

Abstract
High field asymmetric waveform ion mobility spectrometry (FAIMS), also known as differential ion mobility spectrometry, coupled with liquid chromatography tandem mass spectrometry (LC-MS/MS) offers benefits for the analysis of complex proteomics samples. Advantages include increased dynamic range, increased signal-to-noise, and reduced interference from ions of similar m/z. FAIMS also separates isomers and positional variants. An alternative, and more established, method of reducing sample complexity is prefractionation by use of strong cation exchange chromatography. Here, we have compared SCX-LC-MS/MS with LC-FAIMS-MS/MS for the identification of peptides and proteins from whole cell lysates from the breast carcinoma SUM52 cell line. Two FAIMS approaches are considered: (1) multiple compensation voltages within a single LC-MS/MS analysis (internal stepping) and (2) repeat LC-MS/MS analyses at different and fixed compensation voltages (external stepping). We also consider the consequence of the fragmentation method (electron transfer dissociation or collision-induced dissociation) on the workflow performance. The external stepping approach resulted in a greater number of protein and peptide identifications than the internal stepping approach for both ETD and CID MS/MS, suggesting that this should be the method of choice for FAIMS proteomics experiments. The overlap in protein identifications from the SCX method and the external FAIMS method was ~25 % for both ETD and CID, and for peptides was less than 20 %. The lack of overlap between FAIMS and SCX highlights the complementarity of the two techniques. Charge state analysis of the peptide assignments showed that the FAIMS approach identified a much greater proportion of triply-charged ions.
Electronic supplementary material
The online version of this article (doi:10.1007/s13361-012-0544-2) contains supplementary material, which is available to authorized users.
Key words: FAIMS, High-field asymmetric waveform ion mobility spectrometry, Differential ion mobility, Liquid chromatography, Strong cation exchange, Collision induced dissociation, Electron transfer dissociation, Proteomics
Introduction
Recent work by Mann and coworkers [1] revealed that while >100,000 detectable peptides were present in a typical proteomics liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis, only ~16 % were actually selected for fragmentation. They found the greatest constraint to be precursor ion isolation: just 14 % of the ion current in the isolation window derived from the precursor ion. That is, co-fragmentation of peptides would occur in virtually all cases. A further challenge for comprehensive proteome coverage is the dynamic range [2, 3]. These limitations can be addressed to some extent by prefractionation of the sample, for example, by gel electrophoresis [4] or by strong cation exchange chromatography [5, 6]. Nevertheless, prefractionation is associated with additional sample clean-up steps and inevitable sample loss.
High field asymmetric waveform ion mobility spectrometry (FAIMS), or differential ion mobility, coupled with LC MS/MS has the potential to circumvent the restrictions of prefractionation. FAIMS separates gas-phase ions at atmospheric pressure on the basis of differences in their ion mobility in high and low electric fields [7, 8]. FAIMS coupled with electrospray ionization has shown advantages for peptide analysis by reducing chemical noise and improving signal-to-noise [9–12], and enabling the separation of localization isomers and sequence variants [13–16]. Saba et al. [17] demonstrated the benefits of FAIMS for proteomics by comparing LC collision-induced dissociation (CID) MS/MS with LC FAIMS CID MS/MS for the analysis of tryptic digests of simple protein mixtures and whole cell lysates from human U937 monocytic cells. The results showed a 10-fold improvement in limits of detection with a corresponding increase of 55 % in the number of assigned MS/MS spectra (i.e., protein identification and sequence coverage were significantly improved as a result of implementation of FAIMS in the proteomics workflow). More recent work from that laboratory [18] applied LC FAIMS MS/MS to the analysis of the Drosophila melanogaster phosphoproteome, again resulting in a 50 % increase in peptide identifications. Swearingen et al. [19] have also shown that incorporation of FAIMS in the workflow improves proteome coverage. Using a modified electrospray source, they observed an increase of 50 % in peptide identifications and 64 % in protein identifications compared with LC MS/MS.
In a FAIMS analysis, ions are transported by a carrier gas between two electrodes to which an asymmetric waveform is applied. As a consequence of their differential ion mobility, the ions travel a greater distance towards one electrode than the other, and will eventually collide with the electrode. To prevent that occurrence, a compensation voltage (CV) is applied to one electrode. By scanning the compensation voltage, it is possible to selectively transmit ions through the FAIMS device. In terms of a proteomic analysis, there are two possible approaches for CV scanning: the CV may be scanned within the LC MS/MS analysis as described by Thibault and co-workers [18] and referred to herein as “internal CV stepping,” or each LC MS/MS analysis is performed at a fixed CV with multiple analyses at different CVs, as described by Swearingen et al. [19] and referred to herein as “external CV stepping.” We have evaluated the two approaches by comparing the results obtained from analyses of whole cell lysates from human SUM52 cells. Our results show that the external CV stepping method results in greater proteome coverage. In addition, we have compared the gas-phase fractionation afforded by FAIMS with prefractionation by strong cation exchange (SCX) chromatography. Our findings support those of Bridon et al. [18] in their comparison of 2D SCX-LC MS/MS with the internal stepping method for the analysis of phosphopeptides. Finally, we have considered the relationship between fragmentation method and FAIMS, in terms of proteome coverage obtained. Replicate analyses were performed in which either CID or electron transfer dissociation (ETD) was the fragmentation method. That approach avoids any bias that may be inherent in a decision tree method in which ETD or CID is triggered depending on the precursor ion m/z and charge.
Experimental
SUM52 Cell Culture
SUM52 breast cancer carcinoma cells were cultured in HPMi-1640 formulation, supplemented with 2 mM L-glutamine, 1 % Pen-Strep and 10 % PBS at 37 °C in a 5 % CO2 atmosphere. When confluent, cells were washed in PBS twice and lysis buffer added on ice for 30 min. Lysis buffer contained Triton X-100 (0.5 %), NaCl (0.15 M), PhosphoStop phosphatase inhibitor tablet (Roche, Indianapolis, IN, USA) and Mini-Complete protease inhibitor (Roche). The lysed cells were removed from the flask with a cell scrapper. Total protein concentrations of the cleared lysates were then determined by Coomassie (Bradford) Protein Assay kit (Pierce, Thermo Fisher Scientific, Rockford, IL, USA) according to the manufacturer’s instructions.
In-Solution Digestion
Proteins were reduced with 50 mM dithiothreitol (Sigma Aldrich, Gillingham, UK) and alkylated with 20 mM iodoacetamide (Sigma Aldrich). SUM52 proteins were digested overnight at 37 °C with trypsin (Trypsin Gold, Promega, Madison, WI, USA) (50:1 wt/wt). The digested sample was centrifuged at 12,000 RPM to remove cell debris. The peptide mixture was acidified after digestion with 0.5 % TFA. Prior to analysis the digest was desalted (C8 cartridge; Michrom, Auburn, CA, USA).
Strong Cation Exchange (SCX) Chromatography
Desalted and dried peptides from 200 μg of lysate (as measured prior to digestion) were resuspended in 100 μL mobile phase A (10 mM KH3PO4, 25 % acetonitrile, pH 3) and loaded onto a 100 × 2.1 mm polysulfoethyl A column (5 μm particle size, 20 nm pore size, PolyLC, Columbia, MD, USA) at a flow rate of 200 μL/min. Peptides were separated with a gradient from 0 %–50 % mobile phase B (10 mM KH3PO4, 25 % acetonitrile, 500 mM KCl, pH 3) over 40 min, increasing to 70 % B over 5 min before returning to 100 % A. Fifteen fractions were collected over 54 min. Fractions were combined as follows: 1, 14 and 15, 2, 12 and 13, 3, 10 and 11, 4 and 9, 5 and 8, 6 and 7 for a total of 6 fractions. The combined fractions were desalted as above.
Liquid Chromatography
Peptides (1.66 μg) were loaded onto a 150 mm Acclaim PepMap100 C18 column (LC Packings, Sunnyvale, CA, USA) in mobile phase A (0.1 % formic acid; JT Baker, Holland Sigma Aldrich, Deventer, Holland). Peptides were separated over a linear gradient from 3.2 % to 44 % mobile phase B (acetonitrile + 0.1 % formic acid, JT Baker, Sigma Aldrich, Deventer, Holland) with a flow rate of 350 nL/min. The column was then washed with 90 % mobile phase B before re-equilibrating at 3.2 % mobile phase B. The column oven was heated to 35 °C. For standard (non-FAIMS) LC-MS/MS the LC system was coupled to an Advion Triversa Nanomate (Advion, Ithaca, NY, USA), which infused the peptides with a spray voltage of 1.7 kV. In FAIMS analyses, the LC system was coupled to an ADPC-IMS PicoFrit nano-ESI probe (New Objective, Woburn, MA, USA). The spray voltage was 2.95 kV. Peptides were infused directly into the LTQ-Orbitrap Velos ETD (Thermo Fischer Scientific, Bremen, Germany).
Tandem Mass Spectrometry MS/MS
ETD The mass spectrometer performed a full FT-MS scan (m/z 380–1600) and subsequent ETD MS/MS scans of the three most abundant ions above a threshold of 1000. To facilitate CV scanning in the internal stepping method, and to ensure consistency between methods, a top 3 method was utilized. Survey scans were acquired in the Orbitrap with a resolution of 15,000 at m/z 400. Precursor ions were subjected to supplemental activation (sa) ETD in the linear ion trap. Width of the precursor isolation window was 3 m/z and only multiply charged precursor ions were subjected to saETD. saETD was performed with fluoranthene ions. Automatic gain control (AGC) was used to accumulate sufficient ions (fluoranthene, target 1 × 105, maximum fill time 50 ms. Precursor ions, target 5 × 104, maximum fill time 100 ms) precursor ions were activated for 100 ms (charge dependent activation time was enabled). Dynamic exclusion repeat count was set to 1 with duration of 60 s. Data acquisition was controlled by Xcalibur 2.1 (Thermo Fisher Scientific). The mass exclusion window was m/z ±0.05 and the exclusion list was set to 500.
CID The mass spectrometer performed a full FT-MS scan (m/z 380–1600) and subsequent CID MS/MS scans of the three most abundant ions above a threshold of 1000. To facilitate CV scanning in the internal stepping method, and to ensure consistency between methods, a top 3 method was utilized. Survey scans were acquired in the Orbitrap with a resolution of 15,000 at m/z 400. CID was performed in the linear ion trap with helium gas at a normalized collision energy of 35 % (target 5 × 104, maximum fill time 100 ms). CID activation was performed for 10 ms. Width of the precursor isolation window was 2 m/z and only multiply charged precursor ions were subjected to CID. The mass exclusion window was m/z ±0.05 and the exclusion list was set to 500.
LC-FAIMS MS/MS Analysis
FAIMS settings were: dispersion voltage (DV) -5000 V, gas flow 2.75 L/min, gas composition 50/50 He/N, inner electrode 70 °C, outer electrode 90 °C. The dwell time was set at 50 ms.
Internal CV Stepping The mass spectrometer performed a full FT-MS scan (m/z 380–1600, resolution 15,000) at compensation voltage (CV) of −25 V and subsequent CID or ETD MS/MS events of the three most abundant ions (MS/MS parameters as described above) at the same CV value. The sequence was repeated for CVs of −30 V, –35 V, –40 V, –45 V, and −50 V, before cycling back to CV = −25 V. Replicate (n = 6) analyses were performed.
External CV Stepping The mass spectrometer performed a full FT-MS scan and subsequent MS/MS of the three most abundant ions (MS/MS parameters as described above). Six analyses were performed and for each the CV remained constant throughout (CV = −25, –30, –35, –40, –45, and −50 V).
Database Search Parameters
All data were searched against IPI Human database (V 3.81) containing common contaminants and concatenated with a reverse database (184746 sequences). The data were searched using both the SEQUEST and Mascot algorithms (controlled through Proteome Discoverer ver. 1.2, mascot ver. 2.2.0). In both SEQUEST and Mascot searches, the following parameters were used: no spectral grouping; total intensity threshold, 0; minimum peak count, 1; precursor ion m/z tolerance, ±5 ppm; fragment ion m/z tolerance, ±0.5 Da; fully tryptic, 2 missed cleavages allowed; Cys carboxyamidomethylation was set as a fixed modification; N-terminal acetylation, deamidation of Asn and Gln, oxidation of Met, and phosphorylation of Ser, Thr, and Tyr were set as variable modifications. Product ion types for CID data were b and y, for ETD c, y, and z ions were accepted. Data were filtered to a protein FDR of 1 % (peptide FDR was also 1 % or lower) using the Discoverer software (Exp values and XCorr values for filter are detailed in Supplemental Table 1). Protein and peptide false discovery rates were calculated by dividing number of reverse hits by the total number of proteins/peptides identified. Protein grouping was performed by the Proteome Discoverer software. One peptide was required for a positive protein identification.
Results
Whole cell lysates (WCL) from SUM52 cells were digested with trypsin. Equal amounts (~30 μg) of the digest were analyzed by one of three proteomic workflows: (1) on-line reversed-phase liquid chromatography FAIMS MS/MS in which the compensation voltage remained constant for the entire analysis. Analyses were performed at CVs of −25, –30, –35, –40, –45, and –50 V, for a total of six analyses (5 μg each). We refer to this method as “external CV stepping;” (2) on-line reversed-phase liquid chromatography FAIMS MS/MS in which the compensation voltage was cycled (CV = −25, –30, –35, –40, –45, and –50 V) during the analysis. Six repeats were performed. We refer to this method as “internal CV stepping;” (The CV values were selected in order to both maximize peptide ion transmission and minimize duty cycle. Preliminary direct infusion FAIMS experiments (data not shown) on a set of 15 tryptic peptides from alcohol dehydrogenase, cytochrome c, and bovine serum albumin suggest that the majority of 2+ and 3+ peptide ions are transmitted over this CV range); (3) the digest was separated by strong cation exchange chromatography (SCX) and 6 fractions collected. Each fraction was subjected to on-line reversed-phase liquid chromatography MS/MS. Each workflow was repeated for CID MS/MS and ETD MS/MS. Each experiment required ~6 hours of instrument time (6 LC MS/MS runs), resulting in ~36 hours of instrument time. The data were searched against the human IPI database, ver. 3.81 (concatenated with reversed entries) using both the Mascot and SEQUEST algorithms and filtered to give a 1 % false discovery rate. The database results were combined and redundancy removed.
LC-ETD-MS/MS
The number of proteins identified from the three workflows is summarized in Figure 1 (top). A total of 407 proteins were identified by external CV stepping, 302 by internal CV stepping and 463 by the SCX method (see Supplemental Table 2 for proteins identified). Sixty percent of the proteins identified by internal CV stepping were also identified by external CV stepping, and 44 % were also identified by the SCX method. Figure 1 (middle) summarizes the proteins identified by external CV stepping and the SCX method. Only 22 % of the total proteins identified are identified by both methods. That result demonstrates the complementarity of SCX-based 2D-LC-MS/MS analysis and LC-FAIMS-MS/MS analysis. To probe the origin of this complementarity, we analyzed the protein identifications in terms of the number of non-redundant peptide assignments. Figure 1, bottom, shows that the majority of the proteins identified by external CV stepping only (74 %) were identified by a single peptide (left histogram). The same is true for those identified by the SCX method alone (58 %, right histogram). The proteins identified by both methods had a higher proportion of multiple peptide assignments (45 % for external CV stepping and 61 % for the SCX method, middle histogram). These findings may indicate that those proteins identified by both methods are more abundant and, therefore, their peptides are more likely to be selected for MS/MS and produce higher quality spectra. It is also possible that the proteins identified by both methods may be identified by different peptides and that the orthogonality between the two methods may be even greater at the peptide level. Analysis of the peptides responsible for the assignments of the proteins identified by both methods reveals that 27 % were unique to the external stepping analyses and 44 % to the SCX analyses.
Figure 1.
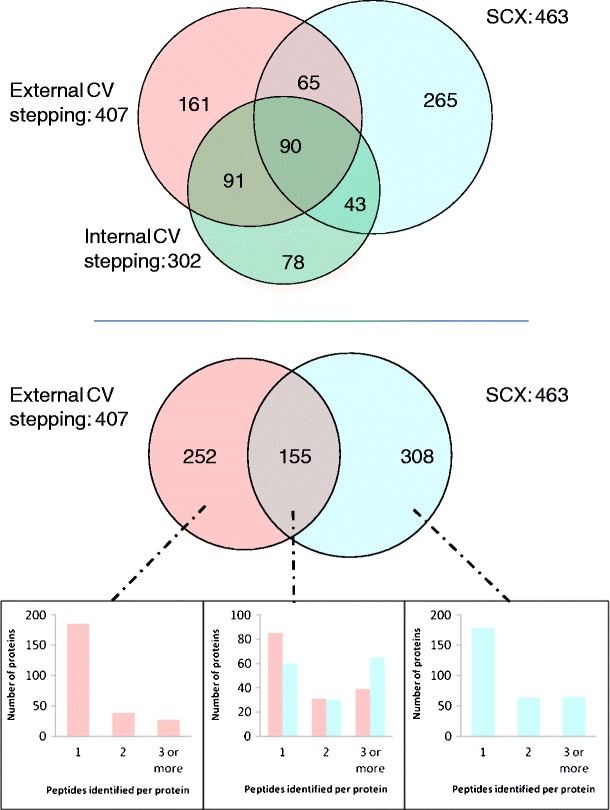
Top: protein identifications resulting from LC FAIMS ETD MS/MS with external CV stepping (pink), internal CV stepping (green), and LC ETD MS/MS with SCX prefractionation (blue); middle: protein identifications resulting from external CV stepping (pink) and SCX prefractionation (blue); bottom: number of peptides identified per protein for each of the sections of Figure 1 middle (external CV stepping only, both external CV stepping and SCX prefractionation, SCX prefractionation only)
The number of peptides identified by the external CV stepping and the SCX method are summarized in the Venn diagram in Figure 2 (top). Peptides with identical sequence but differing charge state are treated as unique assignments. Clearly, the SCX method results in a greater number of peptide identifications; however, 25 % of the total identifications arise from external CV stepping alone. The overlap in peptide identifications between the two methods is 13 % of the total peptides identified. The peptides identified were analyzed in terms of charge state distribution (Figure 2, middle). The key difference between the two methods is the proportion of identifications for the 3+ charge state: 67 % for external CV stepping versus 40 % for the SCX method. The number of non-redundant peptide identifications (i.e., peptides with multiple charge states treated as single assignments) obtained via the three workflows is shown in Figure 2 (bottom). The peptide identifications are summarized in Supplemental Table 3.
Figure 2.
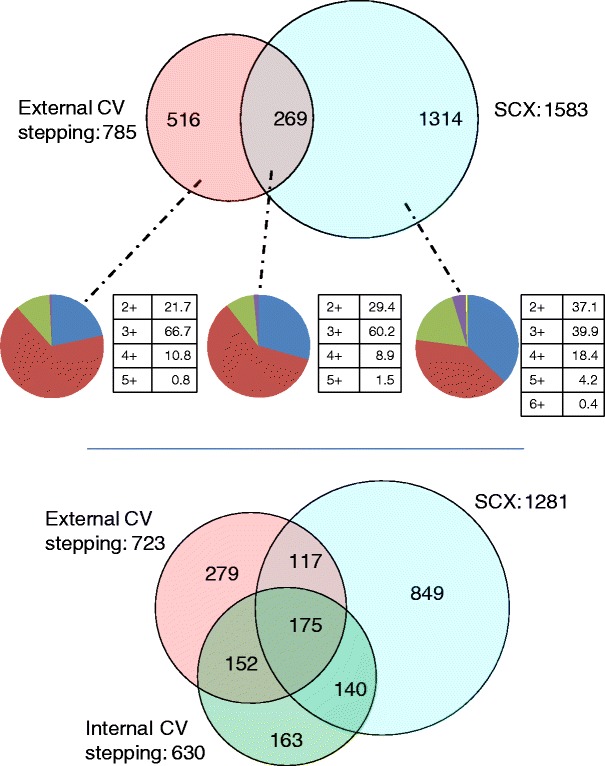
Top: peptide identifications resulting from ETD MS/MS with the external CV stepping (pink) and SCX methods (blue). Peptides with identical sequence but differing charge states are treated as unique assignments; middle: charge state analysis of peptides identified by external CV stepping only (right), SCX method only (left) and both methods (centre) (blue = 2+, red = 3+, green = 4+, purple = 5+, and yellow = 6+); bottom: non-redundant peptide identifications resulting from LC FAIMS ETD MS/MS with external CV stepping (pink), internal CV stepping (green), and LC ETD MS/MS with SCX prefractionation (blue)
LC-CID-MS/MS Analysis
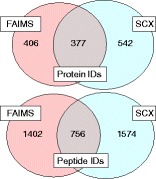
Figure 3 (top) summarizes the proteins identified by CID MS/MS for the three workflows (see also Supplemental Table 4). A total of 783 proteins were identified by external CV stepping, 311 by internal CV stepping, and 919 by the SCX method. There is greater overlap in the proteins identified by each of the workflows than was observed for the ETD data. Of the proteins identified via internal CV stepping, 69 % were also identified via external CV stepping and 54 % were also identified via the SCX method. Figure 3 (middle) compares the proteins identified by external CV stepping and the SCX method; 28 % of the total proteins identified were identified by both methods. The proteins identified were analyzed in terms of the number of peptide assignments (Figure 3, bottom). As seen for the ETD data, the majority of the proteins unique to a particular workflow were identified by a single peptide: 63 % of those proteins identified by external CV stepping alone and 64 % of those identified solely by the SCX method; 65 % and 66 % of those proteins identified by both methods (external CV stepping and SCX prefractionation, respectively) had 2 or more peptide assignments. Of the peptide assignments for the proteins identified by both methods, 36 % were unique to the external CV stepping method and 25 % were unique to the SCX prefractionation method.
Figure 3.
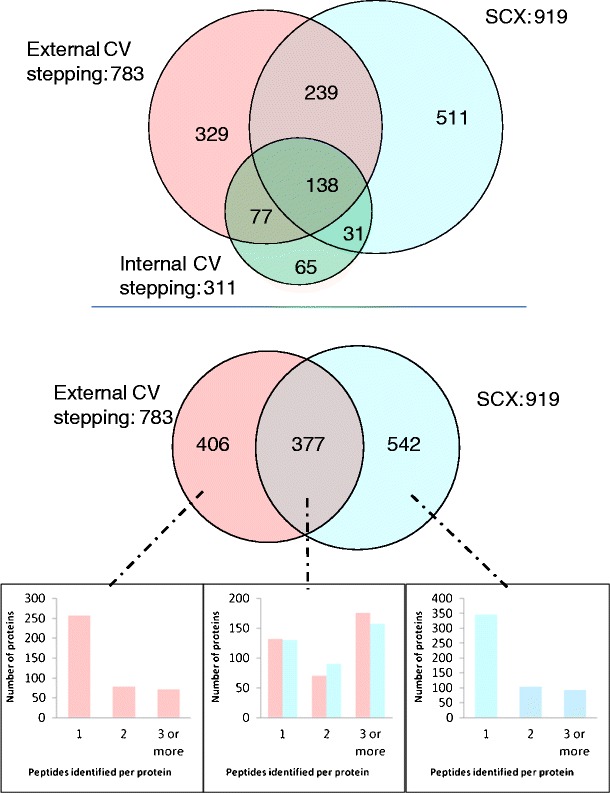
Top: protein identifications resulting from LC FAIMS CID MS/MS with external CV stepping (pink), internal CV stepping (green), and LC CID MS/MS with SCX prefractionation (blue); middle: protein identifications resulting from external CV stepping (pink) and SCX prefractionation (blue); bottom: number of peptides identified per protein for each of the sections of Figure 3 middle (external CV stepping only, both external CV stepping and SCX prefractionation, SCX prefractionation only)
The number of peptides identified by external CV stepping and the SCX methods is summarized in Figure 4 (top). As in the ETD analysis, peptides with identical sequence but differing charge state are treated as unique assignments. Thirty-eight percent of the peptides were identified by external CV stepping alone and 42 % by the SCX method alone. The overlap in peptide identifications between the two methods is 20 % of the total peptides identified. The peptide identifications were analyzed for distribution of charge states (Figure 4, middle). As seen for the ETD data, external CV stepping resulted in a higher percentage of 3+ peptide identifications than the SCX method (38 % versus 24 %); however, external CV stepping with CID resulted in fewer 3+ identifications than with ETD (see Figure 2, middle). Figure 4 (bottom) shows the non-redundant peptide identifications obtained via the three workflows (see Supplemental Table 5). The overlap between the peptides identified by both external CV stepping and internal CV stepping is high: 83 % of the “internal” identifications were also “external” identifications.
Figure 4.
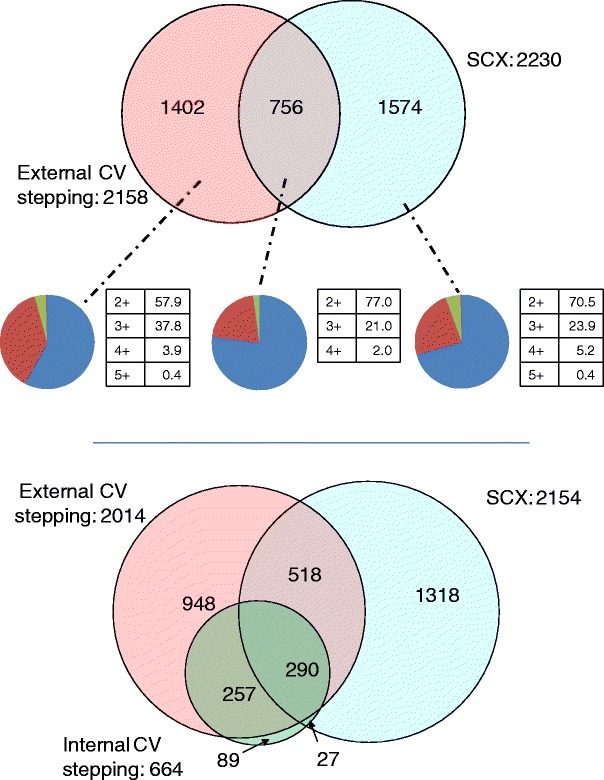
Top: Peptide identifications resulting from CID MS/MS with the external CV stepping (pink) and SCX methods (blue). Peptides with identical sequence but differing charge states are treated as unique assignments; middle: charge state analysis of peptides identified by external CV stepping only (right), SCX method only (left), and both methods (center) (blue = 2+, red = 3+, green = 4+, and purple = 5+); bottom: non-redundant peptide Identifications resulting from LC FAIMS CID MS/MS with external CV stepping (pink), internal CV stepping (green), and LC CID MS/MS with SCX prefractionation (blue)
Discussion
The results clearly demonstrate that coupling of FAIMS, and particularly using an external CV stepping method, with LC MS/MS extends proteome coverage. This finding is in agreement with the findings of Swearingen et al. [19], who compared external CV stepping with repeat reversed-phase LC MS/MS injections of the same sample. In the ETD dataset, use of external CV stepping resulted in identification of 252 additional proteins (i.e., an increase of 54 % over those identified by SCX LC ETD MS/MS. At the peptide level, external CV stepping generated 516 additional peptide assignments, an increase of 33 %. In the CID dataset, external CV stepping gave 406 additional proteins and 1402 additional peptides, i.e., 44 % and 60 % increases over SCX LC CID MS/MS alone. Overall, the number of identifications is quite low. It is possible that by using a longer LC gradient or by increasing the number of MS/MS events per survey scan (e.g., by use of a top 7 or top 20 method) more identifications might be made. The latter possibility would not be suitable for the internal stepping method because the time for the CV cycle needs to be kept to a minimum. For the FAIMS analyses, use of a source such as that described by Swearingen et al. [19] should improve the number of identifications.
CID outperformed ETD in terms of number of identifications and there are two possible explanations for this observation. First, the protein database search algorithms were originally designed for CID data [6, 20] and, consequently, ETD (which produces very different spectra) generally results in lower scores and fewer identifications [21–24]. Secondly, trypsin cleaves proteins at Lys and Arg residues meaning that the majority of tryptic peptides favour the 2+ charge state. It is well known that ETD of doubly-charged ions, even with the aid of supplemental activation, produces fewer fragment ions than for higher charge states resulting in lower search scores [25, 26]. Nevertheless, the two fragmentation techniques do provide complementary information: in the external stepping method, a total of 2266 non-redundant peptides were identified, 252 of which were unique to ETD and 1543 unique to CID. For the internal method, 934 non-redundant peptides were identified, 270 unique to ETD and 304 unique to CID. For the SCX analyses, 492 out of a total of 2646 were unique to ETD and 1365 were unique to CID. Clearly, the number of identifications is increased by incorporating ETD in the workflow.
Figures 2 and 4 (middle) show the distribution of charge states of the peptide identifications by the external CV stepping method and the SCX MS/MS methods. There is a notable increase in the proportion of 3+ identifications when ETD is employed, compared with CID. Within the ETD data set, the proportion of 3+ and 2+ identifications by SCX MS/MS is approximately equal (39.9 % versus 37.1 %) and the overlap in identifications between the two charge states is 32 %. The proportion of 3+ to 2+ identifications by external stepping, however, is approximately threefold (66.7 % to 21.7 %), with an overlap of 6 %. As described above, electrospray of tryptic peptides tends to produce predominantly 2+ ions, with 3+ ions forming a minor component. In a typical LC MS/MS proteomics workflow, the survey scan will reveal both charge states and the more abundant 2+ ions will be selected for fragmentation. FAIMS, however, transmits 2+ and 3+ ions at different compensation voltages, thereby increasing the likelihood that a 3+ ion will be selected for fragmentation at the compensation voltages applied here. This is especially valuable for ETD, which is known to be more efficient for 3+ and higher charge states. It is possible that the CV values used in these experiments lead to underrepresentation of 2+ ions; however, preliminary direct infusion FAIMS data (not shown) from a set of tryptic peptides revealed that 10/11 of the 2+ ions were transmitted between −20 and −35 V, and 3/4 of the 3+ ions were transmitted between −30 and –50 V.
External CV stepping resulted in a greater number of identifications than internal CV stepping: 407 versus 302 proteins, and 723 versus 630 non-redundant peptides for ETD; 783 versus 311 proteins, and 2014 versus 664 non-redundant peptides for CID. This might be expected as internal CV stepping has a much longer duty cycle than external CV stepping: for a particular CV in the internal stepping method, the method will cycle through five other CV values before recording the next survey scan. For many peptides, the top of the LC chromatographic peak for a particular CV will not coincide with mass spectral recording. This will become problematic if, during the survey scan at a particular CV, the ions are not sufficiently abundant to either produce high quality MS/MS spectra or to trigger MS/MS. In the CID analyses, the total numbers of MS/MS events were 13,613 (internal stepping), and 18,195 (external stepping). Conversely, for the ETD analyses, the numbers were 9528 (external stepping) and 10,313 (internal stepping). ETD has a longer duty cycle than CID; the target number of ions for CID is half that for ETD, and the activation time is 10-fold shorter. In addition, although dynamic exclusion was applied within each analysis, the software is not currently available to apply between analyses and so it was not used between the six repeats of the internal CV stepping analyses. For the CID data, 83 % of the peptides identified by internal CV stepping were also identified by external CV stepping; however, for the ETD data, that figure is just 69 %. This unexpected observation is explored further below.
In order to explore the origin of complementarity between the three workflows, we considered the top five unique peptide assignments obtained from each for both ETD and CID. The findings are summarized in Table 1. Of the five highest scoring peptides identified by external CV stepping only, three (LKKEDIYAVEIVGGATR 3+, LLKIPVDTYNNILTVLK 3+, VPVITGSFVDLSVELK 3+) were also observed in the internal stepping survey scans (mass error <3 ppm); however, they were not selected for fragmentation. Figure 5 shows the survey scans and ETD spectra (inset) of those peptides identified in the external CV stepping method, and the equivalent survey scans from the internal CV stepping method( i.e., the survey scans obtained at the same CV value and closest retention time. (Note that differences in the retention times at which the survey scans were collected are due in part to the longer duty cycle, i.e., time between survey scans at a particular CV, of the internal stepping method). None of the five peptides or ions which have the same mass at similar retention times were observed in the SCX data set. (Nor were the peptides observed in different charge states or at different retention times). This observation can be explained by the reduction in chemical noise and singly-charged ions in the FAIMS mass spectra.
Table 1.
Top five unique peptide assignments for the three workflows (external CV stepping, internal CV stepping, and SCX prefractionation) (top ETD, bottom CID). Counterpart datasets were interrogated manually for the presence of equivalent precursor ions and fragmentation spectra. Mascot probability scores for the assigned peptides are given
| ETD Top 5 | Internal | External | SCX | Probability |
|---|---|---|---|---|
| External | ||||
| ILGGSVLHLVLALR | Not present | Identified | Not present | 1.27E-06 |
| LKKEDIYAVEIVGGATR | Present | Identified | Not present | 4.50E-07 |
| LLKIPVDTYNNILTVLK | Present | Identified | Not present | 1.66E-07 |
| VPVITGSFVDLSVELK | Present | Identified | Not present | 2.27E-06 |
| VVVDALSGLKGDLAGR | Not present | Identified | Not present | 2.68E-06 |
| Internal | ||||
| AMGIMNSFVNDIFER | Identified | Fragmented | Not present | 2.51E-07 |
| KPLVIIAEDVDGEALSTLVLNR | Identified | Present | Fragmented | 8.79E-06 |
| TALLDAAGVASLLTTAEVVVTEIPKEEK | Identified | Fragmented | Not present | 9.08E-06 |
| TDEFQLHTNVNDGTEFGGSIYQK | Identified | Present | Not present | 8.74E-06 |
| TPAVEGLTEAEEEELRAELTK | Identified | Fragmented | Not present | 1.61E-07 |
| SCX | ||||
| EGQGEGETQEAAAAAAAAR | Present | Present | Identified | 1.80E-10 |
| GVVPLAGTNGETTTQGLDGLSER | Present | Not present | Identified | 5.50E-11 |
| SAAQAAAQTNSNAAGK | Not present | Not present | Identified | 1.19E-09 |
| SSGNSSSSGSGSGSTSAGSSSPGAR | Not present | Not present | Identified | 2.55E-14 |
| TLAPLLASLLSPGSVLVLSAR | Present | Not present | Identified | 7.29E-11 |
| CID Top 5 | Internal | External | SCX | Probability |
| External | ||||
| LIALSIDSVEDHLAWSK | Present | Identified | Present | 9.49E-11 |
| LGANSLLDLVVFGR | Present | Identified | Present | 2.08E-10 |
| TVAGQDAVIVLLGTR | Present | Identified | Present | 4.71E-10 |
| QVLLSAAEAAEVILR | Present | Identified | Not present | 5.42E-10 |
| LTTDFNVIVEALSK | Present | Identified | Present | 5.78E-10 |
| Internal | ||||
| TAFDEAIAELDTLNEDSYKDSTLIMQLLR | Identified | Fragmented | Present | 4.87E-07 |
| VLFPATGYLSIVWK | Identified | Present | Not present | 5.17E-07 |
| GLGTGTLYIAESR | Identified | Fragmented | Not present | 5.89E-07 |
| LALDIEIATYR | Identified | Present | Present | 7.46E-07 |
| TVLIMELINNVAK | Identified | Present | Present | 1.08E-06 |
| SCX | ||||
| PNSEPASLLELFNSIATQGELVR | Not present | Not present | Identified | 1.53E-12 |
| VDNALQSGNSQESVTEQDSK | Not present | Not present | Identified | 3.89E-12 |
| ASASGSGAQVGGPISSGSSASSVTVTR | Not present | Not present | Identified | 2.81E-11 |
| GLAFIQDPDGYWIEILNPNK | Not present | Not present | Identified | 7.00E-11 |
| TQLEELEDELQATEDAK | Not present | Not present | Identified | 1.65E-10 |
Figure 5.
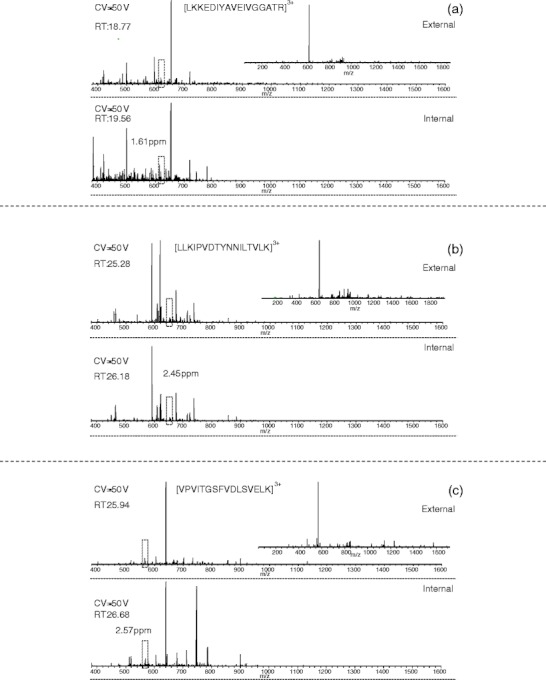
The full MS spectra and ETD mass spectra of (a) LKKEDIYAVEIVGGATR + 3H]3+, (b) [LLKIPVDTYNNILTVLK + 3H]3+, and (c) [VPVITGSFVDLSVELK + 3H]3+ identified in the external CV stepping dataset only (top). Manual analysis of the internal spectra identified precursor ions with similar retention time and less than 3 ppm mass error (bottom). Peaks corresponding to the precursor m/z were not identified in the spectra from the SCX prefractionation dataset
An unexpected observation is the apparent difference between survey scans obtained at the same retention time and CV value for the external and internal stepping methods. This is also evident in Supplemental Figure 1a–e, which show the survey scans and the ETD spectra (inset) of the five highest scoring peptides identified by internal CV stepping only (top), and the equivalent survey scans (and ETD spectra if triggered) from the external CV stepping method (middle), and SCX method (bottom). Some variation between the internal and external stepping methods might be expected as a result of the differing duty cycles: the internal stepping method consists of six survey scans and 18 MS/MS events compared with one survey scan and three MS/MS events for the external stepping method. In addition, there appears to be some variation in the chromatography. Even with a duty cycle of 13.2 s (empirically observed to be the maximum duty cycle), one would expect to match retention times to within 20 s, the typical peak width. Nevertheless, when corrected for retention time differences (e.g., see Figure 5 and Supplemental Figure 1), differences are observed. These differences may be the result of variations in chromatography, instrumental performance, or the stochastic sampling of the chromatogram.
Each of the top five peptide identifications by internal CV stepping alone were also observed in the external survey scans (mass error <3.5 ppm), Supplemental Figure 1 a–e. Two of the five (AMGIMNSFVNDIFER and TPAVEGLTEAEEEELRAELTK) were fragmented in the external stepping analysis, but the database search scores were below the filter cut-off. A third peptide TALLDAAGVASLLTTAEVVVTEIPKEEK was also fragmented but was not identified in the database search. Manual analysis confirmed this as the peptide identified by the internal method. Only one of the peptides (KPLVIIAEDVDGEALSTLVLNR) was observed and fragmented in the SCX analysis. It was identified in the database search but the score fell below the filter cut-off. Peptide sequence was confirmed by manual analysis.
Supplemental Figure 1f–j shows the survey scans and corresponding ETD mass spectra for the five top scoring peptides unique to the SCX analyses. Three of the peptides (EGQGEGTQEAAAAAAAAR, GVVPLAGTNGETTTQGLDGLSER, SSGNSSSSGSGSGSTSAGSSSPGAR) were observed in the internal CV stepping dataset and one (EGQGEGTQEAAAAAAAAR) in the external CV stepping dataset (mass accuracy <3.5 ppm) at the compensation voltages indicated; however, no ETD events were triggered, presumably because of their low abundance. Although incorporation of FAIMS improves signal-to-noise, there is a trade-off with transfer efficiency, which we typically observe to be between 10 % and 20 %. That is, the low transmission efficiency may explain why some peptides identified in the SCX analysis are not identified in the FAIMS analyses. Improving transfer efficiency, for example by modifying the electrospray source as described by Swearingen and co-workers [19], might allow sufficient signal intensity to trigger ETD. Another possible explanation for lack of FAIMS identification of peptides identified in the SCX analysis is that the peptides may not be optimally transmitted at the CV values used here. Typically, the FAIMS peak width is between 5 and 10 V and, therefore, the peptides should be transmitted at one of the compensation voltages employed here. However, if the peak edge coincides with the pre-set CV value, the ions may not be sufficiently abundant to trigger MS/MS. Greater overlap in the FAIMS/SCX identifications may be obtained by reducing the CV step size. The consequence of such an approach for the external stepping method would be increased instrument time, and for the internal stepping method, increased duty cycle to such an extent that is not practical. Similarly, increasing the number of SCX fractions analyzed should improve the overlap; however, this will also increase instrument time.
A similar analysis was performed for the CID datasets. Supplemental Figure 2 a–e show the survey scans and corresponding CID mass spectra for the five top-scoring peptides solely identified by the external CV stepping method. The equivalent survey scans from the internal stepping method (CV and retention time) and SCX method (retention time) are shown in Supplementary Figure 2a–e middle and bottom, respectively. All of the peptides were also observed in the survey scans from the internal stepping analyses with mass accuracies of <2.5 ppm. Four of the peptides were also observed in the SCX survey scans (mass accuracies <2 ppm). None of the peptides were selected for fragmentation.
All of the five top scoring peptides uniquely identified by internal CV stepping coupled with CID (Supplementary Figure 2f–j [top]) were also observed in the equivalent survey scans (CV, retention time) from the external stepping analyses (Supplementary Figure 2f–j [middle]) with mass accuracies <4 ppm. Two of the peptides (TAFDEAIAELDTLNEDSYKDSTLIMQLLR and GLGTGTLYIAESR) were selected for fragmentation and while manual analysis confirmed their identity, they were not assigned in the database searches. In the case of GLGTGTLYIAESR, manual analysis suggests there is a co-eluting peptide, which may explain the failure of the database search. Three of the peptides were observed in the SCX survey scans at equivalent retention times with mass accuracies <3 ppm; however, none were selected for fragmentation. Supplementary Figure 2k–o show the survey scans and corresponding CID spectra for the five top-scoring data from the SCX. None of the peptides were detected by either of the FAIMS methods.
Conclusion
The results demonstrate that inclusion of FAIMS within a proteomics workflow extends proteome coverage. The total number of proteins identified using FAIMS ETD MS/MS (external stepping) was 88 % of the total number identified using SCX ETD MS/MS for the same starting amount of protein. For CID, the total number of proteins identified using FAIMS MS/MS (external stepping) was 85 % of the total number identified by SCX MS/MS. When the internal stepping method is included, the values are 114 % and 96 % for ETD and CID. Crucially, 35 % of the total proteins identified by either external stepping ETD or SCX ETD MS/MS, and 31 % of those identified by CID were identified by the external method alone, thus demonstrating the complementarity of the FAIMS approach. Inclusion of FAIMS looks particularly promising for proteomics workflows utilizing ETD. ETD offers a number of advantages for peptide analysis and, in particular, the characterization of post-translational modifications, however, is inefficient for 2+ ions, which predominate in the electrospray of tryptic digests. FAIMS allows the separation of 2+ and 3+ ions increasing the probability that a 3+ ion will be selected for fragmentation and that the subsequent ETD spectrum will be of sufficient quality to result in a peptide identification. The key challenge now is to improve transmission efficiency through the FAIMS device. In our hands, the maximum transmission efficiency is ~20 %, suggesting the scope for this application in terms of identifiable peptides and proteins could be greatly increased.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
(PDF 831 kb)
(XLSX 90 kb)
(XLSX 329 kb)
(XLSX 147 kb)
(XLSX 635 kb)
Acknowledgments
The authors acknowledge support for this work by the Wellcome Trust (091892) (H.J.C. and J.K.H.) and Cancer Research, UK (A.J.C., N.J.S., K.P.B.L., and J.K.H.). The ThermoFisher Velos Orbitrap ETD mass spectrometer used in this research was obtained through the Birmingham Science City Translational Medicine: Experimental Medicine Network of Excellence project, with support from Advantage West Midlands.
References
- 1.Michalski A, Cox J, Mann M. More than 100,000 detectable peptide species elute in single shotgun proteomics runs but the majority is inaccessible to data-dependent LC-MS/MS. J. Proteome Res. 2011;10:1785–1793. doi: 10.1021/pr101060v. [DOI] [PubMed] [Google Scholar]
- 2.Fonslow BR, Carvalho PC, Academia K, Freeby S, Xu T, Nakorchevsky A, Paulus A, Yates JR. Improvements in proteomic metrics of low abundance proteins through proteome equalization using proteominer prior to MudPIT. J. Proteome Res. 2011;10:3690–3700. doi: 10.1021/pr200304u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Han DK, Wu LF. Overcoming the dynamic range problem in mass spectrometry-based shotgun proteomics. Expert Rev. Proteom. 2006;3:611–619. doi: 10.1586/14789450.3.6.611. [DOI] [PubMed] [Google Scholar]
- 4.de Godoy LM, Olse JV, Cox J, Nielsen ML, Hubner NC, Frohlich F, Walther TC, Mann M. Comprehensive mass spectrometry-based proteome quantification of haploid versus diploid yeast. Nature. 2008;455:1251–1254. doi: 10.1038/nature07341. [DOI] [PubMed] [Google Scholar]
- 5.Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villen J, Li JX, Cohn MA, Cantley LC, Gygi SP. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. U.S.A. 2004;101:12130–12135. doi: 10.1073/pnas.0404720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sweet SMM, Bailey CM, Cunningham DL, Heath JK, Cooper HJ. Large-scale localization of protein phopshorylation by electron capture dissociation mass spectrometry. Mol. Cell. Proteom. 2009;8:904–912. doi: 10.1074/mcp.M800451-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guevremont R. High-field asymmetric waveform ion mobility spectrometry: a new tool for mass spectrometry. J. Chromatogr. A. 2004;1058:3–19. [PubMed] [Google Scholar]
- 8.Purves, R.W., Guevremont,R., Day, S., Pipich, C.W., Matyjaszczyk, M.: Mass spectrometric chracterization of a high-field asymmetric waveform ion mobility spectrometer. Rev. Sci. Instrum. 69, 4094–4105 (1998)
- 9.Barnett DA, Ells B, Guevremont R, Purves RW. Application of ESI-FAIMS-MS to the analysis of tryptic peptides. J. Am. Soc. Mass Spectrom. 2002;13:1282–1291. doi: 10.1016/S1044-0305(02)00527-5. [DOI] [PubMed] [Google Scholar]
- 10.Guevremont R, Barnett DA, Purves RW, Vandermey J. Analysis of a tryptic digest of pig hemaglobin using ESI-FAIMS-MS. Anal. Chem. 2000;72:4577–4584. doi: 10.1021/ac0000271. [DOI] [PubMed] [Google Scholar]
- 11.Venne K, Bonneil E, Eng K, Thibault P. Improvement in peptide detection for proteomics analyses using nanoLC-MS and high-field asymmetry waveform ion mobility mass spectrometry. Anal. Chem. 2005;77:2176–2186. doi: 10.1021/ac048410j. [DOI] [PubMed] [Google Scholar]
- 12.Xia Y-Q, Wu ST, Jemal M. LC-FAIMS-MS/MS for quantification of a peptide in plasma and evaluation of FAIMS global selectivity from plasma components. Anal. Chem. 2008;80:7137–7143. doi: 10.1021/ac8010846. [DOI] [PubMed] [Google Scholar]
- 13.Xuan Y, Creese AJ, Horner JA, Cooper HJ. Separation of isobaric phosphopeptides by high field asymmetric waveform ion mobility spectrometry confirmed by high resolution electron transfer dissociation mass spectrometry. Rapid Commun. Mass Spectrom. 2009;23:1963–1969. doi: 10.1002/rcm.4101. [DOI] [PubMed] [Google Scholar]
- 14.Shvartsburg AA, Creese AJ, Smith RD, Cooper HJ. Complete separation of peptide isomers with variant modified sites by high-resolution differential ion mobility spectrometry. Anal. Chem. 2010;82:8327–8334. doi: 10.1021/ac101878a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shvartsburg AA, Creese AJ, Smith RD, Cooper HJ. Separation of a set of peptide sequence isomers using differential ion mobility spectrometry. Anal. Chem. 2011;83:6918–6923. doi: 10.1021/ac201640d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Creese AJ, Cooper HJ. The separation and identification of isomeric glycopeptides by high field symmetric waveform ion mobility spectrometry. Anal. Chem. 2012;84:2597–2601. doi: 10.1021/ac203321y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saba J, Bonneil E, Pomies C, Eng K, Thibault P. Enhanced sensitivity in proteomics experiments using FAIMS coupled with a hybrid linear ion trap/orbitrap mass spectrometer. J. Proteome Res. 2009;8:3355–3366. doi: 10.1021/pr801106a. [DOI] [PubMed] [Google Scholar]
- 18.Bridon G, Bonneil E, Muratore-Schroeder T, Caron-Lizotte O, Thibault P. Improvement of phosphoproteome analyses using FAIMS and decision tree fragmentation. Application to the Insulin Signaling Pathway in Drosophila melanogaster S2 cells. J. Proteome Res. 2012;11:927–940. doi: 10.1021/pr200722s. [DOI] [PubMed] [Google Scholar]
- 19.Swearingen, K.E., Hoopmann, M.R., Johnson, R.S., Saleem, R.A., Aitchison, J.D., Moritz, R.L.: Nanospray FAIMS fractionation provides significant increases in proteome coverage of unfractionated complex protein digests. Mol. Cell. Proteom. 11, M111.014985 (2012) [DOI] [PMC free article] [PubMed]
- 20.Kim S, Mischerikow N, Bandeira N, Navarro JD, Wich L, Mohammed S, Heck AJR, Pevzner PA. The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search. Mol. Cell. Proteom. 2010;9:2840–2852. doi: 10.1074/mcp.M110.003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sweet SMM, Jones AW, Cunningham DL, Heath JK, Creese AJ, Cooper HJ. Database Search strategies for proteomic data sets generated by electron capture dissociation mass spectrometry. J. Proteome Res. 2009;8:5475–5484. doi: 10.1021/pr9008282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Molina H, Matthiesen R, Kandasamy K, Pandey A. Comprehensive comparison of collision induced dissociation and electron transfer dissociation. Anal. Chem. 2008;80:4825–4835. doi: 10.1021/ac8007785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Good DM, Wirtala M, McAlister GC, Coon JJ. Performance characteristics of electron transfer dissociation mass spectrometry. Mol. Cell. Proteom. 2007;6:1942–1951. doi: 10.1074/mcp.M700073-MCP200. [DOI] [PubMed] [Google Scholar]
- 24.Shen YF, Tolic N, Xie F, Zhao R, Purvine SO, Schepmoes AA, Ronald JM, Anderson GA, Smith RD. Effectiveness of CID, HCD, and ETD with FT MS/MS for degradomic-peptidomic analysis: comparison of peptide identification methods. J. Proteome Res. 2011;10:3929–3943. doi: 10.1021/pr200052c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swaney, D.L., McAlister, G.C., Wirtala, M., Schwartz, J.C., Syka, J.E.P., Coon, J.J.: Supplemental activation method for high‐efficiency electron‐transfer dissociation of doubly protonated peptide precursors. Anal. Chem. 79, 477–485 (2007) [DOI] [PMC free article] [PubMed]
- 26.Frese, C.K., Altelaar, A.F.M., Hennrich, M.L., Nolting, D., Zeller, M., Griep‐Raming, J., Heck, A.J.R., Mohammed, S.: Improved peptide identification by targeted fragmentation using CID, HCD and ETD on an LTQ‐Orbitrap velos. J. Proteome Res.10, 2377–2388 (2011) [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 831 kb)
(XLSX 90 kb)
(XLSX 329 kb)
(XLSX 147 kb)
(XLSX 635 kb)