

Abstract
Orthogonal arrays of particles (OAPs) have been visualized for many years by freeze-fracture electron microscopy. Our laboratory discovered that aquaporin-4 (AQP4) is the protein responsible for OAP formation by demonstrating OAPs in AQP4-transfected cells and absence of OAPs in AQP4 knockout mice. We recently developed live-cell, single-molecule imaging methods to study AQP4 diffusion and interactions in OAPs. The methods include single particle tracking of quantum-dot labeled AQP4, and total internal reflection fluorescence microscopy of green fluorescent protein (GFP) and small fluorophore-labeled AQP4. The full-length (M1) form of AQP4 diffuses freely in membranes and does not form OAPs, whereas the shorter (M23) form of AQP4 forms OAPs and is nearly immobile. Analysis of a series of AQP4 truncations, point mutants and chimeras revealed that OAP formation by AQP4-M23 is stabilized by hydrophobic tetramer-tetramer interactions involving N-terminus residues, and that absence of OAPs in AQP4-M1 results from blocking of this interaction by residues just upstream from Met23. These biophysical methods are being extended to identify the cellular site of AQP4 assembly, AQP4 isoform interactions, OAP size and dynamics, and the determinants of regulated OAP assembly.
Keywords: aquaporin, orthogonal arrays, water channel, single particle tracking, diffusion
Roles of AQP4 in Brain Deduced From Knockout Mice
Aquaporin-4 (AQP4) is the major water channel in the CNS, where it is mainly localized to the end foot processes of pericapillary astrocytes and in ependymal cells lining the ventricles (Nielsen et al., 1997; Rash et al., 1998). Analysis of AQP4 knockout mice, which were originally generated in our laboratory by targeted gene deletion (Ma et al., 1997), have revealed several distinct roles for AQP4 in brain and have been useful in testing proposed new roles of AQP4. AQP4 knockout mice are indistinguishable from wild-type mice at baseline in their appearance, growth and behavior, and in their brain anatomy, structure, cellular composition, compliance and intracranial structure (Manley et al., 2000; Papadopoulos et al., 2004; Saadoun et al., 2009). However, interesting phenotypes were discovered following stresses, revealing AQP4 involvement in brain water balance, neuroexcitation and glial cell migration. Mice lacking AQP4 showed improved outcome and reduced brain water accumulation compared to wild-type mice in models of cytotoxic brain edema, including water intoxication and ischemic stroke (Manley et al., 2000) and bacterial meningitis (Papadopoulos and Verkman, 2005). Improved outcome in AQP4 null mice was also seen following spinal cord compression injury (Saadoun et al., 2008), which was attributed to reduced spinal cord edema. AQP4 thus provides a major route for water entry into the brain through an intact blood–brain barrier. In vasogenic edema, excess water moves into the brain by a bulk fluid flow mechanism through a leaky blood–brain barrier, and exits the brain by movement into the cerebrospinal space through the AQP4-rich glia limitans lining brain ventricles and the brain surface. In obstructive hydrocephalus, water also moves out of the brain back into microvessels through the blood– brain barrier. Mice lacking AQP4 manifest worse clinical outcome and greater brain water accumulation in models of vasogenic brain edema, including cortical-freeze injury and brain tumor (Papadopoulos et al., 2004) and brain abscess (Bloch et al., 2005). AQP4 null mice also show an accelerated course of brain swelling in obstructive hydrocephalus (Bloch et al., 2006). AQP4 thus facilitates removal of excess brain water in vasogenic brain edema and hydrocephalus, though it remains to be resolved how a water-selective channel facilitates apparent bulk fluid outflow.
An unanticipated role of AQP4 in neural function was discovered from findings of impaired neurosensory transduction and altered seizure dynamics in AQP4 knockout mice. AQP4 is expressed in supportive cells adjacent to electrically excitable cells, as in: glia versus neurons in brain, Müller versus bipolar cells in retina, hair versus supportive cells in inner ear, and olfactory receptor neurons versus supportive cells in olfactory epithelium. We found impaired vision, hearing and olfaction in AQP4 null mice, as demonstrated by increased auditory brainstem response thresholds (Li and Verkman, 2001), reduced electroretinogram potentials (Li et al., 2002), and reduced electroolfactogram potentials (Lu et al., 2008). Electroen-cephalographic measurements showed reduced seizure threshold and prolonged seizure duration in AQP4 deficiency (Binder et al., 2006). These phenomena might involve impaired K+ reuptake into glial cells following neuroexcitation and/or extracellular space (ECS) expansion. Delayed K+ uptake from brain ECS in AQP4 deficiency was found (Padmawar et al., 2005; Binder et al., 2006). However, the precise mechanism by which AQP4 deficiency slows glial cell K+ uptake is unclear, as patch-clamp studies showed AQP4-independent function of Kir4.1 K+ channels (Ruiz-Ederra et al., 2007). We also found evidence for an expanded ECS in AQP4 deficiency with reduced ECS solute diffusion (Binder et al., 2004; Zador et al., 2008). Perhaps the increased ECS aqueous volume dilutes K+ exiting from neurons and consequently attenuates changes in ECS K+ concentration.
Another unanticipated role of AQPs is in cell migration, which was discovered following the observation of impaired tumor angiogenesis in AQP1 null mice and impaired migration of aortic endothelial cells from AQP1 knockout mice (Saadoun et al., 2005a). We proposed that actin cleavage and ion uptake at the tip of a lamellipodium create local osmotic gradients that drive water influx, facilitating lamellipodial extension and cell migration (reviewed in Papadopoulos et al., 2008). AQP4-facilitated cell migration was found in brain glial cells, resulting in reduced glial scarring following brain injury (Saadoun et al., 2005b). The migration of AQP4-deficient glial cells was remarkably slowed and lacked directionality when injected into stab-injured mouse brain (Auguste et al., 2007). AQP4-facilitated cell migration may also be important in brain tumor spread, as high-grade glioblastomas strongly express AQP4, and tumor cell migration has been found to be AQP-dependent (reviewed in Verkman et al., 2008).
Orthogonal Arrays of Aqp4 In the Cell Plasma Membrane
Since the 1970s, several groups using freeze-fracture electron microscopy (FFEM) have shown the existence of regular, square arrays of intramembrane particles, known as orthogonal arrays of particles (OAPs), in astrocytes and other cell types (reviewed by Wolburg, 1995). It was first speculated that AQP4 is a major constituent of OAPs based on AQP4 expression in multiple cell types known to exhibit array structures (Frigeri et al., 1995). This conjecture was verified by demonstrating OAP formation in AQP4-transfected cells (Yang et al., 1996) and absence of OAPs in tissues from AQP4 knockout mice (Verbavatz et al., 1997). FFEM combined with immunogold labeling confirmed the presence of AQP4 in OAPs (Rash et al., 1998). The biological relevance of AQP4 assembly into OAPs is currently debated, with data suggesting that OAPs might enhance AQP4 water permeability (Yang et al., 1997; Van Hoek et al., 2000; Silberstein et al., 2004), maintain AQP4 polarization in astrocyte foot processes (Amiry-Moghaddam et al., 2003), and mediate cell-cell adhesion (Hiroaki et al., 2006). Correlations have been reported between OAP abundance in astrocytes and disease processes such as toxic encephalopathies (Hatton and Ellisman, 1984) and muscular dystrophy (Schotland et al., 1981).
AQP4 was initially cloned from lung (Hasegawa et al., 1994), and subsequently from brain, where two distinct isoforms were identified with different translation initiation sites (Fig. 1A) (Jung et al., 1994; Yang et al., 1995; Lu et al., 1996). Western blots from homogenized rat cerebellum show a longer “M1”form of AQP4 (AQP4-M1), visible as a 34 kDa band, along with a shorter “M23” form of AQP4 (AQP4-M23), visible as a 32 kDa band (Lu et al., 1996). Both of these isoforms function as water channels (Jung et al., 1994) and form stable tetramers. The relative abundances of AQP4 isoforms are tissue specific, with rat brain containing an approximate 3:1 ratio of AQP4-M23 to AQP4-M1 (Neelyetal., 1999). A recent study of the rat brain genome suggests the existence of six AQP4 isoforms, one of which (AQP4e) is a 39 kDa functional water channel containing an additional 41 residues at the AQP4-M1 N-terminus. However, AQP4e was detected at very low levels in rat brain by immunoblot analysis (Moe et al., 2008).
Fig. 1.

AQP4 domains and labeling, and static methods to measure OAP formation. (A) AQP4 sequence and topology showing site of Myc or GFP insertion in the second extracellular loop for fluorescence labeling. Black: Met1 and Met23 translation initiation sites; blue: residues where single mutations had no effect on OAP formation or disruption; red: residues where single mutations significantly disrupted OAPs; pink: residues where single mutations mildly disrupted OAPs; yellow: residues where mutation produced loss of plasma membrane expression; green: C-terminal PDZ-binding domains. Horizontal lines indicate tested C-terminal truncation sites. (B) Immunoblots following BN-PAGE of homogenized brain tissues (left) and lysates of COS-7 cells transfected with AQP4-M23 or AQP4-M1 (right). (C) Freeze-fracture electron micrographs of the plasma membrane P-face of COS-7 cells expressing Myc-tagged AQP4-M1 (left) or AQP4-M23 (right).
FFEM in cells transfected with AQP4-M1 vs. AQP4-M23 showed that only the shorter isoform assembles into large OAPs of >100 particles, while AQP4-M1 tetramers are largely dispersed and form only occasional small arrays of <12 particles (Furman et al., 2003). In primary astrocytes, and in cells co-transfected with AQP1-M1 and AQP4-M23, OAPs are considerably smaller on average than those in AQP4-M23-only cells (Furman et al., 2003; Silberstein et al., 2004). This suggests that AQP4-M1 interacts with the array-forming AQP4-M23 in the plasma membrane, likely limiting the size to which arrays assemble in vivo.
Prior to 2008, the only method reported in the literature for identifying OAPs in cell membranes was FFEM. By coupling FFEM with immunogold labeling, investigators have estimated the fraction of natural AQP4 isoforms (Furman et al., 2003) and AQP4 mutants (Suzuki et al., 2008) that associate in OAPs. However, various technical challenges limit the usefulness of FFEM when examining questions regarding the mechanisms involved in OAP formation and regulation. Limitations include fixation artifacts, difficulty in obtaining statistically rigorous information about numbers of AQP4 tetramers in OAPs, and difficulty in identifying OAPs in cells expressing low levels of AQPs. FFEM often requires enrichment of cell populations or creation of stable AQP-overexpressing cell lines. Recently, Blue-native polyacrylamide gel electrophoresis (BN-PAGE) was demonstrated as an alternative to FFEM for OAP identification (Sorbo et al., 2008). In this technique, a non-denaturing detergent is used to separate AQP4 tetramers from higher-order AQP4 complexes on a gel. We found that immunoblotting for AQP4 following BN-PAGE of homogenized brain tissue from mouse, rat, and human shows a distinct band at ∼300 kDa, corresponding to AQP4 tetramers, and a series of higher bands corresponding to array complexes of varying apparent sizes (Fig. 1B, left). BN-PAGE from COS-7 cells transiently transfected with AQP4-M1 or AQP4-M23 showed distinctly different patterns. The immunoblots show a diffuse band migrating at >1200 kDa for AQP4-M23, suggesting large AQP4 complexes, along with a smaller band corresponding to AQP4 tetramers, while AQP4-M1 showed a dense tetramer band and a faint band at ∼600 kDa, but no very high molecular weight bands (Fig. 1B, right). These gel patterns are consistent with large OAPs that are observable by FFEM in transfected COS-7 cells (Fig. 1C). BN-PAGE is technically simple and relatively rapid when compared to FFEM. However, cell lysis by detergents precludes meaningful quantitative determination of AQP4 accumulation in OAPs at the plasma membrane, which limits its effectiveness as a method.
We review here recent advances obtained using newly-developed fluorescence methods for identifying the determinants, dynamics and distribution of AQP4 OAPs in intact live cells under physiological conditions in real time. One method used in our studies has been single particle tracking (SPT), a technique that is technically and conceptually simple. A Myc epitope is engineered into the second extracellular loop of the AQP molecule (Fig. 1A), cells are transiently transfected, and a subset of AQP molecules at the plasma membrane in live cells is labeled via antibody binding to fluorescent nanoparticles known as quantum dots (Fig. 2A, left). The movement, due to AQP diffusion, of individual quantum dots is followed using a fluorescence microscope, and trajectories of individual quantum dots are reconstructed and analyzed (Fig. 2A, right). We initially applied SPT to study AQP1 diffusion, finding long-range free diffusion over a wide variety of conditions, indicating that AQP1 exists in the plasma membrane largely free of specific interactions (Crane and Verkman, 2008). In applying SPT to AQP4, we reasoned that individual AQP4 tetramers should be mobile, whereas AQP4 in large OAPs should be relatively immobile. As expected, in a variety of cell lines and primary astrocytes, we found remarkable immobility of AQP4-M23 and rapid diffusion of AQP4-M1. We have therefore exploited AQP4 diffusion as a “read-out” of OAP assembly in live cells to investigate a series of questions regarding the biophysics and determinants of OAP formation (Crane et al., 2008; Crane and Verkman, 2009). Also, we discuss here results from another fluorescence technique, total internal reflection fluorescence microscopy (TIRFM), using antibody-labeled AQP4 isoforms or green fluorescent protein (GFP)-AQP4 chimeras. This method allows direct visualization of the spatial distribution of OAPs over the entire cell surface, and is useful to study the nature of OAP trafficking and regulation under different physiological conditions.
Fig. 2.
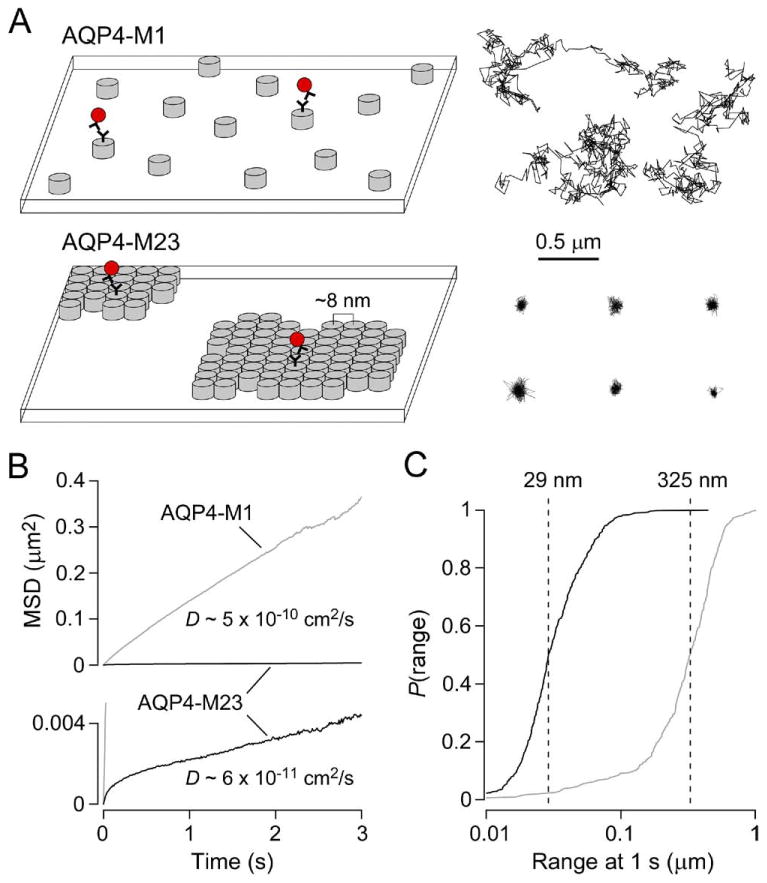
Membrane assembly and diffusional mobility of M1 and M23 isoforms of AQP4. (A) Schematic showing the organization of AQP4 tetramers (left) and representative single particle trajectories (right) of quantum dot-labeled AQP4 molecules in the plasma membrane of COS-7 cells expressing AQP4-M1 (top) or AQP4-M23 (bottom). Each grey cylinder represents one AQP4 tetramer. A subset of AQP4 molecules are labeled with quantum dots (red) for single particle tracking. (B) Combined MSD vs. time plots and averaged diffusion coefficients for AQP4-M1 (grey) and AQP4-M23 (black) in COS-7 cells. (C) Cumulative probability distribution of ranges at 1 s (P(range)) for AQP4-M1 (grey) and AQP4-M23 (black), with dashed lines indicating median range.
Single Particle Tracking Reveals A Wide Range of Diffusive Behaviors of Different Aquaporins
To understand molecular diffusion in cell membranes it is often useful to design SPT experiments that cover a wide range of time frames, thus providing information on both the short- and long-range motions of membrane proteins (such as AQP4-M23 “vibration” within OAPs versus slow, raft-like OAP diffusion). For studying short-range motions of aquaporins, we have used continuous image acquisition at 91 Hz over 6 s. It is immediately obvious from reconstructed trajectories of quantum dot-labeled AQP4-M1 versus AQP4-M23 that OAP assembly greatly restricts diffusion (Fig. 2A, right). The mean-squared displacement (MSD) versus time plot computed from many AQP4-M23 trajectories shows the characteristic negative curvature expected for confined or anomalous diffusion (Fig. 2B), while the MSD for AQP4-M1 diffusion is nearly linear over 3 s. The average diffusion coefficient and range at 1 s for OAP-associated AQP4-M23 is an order of magnitude lower than that of AQP4-M1 (Fig. 2B, C). A more detailed analysis of high-frame rate SPT data is required to rigorously determine the modes of diffusion. We initially used two independent methods of analysis to determine the fraction of AQP1 molecules in live cell membranes that undergo Brownian diffusion (Crane and Verkman, 2008), which were applied to AQP4 as well. We adapted the approach used by Kusumi et al. (1993), which is based on the deviation of the MSD versus time plot from linearity to classify individual trajectories as free versus restricted. We also adapted the method of Schütz et al. (1997), which uses a two-component exponential fit to the cumulative distribution function of square displacements to calculate the Brownian fraction in a population of diffusing molecules. For a detailed description of these methods, the reader is referred to Crane and Verkman (2008). Both types of analyses indicated that ∼75% of AQP1 and AQP4-M1 diffuse in a Brownian manner in COS-7 cells, in contrast to ∼10% for AQP4-M23. While a detailed analysis is required in some cases for comparing subtle changes in molecular diffusion, in the case of OAP assembly the differences are sufficiently large and robust that a simple examination of the distribution ranges (Fig. 2C) allows estimation of the fraction of AQP4 associating in OAPs.
We also characterized the long-range motions of aquaporins using time-lapse image acquisition at 1 Hz over 6 min. In these experiments, COS-7 cells were co-transfected with Myc-tagged AQP1 or AQP4 for SPT, and with GFP in order to identify cell edges. Examples of AQP trajectories from time-lapse SPT are shown in Fig. 3A. These experiments revealed that long-range diffusion is also highly dependent on OAP formation or association. AQP1 and AQP4-M1 diffuse over the entire surface of COS-7 cell membranes, covering an average distance of ∼2 μm in 1 min, with individual AQP1 channels moving up to 18 μm in 5 min. In contrast, AQP4-M23 diffusion was limited to an average distance of ∼110 nm in 1 min. The cumulative MSD versus time plots for AQP1 and AQP4-M1 were approximately linear over 5 min (Fig. 3B). Unlike the MSD from high-frame rate SPT, time-lapse SPT shows no negative curvature for AQP4-M23. This is likely because acquisition at 1 Hz is too slow to identify the individual motions of confined tetramers, but instead describes the diffusion of entire OAPs.
Fig. 3.

Comparison of long and short-range diffusion of full-length AQPs and C-terminal deletion mutants. (A) Representative trajectories superimposed on cellular profiles (white regions) of COS-7 cells expressing AQP1 (left), AQP4-M1 (middle) or AQP4-M23 (right) determined from time-lapse SPT at 1 Hz over 6 min. (B) Combined MSD vs. time plots for AQP1 (grey), AQP4-M1 (black), AQP4-M23 (red), and deletion mutant M23Δ6 (blue). (C) Expansion of Fig. 3B with additional MSD plots for AQP4-M23 after treatment with latrunculin B (green), jasplakinolide (orange), nocodazole (purple) or paraformaldehyde (grey). (D) Average diffusion coefficients of AQP4-M1 (top group), AQP4-M23 (middle group), and AQP1 (bottom group) following indicated treatments or truncations. (E) P(range) for AQP1 (grey), AQP4-M1 (black), AQP4-M23 (red), and deletion mutants M23Δ6 (dark blue) and M1Δ6 (light blue). Data obtained from transiently transfected COS-7 cells. MSD plots (B and C) are results from time-lapse acquisition at 1 Hz over 6 min, while diffusion coefficients and ranges (D and E) were calculated following short-range measurements at 91 Hz over 6 s.
SPT at different rates thus enables resolution of the diffusion of individual AQP4 tetramers from that of whole OAPs. Such analysis, following maneuvers or mutations that alter OAP assembly, allow separation of effects on OAP assembly from possible intermolecular interactions between OAPs and anchoring molecules that may restrict the slow movements of whole OAPs. AQP4 contains a C-terminal PDZ-binding domain, Leu–Ser–Ser–Val (Fig. 1A), which could be involved in intermolecular interactions related to AQP4 polarization or basolateral membrane targeting (Neely et al., 2001). We measured, by time-lapse and continuous SPT, the diffusion of a C-terminal deletion mutant of AQP4-M23 (M23Δ6). By time-lapse SPT, M23Δ6 diffused over an average range of ∼240 nm in 1 min, ∼twofold greater than that of full-length AQP4-M23 (Fig. 3C). However, two lines of evidence suggested that PDZ-domain deletion does not prevent OAP assembly. First, the observed diffusion of M23Δ6 is orders of magnitude lower than that of AQP4-M1. Second, short-range SPT indicated that diffusion of individual M23Δ6 tetramers is identical to that of full-length AQP4-M23 (Fig. 3D, E). Therefore we conclude that the C-terminal PDZ-binding domain of AQP4 is not involved in OAP assembly, but does provide an anchor to assembled OAPs, probably by interacting with structures on the cytoplasmic side of the membrane. Chemical alterations of actin polymerization state or tubulin did not result in an increase in AQP4-M23 diffusion on either the short or long time scales (Fig. 3C, D), indicating that the OAP-anchoring PDZ interactions do not involve the cytoskeleton. Deletion of the PDZ-binding domain in AQP4-M1 (M1Δ6) resulted in a small but significant increase in diffusion, indicating that the diffusion of individual AQP4 tetramers is mildly slowed by PDZ interactions. Diffusion of M1Δ6 was nearly identical to that of AQP1 (Fig. 3D, E).
Molecular Determinants of OAP Formation by AQP4
For the purpose of comparing the fraction of AQP4 variants that are in OAPs, diffusion data are shown graphically in the form of the cumulative distribution of ranges at 1 s (Fig. 2C, fig. 3E, and fig. 4B–E), where P(range) is defined as the probability that a particle's range is less than or equal to a given distance at t=1 second. Based on the finding that ∼95% of AQP4 is present in OAPs in AQP4-M23-expressing cells (Furman et al., 2003; Suzuki et al., 2008), and from the spatial resolution of 18 nm for our SPT system, we estimated the percentage of AQP4 in OAPs for the various mutants as P(79±18 nm), the range at which P(range) = 0.95 for AQP4-M23. Estimates of OAP content using this method for key AQP4 mutants are summarized at the right in Fig. 4A.
Fig. 4.
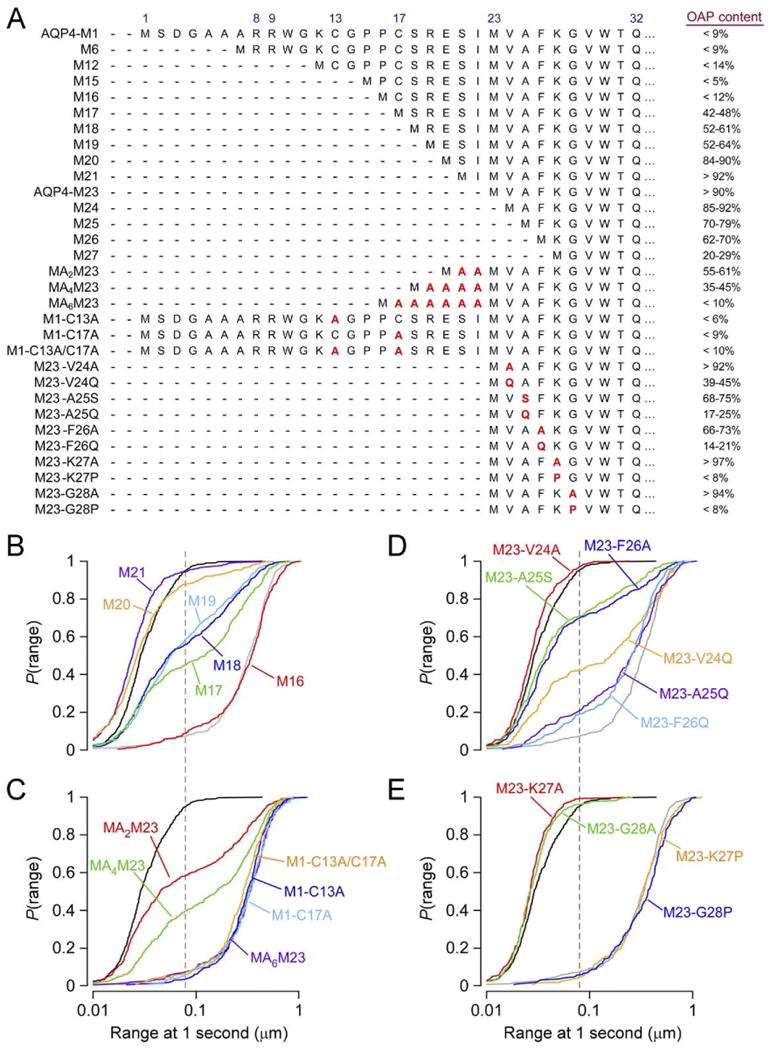
Mutations in the AQP4 N-terminal domain affect OAP formation. (A) Amino acid sequences leading up to the first transmembrane helix at Gln32 of indicated AQP4 mutants (left), and resulting fraction of each mutant found to reside in OAPs (right). Measurements done by SPT in live, transfected COS-7 cells. (B) P(range) for indicated AQP4 truncation mutants upstream of Met23: M16 (red), M17 (green), M18 (dark blue), M19 (light blue), M20 (orange), M21 (purple). (C) P(range) resulting from polyalanine additions upstream of Met23: MA2M23 (red), MA4M23 (green), MA6M23 (purple), and for alanine point mutations in AQP4-M1: M1-C13A (dark blue), M1-C17A (light blue), M1-C13A/C17A (orange). (D) P(range) for point mutations of hydrophobic N-terminus residues in AQP4-M23: M23-V24A (red), M23-V24Q (orange), M23-A25S (green), M23-A25Q (purple), M23-F26A (dark blue), M23-F26Q (light blue). (E) P(range) for point mutations just downstream of the hydrophobic N-terminus of AQP4-M23: M23-K27A (red), M23-K27P (orange), M23-G28A (green), M23-G28P (blue). (B–E) P(range) for AQP4-M23 (black) and AQP4-M1 (grey) are shown for reference. Dashed line indicates 95th percentile of the range of AQP4-M23.
We generated a series of deletion mutants at the AQP4-M1 N-terminus to investigate whether specific residues in AQP4-M1 between Met1 and Met23 are responsible for preventing OAP formation. The deletion mutants were named for the position of the N-terminal methionine relative to Met1 in full-length AQP4-M1. Deletions of up to 16 residues from the N-terminus of AQP4-M1 had little effect on diffusion (Fig. 4A, B), indicating the absence of OAPs formed by mutants of length equal to or greater than M16. Further truncations, however, resulted in a large fraction of AQP4 with restricted diffusion, with >40% of M17 present in OAPs. Continued deletions up to Met23 further increased the percentage of AQP4 in OAPs (Fig. 4A, B). Truncations downstream of Met23 produced progressive loss of OAPs from M25 through M27 (Fig. 4A), with truncations past position 27 resulting in a loss of membrane expression. AQP4 also contains a 73-amino acid cytoplasmic C-terminal domain (Fig. 1B). Deletion of part or all of this domain did not significantly affect the diffusion of AQP4-M1 or AQP4-M23 (Crane and Verkman, 2009).
A recent study by FFEM reported that palmitoylation of Cys13 or Cys17 in AQP4-M1 is required for prevention of OAP formation (Suzuki et al., 2008). We tested this conclusion by measuring diffusion of a double mutant in which both cysteines were changed to alanines (M1-C13A/C17A), which cannot undergo palmitoylation. In contrast to the conclusion of Suzuki et al. (2008) we found no reduction in diffusion, indicating that palmitoylation is not required to prevent OAP formation by AQP4-M1 (Fig. 4A, C). A series of alanine substitutions at Cys17, Ser18, Arg19, Glu20, individually, or at all four positions together, also did not affect AQP4-M1 diffusion (Crane and Verkman, 2009). To further delineate the requirements for OAP prevention, we constructed mutants in which polyalanine appendages of various lengths were added to the N-terminus of AQP4-M23 (Fig. 4A). Unexpectedly, the polyalanine appendages were very effective at reducing OAP content, with MA6M23 having identical diffusion to M16 and AQP4-M1 (Fig. 4A, C). In live cells, prevention of AQP4-M1 OAPs thus occurs independently of palmitoylation, and does not depend on specific AQP4 sequence elements of residues upstream of Met23.
From these data we postulated that the AQP4 residues just upstream of Met23 disrupt OAPs in AQP4-M1 by interfering with interactions involving residues just downstream of Met23. A Kyte–Doolittle hydropathy plot of the N-terminus of AQP4-M23 shows that it is highly hydrophobic, with decreasing hydropathy moving inward toward the beginning of the first transmembrane helix at Gln32. Truncation mutations and site directed mutagenesis of AQP4-M23 suggest that the four most N-terminal residues of AQP4-M23 (Met–Val–Ala–Phe) form a key hydrophobic domain responsible for OAP formation (Crane and Verkman, 2009). Successively reducing hydropathy by mutating Val24 or Phe26 to alanine and then to glutamine, or mutating Ala25 to serine or glutamine, produced successive reductions in OAP content (Fig. 4A, D).
Efficient intermolecular interactions producing OAPs may require the correct positioning of the hydrophobic N-terminus relative to a hydrophobic binding pocket in adjacent AQP4 tetramers. A proline substitution just downstream from the N-terminus of AQP4-M23 might thus prevent OAP forming interactions by disrupting the secondary structure of this domain or by introducing a rigid kink that prevents the OAP-forming interactions. Indeed, proline substitution at Lys27 or Gly28 produced complete disruption of OAP formation, giving diffusion identical to that of AQP4-M1. Therefore, mutations downstream from the hydrophobic N-terminus, which may alter domain structure, are even more effective at OAP prevention than mutations that reduce hydropathy. An intriguing possibility is that the nine residues at the N-terminus domain of AQP4-M23 (Met23–Thr31) form a secondary structural element that is critical to OAP formation. The four-residue spacing between Trp30 and Phe26 suggests the possibility of a one-turn amphipathic helix anchored in the cytoplasmic face of the membrane by these aromatic residues that favor the lipid–water interface (White and Wimley, 1999). The introduction of a proline residue between Trp30 and Phe26 would prevent the formation of a short helix, and may account for the complete loss of OAP formation. However, a recent 1.8 Å resolution crystal structure of human AQP4 (trypsin cleaved at residue Arg19) showed no structure up to Gln32 (Ho et al., 2009). If the AQP4-M23 N-terminus is normally unstructured, the introduction of a proline residue at position 27 or 28 may produce a rigid kink that drives this domain out of the membrane interface, away from its ideal position for hydrophobic interaction.
Prior to publication of the x-ray crystallographic structure mentioned above, a structure of rat AQP4 was published and recently refined to a resolution of 2.8 Å (Hiroaki et al., 2006; Tani et al., 2009). These structures were determined by electron crystallography of AQP4 in 2D bilayer stacks. From the crystal contacts in this system, Hiroaki et al. (2006) hypothesized that a salt bridge between Arg108 and Tyr250 is primarily responsible for OAP formation. To test this interaction, we also generated alanine mutants at these residues. We found that the diffusion of M23-R108A was identical to that of AQP4-M23, while M23-Y250A did not express at the plasma membrane (Crane and Verkman, 2009). Therefore, we conclude that subunit-subunit contacts observed in 2D AQP4 crystals are not the same interactions responsible for OAP formation in live cell membranes.
Visualizing OAPS by Total Internal Reflection Fluorescence
While monitoring the diffusion of sparsely labeled AQP4 has proven to be a powerful technique, we also found it useful to examine the spatial distribution of AQP4 OAPs over entire cell surfaces, which can be accomplished by fluorescence imaging. One approach is to heavily label Myc-tagged AQP4 molecules for TIRFM imaging, using one hundred times more primary and secondary antibody than is used in SPT experiments. In both COS-7 cells and primary astrocytes transfected with AQP4-M23, distinct puncta are visible, while AQP4-M1 appears uniformly distributed in the membrane (Fig. 5A, B). Histograms of cluster pixel intensities in COS-7 cells estimated an average of ∼136 secondary antibodies per cluster, while analysis of FFEM in the same cell line (Fig. 1C) gave an average of 208 tetramers per OAP (assuming one tetramer per intramembrane particle), corresponding to ∼800 AQP4 monomers per OAP. Comparing fluorescence images to FFEM, one secondary antibody was bound for approximately six Myc sites, just under one binding event per AQP4 tetramer (Crane et al., 2008). We hypothesize that steric hindrance prevents access to all available binding sites.
Fig. 5.

Visualization of AQP4 OAPs by total internal reflection fluorescence microscopy. TIRF micrographs showing: (A) Alexa-labeled AQP4-M1.myc (left) and AQP4-M23.myc (right) in transfected primary mouse astrocytes. (B) Alexa-labeled AQP4-M1.myc (left) and AQP4-M23.myc (right) in COS-7 cells. Inset shows an expanded 5×5 μm area. (C) AQP4-M1.GFP (left) and AQP4-M23.GFP (right) in COS-7 cells.
A distinct advantage of FFEM over fluorescent antibody labeling of AQP4 is the ability to examine the size of OAPs, which is not possible with antibodies due to incomplete labeling. Another approach to fluorescent labeling that potentially solves this limitation is genetically encoding fluorescent proteins into the AQP4 sequence, thereby ensuring that every AQP4 monomer is fluorescent. OAP size may then be determined by simply measuring fluorescence intensity of observed clusters. Because genetic modification at the N-terminus, and to some extent the C-terminus, produces alterations in AQP4 diffusion, we concluded that addition of a GFP at these sites could interfere with proper OAP assembly. We instead introduced a GFP into the second extracellular loop, at the same site as the Myc insertion used for antibody labeling (Fig. 1A). Our laboratory and others have previously had success in engineering whole fluorescent proteins into extracellular domains of membrane proteins without altering trafficking or function (Voglmaier et al., 2006; Haggie and Verkman, 2008). AQP4 with extracellular GFP is efficiently trafficked to the plasma membrane in COS-7 and other cell types. Just as with heavy antibody labeling, GFP-tagged AQP4-M23 exhibits distinct diffraction-limited puncta when imaged by TIRFM, while GFP-tagged AQP4-M1 is uniformly distributed over the membrane surface (Fig. 5C). In the antibody labeling study, cells are fixed prior to labeling to prevent AQP4 clustering due to cross-linking that occurs when high concentrations of antibodies are used. Cell fixation is not required with GFP labeling. This advantage potentially enables experiments to analyze the movements of entire OAPs within membranes, and to examine the trafficking and regulation of whole OAPs in live cells under physiological conditions.
Directions
The ability to visualize individual AQP4 molecules and AQP4-containing OAPs in membranes in live cells has allowed analysis of AQP4 diffusion within and outside of OAPs, and study of the molecular determinants of AQP4 assembly. The use of single particle tracking and total internal reflection fluorescence microscopy, together with photobleaching recovery, fluorescence correlation spectroscopy and super-resolution imaging methods, should allow unprecedented analysis of AQP4 and OAP dynamics and interactions. These biophysical tools will allow imaging, under physiologically relevant conditions, of heterotetrameric associations of AQP4 isoforms, OAP size and stability, the cellular site of OAP formation, and the energetics stabilizing OAPs.
Acknowledgments
This work was supported by grants DK35124, EY13574, EB00415, HL59198, DK72517 and HL73856 from the National Institutes of Health and by a grant from the Guthy-Jackson Charitable Foundation.
Abbreviations
- AQP4
aquaporin-4
- BN-PAGE
blue-native polyacryl-amide gel electrophoresis
- ECS
extracellular space
- FFEM
freeze-fracture electron microscopy
- OAPs
orthogonal arrays of particles
- SPT
single particle tracking
- TIRFM
total internal reflection fluorescence microscopy
References
- Amiry-Moghaddam M, Otsuka T, Hurn PD, Traystman RJ, Haug FM, Froehner SC, Adams ME, Neely JD, Agre P, Ottersen OP, Bhardwaj A. An alpha-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc Natl Acad Sci U S A. 2003;100:2106–2111. doi: 10.1073/pnas.0437946100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auguste KI, Jin S, Uchida K, Yan D, Manley GT, Papadopoulos MC, Verkman AS. Greatly impaired migration of implanted aqua-porin-4-deficient astroglial cells in mouse brain toward a site of injury. FASEB J. 2007;21:108–116. doi: 10.1096/fj.06-6848com. [DOI] [PubMed] [Google Scholar]
- Binder DK, Papadopoulos MC, Haggie PM, Verkman AS. In vivo measurement of brain extracellular space diffusion by cortical surface photobleaching. J Neurosci. 2004;24:8049–8056. doi: 10.1523/JNEUROSCI.2294-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DK, Yao X, Zador Z, Sick TJ, Verkman AS, Manley GT. Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia. 2006;53:631–636. doi: 10.1002/glia.20318. [DOI] [PubMed] [Google Scholar]
- Bloch O, Auguste KI, Manley GT, Verkman AS. Accelerated progression of kaolin-induced hydrocephalus in aquaporin-4-deficient mice. J Cereb Blood Flow Metab. 2006;26:1527–1537. doi: 10.1038/sj.jcbfm.9600306. [DOI] [PubMed] [Google Scholar]
- Bloch O, Papadopoulos MC, Manley GT, Verkman AS. Aqua-porin-4 gene deletion in mice increases focal edema associated with staphylococcal brain abscess. J Neurochem. 2005;95:254–262. doi: 10.1111/j.1471-4159.2005.03362.x. [DOI] [PubMed] [Google Scholar]
- Crane JM, Van Hoek AN, Skach WR, Verkman AS. Aquaporin-4 dynamics in orthogonal arrays in live cells visualized by Quantum dot single particle tracking. Mol Biol Cell. 2008;19:3369–3378. doi: 10.1091/mbc.E08-03-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane JM, Verkman AS. Long-range nonanomalous diffusion of quantum dot-labeled aquaporin-1 water channels in the cell plasma membrane. Biophys J. 2008;94:702–713. doi: 10.1529/biophysj.107.115121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane JM, Verkman AS. Determinants of aquaporin-4 assembly in orthogonal arrays revealed by live-cell single-molecule fluorescence imaging. J Cell Sci. 2009;122:813–821. doi: 10.1242/jcs.042341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigeri A, Gropper MA, Umenishi F, Kawashima M, Brown D, Verkman AS. Localization of MIWC and GLIP water channel homologs in neuromuscular, epithelial and glandular tissues. J Cell Sci. 1995;108(9):2993–3002. doi: 10.1242/jcs.108.9.2993. [DOI] [PubMed] [Google Scholar]
- Furman CS, Gorelick-Feldman DA, Davidson KG, Yasumura T, Neely JD, Agre P, Rash JE. Aquaporin-4 square array assembly: opposing actions of M1 and M23 isoforms. Proc Natl Acad Sci U S A. 2003;100:13609–13614. doi: 10.1073/pnas.2235843100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggie PM, Verkman AS. Monomeric CFTR in plasma membranes in live cells revealed by single molecule fluorescence imaging. J Biol Chem. 2008;283:23510–23513. doi: 10.1074/jbc.C800100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa H, Ma T, Skach W, Matthay MA, Verkman AS. Molecular cloning of a mercurial-insensitive water channel expressed in selected water-transporting tissues. J Biol Chem. 1994;269:5497–5500. [PubMed] [Google Scholar]
- Hatton JD, Ellisman MH. Orthogonal arrays are redistributed in the membranes of astroglia from alumina-induced epileptic foci. Epilepsia. 1984;25:145–151. doi: 10.1111/j.1528-1157.1984.tb04170.x. [DOI] [PubMed] [Google Scholar]
- Hiroaki Y, Tani K, Kamegawa A, Gyobu N, Nishikawa K, Suzuki H, Walz T, Sasaki S, Mitsuoka K, Kimura K, Mizoguchi A, Fujiyoshi Y. Implications of the aquaporin-4 structure on array formation and cell adhesion. J Mol Biol. 2006;355:628–639. doi: 10.1016/j.jmb.2005.10.081. [DOI] [PubMed] [Google Scholar]
- Ho JD, Yeh R, Sandstrom A, Chorny I, Harries WE, Robbins RA, Miercke LJ, Stroud RM. Crystal structure of human aquaporin 4 at 1.8 A and its mechanism of conductance. Proc Natl Acad Sci U S A. 2009;106:7437–7442. doi: 10.1073/pnas.0902725106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung JS, Bhat RV, Preston GM, Guggino WB, Baraban JM, Agre P. Molecular characterization of an aquaporin cDNA from brain: candidate osmoreceptor and regulator of water balance. Proc Natl Acad Sci USA. 1994;91:13052–13056. doi: 10.1073/pnas.91.26.13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusumi A, Sako Y, Yamamoto M. Confined lateral diffusion of membrane receptors as studied by single particle tracking (nanovid microscopy). Effects of calcium-induced differentiation in cultured epithelial cells. Biophys J. 1993;65:2021–2040. doi: 10.1016/S0006-3495(93)81253-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Patil RV, Verkman AS. Mildly abnormal retinal function in transgenic mice without Muller cell aquaporin-4 water channels. Invest Ophthalmol Vis Sci. 2002;43:573–579. [PubMed] [Google Scholar]
- Li J, Verkman AS. Impaired hearing in mice lacking aquaporin-4 water channels. J Biol Chem. 2001;276:31233–31237. doi: 10.1074/jbc.M104368200. [DOI] [PubMed] [Google Scholar]
- Lu DC, Zhang H, Zador Z, Verkman AS. Impaired olfaction in mice lacking aquaporin-4 water channels. FASEB J. 2008;22:3216–3223. doi: 10.1096/fj.07-104836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Lee MD, Smith BL, Jung JS, Agre P, Verdijk MA, Merkx G, Rijss JP, Deen PM. The human AQP4 gene: definition of the locus encoding two water channel polypeptides in brain. Proc Natl Acad Sci U S A. 1996;93:10908–10912. doi: 10.1073/pnas.93.20.10908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Yang B, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS. Generation and phenotype of a transgenic knockout mouse lacking the mercurial-insensitive water channel aquaporin-4. J Clin Invest. 1997;100:957–962. doi: 10.1172/JCI231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- Moe SE, Sorbo JG, Sogaard R, Zeuthen T, Petter Ottersen O, Holen T. New isoforms of rat aquaporin-4. Genomics. 2008;91:367–377. doi: 10.1016/j.ygeno.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Neely JD, Amiry-Moghaddam M, Ottersen OP, Froehner SC, Agre P, Adams ME. Syntrophin-dependent expression and localization of aquaporin-4 water channel protein. Proc Natl Acad Sci U S A. 2001;98:14108–14113. doi: 10.1073/pnas.241508198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely JD, Christensen BM, Nielsen S, Agre P. Heterotetrameric composition of aquaporin-4 water channels. Biochemistry. 1999;38:11156–11163. doi: 10.1021/bi990941s. [DOI] [PubMed] [Google Scholar]
- Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. 1997;17:171–180. doi: 10.1523/JNEUROSCI.17-01-00171.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmawar P, Yao X, Bloch O, Manley GT, Verkman AS. K+ waves in brain cortex visualized using a long-wavelength K+-sensing fluorescent indicator. Nat Methods. 2005;2:825–827. doi: 10.1038/nmeth801. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Manley GT, Krishna S, Verkman AS. Aquaporin-4 facilitates reabsorption of excess fluid in vasogenic brain edema. FASEB J. 2004;18:1291–1293. doi: 10.1096/fj.04-1723fje. [DOI] [PubMed] [Google Scholar]
- Papadopoulos MC, Saadoun S, Verkman AS. Aquaporins and cell migration. Pflugers Arch. 2008;456:693–700. doi: 10.1007/s00424-007-0357-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos MC, Verkman AS. Aquaporin-4 gene disruption in mice reduces brain swelling and mortality in pneumococcal meningitis. J Biol Chem. 2005;280:13906–13912. doi: 10.1074/jbc.M413627200. [DOI] [PubMed] [Google Scholar]
- Rash JE, Yasumura T, Hudson CS, Agre P, Nielsen S. Direct immunogold labeling of aquaporin-4 in square arrays of astrocyte and ependymocyte plasma membranes in rat brain and spinal cord. Proc Natl Acad Sci U S A. 1998;95:11981–11986. doi: 10.1073/pnas.95.20.11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Ederra J, Zhang H, Verkman AS. Evidence against functional interaction between aquaporin-4 water channels and Kir4.1 potassium channels in retinal Muller cells. J Biol Chem. 2007;282:21866–21872. doi: 10.1074/jbc.M703236200. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Bell BA, Verkman AS, Papadopoulos MC. Greatly improved neurological outcome after spinal cord compression injury in AQP4-deficient mice. Brain. 2008;131:1087–1098. doi: 10.1093/brain/awn014. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Papadopoulos MC, Hara-Chikuma M, Verkman AS. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature. 2005a;434:786–792. doi: 10.1038/nature03460. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Papadopoulos MC, Watanabe H, Yan D, Manley GT, Verkman AS. Involvement of aquaporin-4 in astroglial cell migration and glial scar formation. J Cell Sci. 2005b;118:5691–5698. doi: 10.1242/jcs.02680. [DOI] [PubMed] [Google Scholar]
- Saadoun S, Tait MJ, Reza A, Davies DC, Bell BA, Verkman AS, Papadopoulos MC. AQP4 gene deletion in mice does not alter blood–brain barrier integrity or brain morphology. Neuroscience. 2009;161:764–772. doi: 10.1016/j.neuroscience.2009.03.069. [DOI] [PubMed] [Google Scholar]
- Schotland DL, Bonilla E, Wakayama Y. Freeze fracture studies of muscle plasma membrane in human muscular dystrophy. Acta Neuropathol. 1981;54:189–197. doi: 10.1007/BF00687741. [DOI] [PubMed] [Google Scholar]
- Schütz GJ, Schindler H, Schmidt T. Single-molecule microscopy on model membranes reveals anomalous diffusion. Biophys J. 1997;73:1073–1080. doi: 10.1016/S0006-3495(97)78139-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberstein C, Bouley R, Huang Y, Fang P, Pastor-Soler N, Brown D, Van Hoek AN. Membrane organization and function of M1 and M23 isoforms of aquaporin-4 in epithelial cells. Am J Physiol Renal Physiol. 2004;287:F501–F511. doi: 10.1152/ajprenal.00439.2003. [DOI] [PubMed] [Google Scholar]
- Sorbo JG, Moe SE, Ottersen OP, Holen T. The molecular composition of square arrays. Biochemistry. 2008;47:2631–2637. doi: 10.1021/bi702146k. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Nishikawa K, Hiroaki Y, Fujiyoshi Y. Formation of aquaporin-4 arrays is inhibited by palmitoylation of N-terminal cysteine residues. Biochim Biophys Acta. 2008;1778:1181–1189. doi: 10.1016/j.bbamem.2007.12.007. [DOI] [PubMed] [Google Scholar]
- Tani K, Mitsuma T, Hiroaki Y, Kamegawa A, Nishikawa K, Tanimura Y, Fujiyoshi Y. Mechanism of aquaporin-4's fast and highly selective water conduction and proton exclusion. J Mol Biol. 2009;389(4):694–706. doi: 10.1016/j.jmb.2009.04.049. [DOI] [PubMed] [Google Scholar]
- Van Hoek AN, Ma T, Yang B, Verkman AS, Brown D. Aqua-porin-4 is expressed in basolateral membranes of proximal tubule S3 segments in mouse kidney. Am J Physiol Renal Physiol. 2000;278:F310–F316. doi: 10.1152/ajprenal.2000.278.2.F310. [DOI] [PubMed] [Google Scholar]
- Verbavatz JM, Ma T, Gobin R, Verkman AS. Absence of orthogonal arrays in kidney, brain and muscle from transgenic knockout mice lacking water channel aquaporin-4. J Cell Sci. 1997;110(22):2855–2860. doi: 10.1242/jcs.110.22.2855. [DOI] [PubMed] [Google Scholar]
- Verkman AS, Hara-Chikuma M, Papadopoulos MC. Aquaporins—new players in cancer biology. J Mol Med. 2008;86:523–529. doi: 10.1007/s00109-008-0303-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voglmaier SM, Kam K, Yang H, Fortin DL, Hua Z, Nicoll RA, Edwards RH. Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron. 2006;51:71–84. doi: 10.1016/j.neuron.2006.05.027. [DOI] [PubMed] [Google Scholar]
- White SH, Wimley WC. Membrane protein folding and stability: physical principles. Annu Rev Biophys Biomol Struct. 1999;28:319–365. doi: 10.1146/annurev.biophys.28.1.319. [DOI] [PubMed] [Google Scholar]
- Wolburg H. Orthogonal arrays of intramembranous particles: a review with special reference to astrocytes. J Hirnforsch. 1995;36:239–258. [PubMed] [Google Scholar]
- Yang B, Brown D, Verkman AS. The mercurial insensitive water channel (AQP-4) forms orthogonal arrays in stably transfected Chinese hamster ovary cells. J Biol Chem. 1996;271:4577–4580. [PubMed] [Google Scholar]
- Yang B, Ma T, Verkman A. cDNA cloning, gene organization, and chromosomal localization of a human mercurial insensitive water channel. Evidence for distinct transcriptional units. J Biol Chem. 1995;270:22907–22913. doi: 10.1074/jbc.270.39.22907. [DOI] [PubMed] [Google Scholar]
- Yang B, van Hoek AN, Verkman AS. Very high single channel water permeability of aquaporin-4 in baculovirus-infected insect cells and liposomes reconstituted with purified aquaporin-4. Biochemistry. 1997;36:7625–7632. doi: 10.1021/bi970231r. [DOI] [PubMed] [Google Scholar]
- Zador Z, Magzoub M, Jin S, Manley GT, Papadopoulos MC, Verkman AS. Microfiberoptic fluorescence photobleaching reveals size-dependent macromolecule diffusion in extracellular space deep in brain. FASEB J. 2008;22:870–879. doi: 10.1096/fj.07-9468com. [DOI] [PubMed] [Google Scholar]