

Abstract
Activation of the neuronal-glial network in the spinal cord dorsal horn (SCDH) mediates various chronic painful conditions. Here, we studied spinal neuronal-astrocyte signaling interactions involved in the maintenance of painful diabetic neuropathy (PDN) in type 2 diabetes. We used the db/db mouse, an animal model for PDN of type 2 diabetes, which develops mechanical allodynia from 6-12 wk of age. In the current study, enhanced substance P (SP) expression was detected in the presynaptic sensory fibers innervating lamina I-III in the lumbar spinal cord (LSC) dorsal horn (LSCDH) of the db/db mouse at 10 wk of age. This phenomenon is associated with enhanced spinal ERK1/2 phosphorylation in projection sensory neurons and regional astrocyte activation. In addition, peak phosphorylation of the NR1 subunit of NMDAR, along with upregulation of neuronal and inducible nitric oxide synthase (nNOS and iNOS) expression were detected in diabetic mice. Expression of nNOS and iNOS was detected in both interneurons and astrocytes in lamina I-III of the LSCDH. Treatment with MK801, a NMDAR inhibitor, inhibited mechanical allodynia, ERK1/2 phosphorylation, as well as n- and iNOS upregulation in diabetic mice. MK801 also reduced astrocytosis and GFAP upregulation in db/db mice. In addition, L-NAME, a nonspecific NOS inhibitor, had similar effects on NMDAR signaling and NOS expression. These results suggest that NO from surrounding interneurons and astrocytes interacts with NMDAR-dependent signaling in the projection neurons of the SCDH during the maintenance of PDN.
Keywords: N-methyl-D-aspartate receptor, nitric oxide synthase, extracellular signal-regulated kinase, neuropathic pain, neuron-glial interactions
Introduction
Diabetic neuropathy affects up to 50% of patients with either type 1 or type 2 diabetes (Boulton et al. 2005; Feldman et al. 2005). Among these patients, approximately 30% develop painful diabetic neuropathy (PDN), (Davies et al. 2006). Although PDN is a common symptom among diabetic patients, its mechanisms remain unclear. PDN is more prevalent in type 2 than type 1 diabetes (Barrett et al. 2007), yet most published studies for PDN use animal models of type 1 diabetes. Understanding the mechanisms of PDN as it develops in the context of type 2 diabetes could lead to development of effective treatments to target the mechanisms behind this devastating disease. We have previously characterized the db/db mouse as a model for PDN of type 2 diabetes (Cheng et al. 2009). The db/db mouse carries a homozygous null mutation of the leptin receptor (Hummel et al. 1966; Sullivan et al. 2007). We reported that the db/db mouse develops features of PDN, including mechanical allodynia at 6-12 wk of age and evident sensory neuropathy at 24 wk of age (Cheng et al. 2009; Sullivan et al. 2007). We reported that the initiation of mechanical allodynia in db/db mice is associated with early peripheral mechanisms, including increased NGF/Trk A receptor signaling in DRG neurons before 8 wk of age. In our current study, we report these initial peripheral mechanisms activate the neuronal-glial interactions in spinal cord dorsal horn (SCDH) to mediate chronic PDN. Chronic pain is associated with enhanced spinal cord neuronal activity known as central sensitization (Latremoliere and Woolf 2009). It is well known that the neuronal N-methyl-D-aspartate receptor (NMDAR) mediates central sensitization in SCDH. In addition, regional microglial and astrocytic activation also contributes to chronic pain (Beggs and Salter 2010; Gao and Ji 2010b). Here, we demonstrated that increased peripheral nociceptive signals could lead to central SC NMDAR-mediated neuron-astrocyte interactions. We tested the activation of NMDAR and related downstream nociceptive events, including ERK1/2 activation and enhanced nitric oxide synthase (NOS) levels in neuron and astrocytes in the LSCDH. These signaling cascades are enhanced by NO. Our findings provide new understanding of the molecular mechanisms of PDN of type 2 diabetes and indicate that blocking NMDAR and NO signaling could be effective treatments for PDN of type 2 diabetes.
Materials and Methods
Animals
Male C57BLKS db/db mice were purchased from Jackson Laboratories (Bar Harbor, Maine; stock number 000642). The homozygous (Leprdb/Leprdb, or db/db) mice were used as a model of type 2 diabetes, while heterozygous mice (Leprdb/+, or db+) served as nondiabetic controls. Analyses and procedures were performed in compliance with protocols established by the Animal Models of Diabetic Complications Consortium (AMDCC) (http://www.amdcc.org) and were approved by the Use and Care of Animals Committee at the University of Michigan.
Mechanical Allodynia
The animals were placed in a Plexiglass cage with mesh flooring and allowed to acclimate for 1 h. A logarithmic series of calibrated monofilaments (Von Frey hairs; Stoelting, Wood Dale, IL) were applied to the mid-plantar surface of the hind paw and pressed to the point of bending. Brisk withdrawal of the stimulated paw was recorded as a positive response. Testing began with the 1 g filament followed by larger filaments, using the up–down method (Dixon 1980). Although all responses were noted, counting of the critical 6 data points did not begin until the response threshold was first crossed. The resulting pattern of the 6 positive and negative responses was tabulated, and the 50% gram threshold was calculated using the formula described previously (Chaplan et al. 1994).
Immunoblots
Following deep anesthesia, L4-6 DRG were dissected from 4 mice per condition (db/db and db+) and homogenized in ice-cold Tissue Protein Extraction Reagent, T-PER (Pierce Biotechnology, Rockford, IL) containing protease inhibitors (1 μM sodium orthovanadate and 1 μM sodium fluoride; Sigma Life Science, St. Louis, MO) and phosphatase inhibitors (1×, Thermo Scientific, Rockford, IL). 50 μg of protein were boiled in 2× sample buffer, separated on a SDS-PAGE gel, and transferred to a PVDF membrane. Membranes were blocked and incubated overnight at 4°C with primary antibodies: phospho-NR1 (pNR1, 1:1000, rabbit polyclonal, Millipore, Temecula, CA), total NR1 (1:1000, rabbit polyclonal, Millipore), phospho-extracellular signal-regulated kinases (pERK1/2, 1:1000, Millipore), total ERK1/2 (1:1000, Millipore), phospho-c-jun N terminal kinase (pJNK, 1:1000, Millipore), total JNK (1:1000, Millipore), nNOS (1:1000, Abcam Inc, Cambridge, MA), iNOS (1:1000, Abcam), and glial acidic fibrillary protein (GFAP, 1:1000, Abcam). Membranes were then rinsed and incubated with HRP-conjugated secondary antibodies for 1 h at 25°C and processed with chemiluminescence substrate (Pierce) before being exposed to Hyperfilm (Amersham, Piscataway, NJ). Densitometry was performed using Image J software, and the results were normalized against actin densities from the same sample.
Immunohistochemistry
Four mice from each group (db+ and db/db) were deeply anesthetized and perfused with 2% paraformaldehyde in phosphate buffered saline (PBS, pH 7.2, 0.1M). L4-L6 DRG were dissected, embedded in mounting media (OCT), and flash-frozen in liquid nitrogen. Tissue sections (15 μm) were cut, rehydrated with PBS, and blocked in 0.1% TX-100, and 5% non-fat dry milk in PBS. For pERK1/2 and nNOS immunohistochemistry, sections were treated with hot citric acid (10mM Sodium Citrate, 0.05% Tween 20, pH 6.0) for 3 min followed by rinsing with PBS. Sections were then incubated at room temperature for 16-24 h with primary antibodies: pERK1/2 (1:100), substance P (1:500, rat monoclonal, Abcam), nNOS (1:200), iNOS (1:100), and GFAP (1:1000, Abcam). Sections were then rinsed 3 times in PBS and incubated with secondary antiserum conjugated with different fluorophores (AlexaFluor 488, 594, or 647, Invitrogen, Carlsbad, CA). Neurons were identified using NeuN antibody (1:250, Neuronal Nuclei, Alexa Fluor 488, Millipore). Fluorescent signals were examined using an Olympus FluoView 500 laser scanning confocal microscope. The percentage of immunopositive cells was analyzed by counting the number of immunopositive neurons and multiplying by (100 / total number of neurons). A total of 6 LSC sections from each animal were measured in a blinded fashion. Sections were incubated with primary antisera alone, and secondary antisera alone to ensure specificity.
Inhibitor treatments
Several reagents were used in the current study: An NMDAR antagonist, (5S,10R)-(–)-5-Methyl-10,11-dihydro-5H-dibenzo[a,d]acyclohepten-5,10-imine hydrogen maleate (MK801, 0.25 mg/kg, Sigma), or saline control were administered intraperitoneally. N-Nitro-L-Arginine Methyl Ester (L-NAME, 30 μg in 5 μl), a NOS inhibitor, or aCSF were administered intrathecally 1 h before the behavior testing at 10 wk of age. The animals were given the second dose the next day, followed by perfusion and tissue collection 1h after the second administration. The dose of the inhibitors used was based on published data [for MK801 (Mao et al. 1992); for L-NAME: (Meller et al. 1994; Zajac et al. 2000)]. None of the drug treatments caused significant motor impairment nor distress to the animals.
Minipump placement
An osmotic minipump (Alzet minipump model 1007D, 100 μl volume with a 0.51 μl/hr infusion rate, Duent Corporation, Cupertino, CA) was used for continuous intrathecal infusion into the lumbar spinal cord region. The minipumps were filled with artificial cerebrospinal fluid (CSF) with 10% dimethyl sulfoxide (DMSO) with or without MAPK kinase (MEK) 1/2 inhibitor, 1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto) butadiene (UO126, 1 μg/μl; Calbiochem, La Jolla, CA) or artificial cerebrospinal fluid (aCSF) (Obata et al. 2007). The minipumps were implanted into the dorsal subcutaneous space between the shoulder blades of each mouse at 7 wk of age under sterile conditions. A caudally directed polyethylene cannula (Becton Dickinson and Company, Sparks, MD) was threaded subcutaneously and inserted into the subarachnoid space at the L5 level. The intrathecal infusion lasted for 1 wk at which time the mice reached 8 wk of age (Cheng et al. 2010).
Data presentation and statistical analyses
All data are presented as group means ± SEM. The data between db+ and db/db mice of the same age were analyzed using the Mann-Whitney test. Statistical comparisons between different age groups were made by one-way ANOVA tests followed by a post hoc Tukey's multiple comparison test. A p-value of less than 0.05 was considered statistically significant.
Results
Enhanced presynaptic SP inputs and astrocytosis in the superficial lamina of the SCDH in db/db mice
We previously determined that db/db mice develop mechanical allodynia from 6-12 wk of age (Cheng et al. 2009). Here, we hypothesized that increased SP release from presynaptic terminals triggers the neuron-glial interactions in the SCDH of db/db mice to maintain chronic PDN. We studied SP immunohistochemistry (IHC) in SC from db+ and db/db mice at 10 wk of age. In addition, we also performed IHC for GFAP to monitor the associated astrocytosis in LSCDH. In db+ mice, SP expression is mostly in presynaptic terminals of lamina I (Fig. 1A, arrowheads). In db/db mice, there is more extensive SP immunoreactivity in lamina I, II (Fig 1D, arrowheads) and lamina III (Fig. 1D, asterisks). In addition, increased numbers of GFAP-positive astrocytes were detected in lamina I-III in db/db mice (compare 1B to 1E, arrows). These astrocytes were larger and with long processes, indicating they were activated astrocytes (Fig. 1E, arrows).
Fig. 1.
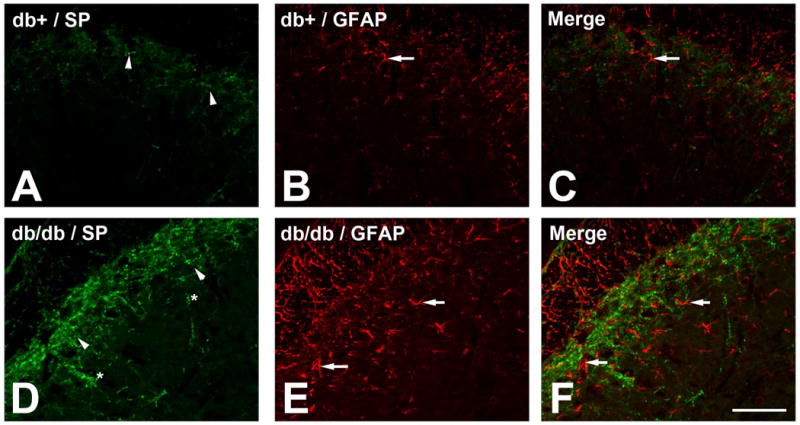
Increased peripheral SP input and astrocytosis in the LSC of db/db mice.
A : SP immunoreactivity was detected in lamina I (arrowheads) in db+ mice. D: SP immunoreactivity extended into lamina II (arrowheads) and lamina III (asterisks) in db/db mice. B, E: GFAP-positive astrocytes were detected in lamina I-III in the LSCDH of db/db mice (compare B to E, arrows). C, F: Merged images demonstrated the enhanced astrocyte aggregation to SP-positive fibers in db/db mice. Bar = 50 μm. N = 4.
Increased pERK1/2-positive neurons in superficial lamina of SCDH in db/db mice
To demonstrate the activation of postsynaptic SCDH neurons at the same stage, we performed double immunohistochemistry for SP and pERK1/2 using LSC sections from 10 wk old mice (Fig. 2). SP immunohistochemistry demonstrated presynaptic nociceptive fibers in SCDH (Fig. 2A, D). Only minimal levels of pERK1/2 expressed in the LSCDH of db+ mice was observed (Fig. 2B, arrow). In contrast, intense pERK1/2 immunoreactivity was detected in in neurons of lamina I-III in db/db mice (Fig. 2E-F, arrows).
Fig. 2.
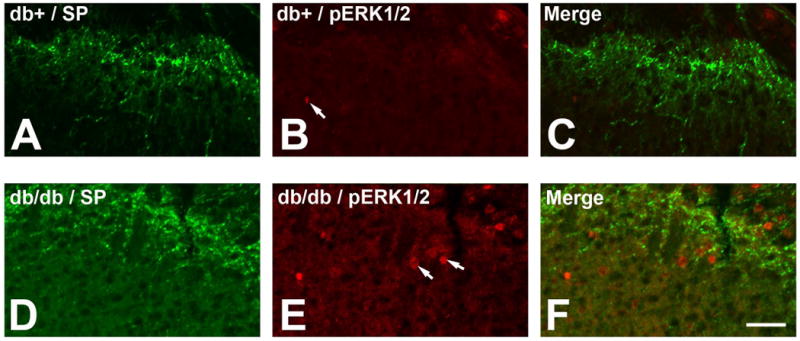
Increased pERK1/2-positive neurons in the LSCDH of db/db mice. A-C: SP (A) and pERK1/2 (B) immunohistochemistry in db+ mice. Minimal pERK1/2 positive neurons were detected in db+ mice (B, arrow). D-F: SP (D) and pERK1/2 (E) immunohistochemistry in db/db mice. In comparison to db+ mice, an increase in pERK1/2-positive neurons was observed in lamina I-III of db/db mice (E, arrows). Bar = 20 μm. N = 4.
Phosphorylation of NR1 and ERK1/2 during the period of mechanical allodynia
We hypothesized that NMDAR signaling is the key mechanism for the neuron-glial activation in SCDH. To test our hypothesis, we first studied the activation of NMDAR during the period of mechanical allodynia in db/db mice. NMDA activation is determined using immunoblots of pNR1. As demonstrated in Fig. 3A, enhanced NR1 phosphorylation was detected at 8 and 10 wk of age in db/db mice, compared to db+ mice of the same age. The total NR1 signals on the same immunoblots demonstrated unchanged total NR1 protein levels and actin signals were used as corresponding loading controls. Densitometric studies determined that there were enhanced pNR1 levels at 8-12 wk of age with the maximal NR1 phosphorylation detected at 10 wk of age with a 2.5 fold increase over the control db+ mice (Fig. 3B). NR1 phosphorylation was not detected before diabetes (at 5 wk). Although its levels were reduced, the pNR1 signal remained above the control level after the period of mechanical allodynia (at 16 wk). The levels of ERK1/2 phosphorylation were determined by pERK1/2 immunoblots (Fig. 3C). In accordance with NR1 phosphorylation, enhanced pERK1/2 levels in SC were detected at 8 to 16 wk of age, with maximal levels of phosphorylation detected at 10 wk of age (Fig. 3C). ERK1/2 immunoblots demonstrated unchanged total protein expression, and actin immunoblots served as loading controls. Densitometric analysis demonstrated that enhanced pERK1/2 levels were detected from 8-16 wk of age with a peak at 10 wk of age in db/db mice. Enhanced pERK1/2 levels were not detected at 5 wk of age, before onset of diabetes. Nevertheless, significant ERK1/2 phosphorylation was still detectable at 16 wk of age (Fig. 3D). Unlike NR1 and ERK1/2, there was no change in JNK phosphorylation in the LSC of db/db mice compared to db+ controls during the tested periods (Fig. 3E, F).
Fig. 3.
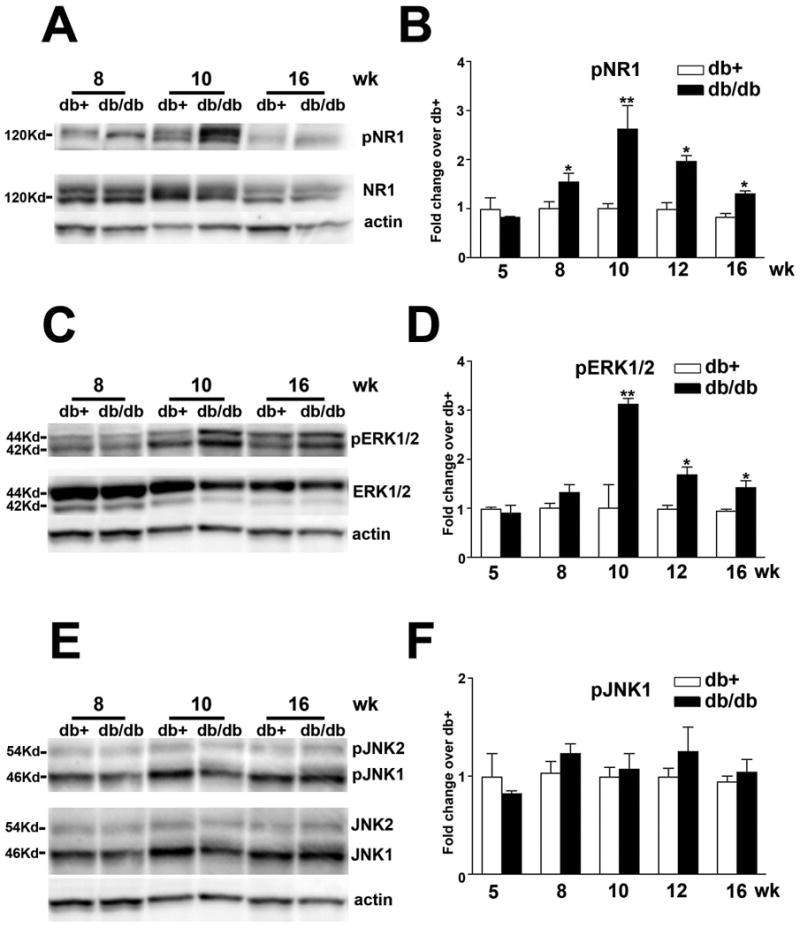
Increased phosphorylation of NR1 and ERK1/2, but not JNK in the LSC of db/db mice.
A: Representative immunoblots for pNR1 and total NR1. Increased NR1 phosphorylation was detected from 8-16 wk of age with maximal pNR1 levels detected at 10 wk of age. B: Densitometric analysis of NR1 phosphorylation. A 2.5-fold increase in NR1 phosphorylation was detected at 10 wk of age. C: Representative immunoblots for pERK1/2 and total ERK1/2. Increased ERK1/2 phosphorylation was detected from 8-16 wk of age with maximum pERK1/2 levels detected at 10 wk of age. D: Densitometric analysis of ERK1/2 phosphorylation. A 3-fold increase in ERK1/2 phosphorylation was detected at 10 wk of age. E: Representative immunoblots for pJNK and total JNK. F: Densitometric analysis of JNK phosphorylation. No significant change of JNK phosphorylation was detected in db/db mice. *, p < 0.05; **, p < 0.01. N = 4.
Increased nNOS expression in neurons and astrocytes of the SCDH in db/db mice
Another NMDA receptor-dependent nociceptive pathway is nNOS signaling (Freire et al. 2009). As the second step, we studied nNOS expression during the period of mechanical allodynia using immunoblots (Fig. 4A, B). A significant elevation of nNOS levels was detected at 10 wk of age. In parallel, the cell-specific distribution of nNOS was examined by immunohistochemistry, demonstrated in Fig. 4C-H. Increased nNOS immunoreaction was detected in neurons of lamina II and III in SCDH of db/db mice (Fig. 4G, H). Likewise, nNOS was detected in GFAP-positive astrocytes (Fig. 4I-K).
Fig. 4.
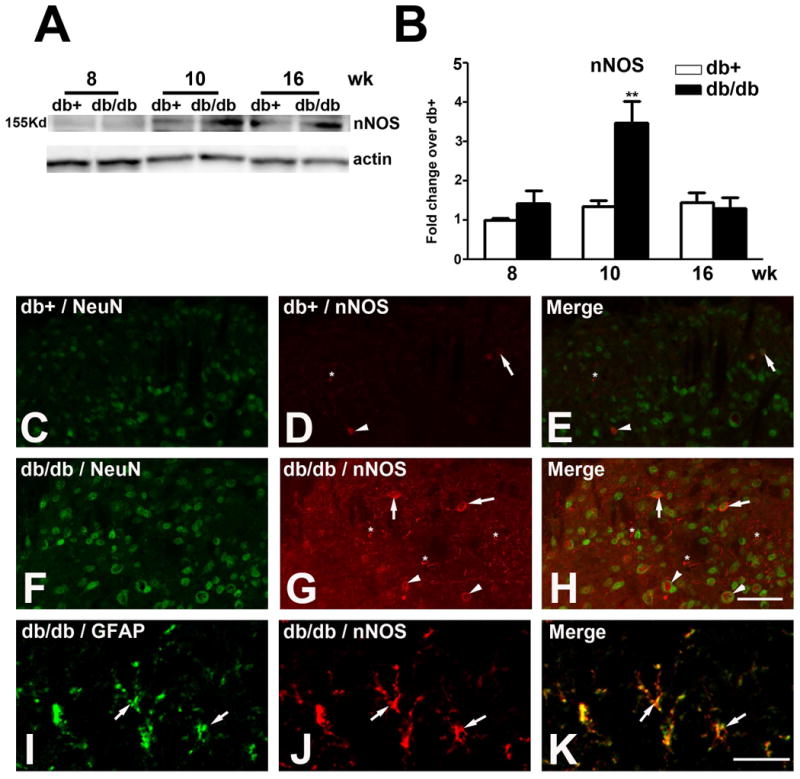
Upregulation of nNOS expression in db/db mice.
A: Representative nNOS immunoblots used proteins from the LSCDH of db+ and db/db mice. Increased nNOS expression was detected in db/db mice at 10 wk of age. B: Densitometric analysis of nNOS immunoblots. There was a 1.8-fold increase of nNOS expression in db/db mice compared to db+ mice at 10 wk of age. C-K: Increased nNOS immunoreactivity was detected in neurons and astrocytes in db/db mice. C-E: Few nNOS positive neurons were detected in lamina II (arrows) and III (arrowheads) in db+ mice. F-H: Increased numbers of nNOS-positive neurons were detected in laminas II (arrows) and III (arrowheads) of LSCDH of db/db mice. In addition, glial cell processes (G, asterisks) were observed to be nNOS-positive. I-K: High power confocal images identified nNOS-positive astrocytes. The nNOS-positive glial cells (J, arrows) expressed GFAP (I, arrows) and were identified as astrocytes (K, arrows). **, p < 0.01. N = 4. C-H, Bar = 20 μm; I-K, Bar = 5 μm.
Increased iNOS expression in astrocytes of the SCDH in db/db mice
We examined the expression of iNOS during the period of mechanical allodynia in the LSCDH of db+ and db/db mice by immunoblots (Fig. 5A). iNOS expression was upregulated at 10 wk of age, with a 3.5 fold increase in db/db mice over that of db+ controls (Fig. 5A, B). In contrast to nNOS, immunoreactivity of iNOS was detected only in astrocytes and not in neurons on lamina I-III of the SCDH (Fig. 5C-H).
Fig. 5.
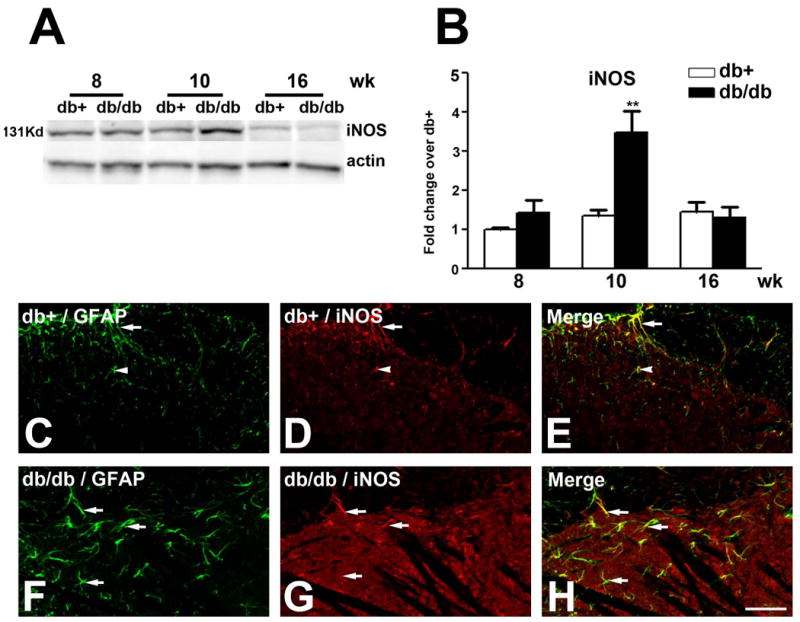
Upregulation of iNOS expression in db/db mice.
A: Representative iNOS immunoblots used proteins from LSCDH of db+ and db/db mice. Increased iNOS expression was detected in db/db mice at 10 wk of age. B: Densitometric analysis of iNOS immunoblots. There was a 3.3-fold increase of iNOS expression in db/db mice compared to db+ at 10 wk of age. C-K: Increased iNOS immunoreactivity was detected in astrocytes of lamina I-III in db/db mice. C-E: iNOS positive astrocytes were detected in lamina I (C, D, arrows) and their processes extended into II (C, D, arrowheads) in db+ mice. F-H: Increased numbers of iNOS positive astrocytes were detected in lamina I- III (F-H, arrows) of the LSCDH of db/db mice. **, p < 0.01. N = 4. Bar = 20 μm.
Effects of MK801, UO126, and L-NAME treatments on mechanical allodynia in db/db mice
To determine if NR1, ERK1/2, nNOS, and iNOS are important for the development of mechanical allodynia at 10 wk of age, we treated db/db mice and control mice with inhibitors for each pathway. MK801, a NMDAR inhibitor, was administered to both db+ and db/db mice at 10 wk of age. MK801 treatment significantly reduced the degree of decreased mechanical thresholds in db/db mice, reversing mechanical allodynia in db/db mice (Fig. 6A). Secondly, UO126, a MAPK kinase inhibitor, was administered intrathecally for 7 d to both db+ and db/db mice at 9 wk of age by using implanted minipumps. Similar to MK801 treatment, UO126 treatment significantly reversed mechanical allodynia in db/db mice (Fig. 6B). Lastly, L-NAME, a nonspecific NOS inhibitor that blocks actions of both nNOS and iNOS, was injected intrathecally at 10 wk of age. In addition to MK801 and UO126, L-NAME treatment significantly reduced mechanical allodynia in db/db mice (Fig. 6C).
Fig. 6.
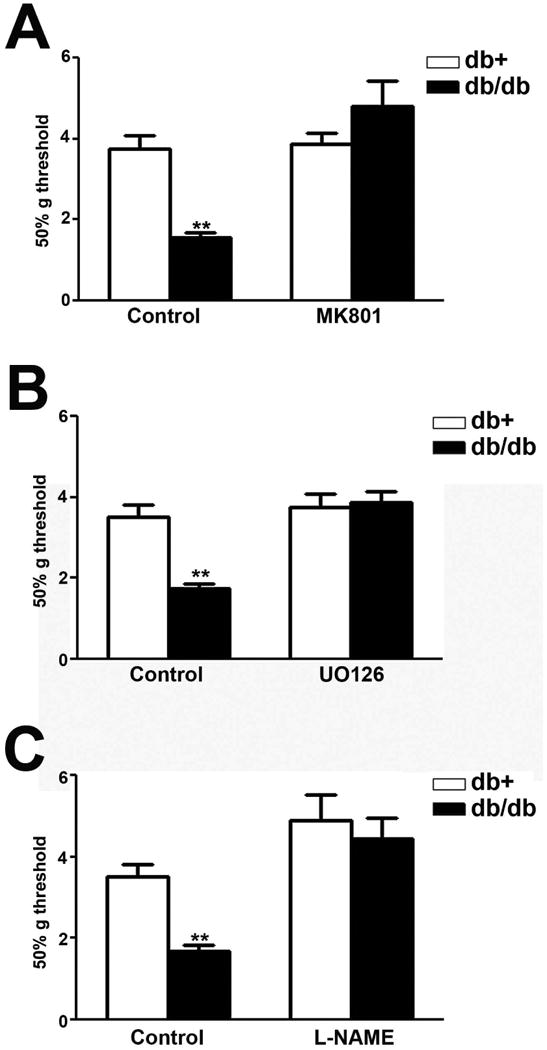
MK801, UO126, and L-NAME inhibit mechanical allodynia in db/db mice.
A: MK801 treatment significantly reversed decreased mechanical thresholds in db/db mice to control levels. B: UO126 treatment significantly reversed decreased mechanical thresholds in db/db mice to the levels of control db+ mice. C: L-NAME treatment significantly reversed decreased mechanical thresholds in db/db mice to the control levels. **, p < 0.01. N= 4.
The effects of MK801 on enhanced pNR1, pERK, nNOS and iNOS levels in the LSC of db/db mice
To determine how these signaling events contribute to the maintenance of mechanical allodynia in db/db mice, we studied the effects of these three inhibitors on NR1 and ERK1/2 phosphorylation as well as nNOS and iNOS upregulation. First, we tested the effects of MK801. Immunoblot analysis demonstrated that MK801 treatment significantly reduced NR1 (Fig. 7A, B) and ERK1/2 phosphorylation (7C, D). In addition, MK801 treatment inhibited the upregulation of nNOS (Fig. 7E, F) and iNOS (Fig. 7G, H) expression in db/db mice. In contrast, MK801 treatment did not affect pERK1/2 and NOS levels in db+ mice.
Fig. 7.
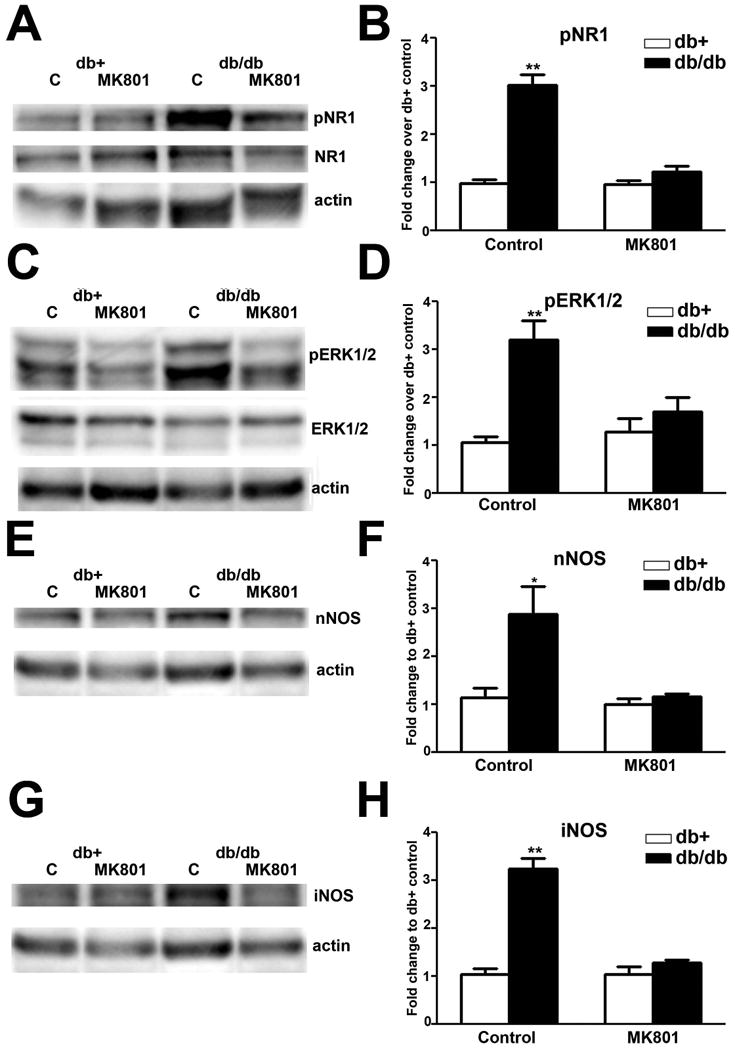
MK801 inhibits NR1 and ERK1/2 phosphorylation, as well as nNOS and iNOS upregulation in db/db mice.
A: Representative immunoblots of pNR1, total NR1, and actin from the LSC of db+ and db/db mice treated with control solution (C) or MK801. MK801 treatment decreased NR1 phosphorylation in db/db LSC, and had no effect on db+ mice. B: Densitometric analysis of pNR1 immunoblots. MK801 treatment decreased levels of NR1 phosphorylation back to control levels. C: Representative immunoblots of pERK1/2, total ERK1/2, and actin from LSC of db+ and db/db mice treated with control solution (C) or MK801. MK801 treatment decreased ERK1/2 phosphorylation in db/db but not in db+ mice. D: Densitometric analysis of pERK1/2 immunoblots. MK801 treatment decreased the levels of ERK1/2 phosphorylation back to control levels. E: nNOS immunoblots revealed that MK801 treatment reduced nNOS upregulation in db/db mice. F: Densitometric analysis of nNOS immunoblots demonstrated MK801 treatment decreased nNOS levels in db/db mice back to control levels. G: iNOS immunoblots showed that MK801 treatment reduced iNOS upregulation in db/db mice. H: Densitometric analysis of iNOS immunoblots demonstrated MK801 treatment decreased iNOS levels in db/db mice back to control levels. **, p < 0.01. N = 4.
Furthermore, MK801 treatment significantly reduced the number of pERK1/2- and nNOS-positive neurons in the LSCDH of db/db mice (Fig. 8). Consistent with Fig. 3 and Fig. 4, pERK1/2 and nNOS were detected in neurons in the corresponding lamina of the LSCDH in db/db mice (Fig. 8B, C). Although MK801 treatment did not affect the SP immunoreactivity in db/db mice (compare Fig. 8A and D), it significantly reduced the number of pERK1/2-(compare Fig. 8B and E), and nNOS-(compare Fig. 8C and F) positive neurons in LSCDH of db/db mice. Quantitative analyses revealed significant reductions of pERK1/2- (Fig. 8G) and nNOS- (Fig. 8H) positive neurons after MK801 treatment in the LSCDH of db/db mice.
Fig. 8.
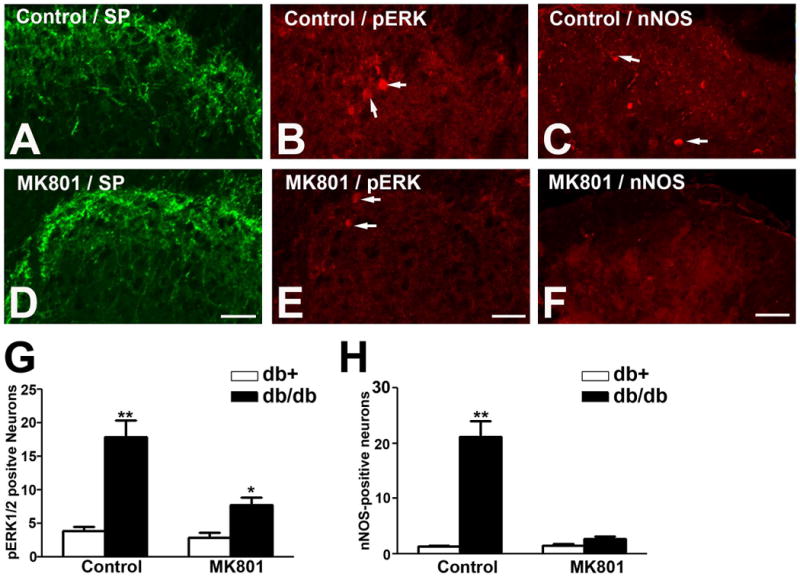
MK801 treatment significantly reduces the numbers pERK1/2 and nNOS-positive neurons in the LSCDH of db/db mice.
A, D: MK801 treatment had no effect on SP expression in the LSCDH of db/db mice. B, E: MK801 treatment reduced the number of pERK1/2-positive neurons in the LSCDH of db/db mice. C, F: MK801 treatment reduced the number of nNOS-positive neurons in the LSCDH of db/db mice. G: MK801 treatment reduced the number of pERK1/2-positive neurons in the LSCDH in db/db mice back to control levels. H: MK801 treatment reduced the numbers of nNOS-positive neurons in the LSCDH in db/db mice back to control levels. **, p < 0.01. N = 4. Bar = 20 μm.
The effects of MK801 on astrocytosis in the LSC of db/db mice
In addition to inhibition of neuronal activity, MK801 treatment reduced astrocytosis in the LSCDH of db/db mice (Fig. 9). GFAP immunoblot analysis detected a 3-fold increase of GFAP expression in the LSC of control-treated db/db mice in comparison with db+ mice of the same condition (Fig. 9A, B). MK801 treatment significantly reduced the GFAP levels of db/db mice to the db+ levels (Fig. 9A, B). In parallel, GFAP immunohistochemistry also demonstrated reduced numbers of astrocytes in the LSCDH of db/db after MK801 treatment (compare Fig. 9E, F).
Fig. 9.
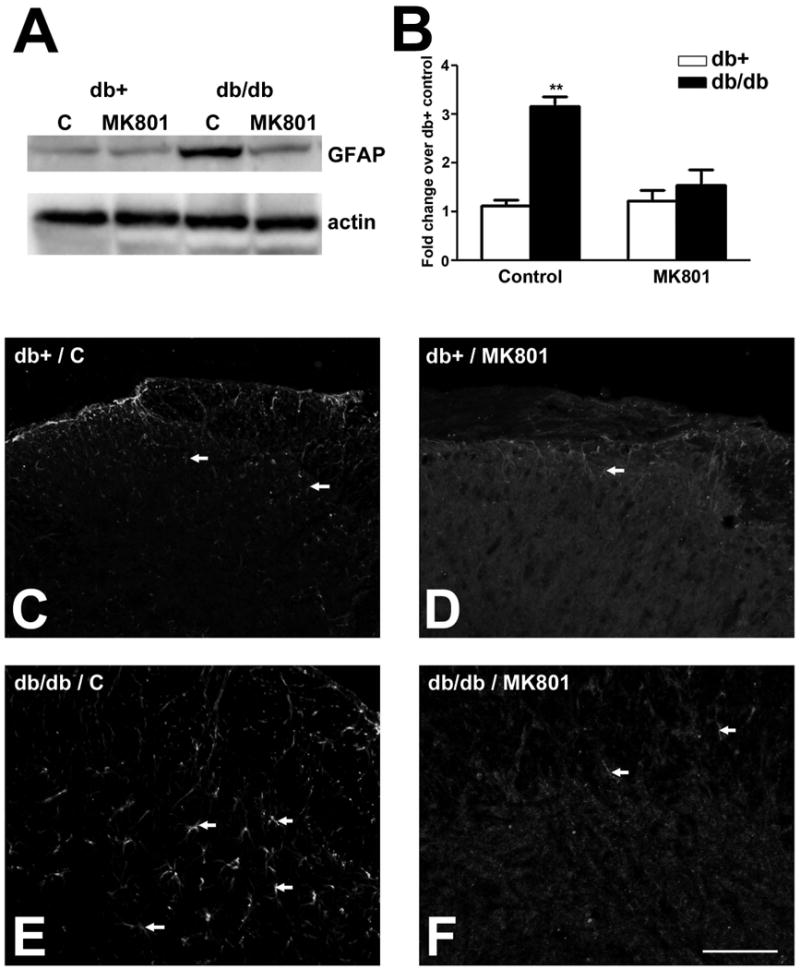
MK801 inhibits GFAP expression and astrocytosis in db/db mice.
A: GFAP immunoblots used protein samples from the LSC of db+ and db/db mice treated with control or MK801. Increased amount of GFAP protein was detected in control-treated db/db mice. The GFAP upregulation in db/db mice was inhibited by MK801 treatment. B: Densitometric analysis of GFAP immunoblots demonstrated MK801 treatment decreased GFAP levels in db/db mice back that of db+ mice. **, p < 0.01. N = 4. C: MK801 treatment reduced the degrees of astrocytosis in db/db mice. Increased numbers of astrocytes in the LSCDH in control-treated db/db mice (arrows, compare C and E) was reduced by MK801 treatment (arrows, compare E and F). Bar = 20 μm, N= 4.
The effects of UO126 on enhanced pERK, nNOS, and iNOS levels in the LSC of db/db mice
As expected, intrathecal UO126 treatment significantly inhibited ERK1/2 phosphorylation in db/db mice (Fig. 10A, B). However, UO126 treatment had no effect on increased levels of nNOS (Fig. 10C, D) and iNOS (Fig. 10E, F) in db/db mice. In addition, UO126 treatment had no significant effects on pERK1/2, nNOS, or iNOS levels in db+ mice.
Fig. 10.
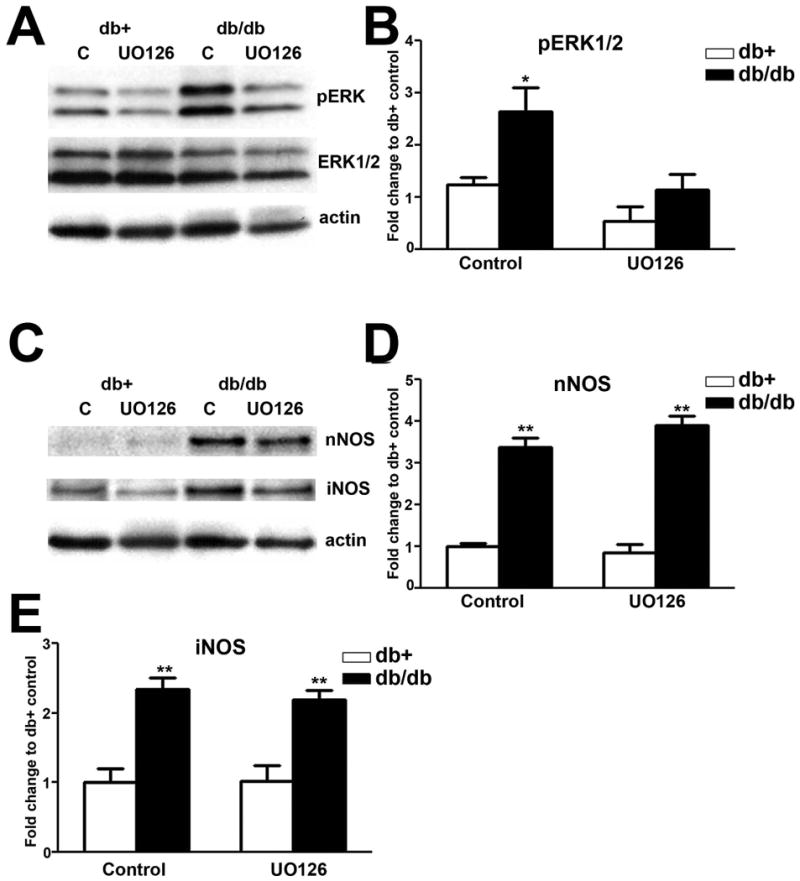
UO126 has no effect on nNOS and iNOS expression.
A: Representative immunoblots of pERK1/2, total ERK1/2, and actin from the LSC of db+ and db/db mice intrathecally treated with control aCSF (C) or aCSF with UO126. UO126 treatment decreased ERK1/2 phosphorylation in db/db, but not db+ mice. B: Densitometric analysis of pERK1/2 immunoblots. UO126 treatment decreased the levels of ERK1/2 phosphorylation back to control levels. C: Representative immunoblots of nNOS, iNOS, and actin from the LSC of db+ and db/db mice treated with control solution (C) or UO126. UO126 treatment had no effect on nNOS and iNOS levels in db/db mice. D,E: Densitometric analysis revealed UO126 treatment did not affect nNOS and iNOS levels in db/db mice. **, p < 0.01. N = 4.
The effects of L-NAME on enhanced pNR1, pERK, nNOS and iNOS levels in LSC of db/db mice
Similar to MK801, L-NAME treatment significantly reduced NR1 (Fig. 11A, B) and ERK1/2 phosphorylation (Fig. 11C, D) in db/db mice. L-NAME treatment had no effects on control levels of pNR1 and pERK1/2 in db+ mice. In addition, L-NAME treatment reduced levels of nNOS (Fig. 11E, F) and iNOS (Fig. 11G, H) in db/db, but not db+ mice.
Fig. 11.
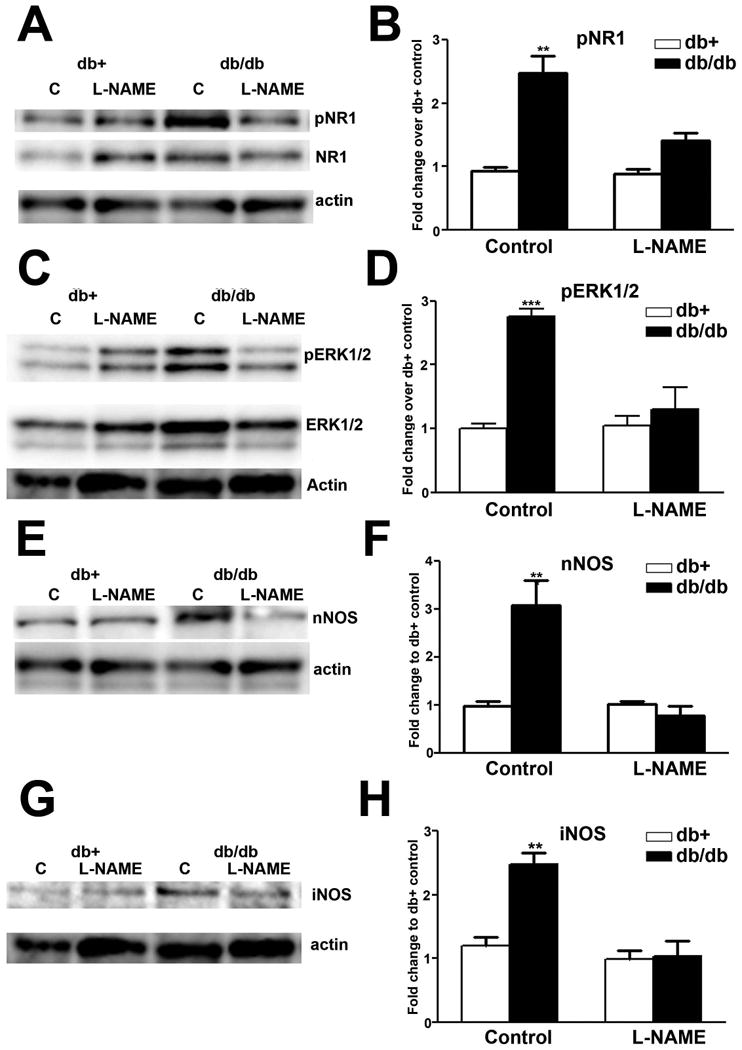
L-NAME inhibits NR1 and ERK1/2 phosphorylation, as well as nNOS and iNOS upregulation in db/db mice.
A: Representative immunoblots of pNR1, total NR1, and actin from the LSC of db+ and db/db mice intrathecally treated with control aCSF (C) or aCSF with L-NAME. L-NAME treatment decreased NR1 phosphorylation in db/db, but not db+ mice. B: Densitometric analysis of pNR1 immunoblots. L-NAME treatment decreased the levels of NR1 phosphorylation back to that of control db+ mice. C: Representative immunoblots of pERK1/2, ERK1/2, and actin from the LSC of db+ and db/db mice treated with control solution (C) or L-NAME. L-NAME treatment decreased ERK1/2 phosphorylation in db/db, but not db+ mice. D: Densitometric analysis of pERK1/2 immunoblots. L-NAME treatment decreased the levels of ERK1/2 phosphorylation back to control levels. E: nNOS immunoblots revealed that L-NAME treatment reduced nNOS upregulation in db/db mice. F: Densitometric analysis of nNOS immunoblots demonstrated L-NAME treatment decreased nNOS levels in db/db mice back to control levels. G: iNOS immunoblots showed that L-NAME treatment reduced iNOS upregulation in db/db mice. H: Densitometric analysis of iNOS immunoblots demonstrated iNOS treatment decreased iNOS levels in db/db mice back to control levels. **, p < 0.01; ***, p < 0.001. N = 4.
Discussion
PDN is a prevalent cause of chronic neuropathic pain in patients with type 2 diabetes. In our current report, we study the involvement of spinal neuron-astrocyte signaling for enhanced nociception in PDN of type 2 diabetes. Our current findings suggest that increased peripheral nociceptive input is essential for the subsequent development of SC mechanisms for mechanical allodynia (Cheng et al. 2009). This notion is supported by Latremoliere and colleagues, who reviewed the literature and recognized that increased levels of subthreshold synaptic inputs to SCDH nociceptive neurons could induce central neural plasticity (Latremoliere and Woolf 2009).
Our results indicate that ERK1/2 phosphorylation occurred in neurons in lamina I- III of the LSC during the maintenance phase of mechanical allodynia in db/db mice. These results are consistent with published data from models of inflammatory pain (Cheng et al. 2008; Gao and Ji 2010a), as well as pain from electric stimulation (Lever et al. 2003), capsaicin treatment (Ji et al. 1999), and nerve injuries (Obata et al. 2004). Here, our data suggest increased ERK phosphorylation is associated with increased presynaptic SP secretion. In support of our hypothesis, Ji and colleagues reported that NK1, the SP receptor, is co-localized with activated ERK in painful conditions (Ji et al. 2002). In addition, Kawasaki and colleagues directly applied SP to SC, which in turn induced spinal ERK activation (Kawasaki et al. 2004).
In the current report, we discovered increased astrocytosis in the LSCDH, suggesting increased neuronal-astrocyte interaction during the maintenance phase of mechanical allodynia in db/db mice. Comparable to our data, Liao and colleagues reported astrocyte activation in LSCDH of db/db mice at the same age (Liao et al. 2011). In addition, astrocyte activation has been reviewed in various animal models for both inflammatory and neuropathic pain (Gao and Ji 2010b). In contrast to astrocyte activation, we failed to detect microglial activation using either OX42 or Iba1 immunohistochemistry (data not shown) consistent with Liao's findings that minocycline did not affect the mechanical allodynia in db/db mice (Liao et al. 2011).
NMDAR activation is a well-known mechanism to mediate central sensitization for prolonged and enhanced nociception in animal models of inflammatory and neuropathic pain (Woolf 2007). However, evidence of spinal NMDAR activation in PDN of type 2 diabetes is still lacking. Based on a plethora of published evidence in other pain models (Mao et al. 1992; Persson et al. 1995; Ren et al. 1992), we studied the roles of NMDAR and its downstream signaling pathways for the maintenance of mechanical allodynia in the db/db mouse, an animal model of type 2 diabetes. In addition to the current study, Liao and colleagues reported a similar time course of NR1 phosphorylation in the db/db mouse model (Liao et al. 2011). A majority of other published studies for spinal mechanisms in PDN have been performed in models of type 1 diabetes. Daulhac and colleagues reported that there are NMDAR-dependent activation of MAPKs, including ERK1/2, p38, and JNK, in the LSC at 21 days after induction of type 1 diabetes by strepzotocin (STZ) treatment (Daulhac et al. 2006). In addition, Rodon and colleagues detected NR1 phosphorylation in the LSC in the STZ rat model 4 wk after diabetes (Rondon et al. 2010). In their report, magnesium sulfate treatment, which served as an inhibitor for NMDAR, not only suppressed NR1 phosphorylation, but also decreased pain behaviors in diabetic animals. These reports indicate that NMDAR activation could be a mechanism for PDN in both type 1 and type 2 diabetes.
Our current results demonstrated upregulation of nNOS in the SCDH. We observed nNOS expression in neurons and astrocytes of lamina II and III. These results are consistent with a previous report by Ruscheweyh et al (Ruscheweyh et al. 2006). In their report, nNOS-positive neurons are mostly interneurons, similar to our current findings. Interestingly, neurons that contain soluble guanylyl cyclase, the target molecule of NO, are distributed mostly in lamina I and II. They concluded that NO diffused from the nNOS-positive interneurons to act on projection neurons in lamina I. Based on immunohistochemical distribution of nNOS in db/db mice, we speculate that similar mechanisms might be involved in PDN of type 2 diabetes. However, these mechanisms might not be associated with PDN of type 1 diabetes. For example, Vareniuk and colleagues detected reduced thermal hyperalgesia, but not mechanical allodynia in STZ-treated nNOS knockout mice compared to diabetic wild type mice (Vareniuk et al. 2009). Unfortunately, iNOS expression was not studied in their report. In addition, Bujalska and colleagues reported that iNOS, but not nNOS, is involved in PDN of STZ rats by using specific NOS inhibitors (Bujalska et al. 2008). In our current report, we detected increased expression of both nNOS and iNOS in db/db mice. In order to block both NOS isoforms, we used L-NAME, a nonspecific NOS inhibitor. We acknowledge that L-NAME treatment only provides information for general NOS-mediated actions. Further studies using specific NOS inhibitors are necessary to elucidate if nNOS and/or iNOS mediate PDN in type 2 diabetes.
In the current report, we detected expression of iNOS in astrocytes of lamina I. Similar to our findings, iNOS expression is reported in a variety of pain models for both inflammatory and neuropathic pain (Freire et al. 2009). In the SCDH, astrocyte-derived iNOS was implicated in presynaptic potentiation, a form of long term potentiation, to enhance nociception (Amitai 2010). The current findings suggest a similar phenomenon could exist in the SCDH of db/db mice to mediate mechanical allodynia.
Our findings indicate that ERK1/2 phosphorylation is a NMDAR-dependent event. Several studies have shown that glutamate transmission via NMDA receptors is essential for ERK activation in SCDH neurons (Ji et al. 1999; Lever et al. 2003). Analogous to our findings, Daulhac and colleagues reported not only ERK1/2, but also p38 and JNK activation in STZ-treated rats (Daulhac et al. 2006). Conversely, we failed to detect the activation of JNK and p38 (data not shown) in the LSC of db/db mice. In addition, we did not detect pERK1/2 immunoreactivity in microglia as reported in their study (Daulhac et al. 2006). This discrepancy could reflect a distinction in the SC mechanisms that mediate PDN in type 1 and type 2 diabetes, or a species-specific difference between these studies. We are currently investigating NMDAR and MAPK activation in PDN of STZ-treated db+ mice to better understand the cause of these discordant observations.
In accord with other studies, our data demonstrated that ERK1/2 phosphorylation is mostly located in the superficial layers (lamina I-III) of the SCDH (Ji et al. 2009)). We speculate that these pERK1/2-positive neurons are projection neurons that provide the spinothalamic tract. In agreement with our hypothesis, Slack and colleagues identified the activation of ERK1/2 in the Trk B-positive SCDH projection neurons that provide the spinothalamic tract after brain-derived neurotrophic factor treatment (Slack et al. 2005). The current findings that demonstrated UO126 inhibition of mechanical allodynia without affecting the increased nNOS and iNOS levels suggest that pERK1/2-positive neurons are likely the projection neurons that integrate the actions of NO from neighboring interneurons and astrocytes in the SCDH and transmit nociceptive signals to the higher levels of the CNS.
Our data suggested MK801 treatment inhibited both nNOS and iNOS expression. Unfortunately, little has been published about NMDAR-mediated NOS expression aside from our current report. In the enteric nervous system, MK801 treatment inhibited the upregulation of nNOS in the enteric plexus after ischemia/reperfusion (Calcina et al. 2005). In addition, MK801 treatment reduced increased SC nNOS levels in morphine tolerance (Wong et al. 2000). After traumatic SC injury, MK801 treatment reduced inflammatory reactions including enhanced iNOS expression and improved function recovery (Esposito et al. 2011). A similar phenomenon was also observed after hypoxic brain injury (Jander et al. 2000). Our results suggest similar NMDAR-mediated NOS expression is involved in interneurons for the mediation of PDN of type 2 diabetes. Specifically, this signaling event occurs in interneurons of lamina II and III, and is ERK-independent.
Our data indicated that MK801 inhibited the enhanced astrocytosis in SCDH of db/db mice. These findings suggest that NMDAR-signaling mediates local astrocyte activation. In the literature, several NMDAR-dependent factors, such as chemokines, and cytokines, are reported to mediate enhanced astrocytosis in a central pain model following spinal cord injury (Gwak et al. 2011). Our studies provide evidence that similar mechanisms could be associated with PDN in type 2 diabetes.
Our results from L-NAME treatment suggest NO is essential for the maintenance of mechanical allodynia in db/db mice. In support of our findings, spinal NO was reported to mediate NMDA-induced hyperalgesia in other pain models. For example, Kitto and colleagues observed inhibition of NMDA-induced hyperalgesia as a result of intrathecal L-NAME treatment (Kitto et al. 1992). Increased NOS expression was also reported in an inflammatory pain model (Infante et al. 2007). In addition, L-NAME inhibits neuropathy-induced thermal hyperalgesia (Inoue et al. 1998; Thomas et al. 1996).
Our data suggest that NO mediates increased NR1 and ERK1/2 phosphorylation. Since pERK1/2 is mostly located in the projection neurons of lamina I-III, distinct from nNOS-positive interneurons, these results indicate that there is NO-mediated NMDAR activation in projection neurons. In support of our hypothesis, Malmberg and colleagues reported that intrathecal L-NAME treatment blocked the second phase of pain from the formalin test and NMDA-induced thermal hyperalgesia, suggesting NO enhances NMDAR actions (Malmberg and Yaksh 1993). In addition, Sorkin and colleagues reported that L-NAME treatment directly inhibits increased levels of glutamate and citrulline, an amino acid co-product of nitric oxide synthesis, in response to spinal administration of NMDA (Sorkin 1993). Other than indirectly enhancing pERK1/2 levels via increased NMDAR activation, NO could also trigger ERK1/2 phosphorylation by mechanisms independent of NMDAR signaling. Komatsu and colleagues reported that NO mediates morphine-3-glucuronide-induced pain via the NO-cGMP-PKG pathway (Komatsu et al. 2009). Since MK801 completely blocks increased pERK1/2 levels in our current study, NO-dependent ERK1/2 activation is less likely to occur in our model. Similar to NO, interleukin (IL)-1β also contributes to enhanced NMDAR phosphorylation in db/db mice, suggesting multiple factors are involved in this action (Liao et al. 2011). Both NO and IL-1β-mediated NMDAR phosphorylation is likely triggered by reactive oxygen species generated from type 2 diabetes (Liao et al. 2011).
In addition to increased nNOS and iNOS expression in the SCDH, we detected evidence that there is NMDAR-mediated ERK1/2 activation during the maintenance phase of mechanical allodynia in db/db mice. Our data suggests increased NMDAR-pERK1/2 signaling in projection neurons is an essential step for the maintenance of mechanical allodynia in db/db mice. Additionally, increased NOS expression in nearby interneurons and astrocytes further serves as a positive feedback mechanism to enhance NMDAR activation by NO (Model 1). This model is different from one proposed by Liao and colleagues, who suggested a one-way mechanism where astrocyte-derived interleukin (IL)1-β induces neuronal NMDAR activation and mechanical allodynia. In contrast, our model suggests a more complicated neuron-astrocyte network to mediate PDN in type 2 diabetes.
Model 1.
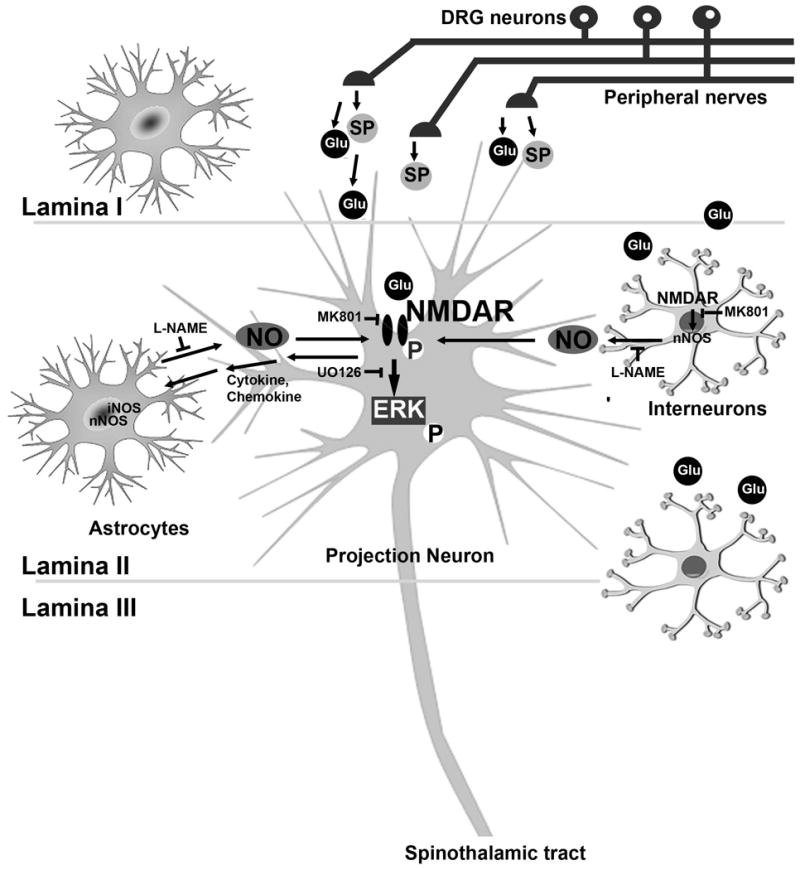
Working hypothesis of neuron-astrocyte interactions during the maintenance phase of mechanical allodynia in db/db mice. During the later period of mechanical allodynia, enhanced presynaptic SP and glutamate (Glu) release induces postsynaptic NR1/ERK1/2 phosphorylation in projection neurons. In addition, NR1 signaling increases nNOS expression in interneurons and both nNOS and iNOS upregulation in astrocytes in the LSCDH. These regulatory mechanisms trigger NO production, which in turn further enhances NR1 signaling in projection neurons.
Acknowledgments
The authors would like to thank Dr. Eva Feldman for her mentorship and support for this study. This work utilized the Morphology and Image Analysis Core of the Michigan Diabetes Research and Training Center funded by NIH (5P60 DK20572) from the National Institute of Diabetes & Digestive & Kidney Diseases.
This study is supported by National Institutes of Health [UO1-DK60994 (ELF); 1K08NS061039 (HTC)] and the Juvenile Diabetes Research Foundation Center for the Study of Complications in Diabetes.
References
- Amitai Y. Physiologic role for “inducible” nitric oxide synthase: a new form of astrocytic-neuronal interface. Glia. 2010;58(15):1775–81. doi: 10.1002/glia.21057. [DOI] [PubMed] [Google Scholar]
- Barrett AM, Lucero MA, Le T, Robinson RL, Dworkin RH, Chappell AS. Epidemiology, public health burden, and treatment of diabetic peripheral neuropathic pain: a review. Pain Med. 2007;8(2):S50–62. doi: 10.1111/j.1526-4637.2006.00179.x. [DOI] [PubMed] [Google Scholar]
- Beggs S, Salter MW. Microglia-neuronal signalling in neuropathic pain hypersensitivity 2.0. Curr Opin Neurobiol. 2010;20(4):474–80. doi: 10.1016/j.conb.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton AJ, Vinik AI, Arezzo JC, Bril V, Feldman EL, Freeman R, Malik RA, Maser RE, Sosenko JM, Ziegler D. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care. 2005;28(4):956–62. doi: 10.2337/diacare.28.4.956. [DOI] [PubMed] [Google Scholar]
- Bujalska M, Tatarkiewicz J, de Corde A, Gumulka SW. Effect of cyclooxygenase and nitric oxide synthase inhibitors on streptozotocin-induced hyperalgesia in rats. Pharmacology. 2008;81(2):151–7. doi: 10.1159/000110787. [DOI] [PubMed] [Google Scholar]
- Calcina F, Barocelli E, Bertoni S, Furukawa O, Kaunitz J, Impicciatore M, Sternini C. Effect of N-methyl-d-aspartate receptor blockade on neuronal plasticity and gastrointestinal transit delay induced by ischemia/reperfusion in rats. Neuroscience. 2005;134(1):39–49. doi: 10.1016/j.neuroscience.2005.03.052. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53(1):55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Cheng HT, Dauch JR, Hayes JM, Hong Y, Feldman EL. Nerve growth factor mediates mechanical allodynia in a mouse model of type 2 diabetes. J Neuropathol Exp Neurol. 2009;68(11):1229–43. doi: 10.1097/NEN.0b013e3181bef710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HT, Dauch JR, Oh SS, Hayes JM, Hong Y, Feldman EL. p38 mediates mechanical allodynia in a mouse model of type 2 diabetes. Mol Pain. 2010;6:28. doi: 10.1186/1744-8069-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HT, Suzuki M, Hegarty DM, Xu Q, Weyerbacher AR, South SM, Ohata M, Inturrisi CE. Inflammatory pain-induced signaling events following a conditional deletion of the N-methyl-D-aspartate receptor in spinal cord dorsal horn. Neuroscience. 2008;155(3):948–58. doi: 10.1016/j.neuroscience.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daulhac L, Mallet C, Courteix C, Etienne M, Duroux E, Privat AM, Eschalier A, Fialip J. Diabetes-induced mechanical hyperalgesia involves spinal mitogen-activated protein kinase activation in neurons and microglia via N-methyl-D-aspartate-dependent mechanisms. Mol Pharmacol. 2006;70(4):1246–54. doi: 10.1124/mol.106.025478. [DOI] [PubMed] [Google Scholar]
- Davies M, Brophy S, Williams R, Taylor A. The prevalence, severity, and impact of painful diabetic peripheral neuropathy in type 2 diabetes. Diabetes Care. 2006;29(7):1518–22. doi: 10.2337/dc05-2228. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–62. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Esposito E, Paterniti I, Mazzon E, Genovese T, Galuppo M, Meli R, Bramanti P, Cuzzocrea S. MK801 attenuates secondary injury in a mouse experimental compression model of spinal cord trauma. BMC Neurosci. 2011;12(1):31. doi: 10.1186/1471-2202-12-31. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Feldman EL, Stevens MJ, Russell JW, Peltier A, Inzucchi S, Porte JD, Sherwin RS, Baron A. The Diabetes Mellitus Manual. McGraw-Hill; 2005. Somatosensory neuropathy; pp. 366–384. [Google Scholar]
- Freire MA, Guimaraes JS, Leal WG, Pereira A. Pain modulation by nitric oxide in the spinal cord. Front Neurosci. 2009;3(2):175–81. doi: 10.3389/neuro.01.024.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Light touch induces ERK activation in superficial dorsal horn neurons after inflammation: involvement of spinal astrocytes and JNK signaling in touch-evoked central sensitization and mechanical allodynia. J Neurochem. 2010a;115(2):505–14. doi: 10.1111/j.1471-4159.2010.06946.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Ji RR. Targeting astrocyte signaling for chronic pain. Neurotherapeutics. 2010b;7(4):482–93. doi: 10.1016/j.nurt.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwak YS, Kang J, Unabia GC, Hulsebosch CE. Spatial and temporal activation of spinal glial cells: Role of gliopathy in central neuropathic pain following spinal cord injury in rats. Exp Neurol. 2011 doi: 10.1016/j.expneurol.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel KP, Dickie MM, Coleman DL. Diabetes, a new mutation in the mouse. Science. 1966;153(740):1127–8. doi: 10.1126/science.153.3740.1127. [DOI] [PubMed] [Google Scholar]
- Infante C, Diaz M, Hernandez A, Constandil L, Pelissier T. Expression of nitric oxide synthase isoforms in the dorsal horn of monoarthritic rats: effects of competitive and uncompetitive N-methyl-D-aspartate antagonists. Arthritis Res Ther. 2007;9(3):R53. doi: 10.1186/ar2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Mashimo T, Shibata M, Shibuta S, Yoshiya I. Rapid development of nitric oxide-induced hyperalgesia depends on an alternate to the cGMP-mediated pathway in the rat neuropathic pain model. Brain Res. 1998;792(2):263–70. doi: 10.1016/s0006-8993(98)00147-4. [DOI] [PubMed] [Google Scholar]
- Jander S, Schroeter M, Stoll G. Role of NMDA receptor signaling in the regulation of inflammatory gene expression after focal brain ischemia. J Neuroimmunol. 2000;109(2):181–7. doi: 10.1016/s0165-5728(00)00317-9. [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2(12):1114–9. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. 2002;22(2):478–85. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Gereau RWt, Malcangio M, Strichartz GR. MAP kinase and pain. Brain Res Rev. 2009;60(1):135–48. doi: 10.1016/j.brainresrev.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki Y, Kohno T, Zhuang ZY, Brenner GJ, Wang H, Van Der Meer C, Befort K, Woolf CJ, Ji RR. Ionotropic and metabotropic receptors, protein kinase A, protein kinase C, and Src contribute to C-fiber-induced ERK activation and cAMP response element-binding protein phosphorylation in dorsal horn neurons, leading to central sensitization. J Neurosci. 2004;24(38):8310–21. doi: 10.1523/JNEUROSCI.2396-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitto KF, Haley JE, Wilcox GL. Involvement of nitric oxide in spinally mediated hyperalgesia in the mouse. Neurosci Lett. 1992;148(1-2):1–5. doi: 10.1016/0304-3940(92)90790-e. [DOI] [PubMed] [Google Scholar]
- Komatsu T, Sakurada S, Kohno K, Shiohira H, Katsuyama S, Sakurada C, Tsuzuki M, Sakurada T. Spinal ERK activation via NO-cGMP pathway contributes to nociceptive behavior induced by morphine-3-glucuronide. Biochem Pharmacol. 2009;78(8):1026–34. doi: 10.1016/j.bcp.2009.06.106. [DOI] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10(9):895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever IJ, Pezet S, McMahon SB, Malcangio M. The signaling components of sensory fiber transmission involved in the activation of ERK MAP kinase in the mouse dorsal horn. Mol Cell Neurosci. 2003;24(2):259–70. doi: 10.1016/s1044-7431(03)00200-8. [DOI] [PubMed] [Google Scholar]
- Liao YH, Zhang GH, Jia D, Wang P, Qian NS, He F, Zeng XT, He Y, Yang YL, Cao DY, et al. Spinal astrocytic activation contributes to mechanical allodynia in a mouse model of type 2 diabetes. Brain Res. 2011;1368:324–35. doi: 10.1016/j.brainres.2010.10.044. [DOI] [PubMed] [Google Scholar]
- Malmberg AB, Yaksh TL. Spinal nitric oxide synthesis inhibition blocks NMDA-induced thermal hyperalgesia and produces antinociception in the formalin test in rats. Pain. 1993;54(3):291–300. doi: 10.1016/0304-3959(93)90028-N. [DOI] [PubMed] [Google Scholar]
- Mao J, Price DD, Mayer DJ, Lu J, Hayes RL. Intrathecal MK-801 and local nerve anesthesia synergistically reduce nociceptive behaviors in rats with experimental peripheral mononeuropathy. Brain Res. 1992;576(2):254–62. doi: 10.1016/0006-8993(92)90688-6. [DOI] [PubMed] [Google Scholar]
- Meller ST, Cummings CP, Traub RJ, Gebhart GF. The role of nitric oxide in the development and maintenance of the hyperalgesia produced by intraplantar injection of carrageenan in the rat. Neuroscience. 1994;60(2):367–74. doi: 10.1016/0306-4522(94)90250-x. [DOI] [PubMed] [Google Scholar]
- Obata K, Katsura H, Mizushima T, Sakurai J, Kobayashi K, Yamanaka H, Dai Y, Fukuoka T, Noguchi K. Roles of extracellular signal-regulated protein kinases 5 in spinal microglia and primary sensory neurons for neuropathic pain. J Neurochem. 2007;102(5):1569–84. doi: 10.1111/j.1471-4159.2007.04656.x. [DOI] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Kobayashi K, Dai Y, Mizushima T, Katsura H, Fukuoka T, Tokunaga A, Noguchi K. Role of mitogen-activated protein kinase activation in injured and intact primary afferent neurons for mechanical and heat hypersensitivity after spinal nerve ligation. J Neurosci. 2004;24(45):10211–22. doi: 10.1523/JNEUROSCI.3388-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson J, Axelsson G, Hallin RG, Gustafsson LL. Beneficial effects of ketamine in a chronic pain state with allodynia, possibly due to central sensitization. Pain. 1995;60(2):217–22. doi: 10.1016/0304-3959(94)00139-6. [DOI] [PubMed] [Google Scholar]
- Ren K, Hylden JL, Williams GM, Ruda MA, Dubner R. The effects of a non-competitive NMDA receptor antagonist, MK-801, on behavioral hyperalgesia and dorsal horn neuronal activity in rats with unilateral inflammation. Pain. 1992;50(3):331–44. doi: 10.1016/0304-3959(92)90039-E. [DOI] [PubMed] [Google Scholar]
- Rondon LJ, Privat AM, Daulhac L, Davin N, Mazur A, Fialip J, Eschalier A, Courteix C. Magnesium attenuates chronic hypersensitivity and spinal cord NMDA receptor phosphorylation in a rat model of diabetic neuropathic pain. J Physiol. 2010;588(Pt 21):4205–15. doi: 10.1113/jphysiol.2010.197004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscheweyh R, Goralczyk A, Wunderbaldinger G, Schober A, Sandkuhler J. Possible sources and sites of action of the nitric oxide involved in synaptic plasticity at spinal lamina I projection neurons. Neuroscience. 2006;141(2):977–88. doi: 10.1016/j.neuroscience.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Slack SE, Grist J, Mac Q, McMahon SB, Pezet S. TrkB expression and phospho-ERK activation by brain-derived neurotrophic factor in rat spinothalamic tract neurons. J Comp Neurol. 2005;489(1):59–68. doi: 10.1002/cne.20606. [DOI] [PubMed] [Google Scholar]
- Sorkin LS. NMDA evokes an L-NAME sensitive spinal release of glutamate and citrulline. Neuroreport. 1993;4(5):479–82. doi: 10.1097/00001756-199305000-00004. [DOI] [PubMed] [Google Scholar]
- Sullivan KA, Hayes JM, Wiggin TD, Backus C, Su Oh S, Lentz SI, Brosius F, 3rd, Feldman EL. Mouse models of diabetic neuropathy. Neurobiol Dis. 2007;28(3):276–85. doi: 10.1016/j.nbd.2007.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DA, Ren K, Besse D, Ruda MA, Dubner R. Application of nitric oxide synthase inhibitor, N omega-nitro-L-arginine methyl ester, on injured nerve attenuates neuropathy-induced thermal hyperalgesia in rats. Neurosci Lett. 1996;210(2):124–6. doi: 10.1016/0304-3940(96)12670-7. [DOI] [PubMed] [Google Scholar]
- Vareniuk I, Pacher P, Pavlov IA, Drel VR, Obrosova IG. Peripheral neuropathy in mice with neuronal nitric oxide synthase gene deficiency. Int J Mol Med. 2009;23(5):571–80. doi: 10.3892/ijmm_00000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong CS, Hsu MM, Chou YY, Tao PL, Tung CS. Morphine tolerance increases [3H]MK-801 binding affinity and constitutive neuronal nitric oxide synthase expression in rat spinal cord. Br J Anaesth. 2000;85(4):587–91. doi: 10.1093/bja/85.4.587. [DOI] [PubMed] [Google Scholar]
- Woolf CJ. Central sensitization: uncovering the relation between pain and plasticity. Anesthesiology. 2007;106(4):864–7. doi: 10.1097/01.anes.0000264769.87038.55. [DOI] [PubMed] [Google Scholar]
- Zajac JM, Latapie JP, Frances B. Opposing interplay between Neuropeptide FF and nitric oxide in antinociception and hypothermia. Peptides. 2000;21(8):1209–13. doi: 10.1016/s0196-9781(00)00261-8. [DOI] [PubMed] [Google Scholar]