

Abstract
C-Jun N-terminal kinase (JNK) is a member of the mitogen-activated protein kinase (MAPK) family and controls essential processes such as inflammation, cell differentiation, and apoptosis. JNK signalling is triggered by extracellular signals such as cytokines and environmental stresses. Macrophage migration inhibitory factor (MIF) is a pleiotropic pro-inflammatory cytokine with chemokine-like functions in leukocyte recruitment and atherosclerosis. MIF promotes MAPK signalling through ERK1/2, while it can either activate or inhibit JNK phosphorylation, depending on the cell type and underlying stimulation context. MIF activities are mediated by non-cognate interactions with the CXC chemokine receptors CXCR2 and CXCR4 or by ligation of CD74, which is the cell surface expressed form of the class II invariant chain. ERK1/2 signalling stimulated by MIF is dependent on CD74, but the receptor pathway involved in MIF activation of the JNK pathway is unknown. Here we comprehensively characterize the stimulatory effect of MIF on the canonical JNK/c-Jun/AP-1 pathway in fibroblasts and T cell lines and identify the upstream signalling components. Physiological concentrations of recombinant MIF triggered the phosphorylation of JNK and c-Jun and rapidly activated AP-1. In T cells, MIF-mediated activation of the JNK pathway led to upregulated gene expression of the inflammatory chemokine CXCL8. Activation of JNK signalling by MIF involved the upstream kinases PI3K and SRC and was found to be dependent on CXCR4 and CD74. Together, these data show that the CXCR4/CD74/SRC/PI3K axis mediates rapid and transient activation of the JNK pathway as triggered by the inflammatory cytokine MIF in T cells and fibroblasts.
Keywords: chemokine, chemokine receptor, cytokine, MAPK, siRNA
1. Introduction
The signalling pathways mediated by mitogen-activated protein (MAP) kinases (MAPK) are important for cellular responses to various growth factors, hormones, cytokines and environmental stresses. Mitogen-activated protein kinases regulate basic functions including proliferation, differentiation, survival and apoptosis. In mammals, 4 distinct MAPK pathways have been identified; namely the extracellular signal-regulated kinase-1/2 (ERK; p42/44), the ERK5, the c-Jun N-terminal kinase (JNK) and the p38 pathway [1-3].
The JNK protein kinases are encoded by three distinct genes. JNK1 and JNK2 are ubiquitously expressed. In contrast, JNK3 is selectively expressed in brain [4-6]. The JNKs also are collectively referred to as stress-activated MAP kinase (SAPKs). In the canonical JNK pathway, JNKs are activated by phosphorylation of specific Thr and Tyr residues by the upstream MAP kinase kinases (MKKs) MKK4 and MKK7, whereas they are inactivated by Ser/Thr and Tyr protein phosphatases [7, 8]. JNK phosphorylation and activation occurs in response to a variety of environmental, developmental, and inflammatory stimuli including the pro-inflammatory cytokine tumour necrosis factor-α (TNF-α) [5, 6, 9] or interleukin-1 β (IL-1 β) [10]. Activated JNK subsequently acts to phosphorylate the transcriptional activation domain of c-Jun, which then forms a homodimer or a heterdimer with c-Fos to constitute the activator protein-1 (AP-1) transcription factor [4].
Binding of the CXC chemokine CXCL12, also termed stromal cell-derived factor-1α (SDF-1α), to its cognate G protein-coupled receptor (GPCR) CXCR4 results in rapid signalling through a pertussis toxin-sensitive G protein-dependent pathway [11]. The CXCL12/CXCR4 axis is involved in homeostatic and inflammatory cell migration processes, including inflammatory and atherogenic T cell recruitment, stem cell homing, and cancer cell metastasis [12, 13]. G-protein-coupled receptors regulate MAPK signalling pathways that result in the expression of specific early as well as late response genes involved in cell proliferation, differentiation and apoptosis [14]. G-protein-coupled receptors have been shown to trigger diverse transcription factors such as AP-1 [15], nuclear factor kappa-B (NFκB), cAMP response element binding protein (CREB), steroid receptor response element binding protein (SREB) [16], or activating transcription factor 1 (ATF1) [17] through activation of JNK, ERK, p38 or ERK5. Interaction of CXCL12 with CXCR4 results in JNK phosphorylation and activation [18, 19], but the details of this signalling pathway remain to be elucidated.
Macrophage migration inhibitory factor (MIF) is a widely expressed and pleiotropic cytokine that functions as a critical upstream mediator of innate immunity but also promotes numerous pathophysiological processes [20, 21]. As such, MIF is a pivotal mediator of acute and chronic inflammatory diseases such as septic shock, rheumatoid arthritis, inflammatory lung diseases, atherosclerosis and cancer [20, 22-27]. MIF is mainly produced by immune cells, but its expression extends to cells outside of the immune system, for example epithelial cells, endothelial cells or various tumour cells. MIF secretion is tightly regulated by immune and stress stimuli and occurs by a non-classic export mechanism [20, 28, 29]. Upon secretion, MIF exhibits a broad range of immune and inflammatory activities, including induction of inflammatory cytokines such as CXCL8 or IL-6, overriding of glucocorticoid-mediated immune suppression, and enhancement of cell proliferation and cell survival, which are often coupled to inhibitory effects on cell apoptosis [20, 30-32].
Recently, the receptors mediating the cellular responses supported by MIF were identified. Depending on the cellular context and stimulation status, MIF can bind to three receptor proteins and trigger several signalling tracks. MIF interacts with CD74, which is the cell surface form of class II invariant chain (Ii) [33]. Following inflammatory stimulation, CD74 also can be found on cells devoid of MHC class II, such as endothelial, stromal, or epithelial cells [34]. Prominent MIF/CD74 interactions were demonstrated on B lymphocytes, tumour cells as well as macrophages, and to a lesser degree on fibroblasts, leading to the activation of ERK1/2 MAPK, which is frequently sustained, and to PI3K/AKT signalling, resulting in cell proliferation, enhanced survival, and CXCL8 gene expression [31, 33, 35-37]. The MIF/CD74 axis has not been shown to stimulate the JNK pathway, although MIF release inhibits signalling through JNK in ischemic cardiomyocytes [38]. MIF-induced MMP-2 production in rheumatoid synovial fibroblasts required activation of protein kinase C, JNK, and SRC signalling pathways [39]. On the other hand, Yu et al. showed that MIF induces MMP-9 expression in murine macrophages mainly via the ERK1/2 MAPK pathway and only to a minor extent through JNK [40]. Direct evidence for a role of MIF in activation of the JNK signalling pathway came from a study on the role of MIF in septic shock, which showed MIF-mediated phosphorylation of JNK [41], but the involved upstream mechanisms have remained elusive. In lung adenocarcinoma cells, MIF and D-dopachrome tautomerase (D-DT), a homolog of MIF, promote JNK-dependent AP-1 transactivation and subsequent CXCL8 transcriptional regulation [42]. In contrast, it appears that when MIF target cells are pre-stimulated by stress, MIF acts to inhibit or attenuate the JNK MAPK pathway. Activation of the JNK pathway in fibroblasts stimulated by TNF or UV irradiation stress is blocked by higher concentrations of MIF [43]. Moreover, JAB1/CSN5, a coactivator of AP-1 activity and subunit of the COP9 signalosome (CSN), promotes JNK and c-Jun phosphorylation in addition to its coactivator effect. Intracellular MIF binds to CSN5 to act as a counter-regulator of CSN5 activities and JNK stimulation following ectopic CSN5 overexpression is down-regulated by MIF [43]. JNK activation in cardiomyocytes by ischemia/reperfusion injury triggers phosphorylation of the pro-apoptotic protein BAD with an ensuing increase in cell death. This effect is elevated in Mif gene-deficient mice, indicating that endogenous MIF inhibits JNK pathway activation during reperfusion in the heart [38].
Not all MIF target cells express CD74. It was thus speculated that additional MIF receptors exist. In fact, MIF was demonstrated to be a non-cognate ligand of the CXC chemokine receptors CXCR2 and CXCR4. MIF promotes the atherogenic and inflammatory recruitment of monocytes/macrophages and T cells through CXCR2 and CXCR4, respectively [44, 45]. The involved signalling pathways have largely remained unknown, but inhibitor studies implicate the AKT pathway in MIF-mediated monocyte chemotaxis and T cell activation.
CXCL12 is the bona fide ligand of CXCR4. In B lymphocytes, CXCL12 treatment was shown to result in a rapid activation of AKT and JNK [46] and in acute lymphoblastic leukaemia T cells, CXCL8 production is regulated by the CXCL12/CXCR4 axis and the NFκB and JNK/AP-1 pathways. MIF has been demonstrated to promote the upregulation of CXCL8 expression in B lymphocytes through CD74, but the role of CXCR4 and the JNK pathway are unknown.
Here we wished to comprehensively study the effect of (patho)physiological concentrations of exogenous MIF on the rapid stimulation of the entire JNK/c-Jun/AP-1 pathway in cell lines, fibroblasts and T cells and to explore the role of upstream kinase mechanisms and that of the MIF receptors CXCR4 and CD74 in JNK activation.
2. Materials and methods
2.1. Chemicals, kinase inhibitors, buffers and antibodies
Oligonucleotide primers and siRNA duplexes were acquired from MWG Biotech AG (Ebersberg, Germany). The Lipofectamin 2000 transfection reagent was obtained from Invitrogen (Karlsruhe, Germany). All other molecular biology reagents were either from MBI Fermentas GmbH (St. Leon-Rot, Germany) or New England Biolabs GmbH (Heidelberg, Germany).
A protease inhibitor cocktail and the protein kinase inhibitors (the PI3K inhibitor Ly294002, the broad spectrum tyrosine kinase inhibitor genistein, and the SRC kinase inhibitors herbimycin and pp2, as well as the JNK inhibitor SP600125) were bought from Merck-Calbiochem (Darmstadt, Germany). The CXCR4 inhibitor AMD3100 was purchased from Sigma-Aldrich Chemicals (Taufkirchen, Germany).
The anti-phospho-c-Jun (KM-1), anti-c-Jun (H-79) and anti-JNK1 (C-17) antibodies were obtained from Santa Cruz Biotechnology Inc. (Heidelberg, Germany). Anti-phospho-SAPK/JNK (Thr183/Tyr185) was purchased from Cell Signalling Technology (New England Biolabs GmbH, Frankfurt a. M., Germany). The anti-CXCR4 antibody was purchased from R&D Systems (Germany). Peroxidase-conjugated sheep anti-mouse (Fab)2 and peroxidase-conjugated donkey anti-rabbit IgG were purchased from GE Healthcare/Amersham Biosciences Europe GmbH (Freiburg, Germany). Neutralizing anti-MIF monoclonal antibodies (mAbs; NIH/III.D9) and isotype control IgG were prepared as described previously [47]. Miscellaneous reagents such as chemicals and salts were from Sigma-Aldrich Chemicals. All reagents were of the highest grade commercially available.
2.2. Recombinant proteins and constructs
Biologically active recombinant human MIF (rMIF) was expressed and purified from the pET11b/BL21-DE3 expression system as described previously [48]. The endotoxin content of rMIF was controlled by the Limulus QCL-1000 kit (Cambrex BioScience, Verviers, Belgium) and was lower than 5 pg LPS/μg rMIF.
2.3. Cell culture
General cell culture reagents such as media, supplements, antibiotics, and serum were from Invitrogen Corporation. Unless stated otherwise, cell lines were obtained from the German Society for Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany) or from the American Type Culture Collections (ATCC).
Wild-type and Mif knock out mouse embryonic fibroblasts (Mif−/− MEFs) were prepared from Mif−/− mice (C57BL/6 background) as described before [49]. MEFs were cultured in Dulbecco’s modified Eagle medium (DMEM), containing 10% fetal calf serum (FCS). For a typical experiment, passage 3 MEFs were plated and cultured for 2 additional passages. Passage 5 MEFs were then used for the JNK activation assays. Cd74−/− MEFs (C57BL/6 background) cells were cultivated in DMEM medium containing 10% FCS.
NIH/3T3 fibroblasts and HEK293 epithelial cells were maintained by routine protocols in DMEM supplemented with 10% FCS, 1% penicillin-streptomycin, and 5 mM L-glutamine. NIH/3T3 and HEK293 cells were sub-cultured 2–3 times a week and passages between 5 and 15 used for the experiments.
Jurkat T cells were cultured in RPMI-1640 medium supplemented with 10% FCS, 1% penicillin-streptomycin, and 2 mM L-glutamine.
2.4. AP-1 transcription factor activation assay
Activation of the AP-1 transcription factor was detected and quantified by the TransAM AP-1 Kit (Active Motif Europe, Rixensart, Belgium) according to the manufacturer’s instructions. Briefly, HEK293 cells were incubated overnight with DMEM medium containing 0.5% FCS and stimulated with rMIF (0 – 100 ng/ml) or 10% FCS as a positive control for 10 min. Nuclear extracts were isolated and incubated in a 96-well plate into which oligonucleotides containing a TPA-responsive element (TRE, 5′-TGA(C/G)TCA-3′ sequence) were immobilized. AP-1 dimers contained in the nuclear extracts specifically bind to these oligonucleotides and were detected through an antibody directed against c-Jun. Addition of a secondary antibody conjugated to horseradish peroxidase (HRP) provided a sensitive colorimetric readout that was readily quantifyable by spectrophotometry.
2.5. Analysis of JNK activation by Western blotting
One million NIH/3T3 fibroblasts were cultured in a 10 cm cell culture plate and incubated in DMEM medium containing 10% FCS for 24 h. Medium was changed and cells cultured in DMEM containing 0.5% FCS for 24–48 h. Cells were treated with the inhibitors for 30 min. Recombinant MIF in 20 mM sodium phosphate buffer, pH 7.2, was added to the cells at a final concentration of 10–12500 ng/ml as indicated. Controls were incubated with buffer alone (negative controls), i.e. obtained from a batch of the final dialysis of MIF renaturation buffer, with the added control buffer volume corresponding to the highest rMIF concentration used in an experimental series, and, where indicated, with 10% FCS (positive controls). Incubations were stopped at the indicated times by washing the cells twice with cold PBS. Cells were then lysed with RIPA buffer (PBS, pH 7.4, containing 1% Igepal CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, 1 x proteinase inhibitor cocktail, and 1 mM sodium orthovanadate), lysates homogenized by pipetting and syringe passage (23 gauge) and cellular proteins analysed in 4–12% NuPAGE gels (Invitrogen). Following transfer to nitrocellulose filters, Western blots were performed essentially as described previously [50]. Blots were probed with anti-phospho-c-Jun or anti-phospho-JNK antibodies and developed with the Super Signal West Dura Extended Duration ECL reagent (Pierce/KMF Laborchemie, St. Augustin, Germany), using a peroxidase-conjugated secondary antibody. For counter-staining and standardisation purposes, membranes were stripped with 0.5% Tween 20 in Tris-buffered saline (TBS; 20 mM Tris–HCl, pH 7.3, 150 mM NaCl) for 30 min and redeveloped with anti-c-Jun or anti-JNK1 antibody to monitor c-Jun and JNK expression levels. Staining was measured with the LAS-3000 imager equipped with a CCD camera with a 16 bit resolution and band intensities quantified using the Aida Image Analyzer Software (Fuji/Raytest Isotopenmessgerät GmbH, Straubenhardt, Germany).
2.7. siRNA knock down of CXCR4
For knock down of CXCR4 in Jurkat T cells, we used small interfering RNA (siRNA) with the human CXCR4-specific sequence 5′CACUCACCUCUGUGAGCAGTT3′ ([51]; Genbank accession no. NM_003467) and the control siRNA sequence ′GGUUUGGCUGGGUGUUAUTT3′. Transfections were performed by electroporation method using the Amaxa nucleofector (Lonza, Cologne, Germany) according to the manufacturer’s instructions with 100 pmol of siRNA per 106 cells. Cells then were incubated for 48 h and total cellular CXCR4 content was determined by anti-CXCR4 western blot to verify knock down efficiency. For JNK stimulation assays, the cells were treated with CXCL12 or MIF and JNK phosphorylation was analysed by western blot.
2.8. Treatment with the CXCR4 inhibitor AMD3100
As indicated in Results, cells were pre-treated with the CXCR4 inhibitor AMD3100 for 30 min before incubation of MIF or CXCL12.
2.9. Quantification of CXCL8 by enzyme-linked immunosorbent Assay (ELISA)
To determine CXCL8 levels in the culture supernatants of Jurkat T cells, cells were plated at a density of 105 cells/ml in RPMI-1640 medium containing 0.5% FCS. The cells were pre-treated with the CXCR4 inhibitor AMD3100 (1 μg/ml) or the JNK inhibitor SP600125 (10 μM) for 30 min and then incubated with 0-100 ng/ml of rMIF or 50 ng/ml of CXCL12 for 24 h. CXCL8 expression and secretion was measured using a human CXCL8 antibody sandwich ELISA kit (RayBiotech, Inc./Hoelzel Diagnostika GmbH, Cologne, Germany) according to the manufacturer’s instructions.
2.10. Statistical analysis
Numerical data are expressed as means ± standard deviation (SD). Statistical differences between groups were evaluated with Origin software (version 7.1) using one-way analysis of variance (ANOVA). Observed differences were considered statistically significant when p values were <0.05.
3. Results
3.1. MIF potently stimulates the rapid and transient phosphorylation of JNK and c-Jun
MIF activates the ERK1/2 pathway, and, depending on the context, both stimulatory and inhibitory effects on JNK signalling have been observed. To begin to comprehensively examine the effects of MIF on the JNK pathway and to define the upstream kinase and receptor mechanisms, we first measured the effect of short term exposure of NIH/3T3 fibroblasts with exogenous recombinant MIF at the physiologically relevant concentration of 50 ng/ml. Fibroblasts were incubated with rMIF for 0 – 180 min and the phosphorylation of c-Jun, the direct substrate of JNK, evaluated by Western blotting. MIF potently upregulated c-Jun phosphorylation within a narrow time window of 30 min upon addition of MIF to the cells (Fig. 1), suggesting that in resting fibroblasts, exogenous MIF is able to rapidly and dramatically trigger JNK signalling. We next verified whether the observed phosphorylation of c-Jun was paralleled by an activation of JNK. In fact, Fig. 2A shows that incubation of NIH/3T3 fibroblasts with rMIF for 15 min led to a marked phosphorylation of JNK. The effect of MIF was dose-dependent with a maximum observed between 25 and 100 ng/ml rMIF. At higher concentrations of MIF, a plateauing of JNK phosphorylation was noted. Quantitative densitometric analysis indicated that rMIF was able to enhance phospho-JNK levels by up to ~2.2-fold. The antibody applied for phospho-JNK detection was a pan-JNK antibody such that in this study, no differentiation was discernable between the three JNK isoforms. As expected from the observed rapid enhancement of c-Jun phosphorylation in Fig. 1, JNK phosphorylation by MIF was even more rapid and peaked at an incubation interval of 5-15 min post rMIF addition (Fig. 2B). Interestingly, the MIF-mediated enhanced phospho-JNK signal continued over a time interval of 4 h, although MIF effects ≥30 min did not reach statistical significance any more. Nevertheless, this may confirm that MIF might more generally support sustained MAPK signalling responses in fibroblasts [36]. We next wished to confirm that exogenous, ‘extracellular’, MIF was responsible for the observed activation of JNK. Thus, fibroblasts genetically deficient in endogenous MIF, Mif −/− MEFs [37, 50], were subjected to a 15 min stimulation pulse with rMIF. As seen in the MIF-expressing NIH/3T3 fibroblasts, rMIF at a concentration of 50 ng/ml or higher triggered JNK phosphorylation in Mif −/− MEFs (Fig. 2C). As a control, WT-MEFs were also subjected to stimulation with rMIF and these cells showed a similar phospho-JNK response as the NIH/3T3 fibroblasts (Supplementary Fig. 1). Since the initial scouting experiments had indicated that the phospho-c-Jun and phospho-JNK signals can be affected by various stress factors such as temperature, volume, pH, and salt, control incubations in the above and all subsequent JNK pathway activation experiments were strictly accompanied by buffer controls with the highest corresponding buffer volume. Buffers (‘zero’ ng/ml MIF or ‘zero’ minute lanes) were always derived from the final dialysis batch used for renaturing rMIF [48], such that identical pH and salt conditions were applied for the rMIF and buffer control samples. Activation of the JNK pathway by rMIF was further underscored by experiments in which we added a 16-meric MIF-agonistic peptide, MIF50-65, which had previously been demonstrated to promote ERK1/2 activation in a MIF-like manner [52], to NIH/3T3 cells. MIF50-65, albeit at 10-fold higher concentrations, triggered the rapid phosphorylation of c-Jun and JNK with a maximum effect seen at around 10 min (Supplementary Fig. 2).
Fig. 1.
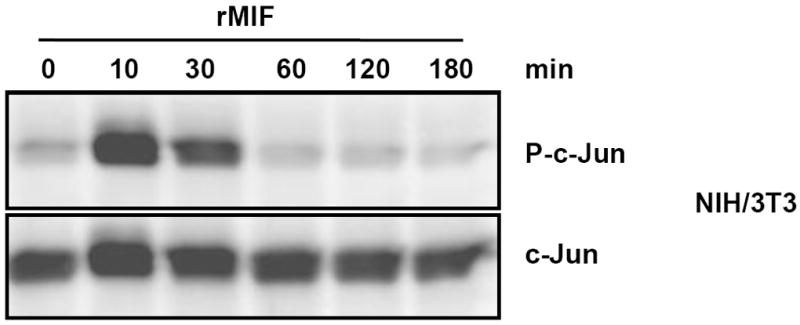
MIF promotes rapid phosphorylation of c-Jun. MIF promotes rapid and transient phosphorylation of c-Jun in NIH/3T3 fibroblasts. A time course from 0-180 min is shown. Phospho-c-Jun levels were determined by Western blot analysis and c-Jun expression levels were used for standardization. The blot shown is representative of three independent experiments.
Fig. 2.
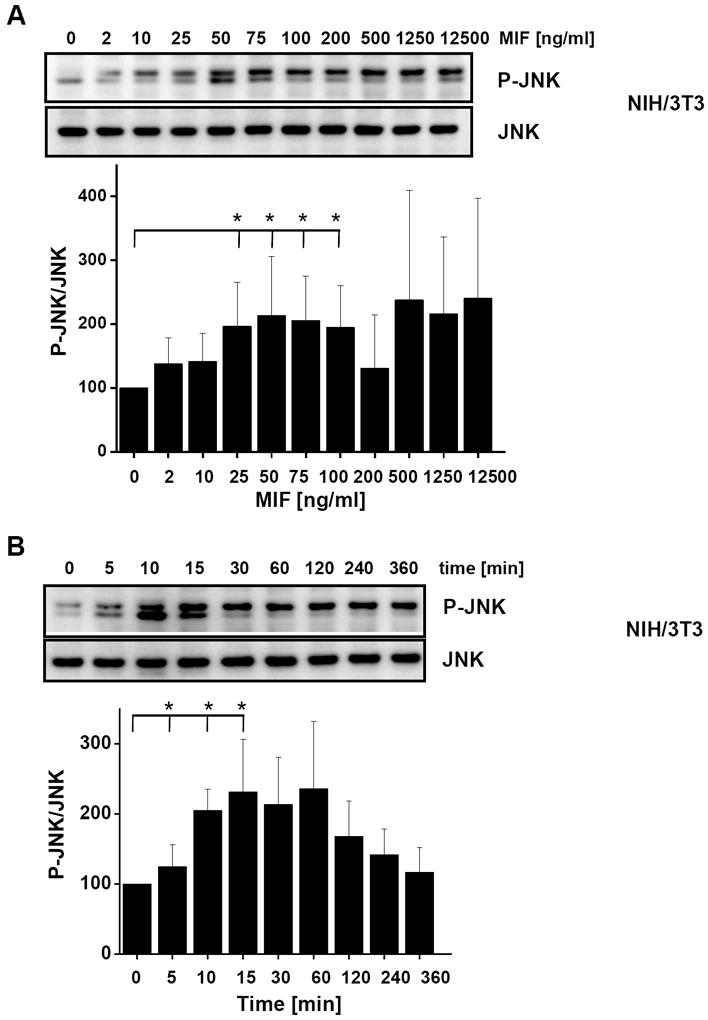
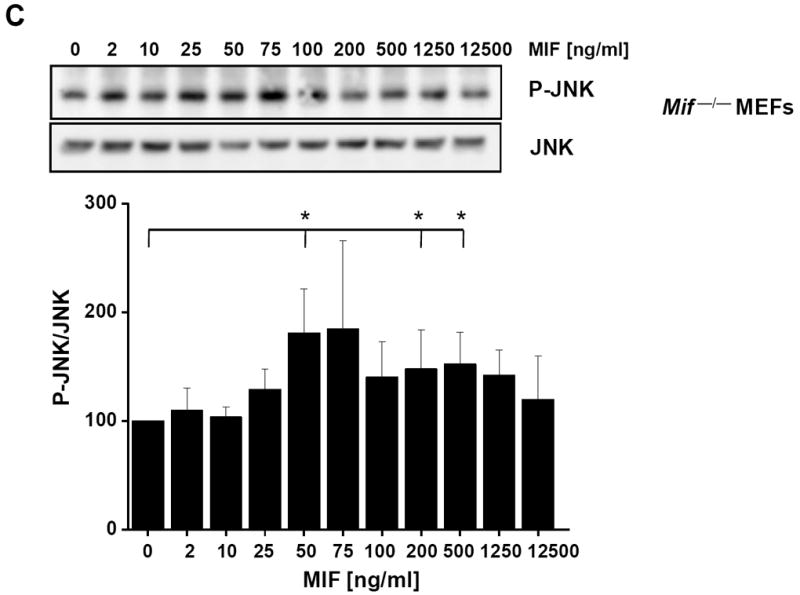
MIF promotes JNK activation in fibroblasts in a concentration- and time-dependent manner. (A) Concentration-dependent phosphorylation of JNK by rMIF in NIH/3T3 fibroblasts. Maximum activation occurs at 50 ng/ml MIF. Upper panel, JNK phosphorylation was measured by Western blot analysis using a pan-phospho-JNK antibody. Lower panel, band densitometry was performed from 4 independent experiments (means ± SD) to quantify MIF-mediated JNK phosphorylation using JNK levels for normalization. (B) JNK phosphorylation by MIF is time-dependent and transient and peaks at an early time point of 5-15 min. MIF-mediated JNK phosphorylation was measured and quantified as in (A). Data are means ± SD from three independent experiments. (C) Concentration-dependent phosphorylation of JNK by rMIF in mouse embryonal fibroblasts genetically deficient for MIF (Mif −/− MEFs). Maximum activation starts at 50 ng/ml MIF. Quantification of JNK activation as in (A). Data are means ± SD from three independent experiments. Asterisks (*) indicate statistical significance (p<0.05) between the control value (0 ng/ml MIF or 0 min) and the corresponding incubations.
3.3 MIF-stimulated JNK activation is dependent on PI3K and SRC-type kinases
We previously demonstrated that MIF promotes AKT phosphorylation at Ser473 and Thr308 in a dose- and time-dependent manner and that this effect is associated with SRC kinase activation [37]. Moreover, activation of PI3K and SRC frequently is upstream of JNK signalling [53].
To test whether MIF-mediated JNK activation is dependent on upstream SRC/PI3K pathways, we treated NIH/3T3 fibroblasts with the small molecule PI3K inhibitor Ly294002 prior to short term incubation of the cells with rMIF. Fig. 3A indicates MIF-mediated JNK phosphorylation was abolished by interfering with upstream PI3K activity as compared with cells treated with a DMSO solvent control. Moreover, also SRC family kinases appear to be located upstream in the MIF-triggered JNK pathway, because not only the broad-spectrum tyrosine kinase inhibitor genistein inhibited MIF-stimulated JNK phosphorylation, but also the SRC family-specific kinase inhibitor PP2, whereas DMSO control solvent only had a minor effect. Herbimycin, another SRC kinase inhibitor led to an overall enhancement of the phospho-JNK signal, but again no MIF-triggered enhancement of JNK activation was observed (Fig. 3B). Inhibition of the MIF-triggered effect on JNK activation by PI3K and SRC kinase inhibitors also was observed in Mif −/− MEFs (data not shown).
Fig. 3.
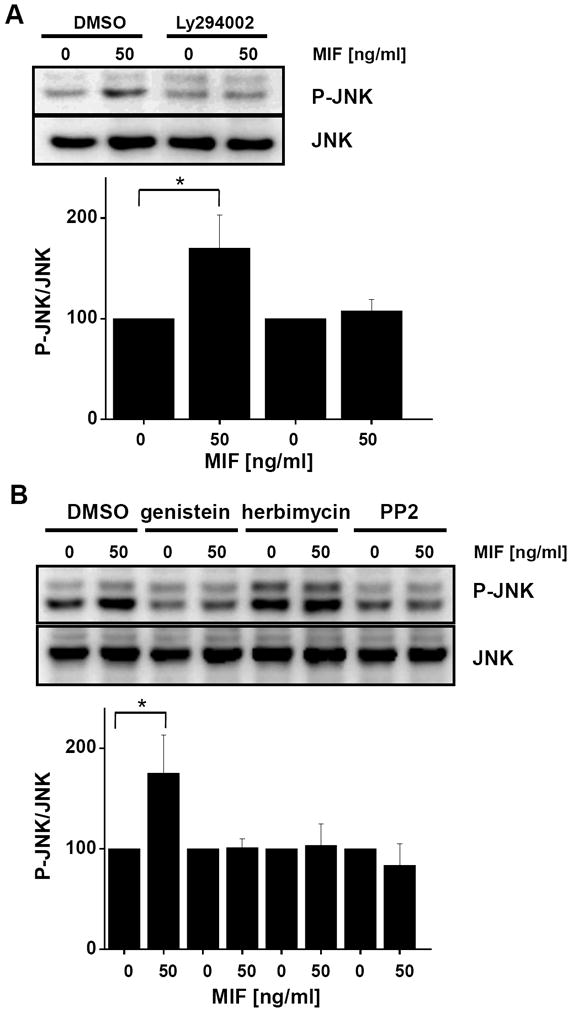
MIF-stimulated JNK phosphorylation is dependent on the upstream activation of PI3K and SRC-type kinases. (A) Activation of transient JNK phosphorylation by MIF is inhibited by the PI3K inhibitor Ly294002. NIH/3T3 fibroblasts were incubated with rMIF for 10 min in the presence of Ly294002 or solvent control (DMSO). Upper panel: JNK phosphorylation was measured by Western blot analysis using a pan-phospho-JNK antibody; lower panel: Statistical analysis of the blots was performed by band densitometry to quantify MIF-mediated JNK phosphorylation using JNK levels for normalization (means ± SD of three independent experiments). (B) MIF-induced JNK phosphorylation in fibroblasts is inhibited by the broad spectrum tyrosine kinase inhibitor genistein or the SRC kinase-specific inhibitors herbimycin or PP2. NIH/3T3 cells were stimulated with rMIF for 10 min in the presence of genistein, herbimycin, PP2, or solvent control (DMSO). Western blot (upper panel) and statistical analysis (lower panel; means ± SD of three independent experiments) was performed as in (A).
3.4 MIF-triggered JNK activation is diminished in fibroblasts genetically deficient in Cd74
Given that activation of the ERK1/2, AKT, and SRC kinase signalling pathways has been demonstrated to be triggered by the MIF/CD74 axis [35, 37], we next asked whether MIF-mediated JNK activation involved CD74. Wildtype MEFs immortalised with large T antigen were compared with corresponding Cd74−/− MEFs in their capacity to upregulate JNK signalling in response to exogenous MIF. Fig. 4A shows that MIF at a concentration of 50 and 100 ng/ml significantly promoted JNK phosphorylation in a dose-dependent manner in the WT MEFs. In contrast in the Cd74−/− MEFs, JNK phosphorylation as stimulated by MIF did not reach statistical significance. In addition, the obtained MIF dose response curves differed between the WT MEFs and the Cd74−/− MEFs. Moreover, the maximum phospho-JNK response in MEFs deficient in Cd74 did not reach the phospho-JNK baseline levels in WT MEFs (Fig. 4B), arguing together for a strict dependence of the MIF-mediated JNK response on Cd74.
Fig. 4.
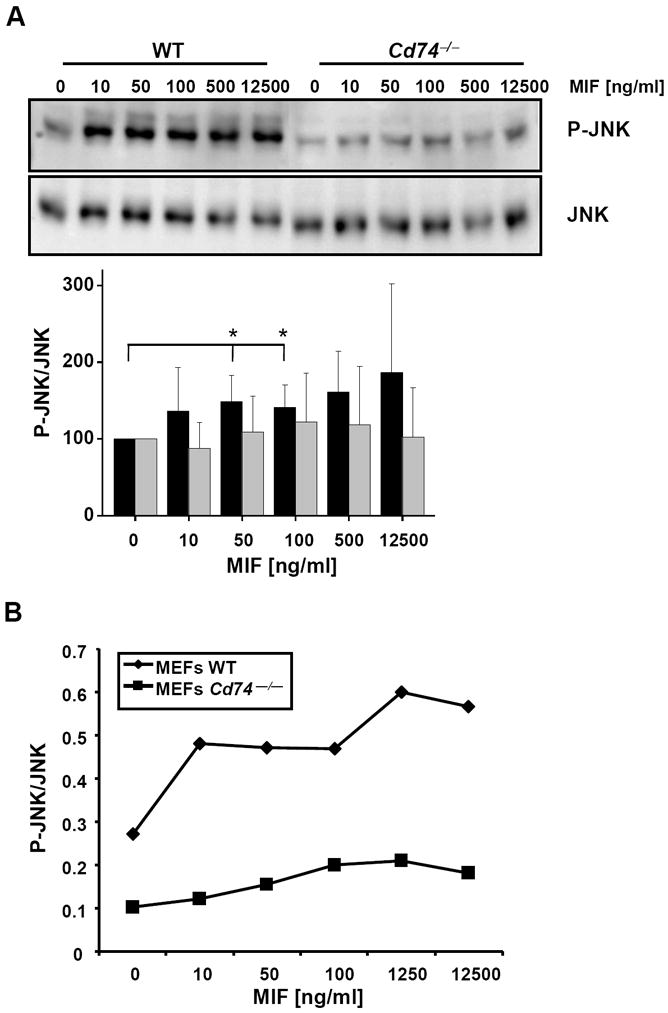
MIF-induced JNK phosphorylation is partially dependent on CD74. (A) Phospho-JNK Western blot of lysates from MIF-stimulated wildtype (WT) and Cd74−/− MEFs. Various concentrations of rMIF were added as indicated. JNK phosphorylation was measured by phospho-JNK-specific Western blot (upper panel) and quantified as before (lower panel; means ± SD of 5 independent experiments). (B) Line graph correlating the phosphorylation ratios as obtained in (A) over the concentration of rMIF and comparison between the effect of MIF on JNK phosphorylation in WT versus Cd74−/− MEFs.
3.5 Blockade or silencing of CXCR4 abolishes MIF-triggered JNK activation
Complexes between CD74 and CXCR4 have previously been shown to mediate MIF-driven signalling responses [45]. Therefore, we next asked whether an interaction between MIF and CXCR4 may be involved in MIF-stimulated activation of the JNK pathway. Scouting experiments in HEK293 cells, which express CXCR4 but not CD74 nor the third MIF receptor protein CXCR2 indicated that rMIF was even able to promote JNK phosphorylation if only the CXCR4 receptor is expressed (data not shown). However, robust effects could not be obtained probably due to the relatively low CXCR4 expression levels in HEK293 cells. To confirm these initial observations, the following experiments were performed in Jurkat T lymphocytes. Jurkat cells express substantial amounts of CXCR4 on their cell surface and T cells have been shown to prominently upregulate the JNK pathway through CXCR4 [18]. Exogenous rMIF markedly and dose-dependently upregulated JNK phosphorylation in Jurkat T cells following short exposure with a maximum observed at 50-100 ng/ml MIF (Fig. 5A, left). Importantly, this response was completely blocked when the cells were preincubated with a neutralizing monoclonal anti-CXCR4 antibody but not when pretreated with an isotype IgG control (Fig. 5A, compare right with left panel). Of note, blockade with the anti-CXCR4 mAb also fully blocked the effect of CXCL12-mediated JNK phosphorylation which peaked at a concentration of 50 ng/ml CXCL12 under control conditions (Fig. 5B). Together, these data strongly indicated that MIF-mediated JNK activation is CXCR4-dependent.
Fig. 5.
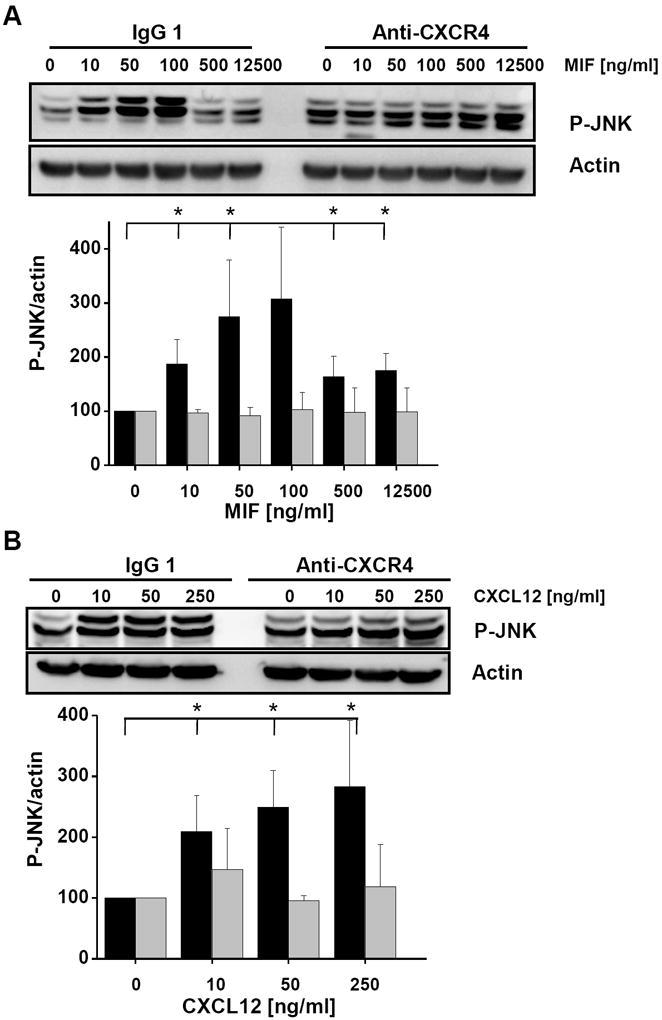
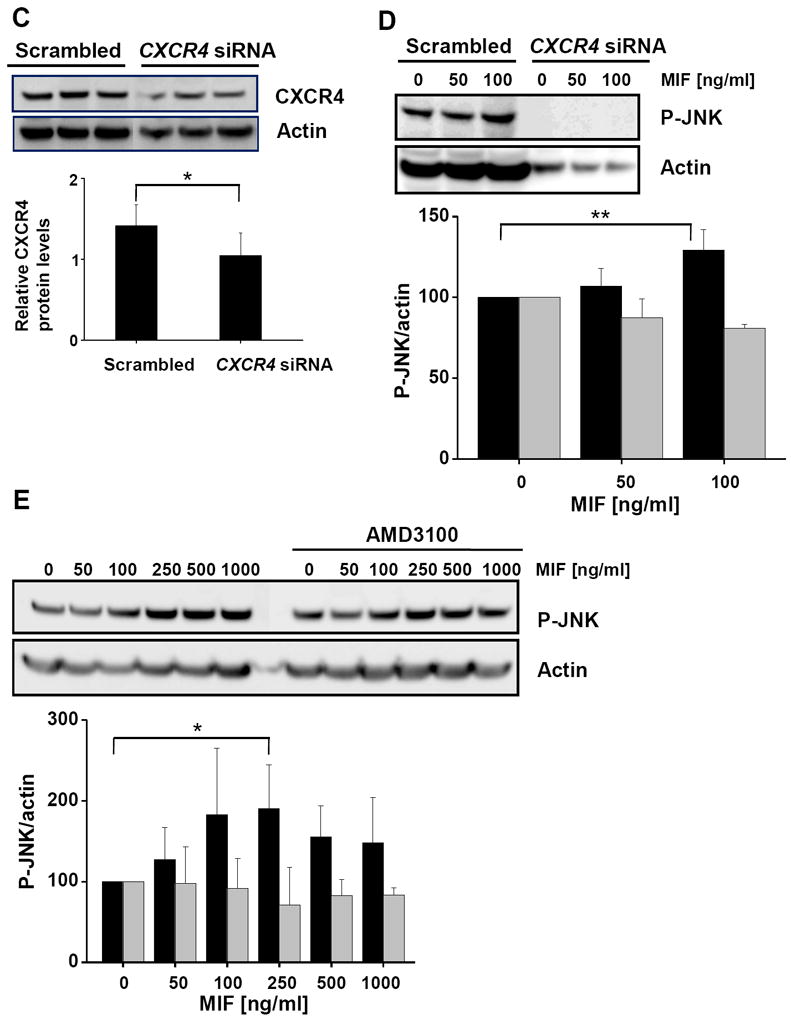
JNK activation stimulated by MIF is dependent on CXCR4. (A) Activation of JNK phosphorylation by MIF in Jurkat T cells is abolished by anti-CXCR4 antibodies. Jurkat cells were incubated with various concentrations of rMIF for 10 min in the presence of anti-CXCR4 or isotype control antibodies. A quantification of MIF-induced JNK phosphorylation was determined by Western blotting using a pan-phospho-JNK antibody and actin for normalization (upper panel) and statistical analysis of 4 independent experiments (means ± SD; lower panel) as before. (B) Activation of JNK phosphorylation by CXCL12 in Jurkat T cells is blocked by anti-CXCR4 antibodies; the data shown are means ± SD from 4 independent experiments; otherwise, see under (A). (C) Knock down of CXCR4 in Jurkat T cells by siRNA-based gene silencing. Top panel, Western blot against CXCR4 from lysates of Jurkat cells treated with CXCR4 siRNA or scrambled control RNA. Actin was used for standardization. Knock down data are from 4 independent experiments. Bottom panel, quantification by band densitometry using actin as standard (bars represent means ± SD of n=4 experiments). (D) Knock down of CXCR4 abolishes phosphorylation of JNK by MIF in Jurkat cells. Comparison between the effect of CXCR4 siRNA and scrambled RNA (upper panel: Western blot using a pan-phospho-JNK antibody and actin as a standard; lower panel: statistical analysis of 4 independent experiments; means ± SD). (E) The CXCR4-specific inhibitor AMD3100 blocks MIF-induced JNK phosphorylation. Jurkat T cells were treated with indicated concentrations of rMIF in the presence or absence of 1 μg/ml AMD3100. Analysis as before (means ± SD of three independent experiments each). Asterisks indicate statistical significance: * p<0.05; ** p<0.01.
To further confirm a role for the MIF/CXCR4 ligand/receptor axis in activation of the JNK pathway, we down-regulated CXCR4 expression levels in Jurkat cells by gene silencing using CXCR4-specific siRNA duplexes. Although only knock down levels for CXCR4 of 50% were achieved (Fig. 5C) and actin levels were somewhat affected by the CXCR4-siRNA treatment, this reduction in CXCR4 lowered baseline JNK activation in the Jurkat cells (compare 0 ng/ml rMIF lanes in Fig. 5D). Importantly, whereas in Jurkat T cells pretreated with scrambled control RNA, rMIF led to an upregulation of phospho-JNK, no enhancement of the phospho-JNK signal by rMIF was seen in Jurkat cells treated with CXCR4 siRNA.
CXCR4 activation by CXCL12 or MIF can be potently inhibited by the antagonistic pharmacological inhibitor AMD3100 [44]. Thus, we next pretreated Jurkat T cells with AMD3100 before addition of rMIF. Fig. 5E demonstrates that AMD3100 reduced the increase in JNK activation that was seen in control-treated cells with MIF concentrations of 100-1000 ng/ml by about three-fold. In aggregate, the data in Fig. 5 confirm the role of exogenous MIF in rapid JNK activation and suggest that an MIF/CXCR4 interaction is also critical for activation of the JNK pathway at least in T cells.
3.6 JNK activation by MIF/CXCR4 leads to downstream transcriptional activation and CXCL8 expression
We next examined whether activation of the JNK pathway by MIF/CXCR4 led to downstream activation of the AP-1 transcription factor and to AP-1-dependent gene expression responses. AP-1 activity was measured from nuclear extracts of HEK293 cells using an AP-1/promoter oligonucleotide-specific ELISA. Stimulation of cells with 50-100 ng/ml rMIF led to a 1.5-fold increase in AP-1 activity, a stimulatory effect comparable to that of 10% FCS, which was used as a positive control (Fig. 6).
Fig. 6.
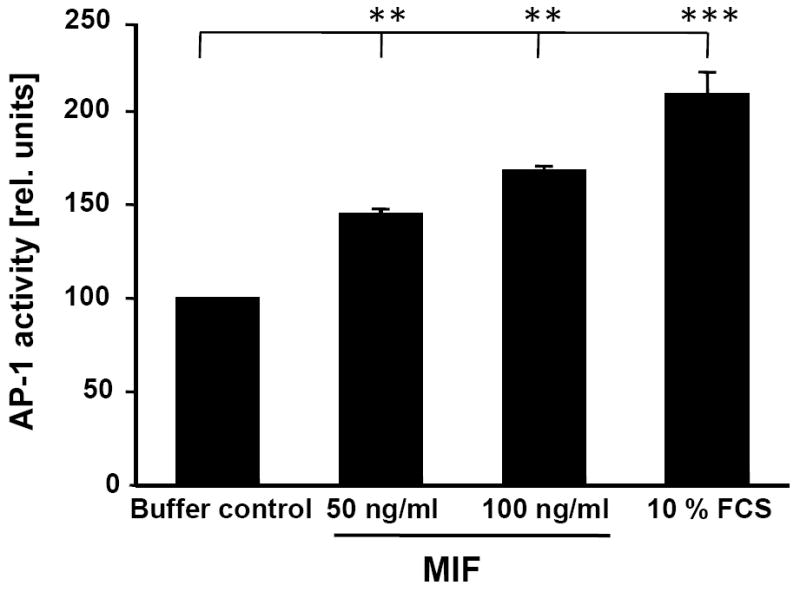
Rapid activation of the AP-1 transcription factor by MIF. HEK293 cells were treated with indicated concentrations of rMIF for 10 min, nuclear extracts isolated and analyzed by AP-1/promoter-specific ELISA using the Trans-AM AP-1 kit with an immobilized consensus oligonucleotide containing the TPA response element (TRE) site (5′-TGAGTCA-3′). Relative AP-1 activity units are shown determined by ELISA using a phospho-c-Jun specific antibody (Ser-73) and HRP-conjugated secondary antibody and colorimetric development (450 nm). Values shown were normalized over the protein concentration of the nuclear extracts and represent means ± SD of triplicate measurements. Asterisks indicate statistical significance: ** p<0.01; *** p<0.005.
CXCL8 transcription is dependent on the activation of an AP-1 responsive element in the gene promoter region. Because Jurkat cells were described to prominently produce CXCL8 upon activation of the JNK pathway and because MIF was shown previously to induce CXCL8 in another lymphocyte cell type, the B lymphocyte [46], we asked whether stimulation of Jurkat cells with exogenous MIF would lead to an upregulation of CXCL8. Jurkat cells were stimulated with 50 ng/ml rMIF for 0-24 h. MIF-induced CXCL8 secretion was first measurable after 8 h (1.5-fold), and was substantial after 24 h (4-fold) (Fig. 7A). Dose response analysis showed that the effect of MIF on CXCL8 secretion was comparable to that of CXCL12 and endotoxin/lipopolysaccharide (LPS) (Fig. 7B).
Fig. 7.
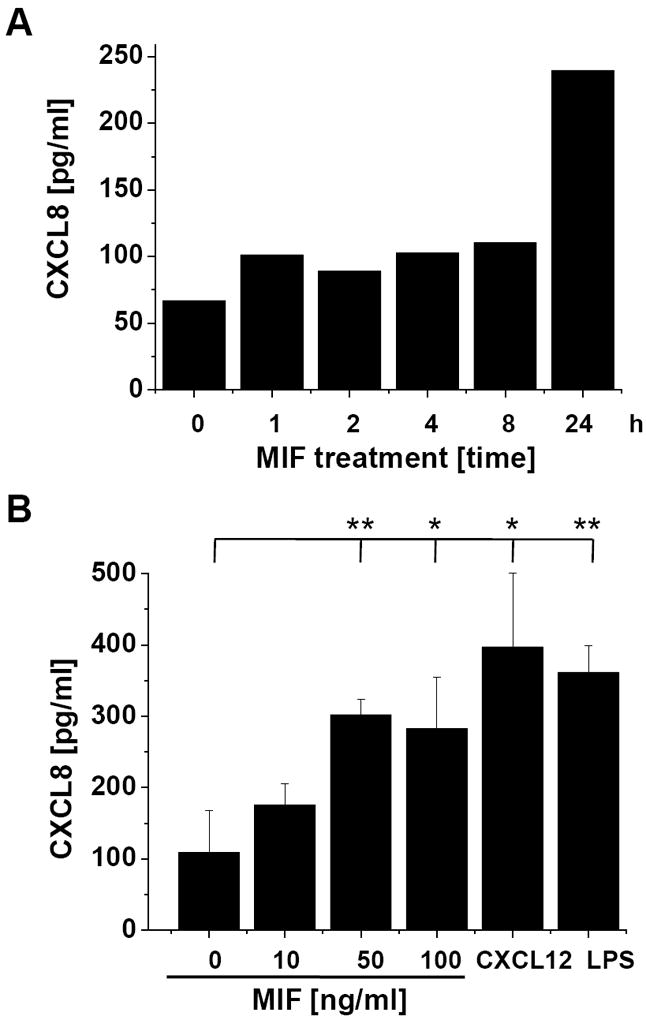
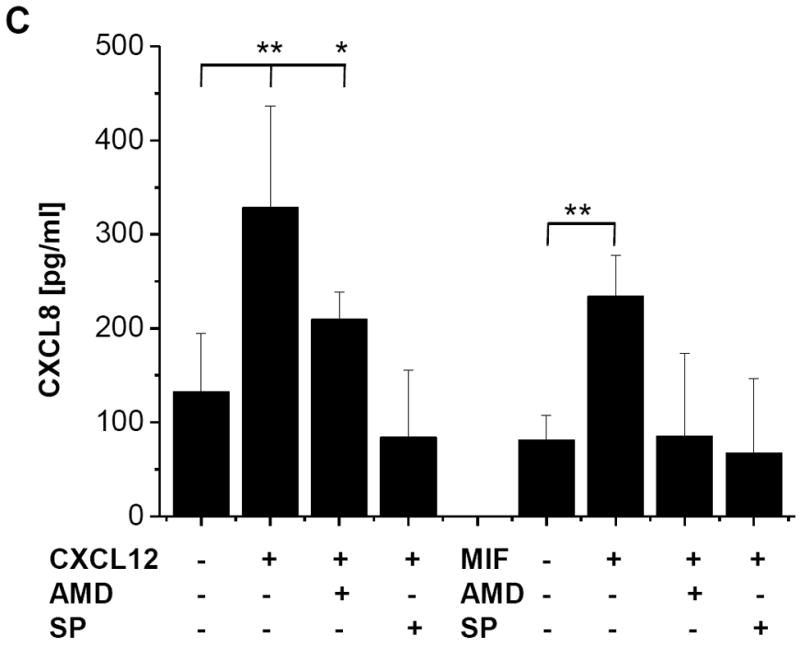
MIF-induced CXCL8 secretion from Jurkat T cells is dependent on CXCR4 and JNK. (A) Time course of MIF-induced CXCL8 secretion in Jurkat cells. The effect of 50 ng/ml MIF is measurable starting at 8 hours. Data represent mean values of two experiments. (B) CXCL8 induction by MIF is comparable with that by CXCL12 and LPS. The effects of 50 or 100 ng/ml rMIF are comparable to those of 100 ng/ml CXCL12 or 1 μg/ml LPS. Incubations were performed for 16 h. Data are means ± SD of 5 experiments. (C) CXCL8 secretion induced by MIF or CXCL12 is fully blocked by the CXCR4 inhibitor AMD3100 (AMD) or the JNK inhibitor SP600125 (SP). Protein levels of secreted CXCL8 were measured by CXCL8 ELISA from the cell supernatants. Data are means ± SD of 3 experiments. Asterisks in Fig. 7B and C indicate statistical significance: * p<0.05; ** p<0.01.
We next examined the effect of the CXCR4 inhibitor AMD3100 and the JNK inhibitor SP600125 on CXCL12- and MIF-induced CXCL8 secretion. AMD3100 fully reversed the stimulatory effect of CXCL12 on CXCL8 secretion back to baseline levels (Fig. 7C, left panel). Moreover, the JNK inhibitor not only reversed the CXCL12 effect but almost completely abolished CXCL8 secretion indicating that in Jurkat cells, CXCL8 production is strongly dependent on JNK signalling. Importantly, MIF-mediated CXCL8 secretion by Jurkat T cells could be also be potently blocked by AMD3100 and SP600125 (Fig. 7C, right panel). Of note, blockade of CXCR4 by AMD3100 not only reversed the MIF-mediated effect back to baseline, as seen for CXCL12, but reduced CXCL8 secretion below unstimulated baseline, indicating that another (endogenous) mediator induced baseline CXCR4-dependent CXCL8 production. Similarly, the JNK inhibitor SP600125 led to a total abrogation of CXCL8 production. Together, these experiments indicate that CXCL12- and MIF-mediated induction of CXCL8 in Jurkat T cells occurs through a signalling pathway involving CXCR4 and JNK.
4. Discussion
MIF is a widely expressed protein mediator that, following secretion, serves not only as an inflammatory cytokine and pro-atherogenic chemokine, but also fulfils intracellular functions as a modulator of cell homeostasis and cell cycle progress and apoptosis [20, 27, 54]. MIF activates rapid and sustained ERK1/2-MAPK signalling [36, 50], but its role in regulating the JNK-MAPK pathway has been less clear (Scheme 1). Depending on the cell type studied and the status of the cells, MIF has been reported to either activate or inhibit JNK signalling. The upstream kinases and the MIF receptor mechanisms involved have been unknown. Here, we have comprehensively characterized the rapid stimulatory effect that MIF has on resting fibroblasts and T cells. We also show for the first time that MIF-mediated triggering of the canonical JNK/c-Jun/AP-1 pathway is co-dependent on the interaction of MIF with its receptors CD74 and CXCR4 and involves an upstream activation of a SRC-type kinase and PI3K.
Scheme 1.
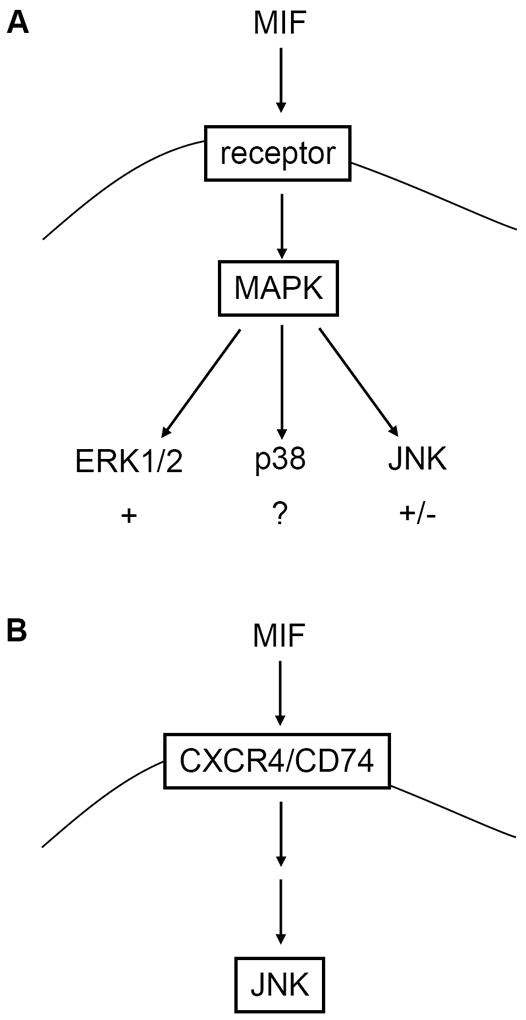
Schematic summarizing the effects of MIF on the canonical MAPK pathways. (A) MIF activates signalling through ERK1/2, whereas evidence for effects on the p38 pathway has been scarce. Both stimulatory and inhibitory effects of MIF on JNK signalling have been observed. (B) Rapid stimulation of the JNK pathway by exogenous MIF is dependent on CXCR4 and CD74.
Depending on whether pre-stimulated or resting cells were investigated, opposing effects of MIF on the JNK pathway have been observed. Initial studies on connections between MIF and JNK signalling focussed on inhibitory effects exerted by MIF on the JNK pathway. Pre-stimulation of fibroblasts or HEK293 cells by overexpression of CSN5/JAB1 led to activation of JNK activity and enhanced endogenous phospho-c-Jun levels. MIF inhibits these effects and also reduces JNK activity triggered by UV- or TNF-stress [55]. It appeared that only relatively high concentrations of MIF (μg/ml range) were sufficient for these inhibitory effects. Similarly, overexpression of MIF inhibited phosphorylation of endogenous c-Jun induced by thiol starvation in HeLa cells, indicating that MIF-based suppression of apoptosis is mediated through reduction of JNK activity [56]. An inhibition by MIF of JNK-mediated apoptotic processes was also observed in a recent study performed on the ischemic heart. Cardiomyocytes challenged by ischemia/reperfusion injury upregulated JNK-dependent apoptotic programs, a response which was blocked by cardiac MIF. It has remained undefined whether autocrine/paracrine or intracrine MIF pathways were involved [38].
In contrast, Watanabe and colleagues noticed that UVA-induced production of matrix metalloproteinase-1 (MMP-1) was enhanced by exogenous MIF in human dermal fibroblasts and was dependent on a plethora of activated signalling pathways including SRC kinase/AP1 [57]. MMP-2 induction by MIF in arthritic synovial fibroblasts was inhibitable, among other kinase pathway blockers, by a JNK I small molecule inhibitor [39]. MMP-9 and -13 induction in osteoblasts is also enhanced by activation of MIF and AP1, but MIF-mediated activation of c-Jun and c-Fos in these cells was independent of JNK and rather seemed to be due to an MIF/ERK1/2-dependent induction of c-Fos/c-Jun gene transcription [58]. In a study investigating effects of MIF released from the lung on myocardial dysfunction during sepsis, Lin et al. noticed that freshly isolated, but otherwise resting, neonatal rat ventricular cardiomyocytes upregulated JNK phosphorylation within 7-15 min when treated with 400 ng/ml of rMIF [41]. Furthermore, rapid JNK activation by MIF was linked to cardiomyocyte apoptosis [59]. Our experiments performed in resting fibroblasts and a human T cell line confirm that MIF can rapidly trigger JNK phosphorylation and in addition indicate that MIF initiates rapid activation of the entire JNK/c-Jun/AP1 pathway. We noticed that optimal activation of the JNK pathway occurred at concentrations of MIF between 50 and 100 ng/ml, while at higher concentrations of MIF, the phospho-JNK signal was attenuated. Differences between our data and the results by Lin et al. may be explained by the different cell types studied and variations between recombinant MIF preparations may be observed between labs.
Interestingly, JNK-dependent induction of CXCL8 gene transcription by MIF or its homolog D-DT was previously observed in A549 lung adenocarcinoma cells [60]. However, this effect was due to endogenous MIF/D-DT expressed in these cells as it could be down-regulated by MIF- or D-DT-siRNA and restored by adenoviral overexpression of MIF/D-DT [60]. Attenuation of this signal by gene knock out of the MIF receptor CD74 in MEFs suggested that endogenously expressed MIF can be released to act in an autocrine, CD74-dependent fashion. In line with those observations, we noted a dependence of rapid and transient MIF-triggered JNK responses on CD74. In conjunction with the results of our study, the study by Coleman and colleagues [60] indicates that both exogenous MIF, acting in a rapid and transient manner, as well as endogenous MIF acting in a sustained, cell-systemic, and possibly autocrine fashion can lead to an upregulation of the JNK pathway and subsequent CXCL8 induction. Together with previously obtained indirect evidence [57], our data also strongly suggest that MIF-directed rapid JNK stimulation involves upstream activation of a SRC family kinase. In addition, we offer evidence that MIF-mediated JNK activation is PI3K/AKT-dependent. Interestingly, this pathway mirrors the mechanism of rapid MIF-mediated ERK1/2 phosphorylation which also encompasses an upstream activation of SRC and PI3K [50]. The two pathways appear to differ though in that ERK1/2 activation by MIF depends on CD74, while MIF-triggered JNK activation as uncovered in our current study appears to also require CXCR4. It is noteworthy that a physical complex between CXCR4 and CD74 has been identified in cells that functions to affect MIF-dependent activation responses, as shown for MIF effects on the PI3K/AKT pathway [45]. Interestingly, Jurkat T cells express high concentrations of surface CXCR4 and also appreciable levels of CD74, and functional complexes between these MIF receptors have been suggested to form in these cells [45].
The critical role for CXCR4 in mediating JNK-dependent MIF responses in Jurkat T cells was concluded on by the following findings. Neutralizing CXCR4 mAbs ablate JNK phosphorylation induced by rMIF to a similar extent as observed for CXCL12 effects on JNK. Gene silencing of CXCR4 not only attenuates baseline JNK signalling activity, but also fully inhibits MIF-triggered JNK responses. In addition, the CXCR4-specific pharmacological compound AMD3100 blocks MIF-mediated JNK activation as well as MIF/JNK-dependent CXCL8 production.
Signalling pathways activated by CXCL12 and CXCR4 not only involve Gαi stimulation and reduction in cellular cAMP levels but also lead to SRC family tyrosine kinase and Gβγ activation, upregulation of Ca2+ fluxes through phospholipase Cβ (PLCβ), and activation of PI3K [61]. It also has been demonstrated that CXCL12 binding to CXCR4 upregulates CXCL8 expression and secretion from endothelial [62], glioblastoma [63], and B-cell chronic lymphatic leukaemia (B-CLL) [64] as well as human T-cell acute lymphoblastic leukaemia cells [18]. The signaling involved in CXCL12-induced CXCL8 production includes PI3K, ERK1/2, NF-κB, and JNK/AP-1 pathways. Modulation of CXCL8 secretion in Jurkat T cells through a MIF/CXCR4 axis is thus in line with CXCL12 effects on CXCL8 induction. Matsuo and colleagues showed that CXCL12 and CXCL8 cooperatively promote invasiveness and angiogenesis in pancreatic cancer [65].
In line with these CXCR4-mediated signalling patterns, we noticed the involvement of SRC and PI3K in the MIF/CXCR4/CD74 pathway of JNK activation, as the broad spectrum kinase inhibitor genistein, the SRC-type kinase inhibitor PP2, and the PI3K inhibitor Ly294002 reduced MIF-triggered JNK phosphorylation as well as CXCL8 secretion (data not shown). Whereas MIF has no effect on the short term release of CXCL8 from endothelial cells [44], CXCL8 gene induction and secretion by MIF was previously reported in B lymphocytes and was implied to contribute to B cell survival and B-CLL tumor progression [31]. B cells are MHC class II-positive cells and express high concentrations of surface CD74 and it was thus not surprising that MIF-mediated CXCL8 secretion was strongly dependent on CD74. T cells express abundant CXCR4 levels but only medium concentrations of CD74 which in addition is only partially localized on the cell surface. This cell-specific difference in MIF receptor expression might explain the shared role of CXCR4 and CD74 observed in our study.
The current study was directed at an en detail characterization of MIF effects on the JNK signalling pathway in fibroblasts and T cells in vitro. MIF rapidly triggered activation of the canonical JNK/c-Jun/AP-1 pathway through the upstream CXCR4/CD74/SRC/PI3K axis, a signalling route that in T cells led to the induction and secretion of CXCL8. Whereas MIF blocks JNK-mediated apoptotic responses in stressed cells, it may be concluded from our current study that the inflammatory cytokine MIF utilizes the JNK pathway in resting cells, for example T cells, to rapidly promote inflammatory processes by inducing the inflammatory and angiogenic chemokine CXCL8, and possibly other mediators. JNK-mediated MIF responses could thus contribute to MIF’s pro-inflammatory behaviour in numerous inflammatory diseases or cancer.
5. Conclusions
In this paper, we show for the first time that rapid stimulation of the JNK signalling pathway by the inflammatory cytokine MIF is mediated through CXCR4, a non-cognate receptor of MIF. The MIF effect is co-dependent on CD74. MIF shares with CXCL12, the bona fide ligand of CXCR4, the ability to induce CXCL8 secretion in T cells through the CXCR4/JNK pathway. Whereas the mechanisms and implications of MIF-mediated ERK1/2 MAPK activation have been well defined over the last decade, MIF’s role in JNK signalling has remained unclear, with both stimulatory and inhibitory effects observed, depending on the cell type and stimulation context studied. Our study clearly indicates that in resting T cells or fibroblasts, MIF rapidly and transiently triggers signalling through the JNK pathway at physiological or pathophysiological concentrations of this inflammatory mediator. In T cells, this can lead to CXCL8 production as an inflammatory signal. While recent studies indicate that in cardiomyocytes, in which the JNK pathway was upregulated by ischemia/reperfusion stress, MIF blocks JNK and JNK-mediated apoptosis, we conclude that in resting immune cells, such as T cells, MIF can promote inflammatory responses in a JNK-dependent manner. This activity resembles that of CXCL12 with which MIF shares the receptor CXCR4.
Supplementary Material
Acknowledgments
We thank B. Lennartz for technical assistance with the cell culture and mice, C. Weber for helpful discussions on chemokine receptor signalling responses, G. Fingerle-Rowson for initially providing the MIF knock out mouse, and A. Kapurniotu for synthesis of MIF peptide 50-65. This work was supported by grant numbers SFB 542/TP-A7 and BE1977/4-1 of the Deutsche Forschungsgemeinschaft (DFG) to J. Bernhagen, and grants from the NIH to R. Bucala.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chang LF, Karin M. Nature. 2001;410:37. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 2.Morrison DK, Davis RJ. Annu Rev Cell Dev Biol. 2003;19:91. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- 3.Wagner EF, Nebreda AR. Nat Rev Cancer. 2009;9:537. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 4.Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Derijard B, Davis RJ. Embo J. 1996;15:2760. [PMC free article] [PubMed] [Google Scholar]
- 5.Bogoyevitch MA, Ngoei KR, Zhao TT, Yeap YY, Ng DC. Biochim Biophys Acta. 2009;1804:463. doi: 10.1016/j.bbapap.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 6.Bogoyevitch MA. Bioessays. 2006;28:923. doi: 10.1002/bies.20458. [DOI] [PubMed] [Google Scholar]
- 7.Davis RJ. Cell. 2000;103:239. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 8.Ip YT, Davis RJ. Curr Opin Cell Biol. 1998;10:205. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- 9.Rao KMK. J Leukoc Biol. 2001;69:3. [PubMed] [Google Scholar]
- 10.Weber A, Wasiliew P, Kracht M. Sci Signal. 2010;3:cm1. doi: 10.1126/scisignal.3105cm2. [DOI] [PubMed] [Google Scholar]
- 11.Moepps B, Frodl R, Rodewald HR, Baggiolini M, Gierschik P. Eur J Immunol. 1997;27:2102. doi: 10.1002/eji.1830270839. [DOI] [PubMed] [Google Scholar]
- 12.Charo IF, Ransohoff RM. N Engl J Med. 2006;354:610. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 13.Beider K, Abraham M, Peled A. Front Biosci. 2008;13:6820. doi: 10.2741/3190. [DOI] [PubMed] [Google Scholar]
- 14.Gutkind JS. Sci STKE. 2000;40:re1. doi: 10.1126/stke.2000.40.re1. [DOI] [PubMed] [Google Scholar]
- 15.Araki S, Haneda M, Togawa M, Kikkawa R. Kidney Int. 1997;51:631. doi: 10.1038/ki.1997.92. [DOI] [PubMed] [Google Scholar]
- 16.Takeuchi Y, Fukunaga K. J Neurochem. 2003;85:1064. doi: 10.1046/j.1471-4159.2003.01763.x. [DOI] [PubMed] [Google Scholar]
- 17.Ghosh SK, Gadiparthi L, Zeng ZZ, Bhanoori M, Tellez C, Bar-Eli M, Rao GN. J Biol Chem. 2002;277:21325. doi: 10.1074/jbc.M201608200. [DOI] [PubMed] [Google Scholar]
- 18.Scupoli MT, Donadelli M, Cioffi F, Rossi M, Perbellini O, Malpeli G, Corbioli S, Vinante F, Krampera M, Palmieri M, Scarpa A, Ariola C, Foa R, Pizzolo G. Haematologica. 2008;93:524. doi: 10.3324/haematol.12098. [DOI] [PubMed] [Google Scholar]
- 19.Jones J, Marian D, Weich E, Engl T, Wedel S, Relja B, Jonas D, Blaheta RA. Exp Cell Res. 2007;313:4051. doi: 10.1016/j.yexcr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 20.Calandra T, Roger T. Nat Rev Immunol. 2003;3:791. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lue H, Kleemann R, Calandra T, Roger T, Bernhagen J. Microb Infect. 2002;4:449. doi: 10.1016/s1286-4579(02)01560-5. [DOI] [PubMed] [Google Scholar]
- 22.Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, Manogue KR, Cerami A, Bucala R. Nature. 1993;365:756. doi: 10.1038/365756a0. [DOI] [PubMed] [Google Scholar]
- 23.Calandra T, Echtenacher B, Le Roy D, Pugin J, Metz CN, Hültner L, Heumann D, Männel D, Bucala R, Glauser M. Nat Med. 2000;6:164. doi: 10.1038/72262. [DOI] [PubMed] [Google Scholar]
- 24.Donnelly SC, Haslett C, Reid PT, Grant IS, Wallace WA, Metz CN, Bruce LJ, Bucala R. Nat Med. 1997;3:320. doi: 10.1038/nm0397-320. [DOI] [PubMed] [Google Scholar]
- 25.Mitchell RA, Bucala R. Semin Cancer Biol. 2000;10:359. doi: 10.1006/scbi.2000.0328. [DOI] [PubMed] [Google Scholar]
- 26.Morand EF, Leech M, Bernhagen J. Nat Rev Drug Discov. 2006;5:399. doi: 10.1038/nrd2029. [DOI] [PubMed] [Google Scholar]
- 27.Zernecke A, Bernhagen J, Weber C. Circulation. 2008;117:1594. doi: 10.1161/CIRCULATIONAHA.107.729125. [DOI] [PubMed] [Google Scholar]
- 28.Merk M, Baugh J, Zierow S, Leng L, Pal U, Lee SJ, Ebert AD, Mizue Y, Trent JO, Mitchell R, Nickel W, Kavathas PB, Bernhagen J, Bucala R. J Immunol. 2009;182:6896. doi: 10.4049/jimmunol.0803710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flieger O, Engling A, Bucala R, Lue H, Nickel W. J Bernhagen FEBS Lett. 2003;551:78. doi: 10.1016/s0014-5793(03)00900-1. [DOI] [PubMed] [Google Scholar]
- 30.Calandra T, Bernhagen J, Metz CN, Spiegel LA, Bacher M, Donnelly T, Cerami A, Bucala R. Nature. 1995;377:68. doi: 10.1038/377068a0. [DOI] [PubMed] [Google Scholar]
- 31.Binsky I, Haran M, Starlets D, Gore Y, Lantner F, Harpaz N, Leng L, Goldenberg DM, Shvidel L, Berrebi A, Bucala R, Shachar I. Proc Natl Acad Sci USA. 2007;104:13408. doi: 10.1073/pnas.0701553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, Bucala R. Proc Natl Acad Sci USA. 2002;99:345. doi: 10.1073/pnas.012511599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leng L, Metz CN, Fang YXJ, Donnelly S, Baugh J, Delohery T, Chen Y, Mitchell RA, Bucala R. J Exp Med. 2003;197:1467. doi: 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beswick EJ, Pinchuk IV, Suarez G, Sierra JC, Reyes VE. J Immunol. 2006;176:6794. doi: 10.4049/jimmunol.176.11.6794. [DOI] [PubMed] [Google Scholar]
- 35.Shi X, Leng L, Wang T, Wang W, Du X, Li J, McDonald C, Chen Z, Murphy JW, Lolis E, Noble P, Knudson W, Bucala R. Immunity. 2006;25:595. doi: 10.1016/j.immuni.2006.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitchell RA, Metz CN, Peng T, Bucala R. J Biol Chem. 1999;274:18100. doi: 10.1074/jbc.274.25.18100. [DOI] [PubMed] [Google Scholar]
- 37.Lue H, Thiele M, Franz J, Dahl E, Speckgens S, Leng L, Fingerle-Rowson G, Bucala R, Luscher B, Bernhagen J. Oncogene. 2007;26:5046. doi: 10.1038/sj.onc.1210318. [DOI] [PubMed] [Google Scholar]
- 38.Qi D, Hu X, Wu X, Merk M, Leng L, Bucala R, Young LH. J Clin Invest. 2009 doi: 10.1172/JCI39738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pakozdi A, Amin MA, Haas CS, Martinez RJ, Haines GK, 3rd, Santos LL, Morand EF, David JR, Koch AE. Arthritis Res Ther. 2006;8:R132. doi: 10.1186/ar2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu XY, Lin SG, Huang XR, Bacher M, Leng L, Bucala R, Lan HY. J Interferon Cytokine Res. 2007;27:103. doi: 10.1089/jir.2006.0054. [DOI] [PubMed] [Google Scholar]
- 41.Lin X, Sakuragi T, Metz CN, Ojamaa K, Skopicki HA, Wang P, Al-Abed Y, Miller EJ. Shock. 2005;24:556. doi: 10.1097/01.shk.0000183238.70374.a8. [DOI] [PubMed] [Google Scholar]
- 42.Coleman AM, Rendon BE, Zhao M, Qian MW, Bucala R, Xin D, Mitchell RA. J Immunol. 2008;181:2330. doi: 10.4049/jimmunol.181.4.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kleemann R, Hausser A, Geiger G, Mischke R, Burger-Kentischer A, Flieger O, Johannes FJ, Roger T, Calandra T, Kapurniotu A, Grell M, Finkelmeier D, Brunner H, Bernhagen J. Nature. 2000;408:211. doi: 10.1038/35041591. [DOI] [PubMed] [Google Scholar]
- 44.Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, Weber C. Nat Med. 2007;13:587. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 45.Schwartz V, Lue H, Kraemer S, Korbiel J, Krohn R, Ohl K, Bucala R, Weber C, Bernhagen J. FEBS Lett. 2009;583:2749. doi: 10.1016/j.febslet.2009.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ortolano S, Hwang IY, Han SB, Kehrl JH. Eur J Immunol. 2006;36:1285. doi: 10.1002/eji.200535799. [DOI] [PubMed] [Google Scholar]
- 47.Schober A, Bernhagen J, Thiele M, Zeiffer U, Knarren S, Roller M, Bucala R, Weber C. Circulation. 2004;109:380. doi: 10.1161/01.CIR.0000109201.72441.09. [DOI] [PubMed] [Google Scholar]
- 48.Mischke R, Gessner A, Kapurniotu A, Jüttner S, Kleemann R, Brunner H, Bernhagen J. FEBS Lett. 1997;414:226. doi: 10.1016/s0014-5793(97)01039-9. [DOI] [PubMed] [Google Scholar]
- 49.Fingerle-Rowson G, Petrenko O, Metz CN, Forsthuber TG, Mitchell R, Huss R, Moll U, Muller W, Bucala R. Proc Natl Acad Sci USA. 2003;100:9354. doi: 10.1073/pnas.1533295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lue H, Kapurniotu A, Fingerle-Rowson G, Roger T, Leng L, Thiele M, Calandra T, Bucala R, Bernhagen J. Cell Signal. 2006;18:688. doi: 10.1016/j.cellsig.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 51.Yu KM, Zhuang J, Kaminski JM, Ambati B, Gao QY, Ma P, Liao DJ, Li F, Liu XA, Ge JA. Biochem Biophys Res Commun. 2007;358:990. doi: 10.1016/j.bbrc.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 52.Nguyen MT, Beck J, Lue H, Fünfzig H, Kleemann R, Koolwijk P, Kapurniotu A, Bernhagen J. J Biol Chem. 2003;278:33654. doi: 10.1074/jbc.M301735200. [DOI] [PubMed] [Google Scholar]
- 53.Chandrasekar B, Mummidi S, Valente AJ, Patel DN, Bailey SR, Freeman GL, Hatano M, Tokuhisa T, Jensen LE. J Biol Chem. 2005;280:26263. doi: 10.1074/jbc.M502586200. [DOI] [PubMed] [Google Scholar]
- 54.Bucala R. Nature. 2000;408:167. doi: 10.1038/35041654. [DOI] [PubMed] [Google Scholar]
- 55.Kleemann R, Rorsman H, Rosengren E, Mischke R, Mai NT, Bernhagen J. Eur J Biochem. 2000;267:7183. doi: 10.1046/j.1432-1327.2000.01823.x. [DOI] [PubMed] [Google Scholar]
- 56.Nguyen M, Lue H, Kleemann R, Thiele M, Tolle G, Finkelmeier D, Wagner E, Braun A, Bernhagen J. J Immunol. 2003;170:3337. doi: 10.4049/jimmunol.170.6.3337. [DOI] [PubMed] [Google Scholar]
- 57.Watanabe H, Shimizu T, Nishihira J, Abe R, Nakayama T, Taniguchi M, Sabe H, Ishibashi T, Shimizu H. J Biol Chem. 2004;279:1676. doi: 10.1074/jbc.M303650200. [DOI] [PubMed] [Google Scholar]
- 58.Onodera S, Nishihira J, Iwabuchi K, Koyama Y, Yoshida K, Tanaka S, Minami A. J Biol Chem. 2002;277:7865. doi: 10.1074/jbc.M106020200. [DOI] [PubMed] [Google Scholar]
- 59.Dhanantwari P, Nadaraj S, Kenessey A, Chowdhury D, Al-Abed Y, Miller EJ, Ojamaa K. Biochem Biophys Res Commun. 2008;371:298. doi: 10.1016/j.bbrc.2008.04.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coleman AM, Rendon BE, Zhao M, Qian MW, Bucala R, Xin D, Mitchell RA. J Immunol. 2008;181:2330. doi: 10.4049/jimmunol.181.4.2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Busillo JM, Benovic JL. Biochim Biophys Acta. 2007;1768:952. doi: 10.1016/j.bbamem.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Calderon TM, Eugenin EA, Lopez L, Kumar SS, Hesselgesser J, Raine CS, Berman JW. J Neuroimmunol. 2006;177:27. doi: 10.1016/j.jneuroim.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 63.Ping YF, Yao XH, Chen JH, Liu H, Chen DL, Zhou XD, Wang JM, Bian XW. J Neuro-Oncol. 2007;84:21. doi: 10.1007/s11060-007-9349-8. [DOI] [PubMed] [Google Scholar]
- 64.Perbellini O, Cioffi F, Malpeli G, Lovato O, Zanotti R, Scarpa A, Pizzolo G, Scupoli MT. Haematologica. 2008;93:S99. doi: 10.3324/haematol.12098. [DOI] [PubMed] [Google Scholar]
- 65.Matsuo Y, Ochi N, Sawai H, Yasuda A, Takahashi H, Fumahashi H, Takeyama H, Tong ZM, Guha S. Int J Cancer. 2009;124:853. doi: 10.1002/ijc.24040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.