

Abstract
Growing evidence suggests that soluble Aβ species can drive Alzheimer disease (AD) pathogenesis by inducing a cascade of events including tau hyperphosphorylation, proteasome impairment, and synaptic dysfunction. However, these studies have relied largely on in vitro approaches to examine the role of soluble Aβ in AD. In particular, it remains unknown whether soluble Aβ oligomers can facilitate the development of human wild-type tau pathology in vivo. To address this question, we developed a novel transgenic model that expresses low levels of APP with the Arctic familial AD mutation to enhance soluble Aβ oligomer formation in conjunction with wild-type human tau. Using a genetic approach, we show that reduction of β-site APP cleaving enzyme (BACE) in these ArcTau mice decreases soluble Aβ oligomers, rescues cognition, and, more importantly, reduces tau accumulation and phosphorylation. Notably, BACE reduction decreases the postsynaptic mislocalization of tau in ArcTau mice and reduces the association between NMDA receptors and PSD-95. These studies provide critical in vivo evidence for a strong mechanistic link between soluble Aβ, wild-type tau, and synaptic pathology.
Introduction
Alzheimer disease (AD) is characterized by the accumulation of two hallmark pathologies, Aβ plaques and neurofibrillary tangles (Querfurth and LaFerla, 2010). Though studies show that Aβ promotes tau pathology in vitro, a key question that remains is whether soluble Aβ species can promote the accumulation and phosphorylation of wild-type tau in vivo.
The past several years of research have revealed an important role for soluble, oligomeric Aβ in cognitive and physiological dysfunction (Glabe, 2008). Aβ oligomers have been detected in both human AD brain and transgenic mouse models of AD and correlate better with disease progression than insoluble fibrillar plaques (McLean et al., 1999; Näslund et al., 2000; Lacor et al., 2004; Billings et al., 2005; Lesné et al., 2006). Interestingly, Aβ oligomers can induce tau mislocalization and phosphorylation in vitro, providing intriguing evidence that soluble Aβ modulates the development of tau pathology (Deshpande et al., 2006; Zempel et al., 2010). However, few studies have examined whether soluble Aβ oligomers influence wild-type human tau pathology in vivo.
To further investigate this question, we designed a double-transgenic mouse model that produces low levels of Arctic mutant Aβ (E22G) to preferentially drive the production of soluble oligomers and protofibrils without producing insoluble Aβ fibrils. By combining Arctic Aβ with the expression of wild-type human tau, this new model provides a unique tool to examine the effects of soluble, near physiological levels of oligomeric Aβ on tau pathology and cognition. To examine this question, hemizygous ArcTau mice were crossed with heterozygous BACE knock-out (BACE+/−) mice. The resulting ArcTau+/BACE+/− mice were then compared with ArcTau+/BACE+/+ littermates. As expected, heterozygous deletion of BACE reduces soluble Aβ and Aβ oligomers. More importantly, the reduction in soluble Aβ oligomers is accompanied by a decrease in human tau pathology, including reduced association of tau with PSD-95, and a rescue of learning and memory deficits. Our data therefore indicate that soluble Aβ, particularly soluble Aβ fibrillar oligomers, facilitate wild-type tau pathology in vivo. Importantly the reduction in soluble Aβ and tau pathology are accompanied by improved cognition. Thus, soluble Aβ fibrillar oligomers represent a viable therapeutic target for early disease modification.
Materials and Methods
Generation of ArcTau transgenic mice.
APP and tau constructs were subcloned into the Thy1.2 expression cassette via homologous recombination (In-Fusion; Clonetech). APP695 with Arctic and Swedish mutations and wild-type human 2N/4R Tau cDNAs [gifts from Drs. Lars Lannfelt (Uppsala University, Uppsala, Sweden) and Michael Vitek (Duke University, Durham, NC)] were amplified by proof-checking PCR using primers with 15 bp homology to the insertion site and a Kozac sequence: APP forward: 5′-GCGTCGACGTGGCTAGCCACCATGCTGCCCGGTTT-3′ and APP reverse: 5′-CGAGAACCGCGGAATCGATCTAGTTCTGCATCTGCTCAAAGAAC-3′, Tau forward: 5′-GCGTCGACGTGGCTAGCCACCATGGCTGAGCCCCGC-3′ and Tau reverse: 5′-CGAGAACCGCGGAATCGATTCACAAACCCTGCTTGGCC-3′. PCR products and linearized Thy1.2 plasmid were purified by gel extraction and the In-fusion reaction performed. Targeting cassettes were liberated from sequence-verified clones, purified by gel extraction, and co-microinjected into the pronuclei of single-cell C57BL6 embryos by the UC Irvine Transgenic Mouse Facility.
Transgenic genotyping and breeding.
All animal procedures were performed in strict accordance with NIH and University of California guidelines. Mice were housed on a 12 h/12 h light/dark schedule with ad libitum food and water. Transgenic mice were identified by tail PCR, and nontransgenic littermate controls were generated by crossing heterozygous transgenics with wild-type C57BL6 mice (Jax Laboratories). Genotyping demonstrated 100% coinheritance of APP and tau transgenes in all three founder lines, suggesting cointegration of the two transgenes, as previously observed (Oddo et al., 2003). Hemizygous ArcTau mice were crossed with heterozygous BACE knock-out mice (Roberds et al., 2001) to produce three experimental groups: ArcTau/BACE+/−, ArcTau/BACE+/+, and WT/BACE+/−.
Morris water maze.
Hippocampal-dependent learning and memory was examined by a blinded observer, using the Morris water maze (MWM) following standard protocols (Billings et al., 2005). Briefly, male and female 15-month-old animals were habituated to a 1-m-diameter circular pool filled with opaque water maintained at 29°C. During the 8 d of training, mice were placed into the pool and allowed to find a submerged escape platform (4 trials/d). On the ninth day, the platform was removed to assess memory retention for the former platform location.
Tissue processing and biochemical analysis.
Mice were killed by Nembutal overdose and cardiac perfusion with 0.01 m PBS. Brains were removed and cut along the sagittal midline. Half the brain was frozen on dry ice for subsequent biochemical analysis and half was fixed in 4% paraformaldehyde (pH 7.4, 48 h). Fixed half-brains were cut coronally on a Vibratome (50 μm) and stored in PBS with 0.02% NaN3 at 4°C. Half brains (excluding cerebellum) were processed to isolate soluble and insoluble protein; Western blots, Aβ ELISAs, and dot blots were performed as previously described (Blurton-Jones et al., 2009). Coimmunoprecipitation was performed using PSD-95 antibody (Abcam) and DynaBeads (Invitrogen).
Immunofluorescence and confocal microscopy.
Fluorescent labeling followed standard protocols (Blurton-Jones et al., 2009). Primary antibodies included: 6E10 (Signet), CT-20 (Calbiochem), Tau: HT7 (Innogenetics), PHF-1 (gift from P. Davies, Albert Einstein College, Yeshiva University, New York, NY), β-actin (Sigma), and GAPDH (Santa Cruz Biotechnology). Primary antibodies were applied overnight at 4°C and detected with appropriate AlexaFluor-conjugated secondary antibodies (Invitrogen). Specificity of all primary antibodies was confirmed by Western blot and by omission of primary antibody in immunofluorescent labeling. Sections were visualized using a Bio-Rad 2100 confocal system and lambda-strobing mode (Bio-Rad Laboratories).
Statistical analysis.
Comparisons between multiple groups were performed using ANOVA followed by Fischer's PLSD post hoc tests. Analysis of MWM acquisition was examined via repeated-measures ANOVA. Groups were considered significantly different when p < 0.05 for both the ANOVA and post hoc comparisons. Comparisons between two groups used unpaired Student's t test. All statistical analysis was performed using Statview5.01 software.
Results
ArcTau mice develop age-related deficits in learning and memory and increasing pathology
To test whether ArcTau mice develop age-related cognitive dysfunction, transgenic and control mice were tested in the MWM at 6, 12, and 18 months of age. At 6 months, ArcTau mice learn normally during MWM acquisition, but exhibit memory deficits for the platform location during probe trial testing (data not shown). By 12 months, cognition further deteriorates as ArcTau mice exhibit not only probe trial deficits but also take significantly longer to learn the platform location during MWM acquisition versus wild-type (WT) mice (Fig. 1D,E). These impairments become even further pronounced by 18 months (data not shown). Importantly, these differences cannot be attributed to motor deficits, as the swim speeds of both ArcTau and WT mice are equivalent (data not shown), and there are no differences found in Rotarod testing (Fig. 1B). Thus, ArcTau mice develop progressive age-related impairments in learning and memory, recapitulating an important feature of AD pathogenesis.
Figure 1.

ArcTau mice develop age-related Alzheimer-like pathology and cognitive decline. A, Schematic depiction of human APP and tau transgenes co-microinjected to produce ArcTau transgenic mice. B, ArcTau mice do not display motor deficits at 15 months, as shown by the average time spent on Rotarod compared with WT. C, ArcTau transgenic mice express twofold higher levels of APP (22C11 antibody) and equivalent levels of human and endogenous mouse tau (Tau46 antibody). D, Twelve-month-old ArcTau mice and WT littermate controls were trained in the Morris water maze for 8 d. ArcTau mice performed significantly worse on days 7 and 8 of acquisition. E, ArcTau mice also exhibited poor memory for the platform's former location, as evidenced by significantly longer latencies in a 24 h probe trial. F, ArcTau mice show a significant increase in OC+ fibrillar oligomers at 12 months. G, Representative dot blot of ArcTau half-brain samples at 6, 12, and 18 months. H–J, Histological analysis at 6 months reveals intraneuronal Aβ-like pathology. Full-length APP and Aβ is shown in green (H) and C terminus of APP is stained in red (I); thus, Aβ is detected by green-only puncta in the merge (J). K–M, Wild-type total htau (HT7) is evident throughout the hippocampus (K), specifically CA1 (L), and the cortex (M) at 6 months. N, O, Rare diffuse plaques only begin to appear at 18 months (subiculum shown). P, Q, Hyperphosphorylated (PHF+) somatodendritic wild-type tau is detected in the CA1 at 18 months. N = 8–10 per group; *p < 0.05, **p < 0.01. Error bars represent ± SEM.
Although we deliberately chose to express low levels (∼2-fold) of APP in the ArcTau model (Fig. 1C), these mice nevertheless develop age-related deficits in learning and memory. Given the importance of soluble Aβ oligomers in cognitive decline, we next examined these species using conformational-dependent antibodies (Kayed et al., 2003, 2007; Lesné et al., 2006). The A11 antibody labels prefibrillar oligomers, whereas OC antibody specifically labels conformationally distinct fibrillar oligomers (Glabe, 2008). Fibrillar oligomers are identified by 6 months and show a dramatic increase at 12 months, corresponding with the time point that significant learning and memory deficits are first observed (Fig. 1F,G). Intraneuronal Aβ, identified by colabeling with 6E10 (Aβ/APP) and a C-terminal-specific APP antibody, is also observed by 6 months within CA1 neurons, the basolateral nucleus of the amygdala, and cortical pyramidal neurons (Fig. 1H–J). At the same age, ArcTau mice exhibit extensive somatodendritic tau pathology throughout the hippocampus, subiculum, amygdala, and cortex, despite the fact that the mice only harbor a wild-type human tau transgene (Fig. 1K–M). As ArcTau mice age, intraneuronal Aβ pathology increases, yet extracellular Aβ deposition does not occur until 18 months (Fig. 1N,O). Interestingly, the small Aβ plaques observed in aged mice are diffuse, thioflavin-negative, and appear similar to the donut-shaped plaques reported in human patients harboring the Arctic mutation (Basun et al., 2008). This result is consistent with recent findings demonstrating that OC-positive fibrillar oligomers do not seed fibrillar plaque formation (Wu et al., 2010). Biochemical analyses confirm the diffuse soluble nature of these plaques, as detergent-insoluble Aβ is undetectable at any of the ages examined (data not shown). Notably, wild-type tau pathology also increases with age, including the appearance of PHF-1-positive neurons within the hippocampus and subiculum of 18-month-old mice (Fig. 1P,Q). Interestingly, Gallyas-positive insoluble aggregates and neuronal loss are not observed in ArcTau mice, even after 20 months (data not shown). This is consistent with previous wild-type tau models that overexpress human tau in the presence of endogenous mouse tau (Duff et al., 2000; Zhang et al., 2004); however, wild-type human tau can form paired-helical filaments and neurofibrillary tangles in a transgenic model that lacks endogenous tau (Andorfer et al., 2003), suggesting that the presence of murine tau may modulate the development of Gallyas-positive neurofibrillary tangles.
Partial BACE deletion reduces soluble Aβ and prevents ArcTau learning and memory impairments
Genetic reduction of BACE1, the primary enzyme responsible for amyloidogenic cleavage of APP, was achieved by crossing hemizygous ArcTau mice with heterozygous BACE knock-outs. Interestingly, ArcTau/BACE+/− mice perform as well as WT/BACE+/− in MWM at 15 months. BACE+/− mice have normal cognition (Roberds et al., 2001). In contrast, ArcTau littermates with physiological BACE expression (ArcTau/BACE+/+) exhibit significant learning and memory deficits (Fig. 2). The fact that only a partial reduction in BACE is sufficient to prevent cognitive deficits indicates that BACE inhibitors could be effective therapeutics for memory impairment.
Figure 2.
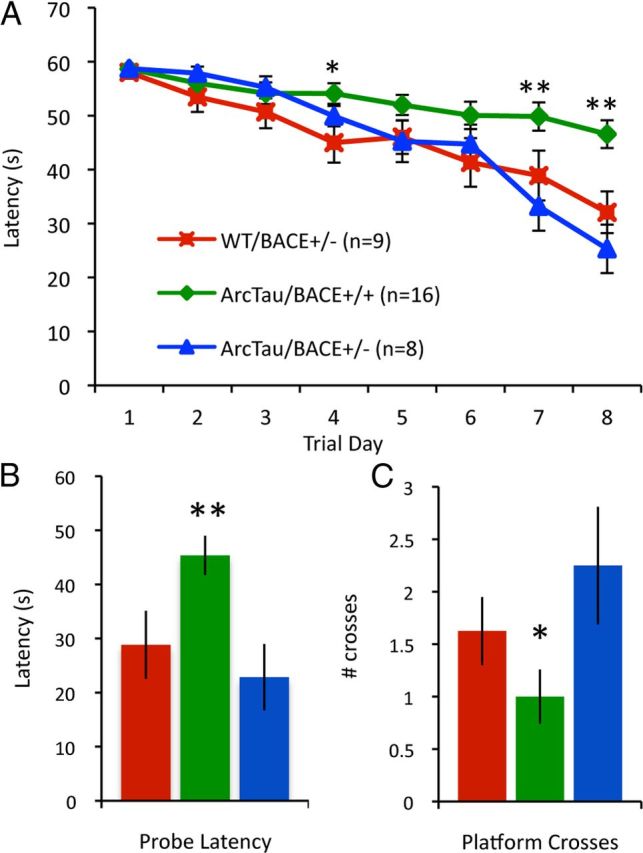
Partial genetic deletion of BACE prevents learning and memory impairments. A, Fifteen-month-old ArcTau, ArcTau/BACE+/−, and WT/BACE+/− were trained in Morris water maze for 8 d. ArcTau/BACE+/− performed similarly to WT/BACE+/− (cognitively normal), while ArcTau/BACE+/+ displayed significant learning deficits on days 4, 7, and 8. B, C, In the 24 h probe trial, ArcTau/BACE+/+ mice took almost twice as long to reach the former platform location and also crossed this area significantly fewer times than ArcTau/BACE+/− and WT/BACE+/− mice. Error bars represent ± SEM; *p < 0.05, **p < 0.01.
We next investigated the effects of BACE reduction on soluble Aβ. As expected, soluble Aβ levels are partially decreased by heterozygous BACE deletion (p = 0.0002; Fig. 3E), consistent with previous studies (McConlogue et al., 2007), while APP expression remains unchanged (Fig. 3A,B). Interestingly, levels of Aβ oligomers, particularly OC-positive fibrillar oligomers, are also significantly decreased in the ArcTau/BACE+/− compared with ArcTau/BACE+/+ (p = 0.01; Fig. 3C,D). Together, these data provide evidence that soluble Aβ oligomers play an important role in ArcTau mice cognitive impairment, as reduction in these species restores cognition to wild-type levels.
Figure 3.

Partial genetic reduction of BACE reduces Arctic Aβ and prefibrillar oligomeric Aβ. A, BACE levels are significantly decreased in ArcTau/BACE+/− compared with ArcTau/BACE+/+ mice, while APP and α-secretase ADAM17 levels are unchanged. B, Quantitation of the Western blots in A by densitometric analysis, shown as percentage of control. C, D, Dot blots from ArcTau animals with BACE knockdown show less fibrillar oligomers, as detected by OC antibody, while levels of prefibrillar oligomers detected by A11 do not vary. E, Levels of Aβ40 are significantly decreased in ArcTau/BACE+/− mice, measured by standard ELISA methods. Aβ42 is undetectable in this model. N = 4–5 per group. Error bars represent ± SEM; *p < 0.05, **p < 0.01.
Reduction of soluble Aβ prevents mislocalization and accumulation of wild-type human tau pathology
Because we demonstrated that partial BACE reduction was sufficient to reduce soluble Aβ, we next sought to investigate whether there was a concomitant decrease in wild-type tau pathology. Biochemical analysis revealed significant reductions in total soluble tau levels (HT7) as well as levels of PHF-1 hyperphosphorylated tau (pS396/S404; Fig. 4A,B). We did not detect differences in levels of murine phosphorylated tau at any of the epitopes examined. Levels of several other human tau phosphorylation epitopes and activity of the major tau kinases, GSK3β and Cdk5, remain unchanged (Fig. 4D,E), suggesting that soluble oligomeric forms of Aβ likely facilitate tau pathology by inhibiting tau degradation, rather than enhancing tau phosphorylation. Additionally, immunohistochemistry analysis demonstrated a lack of somatodendritic localized tau in ArcTau/BACE+/− compared with ArcTau/BACE+/+, indicating that BACE reduction largely prevents tau mislocalization (Fig. 4C). To examine the potential mechanisms by which Aβ may facilitate tau accumulation, we probed for several markers in the proteasome and autophagic degradative pathways. The slight increase in the tau E3 ligase C terminus of heat shock protein 70 interacting protein (CHIP) and decrease in ubiquitinated proteins in ArcTau/BACE+/− mice suggest that there may be improved proteasomal function compared with ArcTau/BACE+/+ mice, but these trends are not statistically significant. LC3-I is significantly increased in both ArcTau/BACE genotypes compared with wild-type, but no further differences are detected between ArcTau/BACE+/+ and ArcTau/BACE+/− (Fig. 4F,G). Additionally, no differences in levels of the presynaptic marker synaptophysin were detected in any genotype, suggesting that altered synaptic number does not contribute to the observed cognitive differences.
Figure 4.

Reduction of soluble Aβ prevents mislocalization and accumulation of wild-type tau. A, B, Total tau levels and tau phosphorylated at pS396/S404 are decreased in ArcTau/BACE+/− mice, whereas other phospho-tau epitopes remain unchanged. C, Somatodendritic wild-type human tau is abundant in ArcTau/ BACE+/+ mice, while barely detectable in the somatodendritic compartments of ArcTau/BACE+/− mice. D, E, Western blots probing for alterations in kinase levels or activity show no differences between ArcTau/BACE+/+ and ArcTau/BACE+/− mice. F, G, There are no statistically significant differences in proteasome activity, as evidenced by levels of CHIP and ubiquitinated proteins, or autophagy (LC3) between ArcTau/BACE+/+ and ArcTau/BACE+/− mice, although there appears to be a buildup of autophagosomes in both ArcTau genotypes compared with wild-type. H, I, Interestingly, coimmunoprecipitation reveals dramatic decreases in the association between PSD-95 and tau, Fyn kinase, and the NR2B subunit of the NMDA receptor in ArcTau/BACE+/− versus ArcTau/BACE+/+ mice. N = 4–5 per group; *p < 0.05 compared with ArcTau/BACE+/+ mice. Error bars represent ± SEM.
To further probe for the mechanisms responsible for the robust cognitive differences observed in ArcTau mice with BACE reduction, we performed coimmunoprecipitation with the presynaptic scaffolding protein, PSD-95. Importantly, we found marked differences in levels of human tau, Fyn kinase, and NR2B between all three genotypes, while levels of Homer remained unchanged (Fig. 4F,G). These data fit well with previous findings that suggest that dendritic mislocalization of tau and Fyn kinase can stabilize the interaction between PSD-95 and NMDA receptor complexes (Ittner et al., 2010), possibly rendering the synapse more susceptible to Aβ-dependent excitotoxicity.
Discussion
We and others previously used mutant forms of tau to study the interaction between Aβ and tau (Götz et al., 2001; Lewis et al., 2001; Oddo et al., 2003, 2004). Although such analyses have revealed important information about the potential relationship between Aβ and tau, some of these interactions likely differ depending on whether mutant or wild-type tau is examined. An important unresolved question is therefore whether Aβ can facilitate wild-type tau in vivo. ArcTau mice develop increasing Aβ oligomer and wild-type human tau pathology, leading to progressive age-related impairments in cognition. More importantly, partial reduction in BACE decreases not only soluble Aβ levels, but also tau pathology and cognitive decline.
In contrast to previous transgenic models using the Arctic mutation, we chose to microinject a low copy number of the Arctic/Swedish APP construct to investigate the effects of near physiological levels of Aβ, pertinent to early events in AD (Cheng et al., 2004; Lord et al., 2006). The aggressive development of insoluble Aβ pathology in these prior models is useful for certain studies but makes it difficult to separate the contributions of soluble versus insoluble Aβ. ArcTau mice do not form insoluble thioflavin-positive Aβ plaques; thus, our data demonstrate that soluble Aβ can drive the development of cognitive dysfunction. Recent clinical data support our findings by showing low Pittsburgh compound B retention in early-onset familial AD patients carrying the Arctic mutation, despite severe cognitive deficits (Schöll et al., 2012). Additionally, our experiments demonstrate that low levels of soluble Aβ oligomers can profoundly influence the development of wild-type human tau pathology.
Notably, BACE reduction resulted in a specific decrease in soluble Aβ levels and OC+ oligomers. In turn, total tau and PHF-1 hyperphosphorylated tau were also significantly reduced. It remains unclear precisely how tau is reduced in ArcTau/BACE+/− mice, although experiments in other models suggest that Aβ-mediated proteasome impairment is likely involved (Oddo et al., 2008; Tseng et al., 2008). Interestingly, we observed a trend for increased expression of the tau E3 ligase, CHIP, and decreased levels of ubiquitinated proteins in ArcTau/BACE+/− mice, suggesting that altered proteasomal function may be involved. However, future studies will be needed to fully decipher the mechanism by which partial BACE deletion reduces tau.
Our examination of synaptic proteins suggest that the cognitive impairment in ArcTau mice is likely due to the accumulation of tau in the synapse and the enhancement of NMDA receptor retention by PSD-95. Notably, each of these findings is partially mitigated by BACE reduction and cognitive function is correspondingly improved. Together, our findings demonstrate that soluble Aβ oligomers drive the development of both cognitive dysfunction and tau pathology. Curiously, a recent study reported that BACE knock-out in 3xTg-AD mice reduced Aβ levels without affecting tau immunoreactivity (Winton et al., 2011). Although biochemical analyses were not included, the conclusions of Winton and colleagues (2011) contradicts several prior studies demonstrating that immunological or genetic reduction of Aβ reduces tau pathology in 3xTg-AD and other AD mouse models (Götz et al., 2001; Oddo et al., 2004, 2008; Pérez et al., 2005). Our current study, using a similar approach to Winton et al. (2011), also contradicts their findings, revealing that reduction of soluble Aβ reduces the development of wild-type human tau pathology. Our data further support the notion that Aβ can drive tau pathogenesis. Importantly, the current study also clearly demonstrates that soluble Aβ is critical for this process. Thus, early targeting of soluble Aβ oligomers may provide an effective method to not only improve memory but also prevent tau-mediated neuronal dysfunction in AD.
Footnotes
This work was supported by NIH Grants PPG AG00538 and AG027544 (to F.M.L.) and AG029378 and AG16573 (to M.B.-J,), and NIA Grant F31AG039968 (to M.A.C.). We thank Drs. Charles Glabe and Peter Davies for antibodies and Drs. Lars Lannfelt and Michael Vitek for Arctic/Swedish APP and 4R/2N hTau cDNAs. We also thank Adam Pabst for technical assistance. We thank the University of California, Irvine, Transgenic Mouse Facility and Tom Fielder for producing the transgenic founders of the ArcTau line.
The authors declare no competing financial interests.
References
- Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- Basun H, Bogdanovic N, Ingelsson M, Almkvist O, Näslund J, Axelman K, Bird TD, Nochlin D, Schellenberg GD, Wahlund LO, Lannfelt L. Clinical and neuropathological features of the arctic APP gene mutation causing early-onset Alzheimer disease. Arch Neurol. 2008;65:499–505. doi: 10.1001/archneur.65.4.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Blurton-Jones M, Kitazawa M, Martinez-Coria H, Castello NA, Müller FJ, Loring JF, Yamasaki TR, Poon WW, Green KN, LaFerla FM. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A. 2009;106:13594–13599. doi: 10.1073/pnas.0901402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng IH, Palop JJ, Esposito LA, Bien-Ly N, Yan F, Mucke L. Aggressive amyloidosis in mice expressing human amyloid peptides with the Arctic mutation. Nat Med. 2004;10:1190–1192. doi: 10.1038/nm1123. [DOI] [PubMed] [Google Scholar]
- Deshpande A, Mina E, Glabe C, Busciglio J. Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci. 2006;26:6011–6018. doi: 10.1523/JNEUROSCI.1189-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff K, Knight H, Refolo LM, Sanders S, Yu X, Picciano M, Malester B, Hutton M, Adamson J, Goedert M, Burki K, Davies P. Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiol Dis. 2000;7:87–98. doi: 10.1006/nbdi.1999.0279. [DOI] [PubMed] [Google Scholar]
- Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, Rasool S, Gurlo T, Butler P, Glabe CG. Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener. 2007;2:18. doi: 10.1186/1750-1326-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer's-related amyloid beta oligomers. J Neurosci. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- Lord A, Kalimo H, Eckman C, Zhang XQ, Lannfelt L, Nilsson LN. The Arctic Alzheimer mutation facilitates early intraneuronal Abeta aggregation and senile plaque formation in transgenic mice. Neurobiol Aging. 2006;27:67–77. doi: 10.1016/j.neurobiolaging.2004.12.007. [DOI] [PubMed] [Google Scholar]
- McConlogue L, Buttini M, Anderson JP, Brigham EF, Chen KS, Freedman SB, Games D, Johnson-Wood K, Lee M, Zeller M, Liu W, Motter R, Sinha S. Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP transgenic mice. J Biol Chem. 2007;282:26326–26334. doi: 10.1074/jbc.M611687200. [DOI] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Näslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Tseng B, Cheng D, Vasilevko V, Cribbs DH, LaFerla FM. Blocking Abeta42 accumulation delays the onset and progression of tau pathology via the C terminus of heat shock protein70-interacting protein: a mechanistic link between Abeta and tau pathology. J Neurosci. 2008;28:12163–12175. doi: 10.1523/JNEUROSCI.2464-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez M, Ribe E, Rubio A, Lim F, Morán MA, Ramos PG, Ferrer I, Isla MT, Avila J. Characterization of a double (amyloid precursor protein-tau) transgenic: tau phosphorylation and aggregation. Neuroscience. 2005;130:339–347. doi: 10.1016/j.neuroscience.2004.09.029. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Roberds SL, Anderson J, Basi G, Bienkowski MJ, Branstetter DG, Chen KS, Freedman SB, Frigon NL, Games D, Hu K, Johnson-Wood K, Kappenman KE, Kawabe TT, Kola I, Kuehn R, Lee M, Liu W, Motter R, Nichols NF, Power M, et al. BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer's disease therapeutics. Hum Mol Genet. 2001;10:1317–1324. doi: 10.1093/hmg/10.12.1317. [DOI] [PubMed] [Google Scholar]
- Schöll M, Wall A, Thordardottir S, Ferreira D, Bogdanovic N, Långström B, Almkvist O, Graff C, Nordberg A. Low PiB PET retention in presence of pathologic CSF biomarkers in Arctic APP mutation carriers. Neurology. 2012;79:229–236. doi: 10.1212/WNL.0b013e31825fdf18. [DOI] [PubMed] [Google Scholar]
- Tseng BP, Green KN, Chan JL, Blurton-Jones M, LaFerla FM. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging. 2008;29:1607–1618. doi: 10.1016/j.neurobiolaging.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winton MJ, Lee EB, Sun E, Wong MM, Leight S, Zhang B, Trojanowski JQ, Lee VM. Intraneuronal APP, not free Abeta peptides in 3xTg-AD mice: implications for tau versus Abeta-mediated Alzheimer neurodegeneration. J Neurosci. 2011;31:7691–7699. doi: 10.1523/JNEUROSCI.6637-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Wu JW, Breydo L, Isas JM, Lee J, Kuznetsov YG, Langen R, Glabe C. Fibrillar oligomers nucleate the oligomerization of monomeric amyloid beta but do not seed fibril formation. J Biol Chem. 2010;285:6071–6079. doi: 10.1074/jbc.M109.069542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous tau into dendrites, tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Higuchi M, Yoshiyama Y, Ishihara T, Forman MS, Martinez D, Joyce S, Trojanowski JQ, Lee VM. Retarded axonal transport of R406W mutant tau in transgenic mice with a neurodegenerative tauopathy. J Neurosci. 2004;24:4657–4667. doi: 10.1523/JNEUROSCI.0797-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]