

Abstract
Comparisons of duration of response (DoR) and duration of clinical benefit (DoCB) within clinical trials are prone to biases. To address these biases, we used new methodology to prospectively analyze expected DoR and expected DoCB. Objective response rate and clinical benefit rate were calculated for fulvestrant 500 and 250 mg, and used to calculate expected DoR and expected DoCB for each dose group. The ratios for expected DoR and expected DoCB (expected DoR500/expected DoR250 and expected DoCB500/expected DoCB250) were then calculated, thereby allowing statistical comparisons of these endpoints between each arm of the COmparisoN of Faslodex In Recurrent or Metastatic breast cancer (CONFIRM) trial. Expected DoRs for fulvestrant 500 and 250 mg were 3.2 and 3.6 months, respectively. The expected DoR ratio between fulvestrant 500 and 250 mg was not statistically significant (0.89; 95 % CI, 0.48–1.67, P = 0.724). The expected DoCBs for fulvestrant 500 and 250 mg were 9.8 and 7.2 months, respectively. The expected DoCB ratio showed that the expected DoCB for fulvestrant 500 mg was significantly improved compared with the expected DoCB for fulvestrant 250 mg (1.36; 95 % CI, 1.07–1.73, P = 0.013). Analysis of the expected DoR and expected DoCB showed fulvestrant 500 mg significantly increased expected DoCB compared with fulvestrant 250 mg in the CONFIRM trial.
Electronic supplementary material
The online version of this article (doi:10.1007/s10549-012-2395-8) contains supplementary material, which is available to authorized users.
Keywords: Duration of response, Duration of clinical benefit, Methodology, Comparison, Advanced breast cancer, Fulvestrant
Introduction
Fulvestrant is an estrogen receptor (ER) antagonist, devoid of agonistic properties, which exerts its effects via downregulation of the estrogen receptor [1, 2]. Historically, fulvestrant 250 mg was indicated for the second-line treatment of postmenopausal women with endocrine-sensitive advanced breast cancer. However, results from several studies suggested that a higher dose of fulvestrant might be more efficacious in this group of patients [2–4]. The COmparisoN of Faslodex In Recurrent or Metastatic breast cancer (CONFIRM) trial was a phase III, randomized, double-blind, parallel-group, multicenter study that evaluated two different doses of fulvestrant in postmenopausal women with locally advanced or metastatic ER-positive breast cancer who had progressed or recurred on endocrine therapy. Results from the CONFIRM trial showed that fulvestrant 500 mg was associated with a clinically meaningful improvement in progression-free survival (PFS) and overall benefit:risk ratio over fulvestrant 250 mg [5]. Based on the findings from this study, fulvestrant 500 mg is now approved by the United States [6], Europe [7], Japan, and many other markets.
Trials investigating agents for the treatment of advanced breast cancer use a variety of efficacy endpoints with which to measure potential benefit. Overall survival (OS) is generally regarded as the endpoint of choice for assessing the efficacy of new therapies in advanced breast cancer, and provides a clear and unbiased measure of clinical benefit as well as ease and reliability of measurement [8, 9]. However, this endpoint requires studies with large patient populations as well as prolonged followup of all patients, and consideration of this endpoint alone can delay the evaluation of novel therapies. In addition, assessment of OS can be confounded by factors such as crossover to active treatment arms within a trial or post-trial treatment with non-experimental active therapies [10]. As such, assessment of a variety of clinical endpoints can be of benefit, allowing a more rapid evaluation of new treatments. Indeed, endpoints such as PFS and time to progression (TTP) have been proposed as potential surrogate endpoints for survival [11]. The primary endpoint in CONFIRM was PFS, but other secondary endpoints included objective response rate (ORR), clinical benefit rate (CBR), duration of response (DoR), and duration of clinical benefit (DoCB) [5].
The proportion of patients who respond to treatment and the DoR and DoCB for patients who respond are widely evaluated during randomized oncology trials and are considered important clinical measures that help to determine a drug’s therapeutic value [12, 13]. However, within a trial, formal comparisons of DoR and DoCB between treatments in responding patients may be prone to biases and may not reflect actual treatment effects. In addition, conflicting results for response rates and DoR, e.g., higher response rate in control therapy versus longer DoR in experimental therapy, can make assessment of preferred treatment options difficult. As such, formal comparison of DoR and DoCB between treatments in responding patients is currently discouraged by the European Medicines Evaluation Agency [13, 14].
New methodology proposed by Ellis et al. enables a formal and unbiased comparison of treatments for expected DoR and expected DoCB across all randomized patients. Here, by this novel methodology, we present a prospective analysis of expected DoR and expected DoCB between the fulvestrant 250 and 500 mg dose groups from the CONFIRM trial.
Methods
Study design
The CONFIRM trial design has been described in detail previously [5]. In brief, CONFIRM was a randomized, phase III, double-blind study in which two different doses of fulvestrant (500 vs 250 mg) were evaluated in postmenopausal patients who had either locally advanced or metastatic ER-positive breast cancer.
Patients who experienced relapse during or within 1 year of completion of adjuvant endocrine therapy were eligible. For patients who experienced relapse after more than 1 year from completion of adjuvant endocrine therapy, or for patients with de novo advanced disease, eligibility required previous treatment with either an anti-estrogen or an aromatase inhibitor as a first-line therapy for metastatic breast cancer. Patients had to have measurable or evaluable disease according to Response Evaluation Criteria In Solid Tumors (RECIST) [5, 15] although patients with lytic or mixed bone lesions were permitted to enter the study without evidence of measurable disease.
Eligible patients were randomly assigned 1:1 to the following two treatment arms: fulvestrant 500 mg administered as two 5-mL intramuscular injections on days 0, 14, and 28, and every 28 (±3) days thereafter; or fulvestrant 250 mg administered as two 5-mL intramuscular injections (one fulvestrant and one placebo) on days 0, 14 (two placebo injections only), and 28, and every 28 (±3) days thereafter [5].
The primary study endpoint was PFS (the time elapsing between the date of randomization and the date of earliest evidence of objective disease progression or death from any cause). Secondary endpoints included ORR, CBR, DoR, DoCB, OS, tolerability, and quality of life [5].
Definitions
In this prospective analysis, ORR and CBR were calculated by modified RECIST (mRECIST) version 1.0 criteria. The RECIST criteria assesses, for each patient, the percentage reduction in tumor mass on a visit-by-visit basis over the course of the trial according to the following definitions: complete response (CR), defined as a disappearance of all measurable lesions and no new lesions; partial response (PR), defined as at least a 30 % reduction in the sum of the longest diameter of the measurable lesions and no progression of non-target lesions and no new lesions; stable disease (SD), defined as disease in which there is neither sufficient shrinkage to qualify for CR or PR, nor sufficient increase to qualify for progressive disease (PD) of measurable lesions, and there is no progression of non-measurable lesions and no new lesions; PD, defined as at least a 20 % increase in the sum of the longest diameter of target lesions or the progression of non-measurable lesions or the appearance of new lesions. Disease for which there was insufficient data to allocate a response was defined as not evaluable (NE).
For each patient, the best overall response achieved during the course of the trial was calculated: an objective responder was defined as any patient who had a best overall response of CR or PR (defined as two visit responses of CR or PR at least 28 days apart); a clinical benefit responder was defined as any patient who had a best overall response of CR, PR, or SD that persisted for ≥24 weeks.
DoR was calculated as the time (in months) from randomization to progression (or death from any cause) in patients who had a best overall response of CR or PR. ORR and, therefore, DoR were calculated out of all randomized patients with measurable disease at baseline (defined as at least one lesion that could be accurately measured in at least one dimension of ≥20 mm with conventional techniques or ≥10 mm with spiral computed tomography scan). Patients with non-measurable disease at baseline were not used for the calculation of DoR, as these patients only had non-targets at baseline: it was decided prospectively to assess only the non-targets for incomplete response/SD, NE, or PD and not for CR or PR.
DoCB was calculated as the time (in months) from randomization to progression (or death from any cause) in patients who had a best overall response of CR, PR, or SD of ≥24 weeks. CBR and, therefore, DoCB were calculated across all randomized patients, as patients could have non-measurable disease at baseline, but still obtain a best overall response of SD of ≥24 weeks.
Derivation of the expected DoR and expected DoCB
Expected DoR and expected DoCB were calculated by the methodology described by Ellis et al. [16]. The formula used to derive the expected DoR is shown in the Supplementary Methods; this formula was similarly used to derive the expected DoCB.
Comparison of expected DoR and expected DoCB between fulvestrant dose groups
The expected DoR is the product of the fraction of patients with a response and the mean DoR in responding patients. The following calculations were, therefore, performed to formally compare the expected DoR for the fulvestrant 500 mg group with that for the 250 mg group:
Calculation of the response rate (i.e., the number of patients who responded to each treatment divided by the total number of patients per group) for each fulvestrant dose group.
Calculation of the mean DoR and associated standard error (SE) for each fulvestrant dose group. For these data, the log Normal distribution was selected with model fitting performed by means of Statistical Analysis Software [SAS], Cary, NC, USA. For the log Normal distribution, the mean duration of response is estimated as
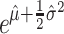 and the variance of the log of the mean is estimated as
and the variance of the log of the mean is estimated as 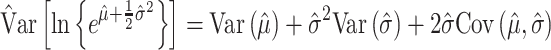
Multiplication of estimates obtained from the above calculations to obtain estimates of the expected DoR for each fulvestrant dose group.
Calculation of the ratio of expected DoR (i.e. R = expected DoR500/expected DoR250) and the variance of [ln(
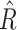 )].
)].- Assessment of the difference between the two fulvestrant treatment groups using
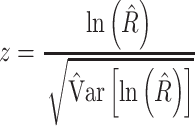
compared with a standard Normal (0,1) distribution.
Similar calculations were used to compare the expected DoCB between fulvestrant dose groups.
Results
Summary of expected DoR
The data used to calculate ORR are shown in Table 1. The ORRs for fulvestrant 500 and 250 mg were 13.8 % (33/240) and 14.6 % (38/261), respectively (odds ratio [OR] 0.94; 95 % confidence interval [CI], 0.57–1.55, P = 0.795) (evaluable for all randomized patients with measurable disease at baseline).
Table 1.
Summary of best objective response (for calculation of percent response)
| Best objective response | Number (%) of patients | |
|---|---|---|
| Fulvestrant 500 mg (n = 240) |
Fulvestrant 250 mg (n = 261) |
|
| CR | 4 (1.7) | 1 (0.4) |
| PR | 29 (12.1) | 37 (14.2) |
| SD | 98 (40.8) | 103 (39.5) |
| PD | 102 (42.5) | 117 (44.8) |
| NE | 7 (2.9) | 3 (1.1) |
Best response derived according to modified RECIST
CR complete response, NE not evaluable, PD progressive disease, PR partial response, RECIST Response Evaluation Criteria In Solid Tumors, SD stable disease
For patients with an objective response, the median DoRs from randomization for fulvestrant 500 and 250 mg were 19.4 and 16.4 months, respectively (Fig. 1). Analysis of DoR from the date of randomization for all patients with measurable disease at baseline is shown in Table 2. The mean DoRs for patients with an objective response were 23.2 and 24.5 months for fulvestrant 500 and 250 mg, respectively. The expected DoRs for all randomized patients with measurable disease at baseline for fulvestrant 500 and 250 mg were 3.2 and 3.6 months, respectively.
Fig. 1.

Duration of response from date of randomization for patients with an objective response (evaluable for all randomized patients with measurable disease at baseline). Patients with a best objective response of complete response or partial response are included
Table 2.
Analysis of duration of response
| Fulvestrant 500 mg (n = 240) |
Fulvestrant 250 mg (n = 261) |
|
|---|---|---|
| Response rate (%) | 13.8 | 14.6 |
| Mean DoRa in months (SE) | 23.2 (3.4) | 24.5 (4.3) |
| Expected DoR (months) | 3.2 | 3.6 |
| Ratio of expected DoR (95 % CI)b | 0.89 (0.48–1.67) | |
| P value | 0.724 | |
CI confidence interval, DoR duration of response, SE standard error
aLimited to patients with an objective response, i.e. a subset of those randomized patients with measurable disease at baseline
bRatios of >1 favor fulvestrant 500 mg
The ratio of expected DoR between the fulvestrant 500 and 250 mg dose groups favored fulvestrant 250 mg; however, this difference was not statistically significant (ratio of expected DoR = 0.89; 95 % CI, 0.48–1.67, P = 0.724).
Summary of expected DoCB
The data used to calculate CBR are shown in Table 3. The CBRs for fulvestrant 500 and 250 mg were 45.6 % (165/362) and 39.6 % (148/374), respectively (OR 1.28; 95 % CI, 0.95–1.71, P = 0.100) (evaluable for all randomized patients).
Table 3.
Summary of best objective response (for calculation of duration of percent response)
| Best objective response | Number (%) of patients | |
|---|---|---|
| Fulvestrant 500 mg (n = 362) |
Fulvestrant 250 mg (n = 374) |
|
| CR | 4 (1.1) | 1 (0.3) |
| PR | 29 (8.0) | 37 (9.9) |
| SD ≥24 weeks | 132 (36.5) | 110 (29.4) |
| SD <24 weeks | 47 (13.0) | 52 (13.9) |
| PD | 140 (38.7) | 167 (44.7) |
| NE | 10 (2.8) | 7 (1.9) |
Best response derived according to modified RECIST
CR complete response, NE not evaluable, PD progressive disease, PR partial response, RECIST Response Evaluation Criteria In Solid Tumors, SD stable disease
For patients with clinical benefit, the median DoCB from randomization was numerically longer for the fulvestrant 500 mg group (16.6 months) compared with the fulvestrant 250 mg group (13.9 months) (Fig. 2). The mean DoCBs for patients with clinical benefit were 21.6 and 18.3 months for fulvestrant 500 and 250 mg, respectively. The expected DoCBs for all randomized patients for fulvestrant 500 and 250 mg were 9.8 and 7.2 months, respectively (Table 4).
Fig. 2.

Duration of clinical benefit in patients with clinical benefit (evaluable for all randomized patients). Patients with a best objective response of complete response, partial response, or stable disease of ≥24 weeks' duration are included
Table 4.
Analysis of duration of clinical benefit
| Fulvestrant 500 mg (n = 362) | Fulvestrant 250 mg (n = 374) | |
|---|---|---|
| Response rate (%) | 45.6 | 39.6 |
| Mean DoCBa in months (SE) | 21.6 (1.3) | 18.3 (1.1) |
| Expected DoCB (months) | 9.8 | 7.2 |
| Ratio of expected DoCB (95 % CI)b | 1.36 (1.07–1.73) | |
| P value | 0.013 | |
CI confidence interval, DoCB duration of clinical benefit, SE standard error
aLimited to patients with clinical benefit, i.e. a subset of all randomized patients
bRatios of >1 favor fulvestrant 500 mg
Expected DoCB for fulvestrant 500 mg was significantly improved compared with the expected DoCB for fulvestrant 250 mg (ratio of expected DoCB = 1.36; 95 % CI, 1.07–1.73, P = 0.013).
Discussion
Our analysis uses new methodology to prospectively analyze expected DoR and expected DoCB from the CONFIRM trial and shows that fulvestrant 500 mg is associated with a clinically meaningful benefit over fulvestrant 250 mg, as demonstrated by the statistically significant improvement in expected DoCB (P = 0.013). Interestingly, although the magnitude of the difference in median DoR is similar to the difference in median DoCB for fulvestrant 500 mg compared with fulvestrant 250 mg (3.0 and 2.7 months, respectively), the expected DoCB analysis is statistically significant while the expected DoR is not. This is likely due to the larger number of patients per group with a clinical benefit response compared with the number with an objective response, as well as the greater proportion of patients with clinical benefit in the 500 mg group.
The evaluation of most endpoints is based on clearly defined criteria and subgroups are predefined before randomization; however, a major concern regarding the comparison of DoR and DoCB between two treatment groups in a randomized trial is that these values are both post-treatment outcomes, calculated using a subset of responding patients defined post-randomization. As a result, the subset of patients who respond in the control arm may not be comparable, in terms of important prognostic factors, with the subset who respond in the experimental arm. Hence, such comparisons may be prone to biases and may not reflect the actual treatment effects. Attempts have been made to overcome these challenges by developing statistical models that combine the fraction of patients responding to treatment and the DoR in responding patients. In 2007, Ellis et al. [16–18] built on earlier work, by a more flexible approach to estimate the expected DoR across all randomized patients. The Ellis methodology was used here to calculate the expected DoR and expected DoCB across all patients with measurable disease and all randomized patients, respectively, in the CONFIRM study, rather than the subset of responding patients. This allows a more formal and unbiased comparison of the treatment effects for fulvestrant 500 versus 250 mg. The improvement in expected DoCB reported here is, therefore, indicative of a treatment effect in favor of fulvestrant 500 versus 250 mg, consistent with the results from the primary CONFIRM study [5].
An example of how to interpret the results is given by the following hypothetical scenario concerning the experimental arm of a given trial: one third of patients did not respond; one third responded for 2 months; and the remaining third responded for 4 months. In this case, the median DoR for patients in the experimental arm who respond to treatment is 3 months; however, for a patient newly randomized to the experimental arm, the expected median DoR is 2 months (calculated as [1/3 × 0] + [1/3 × 2] + [1/3 × 4]). This is a meaningful finding, as before the patient takes the experimental treatment, the physician will not know whether or not an individual patient will respond.
As far as we are aware, there are no other published data that have used this novel approach to prospectively calculate expected DoR and expected DoCB and make statistical comparisons of these endpoints between the experimental and control arms. This methodology provides an avenue by which trials can robustly analyze and report duration data and it may be particularly appropriate for breast cancer trials, given the importance of disease stabilization in these patients.
In conclusion, the analysis of expected DoR and expected DoCB reported here support the primary study conclusion that fulvestrant 500 mg provides a clinically meaningful benefit over fulvestrant 250 mg for the treatment of postmenopausal women with ER-positive advanced breast cancer who have progressed or recurred on endocrine therapy. In our opinion, application of this novel methodology provides an additional efficacy endpoint option for clinical studies where stabilization of disease is a clinically relevant endpoint.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
The CONFIRM study was funded by AstraZeneca. Medical writing assistance was provided by Dr. Varinia Munoz from Complete Medical Communications Ltd., funded by AstraZeneca.
Conflict of interest
SG is an AstraZeneca employee and shareholder. MM has received consultancy fees from and participated in advisory boards for AstraZeneca and Pfizer. GJ has received honoraria and research funding from AstraZeneca. IS has received consultancy fees from and participated in advisory boards for AstraZeneca. JPOL is an AstraZeneca employee and shareholder. ADL has received consultancy fees from, participated in advisory boards for, and received honoraria and research funding from AstraZeneca and Pfizer. All remaining authors have declared no conflicts of interest.
Ethical Standard
The study protocol for the CONFIRM clinical trial was approved by the institutional review board of each participating institution and all patients gave written informed consent before study entry.
Abbreviations
- CBR
Clinical benefit rate
- CI
Confidence interval
- CONFIRM
COmparisoN of Faslodex In Recurrent or Metastatic breast cancer
- CR
Complete response
- DoCB
Duration of clinical benefit
- DoR
Duration of response
- ER
Estrogen receptor
- mRECIST
Modified Response Evaluation Criteria In Solid Tumors
- NE
Not evaluable
- ORR
Objective response rate
- OS
Overall survival
- PFS
Progression-free survival
- PR
Partial response
- RECIST
Response Evaluation Criteria In Solid Tumors
- SD
Stable disease
- SE
Standard error
Footnotes
This study is conducted on behalf of the CONFIRM investigators.
References
- 1.Howell A, Osborne CK, Morris C, Wakeling AE. ICI 182,780 (Faslodex): development of a novel, pure antiestrogen. Cancer. 2000;89:817–825. doi: 10.1002/1097-0142(20000815)89:4<817::AID-CNCR14>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 2.Robertson JF, Nicholson RI, Bundred NJ, Anderson E, Rayter Z, Dowsett M, Fox JN, Gee JM, Webster A, Wakeling AE, Morris C, Dixon M. Comparison of the short-term biological effects of 7alpha-[9-(4,4,5,5,5-pentafluoropentylsulfinyl)-nonyl]estra-1,3,5, (10)-triene-3,17beta-diol (Faslodex) versus tamoxifen in postmenopausal women with primary breast cancer. Cancer Res. 2001;61:6739–6746. [PubMed] [Google Scholar]
- 3.DeFriend DJ, Howell A, Nicholson RI, Anderson E, Dowsett M, Mansel RE, Blamey RW, Bundred NJ, Robertson JF, Saunders C, Baum H, Walton P, Sutcliffe FA, Wakeling AE. Investigation of a new pure antiestrogen (ICI 182780) in women with primary breast cancer. Cancer Res. 1994;54:408–414. [PubMed] [Google Scholar]
- 4.Kuter I, Hegg R, Singer CF, Badwe R, Lowe E, on behalf of the NEWEST investigators Fulvestrant 500 versus 250 mg: first results from NEWEST, a randomized, phase II neoadjuvant trial in postmenopausal women with locally advanced, estrogen receptor-positive breast cancer. Breast Cancer Res Treat. 2008;109:589. [Google Scholar]
- 5.Di Leo A, Jerusalem G, Petruzelka L, Torres R, Bondarenko IN, Khasanov R, Verhoeven D, Pedrini JL, Smirnova I, Lichinitser MR, Pendergrass K, Garnett S, Lindemann JP, Sapunar F, Martin M. Results of the CONFIRM phase III trial comparing fulvestrant 250 mg with fulvestrant 500 mg in postmenopausal women with estrogen receptor-positive advanced breast cancer. J Clin Oncol. 2010;28:4594–4600. doi: 10.1200/JCO.2010.28.8415. [DOI] [PubMed] [Google Scholar]
- 6.AstraZeneca Faslodex prescribing information. Int Agency Res Cancer (IARC) 2010;21:2169–2174. [Google Scholar]
- 7.European Medicines Agency (2009) Faslodex: EPAR summary for the public. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Summary_for_the_public/human/000540/WC500021169.pdf. Accessed 23 July 2011
- 8.Verma S, McLeod D, Batist G, Robidoux A, Martins IR, Mackey JR. In the end what matters most? a review of clinical endpoints in advanced breast cancer. Oncologist. 2011;16:25–35. doi: 10.1634/theoncologist.2010-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Driscoll JJ, Rixe O. Overall survival: still the gold standard: why overall survival remains the definitive end point in cancer clinical trials. Cancer J. 2009;15:401–405. doi: 10.1097/PPO.0b013e3181bdc2e0. [DOI] [PubMed] [Google Scholar]
- 10.Zhuang SH, Xiu L, Elsayed YA. Overall survival: a gold standard in search of a surrogate: the value of progression-free survival and time to progression as end points of drug efficacy. Cancer J. 2009;15:395–400. doi: 10.1097/PPO.0b013e3181be231d. [DOI] [PubMed] [Google Scholar]
- 11.Hurvitz SA. Evolving options for the treatment of metastatic breast cancer: progression-free survival as an endpoint. Cancer Treat Rev. 2011;37(7):495–504. doi: 10.1016/j.ctrv.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Food and Drug Administration (2003) Oncologic drugs advisory committee meeting proceedings. 12–13 March 2003. http://www.fda.gov/ohrms/dockets/ac/cder03.html#OncologicDrugs. Accessed 12 Sept 2011
- 13.European Medicines Agency (Committee for Medicinal Products for Human Use) (2005) Guideline on the evaluation of anticancer medicinal products in man. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/12/WC500017748.pdf. Accessed 12 Sept 2011
- 14.European Medicines Agency (Committee for proprietary medicinal products) (2003) Points to consider on adjustment for baseline covariates. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003639.pdf. Accessed 12 Sept 2011
- 15.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 16.Ellis S, Carroll KJ, Pemberton K. Analysis of duration of response in oncology trials. Contemp Clin Trials. 2008;29:456–465. doi: 10.1016/j.cct.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 17.Begg CB, Larson M. A study of the use of the probability-of-being-in-response function as a summary of tumor response data. Biometrics. 1982;38:59–66. doi: 10.2307/2530288. [DOI] [PubMed] [Google Scholar]
- 18.Temkin NR. An analysis for transient states with application to tumor shrinkage. Biometrics. 1978;34:571–580. doi: 10.2307/2530376. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.