

Abstract
Objective
A workgroup commissioned by the Alzheimer’s Association (AA) and the National Institute on Aging (NIA) recently published research criteria for preclinical Alzheimer’s disease (AD). We performed a preliminary assessment of these guidelines.
Methods
We employed Pittsburgh compound B positron emission tomography (PET) imaging as our biomarker of cerebral amyloidosis and 18fluorodeoxyglucose PET imaging and hippocampal volume as biomarkers of neurodegeneration. A group of 42 clinically diagnosed AD subjects was used to create imaging biomarker cut-points. A group of 450 cognitively normal (CN) subjects from a population based sample was used to develop cognitive cut-points and to assess population frequencies of the different preclinical AD stages using different cut-point criteria.
Results
The new criteria subdivide the preclinical phase of AD into stages 1–3. To classify our CN subjects, two additional categories were needed. Stage 0 denotes subjects with normal AD biomarkers and no evidence of subtle cognitive impairment. Suspected Non-AD Pathophysiology (SNAP) denotes subjects with normal amyloid PET imaging, but abnormal neurodegeneration biomarker studies. At fixed cut-points corresponding to 90% sensitivity for diagnosing AD and the 10th percentile of CN cognitive scores, 43% of our sample was classified as stage 0; 16% stage 1; 12 % stage 2; 3% stage 3; and 23% SNAP.
Interpretation
This cross-sectional evaluation of the NIA-AA criteria for preclinical AD indicates that the 1–3 staging criteria coupled with stage 0 and SNAP categories classify 97% of CN subjects from a population-based sample, leaving just 3% unclassified. Future longitudinal validation of the criteria will be important.
Introduction
Studies of biomarkers for Alzheimer’s disease (AD) have provided clear evidence for the existence of a preclinical phase of the disease. A panel commissioned by the National Institute on Aging and the Alzheimer’s Association (NIA-AA) recently proposed guidelines to define preclinical AD for research purposes [1]. The NIA-AA criteria were based on a conceptual model of the pathophysiology of AD [2] in which the biomarkers of AD become abnormal in an ordered manner [2–6]. Amyloid biomarkers (PET amyloid imaging and CSF Aβ42) become abnormal first, as early as 20 years before significant clinical symptoms appear. Biomarkers of neuronal injury and degeneration (CSF tau, FDG-PET, and anatomic MRI) become abnormal later, closer to the time when individuals become symptomatic. The NIA-AA preclinical criteria are related to this hypothetical biomarker model in Figure 1. The NIA-AA criteria, therefore, identified stage 1 of preclinical AD as one of asymptomatic cerebral amyloidosis, and stage 2 as one in which evidence of synaptic dysfunction and neurodegeneration was also evident. The NIA-AA criteria further assert that subtle cognitive changes will be present in stage 3 and will precede the appearance of overt cognitive impairment.
Figure 1.

Preclinical stages 1–3 of AD (indicated by the yellow highlighted section) in relation to our model of biomarkers of the AD pathological cascade. The horizontal axis indicates clinical stages of AD: cognitively normal, mildly impaired (MCI), and dementia. The vertical axis indicates the changing values of each biomarker – scaled from maximally normal (bottom) to maximally abnormal (top). Aβ amyloid biomarker is PET amyloid imaging (red line). Biomarkers of neuronal injury are FDG-PET or atrophy on MRI (blue line). Onset or worsening of cognitive symptoms is determined from cognitive testing scores (purple line). The horizontal “cut-points” line represents the cut-points used to operationalize preclinical staging.
Although the NIA-AA criteria are based on a conceptual model as well as observational data, they make specific assumptions about relationships among biomarkers and cognitive testing that have not been adequately validated. As a first step in the evaluation process, we examined the distribution of cognitively normal persons in a population-based study of aging in Olmsted County Minnesota who fell into the different stages of the NIA-AA criteria for preclinical AD. To do so, however, we first had to develop an operational approach to implement the criteria.
Methods
Subjects
Two groups of subjects were employed in this study. First, 42 clinically diagnosed AD subjects who had undergone MRI, FDG-PET and PIB-PET were used to create imaging biomarker cut-points. These AD subjects were drawn from our Alzheimer’s Disease Research Center (ADRC) or incident cases in the Mayo Clinic Study of Aging (MCSA). Second, all available cognitively normal (CN) subjects from the MCSA who had undergone MRI, FDG-PET, PIB-PET, and complete neuropsychological testing (n = 450) were used to both develop cognitive cut-points and to assess population frequencies of the different preclinical AD stages using different cut-point criteria.
The MCSA is a population-based study of cognitive aging that was established in Olmsted County, MN starting in October 2004 [7]. All MCSA subjects undergo a clinical and cognitive assessment every 15 months that includes 9 neuropsychological tests [7]. The evaluations of all subjects were reviewed by a consensus panel consisting of physicians (neurologists and geriatricians), neuropsychologists, and study nurses. Subjects in the present study were diagnosed by the consensus panel as being cognitively normal, based on the clinical assessments including mental status examinations and informant interviews as well as the neuropsychological testing battery described below [7, 8].
The neuropsychological battery was constructed as previously described [7, 8]. Domain specific measures are formulated from the Wechsler Adult Intelligence Scale-Revised (WAIS-R), Wechsler Memory Scale-Revised (WMS-R), Auditory Verbal Learning Test (AVLT), Trail Making Test (TMT), category fluency test, and Boston Naming Test (BNT). Four cognitive domains are assessed: Executive (TMT: Part B, WAIS-R Digit Symbol); Language (BNT, category fluency); Memory (WMS-R Logical Memory-II (delayed recall), WMS-R Visual Reproduction-II (delayed recall), AVLT delayed recall); and Visuospatial (WAIS-R Picture Completion, WAIS-R Block Design). Individual test scores were first converted to z-scores using the mean and standard deviation from the MCSA 2004 enrollment visit for subjects that were CN (n=1624). The individual z-scores were averaged to create 4 domain scores which were then also converted to z-scores. A global cognitive summary score was formed from the average of the 4 domain z-scores and then converted to a z-score by subtracting the mean and dividing by the standard deviation. This global summary score was used to assess cognitive impairment in our subjects.
Imaging Methods
MRI was performed at 3T with a 3D-MPRAGE sequence [9] Images were corrected for distortion due to gradient non-linearity and for bias field [10, 11]. Our primary MRI measure was hippocampal volume measured with FreeSurfer software (version 4.5.0) [12]. Each subject’s raw hippocampal volume was adjusted by his/her total intracranial volume [13] to form an adjusted hippocampal volume (HVa). We calculated HVa as the residual from a linear regression of hippocampal volume (y) versus total intracranial volume (x).
PET images [14] were acquired using a PET/CT scanner. The 11C PIB-PET scan consisting of four 5-minute dynamic frames was acquired from 40–60 minutes after injection [15, 16]. 18 Fluorodeoxyglucose (18F-FDG ) PET images were obtained 1 hour after the PIB scan. Subjects were injected with 18F-FDG and imaged after 30–38 minutes, for an 8-minute image acquisition consisting of four 2-minute dynamic frames.
Quantitative image analysis for both PIB and FDG was done using our in-house fully automated image processing pipeline [17]. A global cortical PIB-PET retention ratio was formed by calculating the median uptake over voxels in the prefrontal, orbitofrontal, parietal, temporal, anterior cingulate, and posterior cingulate/precuneus regions of interest (ROIs) for each subject and dividing this by the median uptake over voxels in the cerebellar gray matter ROI of the atlas [18]. FDG-PET scans were analyzed in a similar manner. We used angular gyrus, posterior cingulate, and inferior temporal cortical ROIs, as described in Landau et al [19], normalized to pons uptake.
Methods for developing imaging biomarker and cognitive testing cut-points
While all biomarkers and cognitive tests are continuous measures, the NIA-AA criteria for preclinical AD require that every biomarker and cognitive test is designated normal or abnormal [1]. This requires that cut-points be created in these continuous distributions. The ideal method for selecting biomarker cut-points would be to use autopsy diagnoses as the reference standard of truth [20–22]. Because we do not yet have an adequately large autopsy sample with antemortem 3T MRI, PIB PET and FDG PET, we created cut-points such that a majority of clinically defined AD dementia patients would be deemed abnormal. While we did not have CSF available in our subjects, we had amyloid (PIB-PET) and neurodegenerative (FDG-PET and MRI) biomarkers in all subjects, and were therefore able to stage all subjects in accordance with the NIA-AA criteria (1). We had two sources of data in the neurodegenerative biomarker category (FDG PET and MRI) and we considered a subject positive for evidence of neurodegeneration if either or both fell below the cut-point.
Cut-points were based on estimated percentiles. For example, where higher biomarker values are worse, the cut-point corresponding to 90% sensitivity was the estimated 10th percentile of the AD distribution. Where lower biomarker values are worse, the cut-point was the estimated 90th percentile of the AD distribution. In this example, approximately 90% of ADs are considered abnormal. Biomarker cut-points corresponding to other levels of sensitivity were determined similarly.
Because the cognitive impairment seen in AD dementia or even MCI could not be used as an external reference for evidence of subtle cognitive decline in CN subjects, we used percentiles of the global summary score from the 450 CN subjects in this study. These percentiles can also be thought of as corresponding to a specificity level in that the 10th percentile of the distribution corresponds to a specificity level of 90%. Note that this approach, unlike the approach used for biomarkers, guarantees a certain number of subjects will fall below the subtle “cognitive impairment" cut point.
Results
Subjects
Demographic features of the 42 AD subjects used to develop biomarker cut-points and the 450 MCSA CN subjects used to assess the preclinical staging criteria are shown in Table 1. Table 1 also shows the demographic characteristics of all 2399 CN subjects in the MCSA. The 450 CN subjects who underwent the full battery of imaging studies were largely representative of the larger MCSA CN sample but had slightly higher cognitive scores.
Table 1.
Descriptive characteristics of subjects
| Characteristic | AD (n = 42) |
All MCSA CN (n = 2399) |
MCSA CN with MRI & PET (n = 450) |
|---|---|---|---|
| Age, y, median (IQR) | 80 (76, 84) | 78 (74, 83) | 78 (74, 82) |
| Female gender, no. (%) | 16 (38) | 1195 (50) | 205 (46) |
| Education, y, median (IQR) | 16 (12, 16) | 13 (12, 16) | 14 (12, 16) |
| APOE ε4 genotype, no. (%)* | |||
| ε4 carrier | 26 (68) | 520 (24) | 103 (26) |
| ε4 non-carrier | 12 (32) | 1618 (76) | 287 (74) |
| MMSE, median (IQR)** | 20 (14, 23) | 28 (27, 29) | 28 (27, 29) |
| Cognitive domain Z scores, median (IQR) † | |||
| Global | — | 0.12 (−0.54, 0.81) | 0.47 (−0.16, 1.16) |
| Memory | — | 0.03 (−0.71, 0.77) | 0.45 (−0.34, 1.22) |
| Language | — | 0.13 (−0.53, 0.72) | 0.30 (−0.33, 0.93) |
| Executive | — | 0.21 (−0.49, 0.77) | 0.43 (−0.28, 1.04) |
| Visuospatial | — | 0.12 (−0.60, 0.73) | 0.46 (−0.21, 1.09) |
APOE genotype not available in all subjects
Converted to the MMSE equivalent from the Short Test of Mental Status
Z scores are calculated using the mean and SD of the MCSA 2004 baseline visit of those who were cognitively normal (n = 1624)
MCSA – Mayo Clinic Study of Aging; CN – cognitively normal; IQR – inter-quartile range
Biomarker cut-points based on sensitivity for identifying clinically diagnosed AD dementia
Figure 2 illustrates individual values for PIB, FDG, and HVa for the AD and CN subjects used in this study. In each panel arrows indicate the biomarker cut-points for five levels of diagnostic sensitivity for AD: 80%, 85%, 90%, 95% and 99%. Table 2 contains the numeric cut-points corresponding to these different diagnostic sensitivities for each of the three imaging biomarkers and the proportion of CNs that was abnormal at each cut point. For example, using a cut-point of 90% sensitivity for AD, the proportions of CN subjects that fall into the abnormal range are: 32% for PIB; 28% for FDG; and 19% for HVa.
Figure 2.

Box plots with individual data points for PIB (panel A), FDG (panel B), and HVa (panel C). Arrows in each panel indicate the biomarker cut-points for five levels of AD sensitivity ranging from 80% to 99%.
Table 2.
Biomarker values for various sensitivity based cut-points derived from the clinically diagnosed AD sample (n = 42) and the percent of Cognitively Normal (n = 450) considered abnormal at each cut-point.
| Sensitivity level in AD sample | |||||
|---|---|---|---|---|---|
| 80% | 85% | 90% | 95% | 99% | |
| Biomarker cut-point value | |||||
| PIB PET ratio | 1.91 | 1.82 | 1.50 | 1.41 | 1.34 |
| FDG PET ratio | 1.24 | 1.26 | 1.31 | 1.37 | 1.41 |
| Hippocampal volume* | −0.96 | −0.86 | −0.70 | −0.56 | −0.27 |
| Percent of CN abnormal at corresponding cut-point | |||||
| PIB PET ratio | 16% | 19% | 32% | 41% | 63% |
| FDG PET ratio | 12% | 16% | 28% | 42% | 53% |
| Hippocampal volume * | 12% | 14% | 19% | 23% | 36% |
Adjusted for total intracranial volume as described in the methods section
Cognitive cut-points based on global summary score distributions in 450 CN subjects
The NIA-AA preclinical AD staging criteria require that the presence or absence of subtle cognitive impairment of insufficient severity to qualify for a diagnosis of MCI be established in each subject. We evaluated cut-points at the 5th, 10th and 15th percentile of the distribution of both the global summary score (Figure 3a) and the memory domain composite score (Figure 3b) in the 450 CN subjects.
Figure 3.
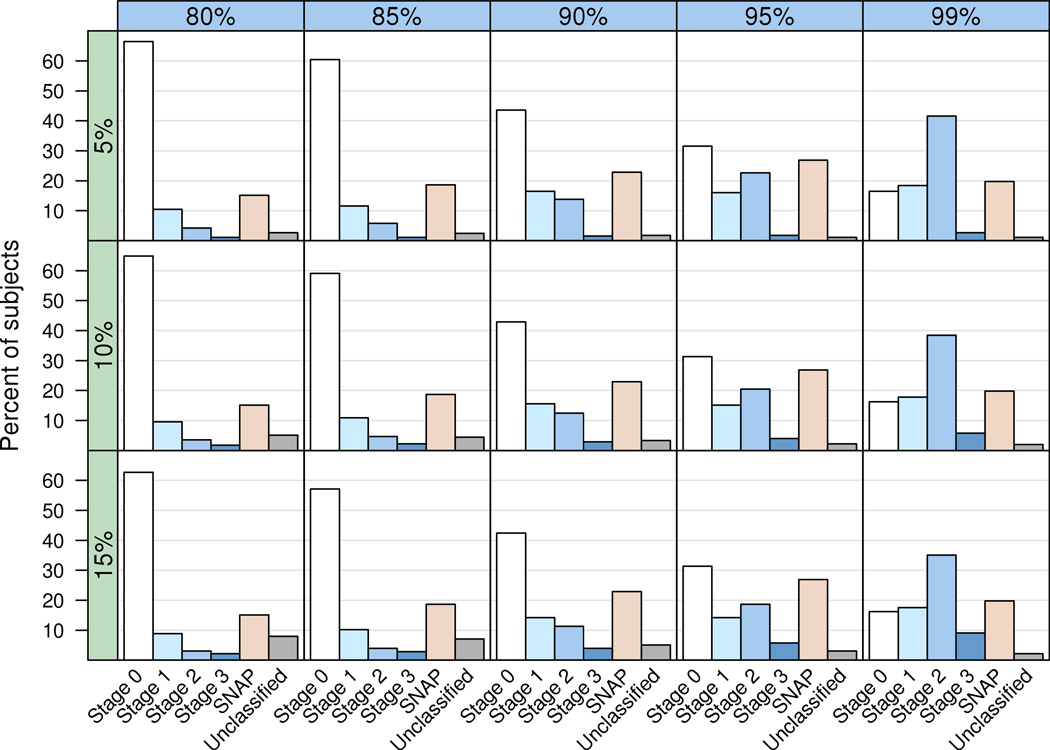

a. Grid of bar plots showing the percent of CN (n = 450) subjects in each category using the global composite measure for cognitive categorization. The columns of the grid (horizontal blue bars) represent different biomarker sensitivities (80% – 99%) used for cut-points while rows of the grid (vertical green bars) represent different global cognitive summary score percentiles (5% – 15%) used for cut-points.
b. Grid of bar plots showing the percent of CN (n = 450) subjects in each category using the memory z-score for cognitive categorization. The columns of the grid (horizontal blue bars) represent different biomarker sensitivities (80% – 99%) used for cut-points while the rows of the grid (vertical green bars) represent different memory z-score cognitive percentiles (5% – 15%) used for cut-points.
Distribution of cognitively normal subjects by pre-clinical stage
Figure 3 shows distributions of subjects that fall into preclinical stages 1–3 in our CN subjects using various combinations of cut-points for biomarkers and cognitive impairment. First, we note that the distribution of subjects among categories were very similar whether the global (Figure 3a) or memory summary scores (Figure 3b) were used. We elected to use the global summary thereafter as our primary measure of subtle cognitive impairment. Next, it is evident that two other categories are necessary to classify all CN subjects in our sample. One we call “stage 0”, those cognitively normal subjects with no biomarker evidence of AD pathophysiology and no evidence of subtle cognitive impairment. The other category, which we refer to as “Suspected Non-AD Pathophysiology (SNAP)”, consists of subjects with normal amyloid PET but one or both neuronal injury biomarkers abnormal. Additionally, there are a small number of subjects that do no fit into any group who we labeled “unclassified.”
Two overall trends are evident in Figure 3a and b. As the biomarker cut-point becomes more lenient (i.e. moves from 80% to 95% sensitivity for AD) subjects move out of stage 0 and into stages 1–3 and SNAP. This is true for all 3 cognitive cut-points evaluated. In contrast, relaxing the cognitive cut-points from 5% to 15% has a much less pronounced effect.
Table 3 illustrates in more detail the breakdown of subject classification at a fixed biomarker cut-point of 90% for all biomarkers, and a fixed global cognitive domain score cut-point of 10%. While any fixed cut-points could be chosen for illustration, with these cut-points, 90% of our AD sample is labeled abnormal on biomarkers and 90% of our CN sample is labeled normal on cognition. A biomarker sensitivity of 90% gives a PIB-PET cut-point of 1.5 (cortical-to-cerebellar ratio) which is commonly used to denote an abnormal scan [23]. With these cut-points, 97% of our CN sample were classified: 43% as stage 0, 16% as stage 1, 12% as stage 2, 3% as stage 3, and 23% as SNAP, and 3% were unclassified. Subjects could arrive at stages 2, 3 and SNAP by various combinations of test results. Subjects classified as SNAP had negative PIB PET plus positive FDG PET, HVa or both, with or without subtle cognitive impairment. Unclassified subjects were those with abnormal cognition alone or abnormal cognition plus positive PIB PET but no neurodegeneration. Demographic information for these groups is in Table 4. Figure 4 shows the distribution of subjects in stages 0–3 and SNAP according to the amyloid biomarker, neurodegeneration biomarker and cognitive status with the same cut points.
Table 3.
Distribution of preclinical AD stage among CN using a biomarker cut-point based on 90% sensitivity for AD and a cognition cut-point based on the 10th percentile of the CN distribution for the global domain.
| PIB PET | HVa | FDG PET | Cognitive Impairment |
Percent | Overall percent |
|
|---|---|---|---|---|---|---|
| Stage 0 | − | − | − | − | 43 | 43 |
| Stage 1 | + | − | − | − | 16 | 16 |
| Stage 2 | + | − | + | − | 6 | 12 |
| + | + | − | − | 3 | ||
| + | + | + | − | 3 | ||
| Stage 3 | + | − | + | + | 1 | 3 |
| + | + | − | + | 0 | ||
| + | + | + | + | 1 | ||
| SNAP | − | − | + | − | 10 | 23 |
| − | − | + | + | 2 | ||
| − | + | − | − | 6 | ||
| − | + | − | + | 1 | ||
| − | + | + | − | 3 | ||
| − | + | + | + | 1 | ||
| Unclassified | − | − | − | + | 2 | 3 |
| + | − | − | + | 2 |
TABLE 4.
DESCRIPTIVE CHARACTERISTICS OF PARTICIPANTS BY CLASSIFICATION GROUP
| Characteristic | Stage 0 (n = 193) |
Stage 1 (n = 70) |
Stage 2 (n = 56) |
Stage 3 (n = 13) |
SNAP (n = 103) |
Unclassified (n = 15) |
|---|---|---|---|---|---|---|
| Age, y, median (IQR) | 76 (73, 79) | 80 (74, 82) | 80 (78, 82) | 82 (80, 84) | 79 (76, 84) | 80 (74, 82) |
| Female gender, no. (%) | 94 (49) | 35 (50) | 19 (34) | 6 (46) | 41 (40) | 10 (67) |
| Education, y, median (IQR) | 13 (12, 16) | 14 (12, 16) | 15 (12, 18) | 13 (12, 15) | 14 (12, 16) | 12 (12, 14) |
| APOE ε4 genotype no. (%)* | ||||||
| ε4 carrier | 38 (24) | 25 (40) | 20 (40) | 7 (58) | 12 (13) | 1 (8) |
| ε4 non-carrier | 120 (76) | 37 (60) | 30 (60) | 5 (42) | 83 (87) | 12 (92) |
| MMSE, median (IQR)** | 29 (28, 29) | 28 (27, 29) | 28 (27, 29) | 27 (27, 28) | 28 (27, 29) | 28 (26, 29) |
| Cognitive domain Z scores, median (IQR)† | ||||||
| Global | 0.84 (0.23, 1.40) | 0.38 (−0.10, 1.29) | 0.35 (−0.26, 0.80) | −1.22 (−1.32, −0.99) | 0.10 (−0.52, 0.96) | −1.24 (−1.35, −0.98) |
| Memory | 0.78 (0.01, 1.43) | 0.50 (−0.40, 1.26) | 0.37 (−0.48, 0.93) | −1.23 (−1.36, −0.75) | 0.25 (−0.39, 1.01) | −0.76 (−0.98, −0.40) |
| Language | 0.58 (0.02, 1.09) | 0.36 (−0.24, 0.82) | 0.16 (−0.35, 0.72) | −0.90 (−1.12, −0.15) | 0.07 (−0.50, 0.83) | −0.81 (−1.12, −0.20) |
| Executive | 0.68 (0.13, 1.17) | 0.44 (−0.16, 1.29) | 0.15 (−0.38, 0.79) | −0.64 (−0.98, −0.29) | 0.03 (−0.59, 0.92) | −0.69 (−1.64, −0.25) |
| Visuospatial | 0.74 (0.10, 1.30) | 0.57 (0.06, 1.27) | 0.53 (−0.16, 0.96) | −1.43 (−1.71, −0.92) | 0.21 (−0.51, 0.72) | −1.48 (−1.91, −1.08) |
| PiB PET ratio | 1.33 (1.29, 1.38) | 1.86 (1.67, 2.07) | 1.93 (1.67, 2.26) | 2.35 (1.91, 2.81) | 1.35 (1.30, 1.39) | 1.43 (1.31, 1.77) |
APOE genotype not available in all subjects
Converted to the MMSE equivalent from the Short Test of Mental Status
Z scores are calculated using the mean and SD of the MCSA 2004 baseline visit for those who were cognitively normal (n = 1624)
IQR - inter-quartile range
Figure 4.

Venn diagram depicting the distribution of all 450 cognitively normal subjects by NIA-AA stage, biomarker and cognitive status
Using these 90% biomarker and 10% cognitive cut-points, the proportion of subjects who were APOE ε4 carriers was: stage 0, 24%; stage 1, 40%; stage 2, 40%; stage 3, 58%; SNAP , 13%, (highly different across groups, P < 0.001).
Discussion
Based on our operational approach to the NIA-AA criteria, at 90% biomarker and 10% cognitive cut-points, 31% of our CN subjects met the NIA-AA criteria for preclinical AD (stages 1–3), 43% were in stage 0 and 23% fell into the SNAP category. Only 3% of subjects could not be classified by our approach. The concept of preclinical AD originated with a literature documenting the presence of AD pathology in approximately a third of elderly CN subjects who came to autopsy [24–28]. Many studies have documented the presence of AD pathophysiological processes in living cognitively normal elderly subjects using PIB-PET imaging [17, 23, 29–39], 18FDG-PET imaging [33, 34, 40, 41], CSF assays [20, 42–48], and structural MR [49–56]. We hypothesize that stage 1–3 subjects have entered the AD pathway and, if they live long enough, will progress to incident MCI and then AD dementia. Subjects in stage 0, as defined, neither have subtle cognitive impairment nor abnormal AD biomarkers now. It is possible that some stage 0 subjects could move to stage 1 or beyond in the future.
Approximately a quarter of our CN subjects, 23%, were designated as SNAP. We believe that SNAP does not represent a stage of pre clinical AD, but rather a distinct biologically-based category where amyloid biomarkers are normal but neuronal injury biomarkers are abnormal. We suspect, but can not prove at this time, that such subjects represent the pre clinical stage of non-AD pathophysiological processes. While most cases of dementia in elderly subjects are found at autopsy to have multiple pathologies that include AD, up to one-third are primarily attributable to pathologies other than AD, primarily cerebrovascular disease and synucleinopathy [57–62]. It is therefore expected that preclinical forms of the non-AD pathologies must exist in elderly CN subjects recruited from a population-based sample. Subjects with predominantly cerebrovascular disease or synuclein pathologies but little or no AD pathology should present with a biomarker profile of normal amyloid PET and abnormalities on MRI and FDG [63, 64]. The low proportion of SNAP APOE ε4 carriers, just 13%, provides strong support for the assertion that many of SNAP subjects are in a different mechanistic pathway from those in stages 1–3 where the proportion of APOE ε4 carriers is 3–4x tiems greater.
In this preliminary analysis, we used the same sensitivity level for all biomarker cut points. In the future, alternative approaches that reflect the underlying biology and longitudinal evolution may be preferred. For example, sensitivity for AD of 90% corresponds to a PIB-PET cut-point of 1.5. While this cut point is widely used to diagnose AD, a lower amyloid PET cut-point may be better suited to answer the question of amyloid positivity in CN subjects [49, 52, 65].
Alternative methods for selecting regions of interest for the neurodegeneration imaging biomarkers may be superior to those we have used here. We chose hippocampal volume as the structural MRI measure for this preliminary analysis because it is the most studied and validated MRI measure at this point in time. However, more complex multi-ROI measures including isocortex might be superior [21, 49, 51, 66]. Alternative ROI combinations might also be useful in FDG-PET [67].
The NIA-AA preclinical criteria specify that in stage 3 subtle cognitive “decline” will be present but do not offer specific guidance on how this should be operationalized. “Decline” is described both in longitudinal and cross-sectional terms – i.e. “evidence of subtle change from baseline level of cognition” or “poor performance on more challenging cognitive tests” [1]. While longitudinal measures of decline have the obvious advantage of controlling for inter-subject variation in baseline performance, implementation of a simple cross-sectional measure of low cognitive performance such as the lowest 10th percentile is more straightforward. Prior longitudinal cognitive test data might not be available in many settings, e.g., preventative therapeutic trials. Somewhat surprisingly, the distribution of subjects among the various categories was similar if the memory domain score vs the composite domain score was used. One possible explanation is that although the pre clinical criteria emphasize memory, a body of literature exists that points to declines in domains other than memory as the initial cognitive signal of impending AD [68, 69]. A cut point based solely on a memory battery would miss these subjects whereas the composite might capture them.
Our approach to cross-sectional cut-point definition for subtle cognitive impairment was different from the approach for cut-point definition for AD biomarkers because the subtle cognitive impairment seen in the preclinical stage of the disease is notably different from that seen in demented subjects or subjects with MCI. In contrast, values for biomarkers, especially amyloid biomarkers, can overlap considerably in CN and demented subjects. By using the cohort under study to define a cognitive cut point, we lacked an independent means of defining abnormality. This meant that a fixed proportion (5%, 10%, 15%) of subjects were designated as cognitively impaired. This has obvious potential for erroneously classifying a few subjects at the margin as abnormal who might not have been with a slightly different definition of abnormal. We suspect that this effect underlies many of our “unclassified” subjects. The problem of using cognitive impairment as both a criterion for preclinical AD and as an outcome in longitudinal observational studies and therapeutic trials will need further exploration. Thus, operationalization of criteria for subtle cognitive impairment is complex, and the definition of the cognitive threshold we have provisionally chosen can likely be improved upon [70].
As a population-based study [7], our cognitively normal subjects differ from those recruited into studies such as the Alzheimer’s Disease Neuroimaging Initiative or other clinic-based samples. Age, education and co-morbidities greatly influence the likelihood and rate of progressing to dementia, and therefore evaluating new diagnostic criteria in samples that approximately reflect these variables as they exist in the general population is essential for generalizability and external validity of results [71, 72]. The high prevalence of SNAP in this preliminary exercise underscores the importance of performing studies in subjects where there are as few implicit inclusion and exclusion criteria as possible. Results might be different from samples drawn from memory clinics where recruitment biases might reduce the number of non-AD etiologies.
While the new NIA-AA preclinical criteria broke new ground conceptually, many operational issues were not addressed. These include standardization of biomarker measures, defining biomarker cut-points, how to address discrepancies within biomarker class (e.g., abnormal FDG but normal hippocampal volume), the definition of subtle cognitive impairment, and how to address the non-AD pathophysiological processes that are present in elderly populations. Some limitations of our study include the fact that our subjects had only MRI and PET imaging biomarkers available, not CSF. The number of cognitive testing sessions, and hence practice effects, varied among subjects in our cohort. Other important options remain to be evaluated including alternative biomarker and cognitive cut points and alternative imaging measures. However with this operational approach to implementation, the NIA-AA preclinical AD guidelines function adequately in a population based sample of elderly subjects and, therefore, should be useful in planning future observational and therapeutic studies.
Acknowledgment
The Alexander Family Alzheimer’s Disease Research Professorship of the Mayo Foundation, USA, and the Robert H. and Clarice Smith Alzheimer’s Disease Research Program of the Mayo Foundation, USA. This study was supported by the NIH/National Institute on Aging (R01 AG11378, U01 AG006786, P50 AG16574, C06 RR018898; C.R.J.).
References
- 1.Sperling RA, et al. Toward defining the preclinical stages of Alzheimer's disease: Recommendations from the National Institute on Aging and the Alzheimer Assocation Workgroup. Alzheimers Dement. 2011 doi: 10.1016/j.jalz.2011.03.003. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jack CR, Jr, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010;9(1):119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ingelsson M, et al. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62(6):925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 4.Mormino EC, et al. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain. 2009;132(Pt 5):1310–1323. doi: 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perrin RJ, Fagan AM, Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer's disease. Nature. 2009;461(7266):916–922. doi: 10.1038/nature08538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jack CR, Jr, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132(Pt 5):1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts RO, et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology. 2008;30(1):58–69. doi: 10.1159/000115751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petersen RC, et al. Prevalence of mild cognitive impairment is higher in men. The Mayo Clinic Study of Aging. Neurology. 2010;75(10):889–897. doi: 10.1212/WNL.0b013e3181f11d85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jack CR, Jr, et al. Update on the magnetic resonance imaging core of the Alzheimer's disease neuroimaging initiative. Alzheimers Dement. 2010;6(3):212–220. doi: 10.1016/j.jalz.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gunter JL, et al. Measurement of MRI scanner performance with the ADNI phantom. Med Phys. 2009;36(6):2193–2205. doi: 10.1118/1.3116776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gunter JL, et al. Is B1-correction for Neuroanatomy necessary? in International Society for Magnetic Resonance in Medicine. Honolulu, HI: 2009. [Google Scholar]
- 12.Fischl B, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33(3):341–355. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- 13.Jack CR, Jr, et al. Anterior temporal lobes and hippocampal formations: normative volumetric measurements from MR images in young adults. Radiology. 1989;172(2):549–554. doi: 10.1148/radiology.172.2.2748838. [DOI] [PubMed] [Google Scholar]
- 14.Mathis CA, et al. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem. 2003;46(13):2740–2754. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- 15.Price JC, et al. Kinetic modeling of amyloid binding in humans using PET imaging and Pittsburgh Compound-B. J Cereb Blood Flow Metab. 2005;25(11):1528–1547. doi: 10.1038/sj.jcbfm.9600146. [DOI] [PubMed] [Google Scholar]
- 16.McNamee RL, et al. Consideration of optimal time window for Pittsburgh compound B PET summed uptake measurements. J Nucl Med. 2009;50(3):348–355. doi: 10.2967/jnumed.108.057612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jack CR, Jr, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain. 2008;131(Pt 3):665–680. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lopresti BJ, et al. Simplified quantification of Pittsburgh Compound B amyloid imaging PET studies: a comparative analysis. J Nucl Med. 2005;46(12):1959–1972. [PubMed] [Google Scholar]
- 19.Landau SM, et al. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology. 2010;75(3):230–238. doi: 10.1212/WNL.0b013e3181e8e8b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shaw LM, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol. 2009;65(4):403–413. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vemuri P, et al. Antemortem MRI based STructural Abnormality iNDex (STAND)-scores correlate with postmortem Braak neurofibrillary tangle stage. Neuroimage. 2008;42(2):559–567. doi: 10.1016/j.neuroimage.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whitwell JL, et al. MRI correlates of neurofibrillary tangle pathology at autopsy: a voxel-based morphometry study. Neurology. 2008;71(10):743–749. doi: 10.1212/01.wnl.0000324924.91351.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rowe CC, et al. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68(20):1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- 24.Knopman DS, et al. Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol. 2003;62(11):1087–1095. doi: 10.1093/jnen/62.11.1087. [DOI] [PubMed] [Google Scholar]
- 25.Price JL, Morris JC. Tangles and plaques in nondemented aging and "preclinical" Alzheimer's disease. Ann Neurol. 1999;45(3):358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 26.Bennett DA, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66(12):1837–1844. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- 27.Schmitt FA, et al. "Preclinical" AD revisited: neuropathology of cognitively normal older adults. Neurology. 2000;55(3):370–376. doi: 10.1212/wnl.55.3.370. [DOI] [PubMed] [Google Scholar]
- 28.Schneider JA, et al. The neuropathology of older persons with and without dementia from community versus clinic cohorts. J Alzheimers Dis. 2009;18(3):691–701. doi: 10.3233/JAD-2009-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klunk WE, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol. 2004;55(3):306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 30.Mintun MA, et al. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67(3):446–452. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- 31.Rowe CC, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging. 2010;31(8):1275–1283. doi: 10.1016/j.neurobiolaging.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 32.Aizenstein HJ, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008;65(11):1509–1517. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lowe VJ, et al. Comparison of 18F-FDG and PiB PET in cognitive impairment. J Nucl Med. 2009;50(6):878–886. doi: 10.2967/jnumed.108.058529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lo RY, et al. Longitudinal Change of Biomarkers in Cognitive Decline. Arch Neurol. 2011 doi: 10.1001/archneurol.2011.123. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Resnick SM, et al. Longitudinal cognitive decline is associated with fibrillar amyloid-beta measured by [11C]PiB. Neurology. 2010;74(10):807–815. doi: 10.1212/WNL.0b013e3181d3e3e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rentz DM, et al. Cognition, reserve, and amyloid deposition in normal aging. Ann Neurol. 2010;67(3):353–364. doi: 10.1002/ana.21904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morris JC, et al. Pittsburgh Compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol. 2009;66(12):1469–1475. doi: 10.1001/archneurol.2009.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pike KE, et al. Cognition and beta-amyloid in preclinical Alzheimer's disease: Data from the AIBL study. Neuropsychologia. 2011 doi: 10.1016/j.neuropsychologia.2011.04.012. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 39.Villemagne VL, et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69(1):181–192. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jagust WJ, et al. The Alzheimer's Disease Neuroimaging Initiative positron emission tomography core. Alzheimers Dement. 2010;6(3):221–229. doi: 10.1016/j.jalz.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reiman EM, et al. Preclinical evidence of Alzheimer's disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med. 1996;334(12):752–758. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- 42.Peskind ER, et al. Age and apolipoprotein E*4 allele effects on cerebrospinal fluid beta-amyloid 42 in adults with normal cognition. Arch Neurol. 2006;63(7):936–939. doi: 10.1001/archneur.63.7.936. [DOI] [PubMed] [Google Scholar]
- 43.Bouwman FH, et al. CSF biomarker levels in early and late onset Alzheimer's disease. Neurobiol Aging. 2009;30(12):1895–1901. doi: 10.1016/j.neurobiolaging.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 44.Stomrud E, et al. Correlation of longitudinal cerebrospinal fluid biomarkers with cognitive decline in healthy older adults. Arch Neurol. 2010;67(2):217–223. doi: 10.1001/archneurol.2009.316. [DOI] [PubMed] [Google Scholar]
- 45.Schott JM, et al. Increased brain atrophy rates in cognitively normal older adults with low cerebrospinal fluid Abeta1-42. Ann Neurol. 2010;68(6):825–834. doi: 10.1002/ana.22315. [DOI] [PubMed] [Google Scholar]
- 46.Fagan AM, et al. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol. 2009;65(2):176–183. doi: 10.1002/ana.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vemuri P, et al. MRI and CSF biomarkers in normal, MCI, and AD subjects: Diagnostic discrimination and cognitive correlations. Neurology. 2009;73(4):287–293. doi: 10.1212/WNL.0b013e3181af79e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vemuri P, et al. Effect of apolipoprotein E on biomarkers of amyloid load and neuronal pathology in Alzheimer disease. Ann Neurol. 2010;67(3):308–316. doi: 10.1002/ana.21953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dickerson BC, et al. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex. 2009;19(3):497–510. doi: 10.1093/cercor/bhn113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fagan AM, et al. Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer's disease. EMBO Mol Med. 2009;1(8-9):371–380. doi: 10.1002/emmm.200900048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dickerson BC, et al. Alzheimer-signature MRI biomarker predicts AD dementia in cognitively normal adults. Neurology. 2011;76(16):1395–1402. doi: 10.1212/WNL.0b013e3182166e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Becker JA, et al. Amyloid-beta associated cortical thinning in clinically normal elderly. Ann Neurol. 2011;69(6):1032–1042. doi: 10.1002/ana.22333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Desikan RS, et al. Selective disruption of the cerebral neocortex in Alzheimer's disease. PLoS One. 2010;5(9):e12853. doi: 10.1371/journal.pone.0012853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chiang GC, et al. Identifying Cognitively Healthy Elderly Individuals with Subsequent Memory Decline by Using Automated MR Temporoparietal Volumes. Radiology. 2011;259(3):844–851. doi: 10.1148/radiol.11101637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Moffat SD, et al. Longitudinal change in hippocampal volume as a function of apolipoprotein E genotype. Neurology. 2000;55(1):134–136. doi: 10.1212/wnl.55.1.134. [DOI] [PubMed] [Google Scholar]
- 56.Schuff N, et al. MRI of hippocampal volume loss in early Alzheimer's disease in relation to ApoE genotype and biomarkers. Brain. 2009;132(Pt 4):1067–1077. doi: 10.1093/brain/awp007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sonnen JA, et al. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol. 2007;62(4):406–413. doi: 10.1002/ana.21208. [DOI] [PubMed] [Google Scholar]
- 58.Schneider JA, et al. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69(24):2197–2204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
- 59.Schneider JA, et al. Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology. 2004;62(7):1148–1155. doi: 10.1212/01.wnl.0000118211.78503.f5. [DOI] [PubMed] [Google Scholar]
- 60.Petrovitch H, et al. AD lesions and infarcts in demented and non-demented Japanese-American men. Ann Neurol. 2005;57(1):98–103. doi: 10.1002/ana.20318. [DOI] [PubMed] [Google Scholar]
- 61.White L, et al. Recent clinical-pathologic research on the causes of dementia in late life: update from the Honolulu-Asia Aging Study. J Geriatr Psychiatry Neurol. 2005;18(4):224–227. doi: 10.1177/0891988705281872. [DOI] [PubMed] [Google Scholar]
- 62.Schneider JA, et al. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol. 2009;66(2):200–208. doi: 10.1002/ana.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Langa KM, Foster NL, Larson EB. Mixed dementia: emerging concepts and therapeutic implications. JAMA. 2004;292(23):2901–2908. doi: 10.1001/jama.292.23.2901. [DOI] [PubMed] [Google Scholar]
- 64.Craft S. The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol. 2009;66(3):300–305. doi: 10.1001/archneurol.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jack CR, Jr, et al. Brain beta-amyloid measure and magnetic resonance imaging atophy both predict time-to-progression from mild cognitive impairment to Alzheimer's disease. Brain. 2010;133(11):3336–3348. doi: 10.1093/brain/awq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vemuri P, et al. Alzheimer's disease diagnosis in individual subjects using structural MR images: validation studies. Neuroimage. 2008;39(3):1186–1197. doi: 10.1016/j.neuroimage.2007.09.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen K, et al. Characterizing Alzheimer's disease using a hypometabolic convergence index. Neuroimage. 2011;56(1):52–60. doi: 10.1016/j.neuroimage.2011.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Albert MS, et al. Preclinical prediction of AD using neuropsychological tests. J Int Neuropsychol Soc. 2001;7(5):631–639. doi: 10.1017/s1355617701755105. [DOI] [PubMed] [Google Scholar]
- 69.Storandt M, et al. Cognitive decline and brain volume loss as signatures of cerebral amyloid-beta peptide deposition identified with Pittsburgh compound B: cognitive decline associated with Abeta deposition. Arch Neurol. 2009;66(12):1476–1481. doi: 10.1001/archneurol.2009.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Reed BR, et al. Measuring cognitive reserve based on the decomposition of episodic memory variance. Brain. 2010 doi: 10.1093/brain/awq154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Glasgow RE, et al. Practical clinical trials for translating research to practice: design and measurement recommendations. Med Care. 2005;43(6):551–557. doi: 10.1097/01.mlr.0000163645.41407.09. [DOI] [PubMed] [Google Scholar]
- 72.Tunis SR, Stryer DB, Clancy CM. Practical clinical trials: increasing the value of clinical research for decision making in clinical and health policy. JAMA. 2003;290(12):1624–1632. doi: 10.1001/jama.290.12.1624. [DOI] [PubMed] [Google Scholar]