

Abstract
Inflammation is a major factor shaping outcome during the early, acute phase of traumatic spinal cord injury (SCI). It is known that pro-inflammatory signaling within the injured spinal cord drives pathological alterations in neurosensory processing and shapes functional outcome early after injury. However, it is unclear whether inflammation persists into the chronic phase of injury or shapes sensory processing long after injury. To investigate these possibilities, we have performed biochemical and behavioral assessments 9 months after moderate thoracic spinal contusion injury in the rat. We have found that levels of the pro-inflammatory lipid mediators leukotriene B4 and prostaglandin E2 are elevated in the chronic spinal cord lesion site. Additionally, using metabolomic profiling, we have detected elevated levels of pro-oxidative and inflammatory metabolites, along with alterations in multiple biological pathways within the chronic lesion site. We found that 28 d treatment of chronically injured rats with the dual COX/5-LOX inhibitor licofelone elevated levels of endogenous anti-oxidant and anti-inflammatory metabolites within the lesion site. Furthermore, licofelone treatment reduced hypersensitivity of hindpaws to mechanical, but not thermal, stimulation, indicating that mechanical sensitivity is modulated by pro-inflammatory signaling in the chronic phase of injury. Together, these findings provide novel evidence of inflammation and oxidative stress within spinal cord tissue far into the chronic phase of SCI, and demonstrate a role for inflammatory modulation of mechanical sensitivity in the chronic phase of injury.
Introduction
Inflammation plays a major role in shaping the pathophysiology of spinal cord injury (SCI). Acute inflammation, occurring hours to days after the initial injury, contributes to glial scar formation (Fitch et al., 1999), cytotoxicity (Alexander and Popovich, 2009), and sensitization of neural pathways (Hulsebosch et al., 2009). Inflammation is also thought to be important for regeneration of injured CNS axons (Benowitz and Popovich, 2011), underscoring the complexity of the inflammatory response within the injured spinal cord. In contrast to what is known about acute-phase inflammation, relatively few studies have focused on characterizing the spinal cord molecular milieu beyond 1–2 months postinjury. Recent findings that peripheral immune cells and activated microglia are present within the spinal cord up to 1 year after injury (Fleming et al., 2006; Beck et al., 2010; Byrnes et al., 2011) raise the possibility that neuroinflammation following SCI may not be transient, but may in fact continue into the chronic phase of injury.
Oxidative arachidonic acid (AA) metabolism is a major hallmark of neuroinflammation (Farooqui et al., 2007). Following its release from the cell membrane by activated neuronal phospholipase A2 (PLA2), AA is metabolized via cyclooxygenase (COX) and 5-lipoxygenase (5-LOX) into the prostaglandins (PGs) and leukotrienes (LTs), respectively (Liu and Xu, 2010). These bioactive lipids are potent mediators of inflammation and secondary injury within the injured spinal cord (Liu and Xu, 2010). Additionally, PGs and LTs contribute to alterations in neurosensory processing within the spinal cord, and are associated with pathological sensory abnormalities such as mechanical allodynia and hyperalgesia (Hains et al., 2001; Zhao et al., 2007; Buczynski et al., 2010; Noguchi and Okubo, 2011), which are often experienced by SCI patients (Baastrup and Finnerup, 2008). Because this pro-inflammatory signaling network contributes to diverse adverse effects after SCI, much work has been done to evaluate the therapeutic potential of inhibiting PLA2 (Huang et al., 2009), COX enzymes (Kwon et al., 2011), and 5-LOX (Genovese et al., 2005, 2008) for promoting recovery of function following SCI.
Whether oxidative AA metabolism occurs within the chronically injured spinal cord and indeed, whether inflammation is present in the chronic phase of injury, has not previously been investigated. We have detected elevated levels of PGs and LTs 9 months after SCI, demonstrating that pro-inflammatory signaling is present even after prolonged periods postinjury. To gain a better understanding of the global metabolic changes within the chronically injured spinal cord, we performed a metabolomic screen to identify biochemical compounds that are altered during chronic SCI. Here, we provide evidence of a pro-inflammatory and pro-oxidative environment within the chronically injured spinal cord. Additionally, we report that prolonged, oral treatment with the new-generation dual COX/5-LOX inhibitor licofelone, a drug with improved gastrointestinal tolerability compared with classical nonsteroidal anti-inflammatory drugs (Cicero and Laghi, 2007), enhances anti-inflammatory and anti-oxidative defenses within the chronic lesion site, and reduces mechanical hypersensitivity in rats several months after SCI.
Materials and Methods
Animal subjects and surgeries
All animal use was approved by the Institute for Animal Care and Use Committee of The University of Texas Health Science Center. A total of 80 adult, female Sprague Dawley rats weighing 225–250 g were used in this study. All SCI surgeries were performed under anesthesia (ketamine, 80 mg/kg; xylazine, 10 mg/kg; and acepromazine, 0.75 mg/kg) at a dose of 0.1 ml/100 g body weight (Herrera et al., 2010). Animals received a laminectomy at spinal cord thoracic level 10 (T10), and the vertebral column was stabilized at T9 and T11. A moderate contusion injury (150 kdyne of force with 1 s dwell) was delivered to the spinal cord at T10 using the Infinite Horizon Impactor (Precision Systems and Instrumentation), without opening the dura mater (Scheff et al., 2003; Dulin et al., 2011). The overlying muscles were immediately sutured and the skin was closed with stainless steel wound clips. Beginning the day after SCI surgery, each animal's urinary bladder was manually expressed twice daily until the animal recovered the ability to void its bladder. As a rule, bladder care was ceased when an individual animal exhibited an empty bladder on two consecutive bladder care sessions. To minimize postsurgical pain, animals were treated twice daily for 5 d post-SCI with buprenorphine (0.02 mg/kg, s.c.). To prevent dehydration, animals also received a 3 ml bolus of 0.9% saline subcutaneously twice daily for the first 72 h after surgery. To prevent infection, animals also received antibiotics (2.5 mg/kg Baytril, i.p.) twice daily for 10 d. Animals were housed in groups of two and maintained under normal veterinary care. Chronically injured animals were housed for 8–9 months following SCI surgery. Age-matched, uninjured rats were used as controls in all experiments.
Exclusion criteria.
Injured subjects were excluded from the study if the actual force of impact to the spinal cord fell outside a range between 150 and 175 kdynes (Scheff et al., 2003). In addition, any injured subjects receiving an impact of force within that range that exhibited a hindlimb locomotor score >2 on postinjury day 1 were also excluded (Basso et al., 1995). Nine animals were excluded from this study based on the above criteria.
Drug treatment
Animals were treated with 50 mg/kg licofelone (Santa Cruz Biotechnology, #sc-207826, lot #J1411) suspended in 1.25 ml vehicle (1.5% carboxymethylcellulose from Sigma-Aldrich in 0.9% saline), or vehicle alone, via oral gavage. Chronically injured animals were treated beginning at 8 months post-SCI with licofelone or vehicle once daily for 28 d. Acutely injured animals were treated beginning at 3 h post-SCI with licofelone or vehicle and again at 18 h post-SCI.
ELISA
ELISA was performed for quantification of prostaglandin E2 (PGE2) and leukotriene B4 (LTB4) in T10 spinal cord tissue of rats at 24 h post-SCI (n = 12 SCI + vehicle; n = 12 SCI + licofelone; n = 12 uninjured controls) or 9 months post-SCI (n = 9 SCI; n = 9 uninjured, age-matched controls). Animals were killed with Beuthanasia (390 mg/ml pentobarbital sodium, 50 mg/ml phenytoin sodium; 75 mg/kg, i.p.). A 10 mm length of tissue centered around the spinal cord lesion site in injured rats, or the corresponding intact tissue in uninjured controls, was immediately harvested and weighed, snap-frozen in liquid nitrogen, and stored at −80°C. Homogenization buffer (0.1 m phosphate, pH 7.4, 1 mm EDTA, 10 μm indomethacin; 1 ml per 100 mg tissue) was added and tissue was homogenized on ice using a Polytron homogenizer. For detection of PGE2, an additional purification step was performed using a PGE2 Affinity Sorbent Kit (Cayman Chemical). Levels of PGE2 and LTB4 in spinal cord tissue were quantified using PGE2 and LTB4 EIA kits (Cayman Chemical). All samples were prepared and analyzed in triplicate, and absorbance at 405 nm was read in a Model 680 Microplate Reader (Bio-Rad). Standard curves were generated and sample concentrations were determined using Cayman EIA data analysis worksheets (www.caymanchem.com/analysis/eia). Data were analyzed by Student's t test (for comparison of two groups) or one-way ANOVA followed by the Student–Newman–Keuls post test (for comparison of more than two groups).
Behavioral testing
Longitudinal behavioral assessments were performed on chronically injured animals (n = 8 SCI + vehicle; n = 9 SCI + licofelone; n = 9 uninjured, age-matched controls).
Hindlimb locomotor function.
Hindlimb locomotor function was assessed using the Basso, Beattie, and Bresnahan (BBB) open-field locomotor scale (Basso et al., 1995). Before SCI surgery, animals were allowed to acclimate to the open-field testing environment (a 40 inch diameter plastic wading pool) in groups of four for 10 min daily until they ceased to exhibit fear-associated behavior (e.g., crouching, cowering away from the examiner, vocalizing) and displayed signs of comfort (e.g., grooming, accepting treats from the examiner's hand, exhibiting continuous locomotion and exploration). No further acclimation to the testing environment was performed. Preinjury testing began once the examiner felt confident that all animals were comfortable in the testing environment. BBB scores were assessed on day 1 post-SCI, then weekly beginning at 8 months post-SCI (before the beginning of drug treatment) until day 28 of treatment. Animals were placed into the open field and allowed to move ad libitum for a period of 4 min. Two independent observers scored each hindlimb using a scoring sheet. Hindlimb BBB scores were rated using the 21 point BBB Open-Field Rating Scale, and inter-rater reliability was confirmed. For each animal, the locomotor scores for both hindpaws were averaged to produce one score per test session. BBB scores were analyzed using a two-way repeated-measures ANOVA (factors: experimental groups and time points with repeated measures on the second factor), followed by a Bonferroni post-test.
Mechanical hypersensitivity.
Mechanical hypersensitivity was defined as a decreased withdrawal threshold when the hindpaw was presented with a series of calibrated von Frey monofilaments (Stoelting), consistent with previous studies (Chaplan et al., 1994). Animals were habituated to the testing environment for 15 min per day, on 3 consecutive days before beginning testing. A series of filaments delivering calibrated amounts of force (ranging from 2 to 15 g) were applied using the Dixon “up–down” method, and 50% mechanical withdrawal threshold was determined as previously described (Dixon, 1991; Chaplan et al., 1994). Differences in individual animals' pretreatment and post-treatment mechanical sensitivity scores were analyzed using a paired Student's t test; effectiveness of pairing was determined by calculating the Pearson correlation coefficient (r).
Thermal hypersensitivity.
Thermal hypersensitivity was defined as a reduction in paw withdrawal latency in response to the radiant heat stimulus, as previously described (Hargreaves et al., 1988; Carlton et al., 2009). Thermal sensitivity of animals' hindpaws was examined using a modified Hargreaves test (Hargreaves et al., 1988; Carlton et al., 2009). Animals were placed on the modified Hargreaves platform, a glass surface maintained at a constant surface temperature of 32°C, within four-sided Plexiglas enclosures. Animals were allowed to acclimate to the device daily for 1 week before testing and for 30 min before each test session. Testing was performed at 8 months post-SCI (before the beginning of drug treatment) and again on day 28 of treatment. Using a 336G Plantar Analgesia Meter (IITC Life Science), radiant heat of a constant intensity (45% active intensity) was focused on the center of the plantar surface of each hindpaw. The withdrawal latency was defined as the time from stimulus initiation until an abrupt paw withdrawal response, and 20 s was used as an automatic cutoff time to avoid tissue damage. For each hindpaw, three measurements were taken with a minimum of 10 min between each measurement. Thermal latency scores were analyzed by finding the mean for the three pretreatment tests and the mean for the three post-treatment tests, from each hindpaw; mean scores from the two hindpaws were averaged for each animal. The overall effect of treatment on individual animals' pretreatment and post-treatment mechanical sensitivity scores was analyzed using a paired Student's t test; effectiveness of pairing was determined by calculating the Pearson correlation coefficient (r).
Metabolomic profiling
The same animals used in behavioral assessments were used for metabolomic profiling. Six hours following the final drug treatment, animals were killed and tissue was immediately harvested and snap-frozen. A 10 mm segment of spinal cord tissue with the lesion site at its center was used for metabolomic analysis. Metabolomic profiling analysis was performed as previously described (Reitman et al., 2011).
Sample accessioning.
Each sample received was accessioned into the Metabolon Laboratory Information Management System (LIMS) and was assigned by the LIMS a unique identifier that was associated with the original source identifier only. This identifier was used to track all sample handling, tasks, results, etc. The samples (and all derived aliquots) were tracked by the LIMS. All portions of any sample were automatically assigned their own unique identifiers by the LIMS when a new task was created; the relationship of these samples was also tracked. All samples were maintained at −80°C until processed.
Sample preparation.
Samples were prepared using the automated Microlab STAR system (Hamilton Robotics). A recovery standard was added before the first step in the extraction process for quality control (QC). Sample preparation was conducted using an aqueous methanol extraction process to remove the protein fraction while allowing maximum recovery of small molecules. The resulting extract was divided into four fractions: one for analysis by ultra performance liquid chromatography tandem mass spectrometry (UPLC/MS/MS) (positive mode), one for UPLC/MS/MS (negative mode), one for gas chromatography/mass spectrometry (GC/MS), and one for backup. Samples were placed briefly on a TurboVap concentration evaporator (Zymark) to remove the organic solvent. Each sample was then frozen and dried under vacuum. Samples were then prepared for the appropriate instrument, either UPLC/MS/MS or GC/MS.
UPLC/MS/MS.
The LC/MS portion of the platform was based on a Waters ACQUITY UPLC system and a Thermo Finnigan linear trap quadrupole mass spectrometer, which consisted of an electrospray ionization source and linear ion-trap mass analyzer. The sample extract was dried then reconstituted in acidic or basic LC-compatible solvents, each of which contained eight or more injection standards at fixed concentrations to ensure injection and chromatographic consistency. One aliquot was analyzed using acidic-positive ion-optimized conditions and the other using basic negative ion-optimized conditions in two independent injections using separate dedicated columns. Extracts reconstituted in acidic conditions were gradient eluted using water and methanol containing 0.1% formic acid, while the basic extracts, which also used water/methanol, contained 6.5 mm ammonium bicarbonate. The MS analysis alternated between MS and data-dependent MS2 scans using dynamic exclusion. Raw data files were archived and extracted as described below.
GC/MS.
The samples destined for GC/MS analysis were re-dried under vacuum desiccation for a minimum of 24 h before being derivatized under dried nitrogen using bis trimethyl-silyl-triflouroacetamide. The GC column was 5% phenyl and the temperature ramp was from 40 to 300°C in a 16 min period. Samples were analyzed on a Thermo Finnigan Trace DSQ fast-scanning single-quadrupole mass spectrometer using electron impact ionization. The instrument was tuned and calibrated for mass resolution and mass accuracy on a daily basis. The information output from the raw data files was automatically extracted as discussed below.
Quality assurance/QC.
For QC purposes, additional samples were included with each day's analysis. These samples included extracts of a pool of well characterized human plasma, extracts of a pool created from a small aliquot of the experimental samples, and process blanks. QC samples were spaced evenly among the injections and all experimental samples were randomly distributed throughout the run. A selection of QC compounds was added to every sample for chromatographic alignment, including experimental samples. These compounds were carefully chosen so as not to interfere with the measurement of the endogenous compounds. A data normalization step was performed to correct variation resulting from instrument interday tuning differences. Each compound was corrected in run-day blocks by registering the median concentration values across all samples to equal one (1.00) and normalizing each data point proportionately (termed the “block correction”).
Data extraction and compound identification.
Raw data were extracted, peak-identified, and QC processed using Metabolon's hardware and software. These systems are built on a Web-service platform using Microsoft's .NET technologies, which run on high-performance application servers and fiber-channel storage arrays in clusters to provide active failover and load-balancing (Dehaven et al., 2010). Compounds were identified by comparison to library entries of purified standards or recurrent unknown entities. Metabolon maintains a library based on authenticated standards containing the retention time/index (RI), mass to charge ratio (m/z), and chromatographic data (including MS/MS spectral data) of all molecules present in the library. Furthermore, biochemical identifications are based on three criteria: RI within a narrow RI window of the proposed identification, nominal mass match to the library ± 0.2 amu, and the MS/MS forward and reverse scores between the experimental data and authentic standards. The MS/MS scores are based on a comparison of the ions present in the experimental spectrum to the ions present in the library spectrum. While there may be similarities between these molecules based on one of these factors, the use of all three data points can be used to distinguish and differentiate biochemicals. At the time of compound identification, >2400 commercially available, purified standard compounds have been acquired and registered into LIMS for distribution to both the LC and GC platforms for determination of their analytical characteristics. Human Metabolome Database (HMDB) identifiers for detected and identified compounds are listed in Tables 1 and 2 (www.hmdb.ca) (Wishart et al., 2009). Compounds without HMDB IDs were not listed in the HMDB at the time of this screen.
Table 1.
Spinal cord metabolites with altered concentrations in chronic SCI, vehicle-treated rats versus uninjured controls
| Class | Subclass | Biochemical name | HMDB ID | Fold change (SCI + vehicle/control) | p value | q value |
|---|---|---|---|---|---|---|
| Amino acid | Glycine, serine, and threonine metabolism | Glycine | HMDB00123 | −1.35 | <0.001 | 0.0003 |
| Homoserine | HMDB00719 | −1.18 | 0.0780 | 0.0650 | ||
| Betaine | HMDB00043 | 2.22 | <0.001 | 0.0000 | ||
| Alanine and aspartate metabolism | Alanine | HMDB00161 | 1.19 | 0.0236 | 0.0276 | |
| Aspartate | HMDB00191 | −1.22 | 0.0091 | 0.0122 | ||
| N-acetylaspartate (NAA) | HMDB00812 | −2.38 | <0.001 | 0.0001 | ||
| Asparagine | HMDB00168 | −1.47 | 0.0557 | 0.0509 | ||
| Glutamate metabolism | Glutamate | HMDB03339 | −1.45 | <0.001 | 0.0000 | |
| Glutamine | HMDB00641 | 1.06 | 0.0982 | 0.0775 | ||
| GABA | HMDB00112 | −1.33 | 0.0650 | 0.0577 | ||
| N-acetylglutamate | HMDB01138 | −1.33 | 0.0220 | 0.0264 | ||
| N-acetyl-aspartyl-glutamate (NAAG) | HMDB01067 | −1.79 | <0.001 | 0.0000 | ||
| Lysine metabolism | 2-Aminoadipate | HMDB00510 | 1.38 | 0.0040 | 0.0059 | |
| N6-acetyllysine | HMDB00206 | 1.47 | 0.0014 | 0.0025 | ||
| Phenylalanine and tyrosine metabolism | Tyrosine | HMDB00158 | 1.15 | 0.0581 | 0.0525 | |
| Valine, leucine, and isoleucine metabolism | Isoleucine | HMDB00172 | 1.40 | <0.001 | 0.0005 | |
| Leucine | HMDB00687 | 1.45 | <0.001 | 0.0013 | ||
| Valine | HMDB00883 | 1.40 | 0.0036 | 0.0054 | ||
| Cysteine, methionine, SAM, taurine metabolism | Taurine | HMDB00251 | 1.52 | 0.0012 | 0.0023 | |
| S-adenosylhomocysteine | HMDB00939 | −1.43 | 0.0012 | 0.0023 | ||
| Methionine | HMDB00696 | 1.18 | 0.0260 | 0.0301 | ||
| N-acetylmethionine | HMDB11745 | 2.53 | <0.001 | 0.0000 | ||
| 2-Hydroxybutyrate | HMDB00008 | 1.65 | <0.001 | 0.0001 | ||
| Urea cycle; arginine-, proline-, metabolism | Citrulline | HMDB00904 | 1.55 | 0.0158 | 0.0206 | |
| Trans-4-hydroxyproline | HMDB00725 | 2.05 | <0.001 | 0.0002 | ||
| Creatine metabolism | Creatine | HMDB00064 | −1.16 | 0.0314 | 0.0332 | |
| Butanoate metabolism | 2-Aminobutyrate | HMDB00650 | 1.72 | 0.0035 | 0.0054 | |
| Glutathione metabolism | Glutathione, oxidized (GSSG) | HMDB03337 | 3.19 | <0.001 | 0.0003 | |
| Cysteine-glutathione disulfide | HMDB00656 | 2.27 | <0.001 | 0.0003 | ||
| Carbohydrate | Fructose, mannose, galactose, starch, and sucrose metabolism | Maltose | HMDB00163 | 1.61 | 0.0062 | 0.0086 |
| Mannose | HMDB00169 | 2.77 | <0.001 | 0.0003 | ||
| Mannose-6-phosphate | HMDB01078 | 2.57 | <0.001 | 0.0012 | ||
| Glycolysis, gluconeogenesis, pyruvate metabolism | Glycerate | HMDB00139 | 1.37 | 0.0313 | 0.0332 | |
| Glucose-6-phosphate | HMDB01401 | 2.53 | <0.001 | 0.0002 | ||
| Glucose | HMDB00122 | 2.73 | <0.001 | 0.0001 | ||
| Fructose-6-phosphate | HMDB00124 | 2.29 | <0.001 | 0.0003 | ||
| 3-Phosphoglycerate | HMDB00807 | −1.35 | 0.0195 | 0.0243 | ||
| 1,3-Dihydroxyacetone | HMDB01882 | 2.22 | <0.001 | 0.0002 | ||
| Lactate | HMDB00190 | 1.23 | <0.001 | 0.0013 | ||
| Nucleotide sugars, pentose metabolism | Arabitol | HMDB01851 | −1.64 | 0.0033 | 0.0054 | |
| Ribulose | HMDB00621, HMDB03371 | 1.40 | 0.0433 | 0.0431 | ||
| Arabinose | HMDB00646 | −5.26 | <0.001 | 0.0005 | ||
| Cofactors and vitamins | Ascorbate and aldarate metabolism | Dehydroascorbate | HMDB01264 | 1.86 | 0.0388 | 0.0393 |
| Nicotinate and nicotinamide metabolism | Nicotinamide | HMDB01406 | 1.32 | 0.0292 | 0.0323 | |
| Nicotinamide adenine dinucleotide (NAD+) | HMDB00902 | 1.45 | <0.001 | 0.0010 | ||
| Pantothenate and Coenzyme A metabolism | Pantothenate | HMDB00210 | 1.69 | <0.001 | 0.0001 | |
| Phosphopantetheine | HMDB01416 | 1.12 | 0.0459 | 0.0444 | ||
| Coenzyme A | HMDB01423 | −2.56 | <0.001 | 0.0000 | ||
| 3′-Dephosphocoenzyme A | HMDB01373 | −2.78 | <0.001 | 0.0007 | ||
| Pyridoxal metabolism | Pyridoxal | HMDB01545 | 2.17 | <0.001 | 0.0001 | |
| Riboflavin metabolism | Flavin adenine dinucleotide (FAD) | HMDB01248 | 1.42 | 0.0036 | 0.0054 | |
| Riboflavin (vitamin B2) | HMDB00244 | 1.78 | <0.001 | 0.0001 | ||
| Tocopherol metabolism | α-Tocopherol | HMDB01893 | −1.43 | <0.001 | 0.0000 | |
| Energy | Krebs cycle | Malate | HMDB00156 | 1.28 | <0.001 | 0.0002 |
| Oxidative phosphorylation | Acetylphosphate | HMDB01494 | −1.28 | 0.0788 | 0.0650 | |
| Phosphate | HMDB01429 | −1.25 | <0.001 | 0.0003 | ||
| Lipid | Medium chain fatty acid | Caproate (6:0) | HMDB00535 | 1.81 | 0.0526 | 0.0489 |
| Pelargonate (9:0) | HMDB00847 | −1.11 | <0.001 | 0.0019 | ||
| Long chain fatty acid | Oleate (18:1n9) | HMDB00207 | −1.23 | 0.0204 | 0.0247 | |
| cis-vaccenate (18:1n7) | — | −1.43 | <0.001 | 0.0006 | ||
| Arachidate (20:0) | HMDB02212 | −1.18 | 0.0658 | 0.0580 | ||
| Eicosenoate (20:1n9 or 11) | HMDB02231 | −1.89 | 0.0016 | 0.0029 | ||
| Dihomo-linoleate (20:2n6) | — | −1.49 | 0.0755 | 0.0642 | ||
| Arachidonate (20:4n6) | HMDB01043 | −1.32 | 0.0776 | 0.0650 | ||
| Fatty acid, monohydroxy | 13-HODE + 9-HODE | — | 1.93 | 0.0051 | 0.0074 | |
| Fatty acid, dicarboxylate | 2-Hydroxyglutarate | HMDB00606 | −1.69 | <0.001 | 0.0001 | |
| Eicosanoid | 15-Hydroxyeicosatetraenoic acid | HMDB02110 | 1.89 | 0.0013 | 0.0025 | |
| Endocannabinoid | Oleic ethanolamide | HMDB02088 | −1.96 | <0.001 | 0.0012 | |
| Palmitoyl ethanolamide | — | −2.22 | <0.001 | 0.0006 | ||
| Fatty acid metabolism (also BCAA metabolism) | Propionylcarnitine | HMDB00824 | 1.42 | 0.0682 | 0.0595 | |
| Carnitine metabolism | Acetylcarnitine | HMDB00201 | 1.24 | 0.0648 | 0.0577 | |
| Myristoylcarnitine | HMDB05066 | −1.56 | 0.0791 | 0.0650 | ||
| Palmitoylcarnitine | HMDB00222 | −1.75 | 0.0236 | 0.0276 | ||
| Stearoylcarnitine | HMDB00848 | −1.47 | 0.0812 | 0.0662 | ||
| Oleoylcarnitine | HMDB05065 | −2.08 | 0.0078 | 0.0106 | ||
| Glycerolipid metabolism | Choline phosphate | HMDB01565 | −1.14 | 0.0076 | 0.0104 | |
| Glycerophosphoethanolamine | HMDB00114 | 1.18 | 0.0558 | 0.0509 | ||
| Glycerol | HMDB00131 | −1.20 | 0.0164 | 0.0212 | ||
| Glycerol 3-phosphate | HMDB00126 | 1.41 | 0.0514 | 0.0482 | ||
| GPC | HMDB00086 | 2.09 | <0.001 | 0.0002 | ||
| Ketone bodies | 3-Hydroxybutyrate | HMDB00357 | 1.50 | <0.001 | 0.0007 | |
| Lysolipid | 2-Docosahexaenoyl GPE | — | −1.27 | 0.0929 | 0.0745 | |
| 2-Palmitoyl GPC | — | −1.52 | 0.0442 | 0.0435 | ||
| 1-Stearoyl GPC | — | −1.37 | 0.0754 | 0.0642 | ||
| 2-Stearoyl GPC | — | −1.45 | 0.0306 | 0.0332 | ||
| 1-Oleoyl GPC | — | −1.52 | 0.0168 | 0.0214 | ||
| 2-Oleoyl GPC | — | −1.35 | 0.0032 | 0.0054 | ||
| 1-Docosahexaenoyl GPC | — | −1.69 | 0.0263 | 0.0301 | ||
| 2-Docosahexaenoyl GPC | — | −1.61 | 0.0286 | 0.0319 | ||
| 1-Palmitoyl-GPI | — | −2.63 | 0.0015 | 0.0026 | ||
| 1-Stearoyl-GPI | — | −2.33 | 0.0314 | 0.0332 | ||
| 1-Arachidonoyl-GPI | — | −2.13 | 0.0011 | 0.0023 | ||
| Monoacylglycerol | 1-Oleoylglycerol (1-monoolein) | HMDB11567 | −2.04 | <0.001 | 0.0003 | |
| 2-Oleoylglycerol (2-monoolein) | — | −1.79 | 0.0049 | 0.0072 | ||
| Sphingolipid | Palmitoyl sphingomyelin | — | 1.60 | <0.001 | 0.0002 | |
| Stearoyl sphingomyelin | HMDB01348 | −1.39 | 0.0342 | 0.0358 | ||
| Mevalonate metabolism | 3-Hydroxy-3-methylglutarate | HMDB00355 | −2.56 | <0.001 | 0.0000 | |
| Sterol/steroid | Cholesterol | HMDB00067 | −1.15 | 0.0011 | 0.0022 | |
| 7-α-Hydroxycholesterol | HMDB01496 | 1.77 | 0.0011 | 0.0022 | ||
| 7-β-Hydroxycholesterol | HMDB06119 | 1.46 | 0.0034 | 0.0054 | ||
| 24(S)-hydroxycholesterol | HMDB01419 | −1.85 | <0.001 | 0.0000 | ||
| Nucleotide | Purine metabolism, (hypo)xanthine/inosine containing | Xanthosine | HMDB00299 | 1.25 | 0.0451 | 0.0440 |
| Inosine | — | −1.06 | 0.0368 | 0.0378 | ||
| Purine metabolism, adenine containing | Adenosine | HMDB00050 | −1.96 | 0.0033 | 0.0054 | |
| Adenosine 2′-monophosphate (2′-AMP) | HMDB11617 | 1.74 | <0.001 | 0.0016 | ||
| Adenosine 3′-monophosphate (3′-AMP) | HMDB03540 | 1.62 | 0.0035 | 0.0054 | ||
| Adenosine 5′-monophosphate (AMP) | HMDB00045 | −1.23 | 0.0198 | 0.0243 | ||
| Purine metabolism, guanine containing | Guanosine | HMDB00133 | −1.89 | <0.001 | 0.0012 | |
| Purine metabolism, urate metabolism | Urate | HMDB00289 | 1.64 | <0.001 | 0.0000 | |
| Pyrimidine metabolism, cytidine containing | Cytidine | HMDB00089 | 1.27 | <0.001 | 0.0001 | |
| 2′-Deoxycytidine | HMDB00014 | 2.49 | <0.001 | 0.0000 | ||
| Uracil | HMDB00300 | 1.54 | 0.0017 | 0.0030 | ||
| 5,6-Dihydrouracil | HMDB00076 | −1.20 | 0.0852 | 0.0689 | ||
| Peptide | Dipeptide derivative | Homocarnosine | HMDB00745 | −1.37 | <0.001 | 0.0003 |
| Anserine | HMDB00194 | 1.83 | 0.0369 | 0.0378 | ||
| γ-Glutamyl | γ-Glutamylglycine | HMDB11667 | −1.33 | 0.0197 | 0.0243 | |
| γ-Glutamylmethionine | — | 1.33 | 0.0481 | 0.0460 | ||
| γ-Glutamylglutamate | — | −1.43 | <0.001 | 0.0012 | ||
| γ-Glutamylglutamine | HMDB11738 | 1.28 | 0.0275 | 0.0311 | ||
| γ-Glutamylphenylalanine | HMDB00594 | −1.16 | 0.0969 | 0.0771 | ||
| γ-Glutamylalanine | — | 1.42 | 0.0012 | 0.0023 | ||
| Xenobiotics | Benzoate metabolism | Hippurate | HMDB00714 | 1.32 | 0.0514 | 0.0482 |
| Chemical | Glycolate (hydroxyacetate) | HMDB00115 | 1.35 | 0.0708 | 0.0612 | |
| Food component/plant | Stachydrine | HMDB04827 | 1.51 | 0.0109 | 0.0144 | |
| Homostachydrine | HMDB04827 | 1.64 | 0.0052 | 0.0074 | ||
| Sugar, sugar substitute, starch | Erythritol | HMDB02994 | −1.30 | 0.0434 | 0.0431 |
Bolded p values ≤0.05 (104 metabolites) and non-bolded p values ≤0.10 (22 metabolites) versus uninjured, age-matched controls. Welch's two-sample t test. 13-HODE + 9-HODE, 13- and 9-Hydroxyoctadecadienoic acid; BCAA, branched-chain amino acid; GPC, glycerophosphocholine; GPE, glycerophosphoethanolamine; GPI, glycerophosphoinositol; SAM, S-adenosylmethionine.
Table 2.
Altered metabolites in spinal cord of chronically injured rats treated with licofelone
| Class | Subclass | Biochemical name | HMDB ID | Fold change (SCI + licofelone/SCI + vehicle) | p value | q value |
|---|---|---|---|---|---|---|
| Amino acid | Alanine and aspartate metabolism | Aspartate | HMDB00191 | 1.10 | 0.0471 | 0.6549 |
| Carbohydrate | Aminosugars metabolism | Erythronate | HMDB00613 | 1.18 | 0.0287 | 0.7033 |
| N-acetylneuraminate | HMDB00230 | 1.22 | 0.0421 | 0.3346 | ||
| Fructose, mannose, galactose, starch, and sucrose metabolism | Maltose | HMDB00163 | 1.28 | 0.0417 | 0.5323 | |
| Glycolysis, gluconeogenesis, pyruvate metabolism | Glucose | HMDB00122 | 1.27 | 0.0633 | 0.6236 | |
| Lactate | HMDB00190 | 1.11 | 0.0223 | 0.7266 | ||
| Nucleotide sugars, pentose metabolism | Arabitol | HMDB01851 | 1.59 | 0.0030 | 0.6236 | |
| Ribose | HMDB00283 | 1.47 | 0.0836 | 0.7033 | ||
| Energy | Krebs cycle | Citrate | HMDB00094 | 1.16 | 0.0877 | 0.4537 |
| Lipid | Long chain fatty acid | Margarate (17:0) | HMDB02259 | 1.22 | 0.0077 | 0.3346 |
| Cis-vaccenate (18:1n7) | — | 1.26 | 0.0183 | 0.7266 | ||
| Behenate (22:0) | HMDB00944 | 1.20 | 0.0615 | 0.7266 | ||
| Eicosanoid | 15-Hydroxyeicosatetraenoic acid | HMDB02110 | 1.42 | 0.0328 | 0.3346 | |
| Bile acid metabolism | Taurocholate | HMDB00036 | −2.44 | 0.0625 | 0.4537 | |
| Glycerolipid metabolism | Glycerol | HMDB00131 | 1.13 | 0.0090 | 0.7033 | |
| Inositol metabolism | Myo-inositol | HMDB00211 | 1.20 | 0.0704 | 0.7266 | |
| Chiro-inositol | — | 1.51 | 0.0224 | 0.5614 | ||
| Inositol 1-phosphate | HMDB00213 | 1.22 | 0.0163 | 0.7033 | ||
| Sterol/steroid | Cholesterol | HMDB00067 | 1.06 | 0.0081 | 0.3346 | |
| 7-α-Hydroxycholesterol | HMDB01496 | 1.26 | 0.0209 | 0.7164 | ||
| 7-β-Hydroxycholesterol | HMDB06119 | 1.19 | 0.0930 | 0.4537 | ||
| 24(S)-Hydroxycholesterol | HMDB01419 | 1.25 | 0.0013 | 0.4537 | ||
| Nucleotide | Purine metabolism, urate metabolism | Allantoin | HMDB00462 | 1.20 | 0.0709 | 0.7266 |
| Peptide | Dipeptide | Glycylglycine | HMDB11733 | 1.24 | 0.0625 | 0.3346 |
| Dipeptide derivative | Homocarnosine | HMDB00745 | 1.14 | 0.0071 | 0.4537 | |
| Xenobiotics | Sugar, sugar substitute, starch | Erythritol | HMDB02994 | 1.30 | 0.0755 | 0.7266 |
Bolded p values ≤0.05 (16 metabolites) and non-bolded p values ≤0.10 (10 metabolites) versus chronic SCI, vehicle-treated group. Welch's two-sample t test.
Statistical analysis.
Missing values (if any) were assumed to be below the level of detection. However, biochemicals detected in all samples from one or more groups but not in samples from other groups were assumed to be near the lower limit of detection in the groups in which they were not detected. In this case, the lowest detected level of these biochemicals was imputed for samples in which that biochemical was not detected. Following log2 transformation and imputation with minimum observed values for each compound, Welch's two-sample t test was used to identify biochemicals that differed significantly between experimental groups. An estimate of the false discovery rate (q value) was calculated to take into account the multiple comparisons that normally occur in metabolomic-based studies (Storey and Tibshirani, 2003); as q values were reasonable for p ≤ 0.05, no q value cutoff was established for this study. Outliers were defined as 1.5* (interquartile range) and removed from analyses. Heat maps of metabolomic data were generated using the open-source software package MultiExperiment Viewer version 4.7.3, part of the TM4 Microarray Software Suite (Dana-Farber Cancer Institute; www.tm4.org/mev/) (Saeed et al., 2006). Scaled, imputed log2-transformed data were used to generate heat maps and to perform clustering analysis. Pearson distances (1 − r, where r is the Pearson correlation coefficient) were used as the pairwise distance between individual replicates for hierarchical clustering by biochemical. Pathways were assigned for each metabolite, allowing examination of over-represented pathways. Network analysis was performed using Ingenuity IPA (Ingenuity Systems; www.ingenuity.com); the statistical likelihood of association between a set of metabolites in the dataset and a related biological function (e.g., lipid peroxidation) were calculated by the Fisher's exact test.
Statistical analysis
All data except metabolomics data were analyzed using SigmaPlot 11 (Systat Software). For all behavioral assessments, post-treatment scores were compared with pretreatment scores on an individual, animal-by-animal basis. Values of p < 0.05 were considered significant. All group data are presented as mean ± SEM.
Results
Chronically injured spinal cord tissue contains elevated levels of PGs and LTs
The PGs and LTs are potent pro-inflammatory mediators that contribute to SCI pathophysiology in the acute phase of injury (Sharma et al., 1993; Genovese et al., 2008; Saiwai et al., 2010), but whether they are present in the chronic phase of injury has not previously been investigated. We performed ELISA to detect presence of the major AA-derived mediators, PGE2 and LTB4, within the spinal cord lesion site of rats 9 months post-SCI. PGE2 was undetectable in uninjured, age-matched controls but significantly elevated in the lesion site of chronically injured animals (87.1 ± 4.91 pg/ml, p < 0.001; Fig. 1A). Likewise, LTB4 was undetectable in controls but significantly elevated within chronic SCI tissue (10.6 ± 1.67 pg/ml, p ≤ 0.001, Fig. 1B).
Figure 1.

Levels of PGs and LTs are elevated in the lesion site of spinal cords 9 months following SCI. PGE2 levels (A, n = 3 per group) and LTB4 levels (B, n = 6 per group) in T10 spinal cord segments of age-matched, uninjured controls (undetectable) and rats 9 months post-SCI (black bars). *p < 0.001, Student's t test. All data are mean ± SEM.
Metabolomic profiling reveals alterations in several metabolic pathways within the chronic spinal cord lesion site
Metabolomics is an analytical method for the identification and quantification of endogenous biochemical compounds within biological samples; it is a useful tool to identify biomarkers and/or metabolic pathways associated with disease or drug treatment (Baker, 2011). To characterize metabolic alterations in the chronic phase of SCI, we performed metabolomic screens on injured spinal cord tissue of vehicle-treated rats 9 months postinjury as well as uninjured, age-matched control rats; 257 named metabolite compounds were detected and identified across all samples. Of these, 126 compounds were significantly (p ≤ 0.05) or near significantly (0.05 < p ≤ 0.10) increased or decreased in chronic SCI (Table 1). These metabolites belonged to diverse molecular classes such as amino acids, carbohydrates, cofactors and vitamins, nucleotides, and lipids. Hierarchical clustering analysis revealed multiple groups of biochemicals that clustered together in chronic SCI samples (Fig. 2). For example, we detected one cluster of glycolytic intermediates that exhibited strong, correlative increases with SCI (Fig. 2, r = 0.88, p < 0.05). We also observed two large clusters of downregulated lipid metabolites that were decreased in the SCI group (Fig. 2, r = 0.72, p < 0.05; Fig. 2, r = 0.53, p < 0.05). Network analysis generated a list of predicted biological functions and/or cellular processes that are likely to be associated with the chronic SCI group based on the metabolic alterations in our dataset; these included lipid peroxidation (p < 0.001), synthesis and release of eicosanoid (p < 0.001), mitochondrial dysfunction (p < 0.001), damage of brain tissue (p < 0.001), and neurodegeneration (p = 0.003).
Figure 2.

Metabolic alterations in the spinal cord lesion site 9 months post-SCI. Heat map illustrates the relative change in levels of 126 metabolites with increased (yellow) or decreased (blue) expression in chronic SCI, vehicle-treated animals (n = 8) versus uninjured, age-matched controls (n = 9). Changes in concentration, relative to the median metabolite level, are indicated by intensity of color (see color scale, upper right). Metabolites are arranged by hierarchical clustering, with representative clusters of metabolites highlighted (A–C). Scale bar (upper left) indicates Pearson correlation coefficient (r). Colored bars (right) indicate super class of each metabolite (amino acids, carbohydrates, cofactors and vitamins, energy metabolites, lipids, nucleotides, peptides, and xenobiotics).
A closer appraisal of the metabolites with increased expression in chronic SCI revealed that several were strong indicators of oxidative stress (Fig. 2, pink lettering). These included oxidized glutathione (+3.19-fold, p < 0.001), cysteine-glutathione disulfide (+2.27-fold, p < 0.001), dehydroascorbate (+1.86-fold, p < 0.05), and 13- and 9-hydroxyoctadecadienoic acid (+1.93-fold, p < 0.05; Fig. 3A). These metabolites are major indicators of reactive oxygen species (ROS)-induced cellular damage (Eriksson and Eriksson, 1967; Schafer and Buettner, 2001; Foyer and Noctor, 2011) and lipid peroxidation (Yoshida et al., 2005). Additionally, levels of several anti-oxidant and anti-inflammatory metabolites were significantly depleted in chronically injured tissue (Fig. 2, green lettering). For example, SCI samples contained decreased levels of the antioxidant α-tocopherol (−1.43-fold, p < 0.05; Fig. 3B), which protects the CNS against pathological lipid peroxidation (Adibhatla and Hatcher, 2010). Levels of the anti-inflammatory endocannabinoids palmitoyl ethanolamide (−2.22-fold, p < 0.001) and oleic ethanolamide (−1.96-fold, p < 0.001) were significantly decreased in chronic SCI. Also decreased was the compound 24(S)-hydroxycholesterol (24-OH-Chol) (−1.85-fold, p < 0.05, Fig. 3B). 24-OH-Chol induces activation of genes through the liver X nuclear receptor (LXR) (Cartagena et al., 2008), which is both anti-inflammatory and neuroprotective (Morales et al., 2008; Cheng et al., 2010).
Figure 3.

Biochemical markers of oxidative stress and inflammation in the chronically injured spinal cord. Box plots illustrate changes in abundance of select metabolites in spinal cord tissue of uninjured, age-matched controls (white, n = 9) and chronically injured animals (gray, n = 8). For each metabolite, concentration values have been rescaled so that samples across all groups have a median value equal to 1. Box legend: horizontal bar inside box represents group median; upper and lower box boundaries represent 75th percentile and 25th percentile, respectively; upper and lower whiskers represent maximum and minimum of distribution; open circles represent extreme data points. A, Several metabolites significantly increased in chronic SCI spinal cords are associated with oxidative stress. B, Levels of anti-inflammatory and anti-oxidative metabolites are decreased in chronic SCI tissue. *p < 0.05, Welch's two-sample t test. 13- + 9-HODE, 13- and 9-hydroxyoctadecadienoic acid; 24-OH-Chol, 24(S)-hydroxycholesterol; α-Toco, α-tocopherol; Cys-S-SG, cysteine-glutathione disulfide; DHA, dehydroascorbate; GSSG, oxidized glutathione; OEA, oleic ethanolamide; PEA, palmitoyl ethanolamide.
Licofelone enhances levels of endogenous anti-oxidative and anti-inflammatory metabolites in the chronically injured spinal cord
We have demonstrated that levels of PGE2 and LTB4 (Fig. 1), as well as other biochemical markers of inflammation and oxidative stress (Table 1, Fig. 3), are elevated in the chronic spinal cord lesion site. Licofelone is a dual COX/5-LOX inhibitor that possesses potent anti-inflammatory properties (Singh et al., 2005, 2006; Wu et al., 2012). We therefore examined the ability of licofelone treatment to combat inflammation and oxidative stress in the chronically injured spinal cord.
We assessed whether licofelone could attenuate production of PGs and LTs within the injured spinal cord. Animals were treated orally with either vehicle or licofelone, once at 3 h post-SCI and again at 18 h post-SCI, and levels of PGE2 and LTB4 in the lesion site were quantified by ELISA at 24 h post-SCI. We detected a significant increase in PGE2 levels at 24 h post-SCI (uninjured = 43.2 ± 4.67 pg/ml, n = 6; SCI + vehicle = 405 ± 47.4 pg/ml, n = 6; p < 0.001). Notably, this SCI-induced increase was completely abolished by licofelone treatment (SCI + licofelone = 16.9 ± 6.87 pg/ml, n = 6; p < 0.001 versus injured, vehicle-treated). However, we failed to detect LTB4 either at 24 h post-SCI (n = 12) or in uninjured controls (n = 6), likely due to the sensitivity of our assay. Thus, we were unable to evaluate the effect of licofelone on LT production within the injured spinal cord.
We next assessed the effects of prolonged licofelone treatment on the metabolomic profile of the chronically injured spinal cord. We performed a metabolomic screen on T10 spinal cord tissue from animals treated with oral licofelone for 1 month, beginning at 8 months post-SCI. Of the 257 metabolites detected and identified across all experimental groups, 26 compounds were either increased or decreased in spinal cord tissue of animals treated with licofelone, compared with vehicle-treated animals (Table 2). Network analysis indicated that biological functions likely to be altered included re-esterification of lipid (p < 0.001), uptake of amino acids (p < 0.001), synthesis of cholesterol (p < 0.001), and efflux of cholesterol (p < 0.001). Hierarchical clustering revealed clusters of biochemicals, primarily lipids and carbohydrates, increased by licofelone treatment (Fig. 4A). These included several anti-inflammatory and anti-oxidative compounds, including 15-hydroxyeicosatetraenoic acid (15-HETE, +1.42-fold, p < 0.05), 24-OH-Chol (+1.25-fold, p < 0.05), chiro-inositol (+1.51-fold, p < 0.05), inositol 1-phosphate (+1.22-fold, p < 0.05), and homocarnosine (+1.14-fold, p < 0.05, Fig. 4B). Interestingly, the only metabolite that decreased with licofelone treatment (−2.44-fold, p = 0.062) was the pro-inflammatory bile acid taurocholate (Table 2, Fig. 4A).
Figure 4.
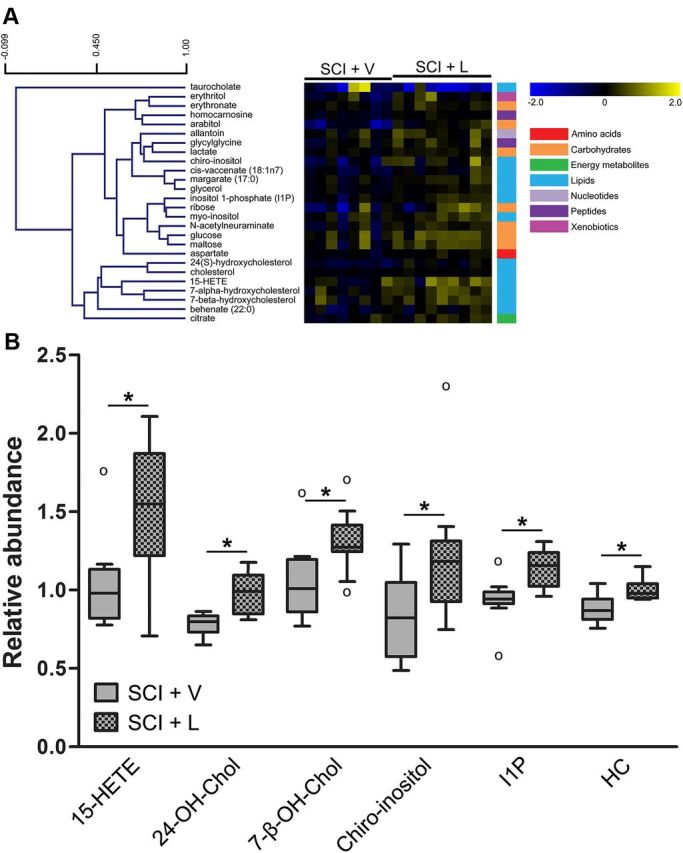
Changes in the metabolomic profile of the chronically injured spinal cord with licofelone treatment. A, Heat map showing 26 metabolites with altered concentrations in chronic SCI, licofelone-treated animals (SCI + L, n = 9), compared with chronic SCI, vehicle-treated animals (SCI + V, n = 8). Metabolites are arranged by hierarchical clustering. Scale bar (upper left) indicates Pearson correlation coefficient (r). Colored bars (right) indicate super class of each metabolite (e.g., amino acids, carbohydrates). Increases (yellow) and decreases (blue) in concentration, relative to the median metabolite level, are indicated by intensity of color (see color scale). B, Box plots illustrating changes in levels of select compounds within the chronic SCI, vehicle-treated (gray) or licofelone-treated (checkered) groups. For box legend, see Figure 3. *p < 0.05, Welch's two-sample t test. 15-HETE, 15-hydroxyeicosatetraenoic acid; 24-OH-Chol, 24(S)-hydroxycholesterol; 7-β-OH-Chol, 7-β-hydroxycholesterol; HC, homocarnosine; I1P, inositol 1-phosphate.
Licofelone reduces mechanical hypersensitivity in rats with chronic SCI
Inflammation is a known modulator of neurosensory function in the acute phase of SCI (Zhao et al., 2007); however, it is not known whether chronic neurosensory alterations are modulated by inflammation. Because we have detected elevated levels of PGE2, LTB4, and other biomarkers of inflammation in the chronic phase of injury, we hypothesized that treatment with licofelone would attenuate neurosensory alterations modulated by inflammation in chronic SCI.
Nesic et al. (2005) have demonstrated that rats up to 9 months following SCI exhibit mechanical hypersensitivity of the hindpaws. We used von Frey hair testing to assess mechanical sensitivity of hindpaws in chronically injured (8 months post-SCI) rats (Fig. 5). Average pretreatment withdrawal thresholds were 5.33 ± 0.94 g for vehicle-treated animals, and 4.09 ± 1.46 g for licofelone-treated animals. Group mean withdrawal scores of injured animals were lower than that of uninjured, age-matched controls (7.59 ± 1.01 g), confirming the presence of mechanical hypersensitivity in these chronically injured animals. Because we observed a considerable extent of interanimal variability in mechanical withdrawal thresholds, we did not compare group means in our analysis; rather, we examined the effects of licofelone on mechanical sensitivity by comparing pretreatment scores to post-treatment scores of individual animals. On the last day of the 28 d treatment period, hindlimb withdrawal thresholds of the licofelone-treated group were significantly increased compared with pretreatment measurements (6.27 ± 1.43 g, p < 0.001, r = 0.955), but there was no significant change in scores of vehicle-treated animals (5.02 ± 1.09 g, p = 0.561, r = 0.884), indicating that licofelone attenuated mechanical sensitivity in chronically injured animals.
Figure 5.

Licofelone attenuates mechanical sensitivity of hindpaws in chronic SCI. Hindpaw mechanical withdrawal thresholds (g) of chronic SCI animals before treatment (Pre) and following 28 d of treatment (Post) with vehicle (n = 8) or licofelone (n = 9). Data points connected by lines represent pretreatment and post-treatment scores for individual subjects. Paired Student's t test.
To assess whether licofelone treatment could also modulate thermal sensitivity, we measured hindpaw withdrawal latency from a thermal stimulus. At 8 months post-SCI, withdrawal latency was lower in injured rats (SCI + V = 4.50 ± 0.20 s; SCI + L = 4.26 ± 0.35 s) than in uninjured, age-matched animals (Uninjured = 5.79 ± 0.48 s) indicating that chronically injured rats were hypersensitive to thermal stimulation (Fig. 6). On day 28 of treatment, there was no significant difference from pretreatment values in vehicle-treated rats (4.78 ± 0.30 s, p = 0.540, r = −0.462) or licofelone-treated rats (4.87 ± 0.44 s, p = 0.431, r = −0.598). Thus, licofelone treatment did not affect thermal sensitivity of hindpaws in chronically injured rats.
Figure 6.

Licofelone does not affect thermal hypersensitivity of hindpaws in chronic SCI. Hindpaw thermal withdrawal latency (s) of chronic SCI animals before treatment (Pre) and following 28 d of treatment (Post) with vehicle (triangles, n = 8) or licofelone (circles, n = 9). Connected data points represent pretreatment and post-treatment scores for individual subjects. Paired Student's t test.
To assess whether licofelone treatment enhanced hindlimb locomotor function, BBB testing was performed weekly throughout the treatment period. We observed no significant difference in BBB scores between SCI groups at any point during treatment; furthermore, neither group experienced any change in BBB scores over time (Fig. 7). Thus, extended licofelone treatment did not affect hindlimb locomotor function of chronically injured animals.
Figure 7.

Licofelone does not affect hindlimb locomotor function in chronic SCI rats. BBB hindlimb locomotor scores beginning at 8 months post-SCI, before treatment (Pre) and weekly throughout the treatment period. Uninjured, uninjured, age-matched controls (n = 9); SCI + V, chronic SCI, vehicle-treated (n = 8); SCI + L, chronic SCI, licofelone-treated (n = 9). All data are mean ± SEM.
Discussion
We have provided novel evidence of pro-inflammatory AA metabolism and oxidative stress within the chronically injured spinal cord. The presence of LTB4 and PGE2 strongly indicates that the inflammatory response is not confined to the earlier, acute phase of SCI, but that neuroinflammation is also present in the chronic phase. Furthermore, using metabolomic profiling, we have shown elevations in oxidative stress markers and depletion of endogenous anti-oxidative and anti-inflammatory compounds within chronically injured tissue. These are significant findings, due to the existing deficiency in knowledge of the molecular environment within the chronically injured spinal cord. Our observations may challenge our understanding of the pathological mechanisms that shape the myriad adverse conditions, including pathologically altered neurosensory function, associated with the chronic phase of SCI (Finnerup et al., 2001).
This is the first study using metabolomic profiling of chronically injured spinal cord tissue. Metabolomics is a relatively new “omics” modality for identification and quantification of biochemical compounds that are universal among organisms, unlike transcripts or proteins. Metabolomic screens are powerful tools to identify biomarkers of disease and/or drug treatment, as the metabolome is a rapid, sensitive indicator of perturbations in the environment (Kaddurah-Daouk et al., 2008). Metabolomics data can be integrated with existing transcriptomics and proteomics datasets for a more comprehensive understanding of complex biological systems (Dunn et al., 2011). However, compound identification in MS-based metabolite detection can be obfuscated by limitations in spatial peak resolution. Furthermore, metabolite databases are not currently comprehensive, and chemical standards for identification are in many cases unavailable (Dunn et al., 2011); hence, metabolomics remains an emerging technique and is not yet a fully comprehensive analytical tool.
Fujieda et al. (2012) recently published metabolomic profiles of spinal cord tissue from 2 to 30 d following thoracic spinal contusion injury. Comparison of this study with our results reveals a number of metabolic differences between 1 and 9 months postinjury. Interestingly, they reported an increase in PGE2 levels (vs shams) at 2 d post-SCI that disappeared by 30 d; in light of the dramatic increases in PGE2 that we have detected by ELISA at both 24 h and 9 months, this may indicate that PG synthesis is a dynamic, multiphasic process that peaks early after injury and undergoes a secondary increase in the chronic phase. (PGE2 was not among the metabolites identified in our metabolomics screen, perhaps due to differences in detection sensitivity between the studies.) Fujieda et al. (2012) also detected significantly increased levels of anti-oxidant compounds (ascorbate and α-tocopherol) and anti-inflammatory endocannabinoids (oleic- and palmitoyl ethanolamide) 30 d post-SCI, all of which were decreased in our study; this may indicate improved anti-oxidant/anti-inflammatory defenses in the earlier weeks post-SCI that diminish with time. Curiously, injury-associated changes in neurotransmitter levels at 30 d post-SCI (glutamate, −1.15-fold; N-acetyl-aspartyl-glutamate, −1.35-fold; GABA, −1.28-fold; aspartate, −1.10-fold; and N-acetylaspartate, −1.85-fold) closely mirror our own observations, suggesting that unlike other pathways, neurotransmitter biosynthesis remains relatively stable as the injury becomes more chronic. Though the injury models differed slightly, comparison of these two datasets suggests that activity of several metabolic pathways may evolve and change over time; if this is true, one month post-SCI does not necessarily represent a “plateau” phase, as the authors of the Fujieda study suggest.
Compared with previous transcriptional profiling studies, our study provides a new layer of insight into the biochemical alterations in chronic SCI. There is upregulation of genes associated with oxidative stress (metallothioneins I and II) and inflammation (IL-6, IL-1β, and TGF-β) at 5 weeks post-SCI (Aimone et al., 2004). At 1–3 months post-SCI, expression of glutathione peroxidase and glutathione S-transferase is increased, suggesting heightened degradation and metabolism of ROS (Velardo et al., 2004). Additionally, Byrnes et al. (2011) recently reported increased expression and activity of NADPH oxidase in spinal microglia/macrophages 6 months postinjury. We have expanded on this limited body of knowledge by showing chronic alterations in multiple pathways associated with oxidative stress, inflammation, and lipid peroxidation. These results demonstrate an ongoing, dynamic inflammatory response in the injured spinal cord, suggesting that the functional consequences of neuroinflammation may extend further into the chronic phase of injury than previously thought. The possible contributions of sustained inflammatory and pro-oxidative signaling pathways to long-term deficits associated with chronic SCI may be of great biological and clinical significance.
We have focused this report on oxidative stress and inflammation; however, our metabolomics data reveals chronic alterations in multiple biological pathways, e.g., glycolysis, neurotransmission, cholesterol metabolism, and amino acid biosynthesis. One particularly striking finding is that a large number of lipid metabolites are decreased in chronic SCI. In this environment of enhanced inflammation and oxidative stress, we speculate that a decreased ability to replenish endogenous lipids may further diminish the spinal cord's ability to undergo repair and/or regeneration, negatively affecting such key issues as cell membrane integrity, myelination/remyelination, and neuronal plasticity, to name only a few. Our metabolomics data may provide important insights for future studies of chronic pathological mechanisms and reveal new therapeutic targets for treatment of chronic SCI. Furthermore, integration of our dataset with other “omics” studies at different time points after injury will allow us to develop a greater understanding of how pathologically altered pathways evolve as spinal cord injuries become more chronic.
Because we have observed elevated levels of PGE2 and LTB4 in the chronic lesion site, we assessed whether treating chronically injured rats with the COX/5-LOX inhibitor licofelone (Cicero and Laghi, 2007) could attenuate neuroinflammation. Though licofelone completely abolished the spike in PGE2 production 24 h post-SCI, we failed to detect LTB4 at this time point. This is likely not due to a lack of LTB4 in the spinal cord, because Xu et al. (1990) have reported elevated LTB4 levels 24 h post-SCI. However, their methods of LTB4 purification and quantification by HPLC were likely more sensitive than the ELISA used here. The effect of licofelone treatment on spinal cord LTB4 production is currently being investigated further in our laboratory. Regardless, our metabolomics data show that licofelone treatment upregulates endogenous anti-inflammatory/anti-oxidative compounds within the chronically injured spinal cord. The precise mechanism by which this occurs is unclear, as AA metabolism is a complex cascade with a plethora of molecular targets. Based on the broad range of metabolic pathways affected, it is possible that licofelone may activate a “master regulator” of the antioxidant response such as Nrf2, which induces transcription of multiple protective anti-oxidant genes (Nguyen et al., 2009). Characterizing the molecular targets of licofelone will require more mechanistic investigations.
The animals in our study exhibited hypersensitivity of the hindpaws to mechanical stimulation, consistent with other chronic studies (Nesic et al., 2005). Licofelone attenuated mechanical, but not thermal, hypersensitivity, suggesting that licofelone specifically modulates mechanisms unique to processing of mechanical stimuli (Caterina et al., 2000). How this occurs is an important topic for future investigation. We have detected neurons within gray matter adjacent to the border of the lesion cavity in chronically injured animals (n = 3, unpublished data). Thus, it is possible that licofelone may attenuate mechanical sensitivity via direct modulation of metabolites in the lesion site and their effects on lesion site neurons. It is also possible that licofelone may act via mechanisms remote from the site of injury. For example, LTs (Okubo et al., 2010) and PGs (Lin et al., 2006) have been shown to modulate mechanical sensitivity at the level of the dorsal root ganglia (DRG). DRG neurons exhibit chronic SCI-associated alterations associated with mechanical sensitivity (Bedi et al., 2010); hence, it is possible that licofelone may attenuate hypersensitivity via an attenuation of PGs/LTs in DRG neurons. Alternatively, licofelone might modulate sensory processing in spinal cord regions remote from the epicenter of injury. Previous work has demonstrated a strong link between below-level PGE2-dependent microglial activation and increased sensitivity to mechanical stimuli (Hains et al., 2003; Hains and Waxman, 2006; Zhao et al., 2007). Thus, inhibition of PGE2 production by licofelone might serve to reduce below-level hyperexcitability. Further investigation will certainly be necessary to understand the effects of licofelone treatment on both peripheral and central components following SCI.
Chronic neuropathic pain plagues a large percentage of SCI patients (Baastrup and Finnerup, 2008). We have observed a pathological sensory alteration that might be attributable to pain. However, we have only performed limited neurosensory assessments and thus cannot make claims about the effect of licofelone on allodynia or other measures of pain. Previous studies have demonstrated that the PGs (Zhao et al., 2007) and LTs (Noguchi and Okubo, 2011) modulate spinally mediated mechanisms of neuropathic pain. We demonstrated that licofelone reduces PGE2 levels and attenuates mechanical hypersensitivity in chronic SCI; however, further studies are needed to evaluate whether these relate to SCI-associated pain.
In summary, we have shown that inflammation and oxidative stress persist within the injured spinal cord long after the initial insult, that inflammation modulates sensory processing in this chronic phase of injury, and that mechanical hypersensitivity can be attenuated with anti-inflammatory therapy. This study provides valuable new information about the chronically injured spinal cord that may help to illuminate the molecular mechanisms of sensory alterations experienced by patients with chronic injuries.
Footnotes
This work is supported by grants from the National Institutes of Health (NIH) (NS 49409 to R.J.G., NS 44174 and MH 70054 to H.W.S.); Paralyzed Veterans of America (#2511 to R.J.G.); The Gillson–Longenbaugh Foundation (R.J.G.); Mission Connect, a project of The Institute for Rehabilitation and Research Foundation (R.J.G.); and the University of Texas Center for Clinical and Translational Sciences, funded by NIH Clinical and Translational Award (TL1 RR024147 to J.N.D.) from the National Center for Research Resources (NCRR). The content of this manuscript is solely the responsibility of the authors and does not necessarily represent the views of the NCRR or the NIH. We gratefully acknowledge Drs. Robyn Crook and Olivera Nesic-Taylor for assistance with behavioral analysis.
The authors declare no competing financial interests.
References
- Adibhatla RM, Hatcher JF. Lipid oxidation and peroxidation in CNS health and disease: from molecular mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2010;12:125–169. doi: 10.1089/ars.2009.2668. [DOI] [PubMed] [Google Scholar]
- Aimone JB, Leasure JL, Perreau VM, Thallmair M. Spatial and temporal gene expression profiling of the contused rat spinal cord. Exp Neurol. 2004;189:204–221. doi: 10.1016/j.expneurol.2004.05.042. [DOI] [PubMed] [Google Scholar]
- Alexander JK, Popovich PG. Neuroinflammation in spinal cord injury: therapeutic targets for neuroprotection and regeneration. Prog Brain Res. 2009;175:125–137. doi: 10.1016/S0079-6123(09)17508-8. [DOI] [PubMed] [Google Scholar]
- Baastrup C, Finnerup NB. Pharmacological management of neuropathic pain following spinal cord injury. CNS Drugs. 2008;22:455–475. doi: 10.2165/00023210-200822060-00002. [DOI] [PubMed] [Google Scholar]
- Baker M. Metabolomics: from small molecules to big ideas. Nat Methods. 2011;8:117–121. [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- Beck KD, Nguyen HX, Galvan MD, Salazar DL, Woodruff TM, Anderson AJ. Quantitative analysis of cellular inflammation after traumatic spinal cord injury: evidence for a multiphasic inflammatory response in the acute to chronic environment. Brain. 2010;133:433–447. doi: 10.1093/brain/awp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedi SS, Yang Q, Crook RJ, Du J, Wu Z, Fishman HM, Grill RJ, Carlton SM, Walters ET. Chronic spontaneous activity generated in the somata of primary nociceptors is associated with pain-related behavior after spinal cord injury. J Neurosci. 2010;30:14870–14882. doi: 10.1523/JNEUROSCI.2428-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz LI, Popovich PG. Inflammation and axon regeneration. Curr Opin Neurol. 2011;24:577–583. doi: 10.1097/WCO.0b013e32834c208d. [DOI] [PubMed] [Google Scholar]
- Buczynski MW, Svensson CI, Dumlao DS, Fitzsimmons BL, Shim JH, Scherbart TJ, Jacobsen FE, Hua XY, Yaksh TL, Dennis EA. Inflammatory hyperalgesia induces essential bioactive lipid production in the spinal cord. J Neurochem. 2010;114:981–993. doi: 10.1111/j.1471-4159.2010.06815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrnes KR, Washington PM, Knoblach SM, Hoffman E, Faden AI. Delayed inflammatory mRNA and protein expression after spinal cord injury. J Neuroinflammation. 2011;8:130. doi: 10.1186/1742-2094-8-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton SM, Du J, Tan HY, Nesic O, Hargett GL, Bopp AC, Yamani A, Lin Q, Willis WD, Hulsebosch CE. Peripheral and central sensitization in remote spinal cord regions contribute to central neuropathic pain after spinal cord injury. Pain. 2009;147:265–276. doi: 10.1016/j.pain.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartagena CM, Ahmed F, Burns MP, Pajoohesh-Ganji A, Pak DT, Faden AI, Rebeck GW. Cortical injury increases cholesterol 24S hydroxylase (Cyp46) levels in the rat brain. J Neurotrauma. 2008;25:1087–1098. doi: 10.1089/neu.2007.0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Cheng O, Ostrowski RP, Liu W, Zhang JH. Activation of liver X receptor reduces global ischemic brain injury by reduction of nuclear factor-kappaB. Neuroscience. 2010;166:1101–1109. doi: 10.1016/j.neuroscience.2010.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicero AF, Laghi L. Activity and potential role of licofelone in the management of osteoarthritis. Clin Interv Aging. 2007;2:73–79. doi: 10.2147/ciia.2007.2.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehaven CD, Evans AM, Dai H, Lawton KA. Organization of GC/MS and LC/MS metabolomics data into chemical libraries. J Cheminform. 2010;2:9. doi: 10.1186/1758-2946-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon WJ. Staircase bioassay: the up-and-down method. Neurosci Biobehav Rev. 1991;15:47–50. doi: 10.1016/s0149-7634(05)80090-9. [DOI] [PubMed] [Google Scholar]
- Dulin JN, Moore ML, Gates KW, Queen JH, Grill RJ. Spinal cord injury causes sustained disruption of the blood-testis barrier in the rat. PLoS One. 2011;6:e16456. doi: 10.1371/journal.pone.0016456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn WB, Broadhurst DI, Atherton HJ, Goodacre R, Griffin JL. Systems level studies of mammalian metabolomes: the roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem Soc Rev. 2011;40:387–426. doi: 10.1039/b906712b. [DOI] [PubMed] [Google Scholar]
- Eriksson B, Eriksson SA. Synthesis and characterization of the L-cysteine-glutathione mixed disulfide. Acta Chem Scand. 1967;21:1304–1312. doi: 10.3891/acta.chem.scand.21-1304. [DOI] [PubMed] [Google Scholar]
- Farooqui AA, Horrocks LA, Farooqui T. Modulation of inflammation in brain: a matter of fat. J Neurochem. 2007;101:577–599. doi: 10.1111/j.1471-4159.2006.04371.x. [DOI] [PubMed] [Google Scholar]
- Finnerup NB, Johannesen IL, Sindrup SH, Bach FW, Jensen TS. Pain and dysesthesia in patients with spinal cord injury: a postal survey. Spinal Cord. 2001;39:256–262. doi: 10.1038/sj.sc.3101161. [DOI] [PubMed] [Google Scholar]
- Fitch MT, Doller C, Combs CK, Landreth GE, Silver J. Cellular and molecular mechanisms of glial scarring and progressive cavitation: in vivo and in vitro analysis of inflammation-induced secondary injury after CNS trauma. J Neurosci. 1999;19:8182–8198. doi: 10.1523/JNEUROSCI.19-19-08182.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming JC, Norenberg MD, Ramsay DA, Dekaban GA, Marcillo AE, Saenz AD, Pasquale-Styles M, Dietrich WD, Weaver LC. The cellular inflammatory response in human spinal cords after injury. Brain. 2006;129:3249–3269. doi: 10.1093/brain/awl296. [DOI] [PubMed] [Google Scholar]
- Foyer CH, Noctor G. Ascorbate and glutathione: the heart of the redox hub. Plant Physiol. 2011;155:2–18. doi: 10.1104/pp.110.167569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujieda Y, Ueno S, Ogino R, Kuroda M, Jönsson TJ, Guo L, Bamba T, Fukusaki E. Metabolite profiles correlate closely with neurobehavioral function in experimental spinal cord injury in rats. PLoS One. 2012;7:e43152. doi: 10.1371/journal.pone.0043152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese T, Mazzon E, Rossi A, Di Paola R, Cannavò G, Muià C, Crisafulli C, Bramanti P, Sautebin L, Cuzzocrea S. Involvement of 5-lipoxygenase in spinal cord injury. J Neuroimmunol. 2005;166:55–64. doi: 10.1016/j.jneuroim.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Genovese T, Rossi A, Mazzon E, Di Paola R, Muià C, Caminiti R, Bramanti P, Sautebin L, Cuzzocrea S. Effects of zileuton and montelukast in mouse experimental spinal cord injury. Br J Pharmacol. 2008;153:568–582. doi: 10.1038/sj.bjp.0707577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26:4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Yucra JA, Hulsebosch CE. Reduction of pathological and behavioral deficits following spinal cord contusion injury with the selective cyclooxygenase-2 inhibitor NS-398. J Neurotrauma. 2001;18:409–423. doi: 10.1089/089771501750170994. [DOI] [PubMed] [Google Scholar]
- Hains BC, Klein JP, Saab CY, Craner MJ, Black JA, Waxman SG. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J Neurosci. 2003;23:8881–8892. doi: 10.1523/JNEUROSCI.23-26-08881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Herrera JJ, Haywood-Watson RJ, 2nd, Grill RJ. Acute and chronic deficits in the urinary bladder after spinal contusion injury in the adult rat. J Neurotrauma. 2010;27:423–431. doi: 10.1089/neu.2009.0997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Bhavsar A, Ward RE, Hall JC, Priestley JV, Michael-Titus AT. Arachidonyl trifluoromethyl ketone is neuroprotective after spinal cord injury. J Neurotrauma. 2009;26:1429–1434. doi: 10.1089/neu.2008.0835. [DOI] [PubMed] [Google Scholar]
- Hulsebosch CE, Hains BC, Crown ED, Carlton SM. Mechanisms of chronic central neuropathic pain after spinal cord injury. Brain Res Rev. 2009;60:202–213. doi: 10.1016/j.brainresrev.2008.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaddurah-Daouk R, Kristal BS, Weinshilboum RM. Metabolomics: a global biochemical approach to drug response and disease. Annu Rev Pharmacol Toxicol. 2008;48:653–683. doi: 10.1146/annurev.pharmtox.48.113006.094715. [DOI] [PubMed] [Google Scholar]
- Kwon BK, Okon E, Hillyer J, Mann C, Baptiste D, Weaver LC, Fehlings MG, Tetzlaff W. A systematic review of non-invasive pharmacologic neuroprotective treatments for acute spinal cord injury. J Neurotrauma. 2011;28:1545–1588. doi: 10.1089/neu.2009.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CR, Amaya F, Barrett L, Wang H, Takada J, Samad TA, Woolf CJ. Prostaglandin E2 receptor EP4 contributes to inflammatory pain hypersensitivity. J Pharmacol Exp Ther. 2006;319:1096–1103. doi: 10.1124/jpet.106.105569. [DOI] [PubMed] [Google Scholar]
- Liu NK, Xu XM. Phospholipase A2 and its molecular mechanism after spinal cord injury. Mol Neurobiol. 2010;41:197–205. doi: 10.1007/s12035-010-8101-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales JR, Ballesteros I, Deniz JM, Hurtado O, Vivancos J, Nombela F, Lizasoain I, Castrillo A, Moro MA. Activation of liver X receptors promotes neuroprotection and reduces brain inflammation in experimental stroke. Circulation. 2008;118:1450–1459. doi: 10.1161/CIRCULATIONAHA.108.782300. [DOI] [PubMed] [Google Scholar]
- Nesic O, Lee J, Johnson KM, Ye Z, Xu GY, Unabia GC, Wood TG, McAdoo DJ, Westlund KN, Hulsebosch CE, Regino Perez-Polo J. Transcriptional profiling of spinal cord injury-induced central neuropathic pain. J Neurochem. 2005;95:998–1014. doi: 10.1111/j.1471-4159.2005.03462.x. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem. 2009;284:13291–13295. doi: 10.1074/jbc.R900010200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi K, Okubo M. Leukotrienes in nociceptive pathway and neuropathic/inflammatory pain. Biol Pharm Bull. 2011;34:1163–1169. doi: 10.1248/bpb.34.1163. [DOI] [PubMed] [Google Scholar]
- Okubo M, Yamanaka H, Kobayashi K, Fukuoka T, Dai Y, Noguchi K. Expression of leukotriene receptors in the rat dorsal root ganglion and the effects on pain behaviors. Mol Pain. 2010;6:57. doi: 10.1186/1744-8069-6-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitman ZJ, Jin G, Karoly ED, Spasojevic I, Yang J, Kinzler KW, He Y, Bigner DD, Vogelstein B, Yan H. Profiling the effects of isocitrate dehydrogenase 1 and 2 mutations on the cellular metabolome. Proc Natl Acad Sci U S A. 2011;108:3270–3275. doi: 10.1073/pnas.1019393108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, Quackenbush J. TM4 microarray software suite. Methods Enzymol. 2006;411:134–193. doi: 10.1016/S0076-6879(06)11009-5. [DOI] [PubMed] [Google Scholar]
- Saiwai H, Ohkawa Y, Yamada H, Kumamaru H, Harada A, Okano H, Yokomizo T, Iwamoto Y, Okada S. The LTB4-BLT1 axis mediates neutrophil infiltration and secondary injury in experimental spinal cord injury. Am J Pathol. 2010;176:2352–2366. doi: 10.2353/ajpath.2010.090839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Rabchevsky AG, Fugaccia I, Main JA, Lumpp JE., Jr Experimental modeling of spinal cord injury: characterization of a force-defined injury device. J Neurotrauma. 2003;20:179–193. doi: 10.1089/08977150360547099. [DOI] [PubMed] [Google Scholar]
- Sharma HS, Olsson Y, Nyberg F, Dey PK. Prostaglandins modulate alterations of microvascular permeability, blood flow, edema and serotonin levels following spinal cord injury: an experimental study in the rat. Neuroscience. 1993;57:443–449. doi: 10.1016/0306-4522(93)90076-r. [DOI] [PubMed] [Google Scholar]
- Singh VP, Patil CS, Kulkarni SK. Effect of licofelone against NSAIDs-induced gastrointestinal ulceration and inflammation. Indian J Exp Biol. 2005;43:247–253. [PubMed] [Google Scholar]
- Singh VP, Patil CS, Kulkarni SK. Anti-inflammatory effect of licofelone against various inflammatory challenges. Fundam Clin Pharmacol. 2006;20:65–71. doi: 10.1111/j.1472-8206.2005.00387.x. [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velardo MJ, Burger C, Williams PR, Baker HV, López MC, Mareci TH, White TE, Muzyczka N, Reier PJ. Patterns of gene expression reveal a temporally orchestrated wound healing response in the injured spinal cord. J Neurosci. 2004;24:8562–8576. doi: 10.1523/JNEUROSCI.3316-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, et al. HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 2009;37:D603–D610. doi: 10.1093/nar/gkn810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YJ, Xue M, Chen H. Licofelone inhibits interleukin-18-induced pro-inflammatory cytokine release and cellular proliferation in human mesangial cells. Basic Clin Pharmacol Toxicol. 2012;111:166–172. doi: 10.1111/j.1742-7843.2012.00882.x. [DOI] [PubMed] [Google Scholar]
- Xu JA, Hsu CY, Liu TH, Hogan EL, Perot PL, Jr, Tai HH. Leukotriene B4 release and polymorphonuclear cell infiltration in spinal cord injury. J Neurochem. 1990;55:907–912. doi: 10.1111/j.1471-4159.1990.tb04577.x. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Hayakawa M, Niki E. Total hydroxyoctadecadienoic acid as a marker for lipid peroxidation in vivo. Biofactors. 2005;24:7–15. doi: 10.1002/biof.5520240102. [DOI] [PubMed] [Google Scholar]
- Zhao P, Waxman SG, Hains BC. Extracellular signal-regulated kinase-regulated microglia-neuron signaling by prostaglandin E2 contributes to pain after spinal cord injury. J Neurosci. 2007;27:2357–2368. doi: 10.1523/JNEUROSCI.0138-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]