRG Expression and the Extent of Autocrine versus Paracrine Expression in Matched Therapy Naïve Primary Specimens and Post Therapy Biopsies Taken at Recurrence.
| Comparison | Rx Naïve | Post-Rx | Significance* |
| HRG | 3.72 (1.99–4.45) | 9.61 (4.6–14.62) | 0.002 |
| HER3 | 4.07 (2.9–4.24) | 4.9 (3.84–5.96) | 0.026 |
| Autocrine (MatLab) | 10.65% (7.12–14.19) | 10.43% (6.62–14.59) | NS |
| Autocrine (Definiens Cells) | 38.04% (31.68–44.41) | 37.4% (30.59–44.21) | NS |
| Autocrine (Definiens Area) | 26.89% (21.01–32.77) | 26.25% (19.65–32.85) | NS |
Wilcoxon rank sign test for paired samples.
Preclinical data from a recent report suggested that identifying tumors with autocrine biology may be important in predicting response to agents targeting HER family receptors [11]. To evaluate this hypothesis, we first examined the performance of a dual-color assay RNA in situ hybridization (ISH) assay in cell lines with previously defined expression levels of HER3 and HRG (Figure S2). Overall there was strong agreement between qRTPCR and ISH for the levels of both HER3 and HRG. Consistent with previously published data, the MCF7 cell line showed abundant levels of HER3 and no detectable HRG, whereas both PCI-6A and CHL-1 showed high levels of both HER3 and HRG [11], [12]. In addition, both PCI-6A and CHL-1 had significant numbers of autocrine cells, whereas H358 was predominantly paracrine (Figure S2).
To determine the extent of autocrine versus paracrine expression of HER3 and HRG expression in SCCHNs we employed a similar (same probes with chromogenic versus fluorescent probes) dual-color chromogenic RNA in situ hybridization assay designed to evaluate HER3 and HRG transcript location and abundance in clinically relevant FFPE tissues. Qualitative examination of benign squamous epithelium suggested that expression of HRG (cyan) was limited to the basal layer, whereas HER3 (red) expression was only seen focally in normal stratified squamous epithelium (Figure 3A). In contrast, in the spiny layer, only HER3 expression was observed. A similar pattern of expression was seen in the pseudostratified upper respiratory tract epithelium where HRG expression was limited to the basal layer and HER3 expression was restricted to the upper layers of the epithelium (Figure 3B; Figure S3A & B). At the single cell level in benign tissue, absent a source of HRG, most cells and tissues express relatively consistent levels of HER3, whereas in the presence of HRG, there is a gradient of HER3 expression that increases the farther away the cells are from the source of HRG. More generally, there is an inverse relationship between the expression of HER3 and HRG both at the level of individual cells as well as in the spatial orientation of cells that express either of these two transcripts.
Figure 3. Dual color in situ hybridization for HER3 and HRG in benign and malignant tissue shows that HER3 and HRG are typically expressed by different cells.

(HER3 = red; HRG = cyan) A) Normal squamous epithelium; B) Normal upper respiratory epithelium; C) well differentiated SCCHN; D) autocrine expression of HRG and HER3. In panels A & B, the yellow arrows point to basal cells and the black arrows point to squamous epithelium or more differentiated cells within the respiratory tract. In panel C, the bracketed structure is a squamous morule, the yellow arrow again points to “basal-like” tumor cells whereas the black arrow identifies spiny cells. Panel D represents anaplastic autocrine cells, characteristic of a poorly differentiated SCCHN. Magnification is 200x for A–C and 400× for part D.
We found clear evidence of paracrine expression of HRG and HER3 in some well-differentiated head and neck squamous cell carcinomas, recapitulating the expression patterns seen in normal tissue (Figure 3C). In other more poorly differentiated squamous cell carcinomas the relationship between basal cell-specific expression of HRG and spiny cell-specific expression of HER3 was lost, consistent with the more disordered architecture, with evidence for both autocrine and paracrine expression patterns (Figures S3E, F, & G). Finally, we identified cases where the majority of HER3/HRG expressing cells appeared to be autocrine (Figure 3D).
To determine if high HRG expression was specifically associated with either autocrine or paracrine expression, we compared qRTPCR with RNA-ISH for HRG and HER3 in the same samples represented in Figure 2B. We employed three different quantitative imaging algorithms to identify autocrine cells (see methods for details). While each algorithm differed in the relative proportion of autocrine component for each tumor, the methods ranked the different tumors similarly (Spearman r: ∼0.75–0.96 for all pairwise combinations, p<0.0001 in all cases). This suggests that the algorithms were differentiating autocrine and paracrine cells in a similar way, albeit with potentially different sensitivities (Table 1).
Next we compared the RNA-ISH data to qRTPCR in tumor tissues. There was a strong, significant positive Spearman correlation between qRTPCR and RNA-ISH for HRG (r = 0.4; p = 0.002) and a lower, near significant, positive correlation between qRTPCR and RNA-ISH for HER3 (ρ = 0.2; p = 0.051) (Figure 4A & B). In contrast to our findings in cell lines (Figure S2 & [11]) we did not observe a strong association between high-level HRG expression as determined by qRTPCR and the proportion of cells with autocrine expression of HRG and HER3 as determined by RNA-ISH (Spearman r: 0.26 (p = 0.22), 0.03 (p = 0.91), respectively; Tables 1 & 2). In addition, while we did observe an increase in autocrine expression of HER3 and HRG between matched therapy-naïve and recurrent SCCHNs in some cases (Figure 4C), unlike HRG expression (p<0.002; Figure 2B), this was not statistically significant overall (Table 2; methods).
Figure 4. Quantitative comparison between RNA-ISH and QRTPCR.

A & B) Pairwise analysis of RNA-ISH and qRTPCR of HRG and HER3 in primary and recurrent SCHNNs. Spearman rank correlations and p-values are shown on the graphs. C) Representative RNA-ISH for a matched primary and recurrent section. The primary therapy naïve specimen (#3) was considered to HRG positive (2+) with frequent positive cells throughout squamous morules, but accentuated in basal cells. The tumor was also considered positive for HER3 (2+) with frequent positive cells throughout squamous morules, but accentuated in differentiated cells. This tumor was considered predominantly paracrine, but harbored occasional cells that expressed both HRG and HER3. Post-chemo Specimen #3 was also considered positive for HRG (2+) with frequent positive cells throughout squamous morules, but accentuated in basal cells. This sample was also positive for HER3 (2+) with frequent positive cells throughout squamous morules, but accentuated in differentiated cells. Her3+/Nrg1− and Her3+/Nrg1+ seen in equal measure. This finding of increased autocrine character was also evident using quantitative imaging (Table 1). (HER3 = red; HRG = cyan). Magnification is 400x.
Discussion
In this report we evaluated the quantitative relationship between HER3 and HRG expression in more than 700 hundred epithelial tumors of diverse tissue origin. We showed that a substantial subset of SCCHNs expressed significantly higher levels of HRG compared to all other tumor types examined. HRG expression has a unique bimodal distribution in SCCHN, with approximately 40% of SCCHN tumors expressing higher levels of HRG than all other tumor types. This pattern of expression is reminiscent of HER2 expression in breast cancer, where HER2 expression levels are at least an order of magnitude higher in HER2 positive breast cancers compared to other types of breast cancers. However, unlike HER2 in breast and gastric cancers, HRG overexpression does not appear to be a function of gene amplification [4], [19], [20].
We further showed that high HRG expression was associated with pHER3, possibly indicating active HER3 signaling in these particular SCCHN tumors. We found that pHER3 was detectable in all cases where HRG was expressed at or near the low point of the bimodal distribution in the SCCHN patients. In some cases pHER3 was detected in samples with HRG levels that were somewhat below the low point of the distribution. Interestingly, the low point of the bimodal distribution was similar in multiple independent data sets, potentially reflecting biologically distinct populations of SCCHN patients. Thus, based on the properties of the distribution of HRG expression and the biochemical association between HRG expression and pHER3 activity, it seems reasonable to hypothesize that a potential cut-off for identifying patients with SCCHN that might benefit from HER3-directed therapeutic intervention could be defined by comparing HRG expression levels with clinical activity in future clinical studies.
Importantly, two of the lowest HRG expressing tumors also had detectable pHER3. It has been shown that HRG-independent phosphorylation of HER3 can occur through heterodimerization with EGFR or other RTKs such as c-Met [1]. Patients with tumors exhibiting HRG-independent activation of HER3 are unlikely to benefit from HER3 directed therapies and thus it is important to note that using HRG as a predictive marker would effectively exclude this patient population [12].
Using a novel dual color ISH assay, we demonstrated that high-level expression of HRG occurs in the context of tumors with both paracrine and autocrine phenotypes, suggesting that a subset of SCCHNs may originate from cell lineages that are programmed to express abundant HRG. To our surprise this lineage appears to be the basal cells in squamous epithelium and not the stroma, whereas HER3 is expressed almost exclusively by the more differentiated squamous epithelium. It will be of interest to determine if other tumor types derived from basal cells within squamous epithelium also express similar phenotypes. As a result of the heterogeneous nature of HRG expression in SCCHNs and the absence of clinical activity to inform this analysis, it is impossible to prospectively define whether a predominantly autocrine biology will be relevant to treatment with therapeutics that inhibit HRG driven HER3 signaling, but it is an important question that should be tested in clinical trials of such agents.
We showed that that HRG expression is higher in recurrent tumor specimens compared to matched and unmatched primary tumors. These findings, taken together with the data above, suggest that HRG expression may be both predictive of response to HER3 inhibitors and prognostic for recurrence of SCCHN. It will be important to consider the evident interaction between the prognostic and predictive properties of HRG expression in the design of future clinical trials.
In conclusion, we suggest that HRG expression levels define a statistically and biologically distinguishable subset of SCCHN patients. We propose that high-level expression of HRG is associated with constitutive activation of HER3 in SCCHN and thus defines an actionable biomarker for drugs that inhibit this important oncogene.
Supporting Information
Analysis of HRG Expression and the effect of MEHD7495A. A) HRG expression depicted as a histogram demonstrates a bimodal distribution in HNSCC. Red bars indicate SCCHN; blue bars indicate all other cancer types examined including NSCLC, CRC, platinum refractory ovarian cancers, tripe negative breast cancers, and melanoma. The percentage of samples within a given in the overall distribution is indicated on the y-axis; transcript level (2−dCt) is indicated on the x-axis. B) Analysis of HER3 expression in the indicated epithelial tumors. NSCLC = non-small cell lung cancer; TI–III = pathological stage I–III; CRC = metastatic colorectal carcinoma; Pl/R Ova = platinum refractory ovarian cancers; SCCHN = squamous cell carcinoma of the head and neck; TNB = triple negative breast cancer. C) MEHD7495A blocks HRG-dependent activation of HER3 signaling. Western blots of HER3, pHER3, AKT, pAKT, ERK, pERK, and HSP90 are shown. Treatment conditions are indicated above each lane. Cells were treated as described in the methods section.
(PPTX)
Validation of dual-color In Situ hybridization assay for HRG and HER3 in the indicated cell lines. A) Fluorescent micrographs of MCF7 cells treated with probes corresponding to the HER3 (ERBB3) and HRG (NRG1) transcript show that MCF7 does not express detectable HRG but has abundant expression of HER3; B) H358; C) PCI-6A; D) CHL-1.
(PPTX)
Additional examples of the colormetric dual color In Situ hybridization assay for HRG and HER3 in benign and malignant head and neck tissues.
(TIF)
Select clinical and demographic information on patient samples evaluated for HRG and HER3 expression.
(XLSX)
Acknowledgments
We would like to thank Drs. Scott Holden, Mark Lackner, and Mark Sliwkowski for critically reading the manuscript. In addition, we would like to thank the MEHD7495A development team, Stephanie Johnson, the Human Tissue Labs, and Pathology Core at Genentech. Finally, we would like to thank the patients who contributed their tissue to this study.
Funding Statement
This study was funded by Genentech (www.gene.com). All authors are employees of Genentech and Genentech played a role in study design, data collection and analysis, decision to publish and preparation of the manuscript.
References
- 1. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, et al. (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316: 1039–1043. [DOI] [PubMed] [Google Scholar]
- 2. Baselga J, Swain SM (2009) Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nature reviews Cancer 9: 463–475. [DOI] [PubMed] [Google Scholar]
- 3. Lefebvre JL (2005) Current clinical outcomes demand new treatment options for SCCHN. Ann Oncol 16 Suppl 6vi7–vi12. [DOI] [PubMed] [Google Scholar]
- 4. Leemans CR, Braakhuis BJ, Brakenhoff RH (2011) The molecular biology of head and neck cancer. Nature reviews Cancer 11: 9–22. [DOI] [PubMed] [Google Scholar]
- 5. Morgan S, Grandis JR (2009) ErbB receptors in the biology and pathology of the aerodigestive tract. Exp Cell Res 315: 572–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kalyankrishna S, Grandis JR (2006) Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol 24: 2666–2672. [DOI] [PubMed] [Google Scholar]
- 7. Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, et al. (2006) Radiotherapy plus Cetuximab for Squamous-Cell Carcinoma of the Head and Neck. New England Journal of Medicine 354: 567–578. [DOI] [PubMed] [Google Scholar]
- 8. Cohen EEW, Davis DW, Karrison TG, Seiwert TY, Wong SJ, et al. (2009) Erlotinib and bevacizumab in patients with recurrent or metastatic squamous-cell carcinoma of the head and neck: a phase I/II study. The Lancet Oncology 10: 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stewart JS, Cohen EE, Licitra L, Van Herpen CM, Khorprasert C, et al. (2009) Phase III study of gefitinib compared with intravenous methotrexate for recurrent squamous cell carcinoma of the head and neck [corrected]. J Clin Oncol 27: 1864–1871. [DOI] [PubMed] [Google Scholar]
- 10. del Campo JM, Hitt R, Sebastian P, Carracedo C, Lokanatha D, et al. (2011) Effects of lapatinib monotherapy: results of a randomised phase II study in therapy-naive patients with locally advanced squamous cell carcinoma of the head and neck. Br J Cancer 105: 618–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wilson TR, Lee DY, Berry L, Shames DS, Settleman J (2011) Neuregulin-1-mediated autocrine signaling underlies sensitivity to HER2 kinase inhibitors in a subset of human cancers. Cancer Cell 20: 158–172. [DOI] [PubMed] [Google Scholar]
- 12. Schaefer G, Haber L, Crocker LM, Shia S, Shao L, et al. (2011) A two-in-one antibody against HER3 and EGFR has superior inhibitory activity compared with monospecific antibodies. Cancer cell 20: 472–486. [DOI] [PubMed] [Google Scholar]
- 13. Holmes WE, Sliwkowski MX, Akita RW, Henzel WJ, Lee J, et al. (1992) Identification of heregulin, a specific activator of p185erbB2. Science 256: 1205–1210. [DOI] [PubMed] [Google Scholar]
- 14. Sliwkowski MX, Schaefer G, Akita RW, Lofgren JA, Fitzpatrick VD, et al. (1994) Coexpression of erbB2 and erbB3 proteins reconstitutes a high affinity receptor for heregulin. The Journal of biological chemistry 269: 14661–14665. [PubMed] [Google Scholar]
- 15. Cervantes-Ruiperez A, Juric D, Hidalgo M, Messersmith WA, Blumenschein GR, et al. (2012) A phase I study of MEHD7945A (MEHD), a first-in-class HER3/EGFR dual-action antibody, in patients (pts) with refractory/recurrent epithelial tumors: Expansion cohorts. ASCO Meeting Abstracts 30: 2568. [Google Scholar]
- 16. Dempster AP, Laird NM, Rubin DB (1977) Maximum Likelihood from Incomplete Data Via Em Algorithm. Journal of the Royal Statistical Society Series B-Methodological 39: 1–38. [Google Scholar]
- 17. Benaglia T, Chaiveau D, Hunter DR, Young D (2009) mixtools: An R Package for Analyzing Finite Mixture Models. Journal of Statistical Software 32: 1–29. [Google Scholar]
- 18. Walter K, Holcomb T, Januario T, Du P, Evangelista M, et al. (2012) DNA methylation profiling defines clinically relevant biological subsets of non-small cell lung cancer. Clin Cancer Res 18: 2360–2373. [DOI] [PubMed] [Google Scholar]
- 19. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, et al. (2011) Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 333: 1154–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, et al. (2011) The mutational landscape of head and neck squamous cell carcinoma. Science 333: 1157–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Analysis of HRG Expression and the effect of MEHD7495A. A) HRG expression depicted as a histogram demonstrates a bimodal distribution in HNSCC. Red bars indicate SCCHN; blue bars indicate all other cancer types examined including NSCLC, CRC, platinum refractory ovarian cancers, tripe negative breast cancers, and melanoma. The percentage of samples within a given in the overall distribution is indicated on the y-axis; transcript level (2−dCt) is indicated on the x-axis. B) Analysis of HER3 expression in the indicated epithelial tumors. NSCLC = non-small cell lung cancer; TI–III = pathological stage I–III; CRC = metastatic colorectal carcinoma; Pl/R Ova = platinum refractory ovarian cancers; SCCHN = squamous cell carcinoma of the head and neck; TNB = triple negative breast cancer. C) MEHD7495A blocks HRG-dependent activation of HER3 signaling. Western blots of HER3, pHER3, AKT, pAKT, ERK, pERK, and HSP90 are shown. Treatment conditions are indicated above each lane. Cells were treated as described in the methods section.
(PPTX)
Validation of dual-color In Situ hybridization assay for HRG and HER3 in the indicated cell lines. A) Fluorescent micrographs of MCF7 cells treated with probes corresponding to the HER3 (ERBB3) and HRG (NRG1) transcript show that MCF7 does not express detectable HRG but has abundant expression of HER3; B) H358; C) PCI-6A; D) CHL-1.
(PPTX)
Additional examples of the colormetric dual color In Situ hybridization assay for HRG and HER3 in benign and malignant head and neck tissues.
(TIF)
Select clinical and demographic information on patient samples evaluated for HRG and HER3 expression.
(XLSX)