

SUMMARY
Vertebrate Hedgehog (Hh) signaling is initiated at primary cilia by the ligand-triggered accumulation of Smoothened (Smo) in the ciliary membrane. The underlying biochemical mechanisms remain unknown. We find that Hh agonists promote the association between Smo and Evc2, a ciliary protein that is defective in two human ciliopathies. The formation of the Smo-Evc2 complex is under strict spatial control, being restricted to a distinct ciliary compartment, the EvC zone. Mutant Evc2 proteins that localize in cilia but are displaced from the EvC zone are dominant inhibitors of Hh signaling. Disabling Evc2 function blocks Hh signaling at a specific step between Smo and the downstream regulators protein kinase A and Suppressor of Fused, preventing activation of the Gli transcription factors. Our data suggest that the Smo-Evc2 signaling complex at the EvC zone is required for Hh signal transmission and elucidate the molecular basis of two human ciliopathies.
INTRODUCTION
Primary cilia have emerged as important signaling centers during development (Goetz and Anderson, 2010). A number of human genetic diseases, called ciliopathies, are caused by defects in cilia structure or function. Patients suffering from these syndromes display pleiotropic phenotypes that affect many organ systems, highlighting the ubiquity of cilia and their profound role in human physiology (Novarino et al., 2011). Although a considerable body of work has focused on the phenotypic consequences of defects in cilia and on the localization of signaling proteins in cilia, the biochemical mechanisms that drive signal transduction at cilia remain poorly understood.
The Hedgehog (Hh) signaling pathway is orchestrated at primary cilia, and many of the phenotypes seen in patients with ciliopathies can be attributed to defective Hh signaling (Huangfu et al., 2003; Goetz and Anderson, 2010). Proper Hh signal transduction critically depends on a set of protein trafficking events at cilia (Corbit et al., 2005; Haycraft et al., 2005; Rohatgi et al., 2007; Kim et al., 2009). When an Hh ligand, such as Sonic Hedgehog (Shh), is received by the cell, the 7-pass transmembrane (TM) protein Smoothened (Smo) accumulates to high levels in the ciliary membrane (Corbit et al., 2005). The mechanism by which Smo concentration in cilia ultimately leads to activation of the Gli family of transcription factors is one of the unsolved mysteries in the vertebrate Hh pathway. Smo has to overcome the two negative regulators of the Gli proteins, Suppressor of Fused (SuFu) and protein kinase A (PKA). Smo signaling promotes the transport of Gli and SuFu to the tip of the cilium, allowing Glis to dissociate from SuFu and enter the nucleus to transcribe target genes (Kim et al., 2009; Humke et al., 2010; Tukachinsky et al., 2010).
Despite the genetic and cell-biological evidence linking the Hh pathway to primary cilia, surprisingly few protein interactions or enzymatic links between ciliopathy proteins and core components of the Hh pathway have been described. An obstacle to dissecting Hh biochemistry at cilia is presented by the fact that many defects seen in ciliopathies compromise the structural integrity of cilia, making it difficult to disentangle direct from indirect effects.
Among the ciliopathies that lead to defects in Hh signaling (Goetz and Anderson, 2010), Ellis–van Creveld syndrome (EvC; MIM 225500) and Weyers Acrofacial Dysostosis (Weyers; MIM 193530) are two related inherited disorders that are uniquely characterized by ultrastructurally normal cilia (Ruiz-Perez et al., 2000, 2003; Galdzicka et al., 2002; Takeda et al., 2002; Blair et al., 2011). Mutations in the EVC2 gene can cause both EvC and Weyers and have been shown to impair Hh signaling in cardiac, skeletal, and orofacial tissues during development (Ruiz-Perez and Goodship, 2009; Blair et al., 2011). However, questions regarding the mechanism of Evc2’s function in Hh signaling, and the crucial issue of whether the effect of Evc2 is direct or indirect, have remained unresolved.
We find a direct role for Evc2 in the signaling step that translates Smo activation to the inhibition of SuFu and PKA. The ciliary accumulation of Smo in response to Hh signaling leads to the physical association of Evc2 with Smo at a spatially distinct ciliary compartment named the EvC zone. The biochemical pathophysiology of EvC and Weyers syndromes is explained by the failure of this signaling complex to assemble at the EvC zone.
RESULTS
Function and Subcellular Localization of Evc2 in Fibroblasts
Although patients with EvC and Weyers have a similar constellation of congenital anomalies, EvC is a recessive disorder and Weyers is a dominant disorder (Ruiz-Perez and Goodship, 2009). EvC is caused by loss-of-function mutations in both alleles of either EVC2 or a second gene called EVC (Ruiz-Perez and Goodship, 2009). These genes encode two single-pass type I TM proteins, Evc2 and Evc, that directly associate with each other (Blair et al., 2011). The small interfering RNA (siRNA)-mediated depletion of Evc2 from either NIH/3T3 fibroblasts or C3H10T1/2 multipotent mesenchymal cells inhibited the Shh-triggered induction of GLI1, a direct Hh target gene (Figure 1A; Figure S1A available online). Evc2 depletion also blocked the Hh-induced differentiation of C3H10T1/2 cell into osteoblasts (Figure 1B). Three controls rigorously established the specificity of the Evc2 siRNA (Figures S1B–S1E).
Figure 1. Evc2 Is a Positive Regulator of Hh Signaling Localized at the EvC Zone in Primary Cilia.
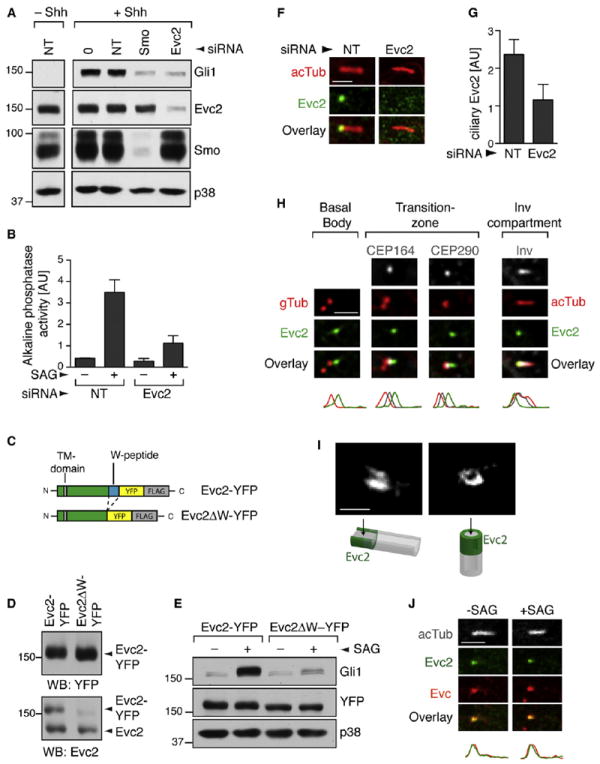
(A) Gli1 induction was used to measure Shh-triggered signaling in NIH/3T3 cells transfected with a nontargeting (NT) siRNA or siRNAs targeting Evc2 and Smo.
(B) The differentiation of C3H10T1/2 cells into osteoblasts was assayed by the increase in alkaline phosphatase activity (mean ± SD) after treatment with the Smo agonist SAG (36 hr). Depletion of Evc2 in this experiment is shown in Figure S1A.
(C) Schematic of the Evc2- and Evc2ΔW-YFP-FLAG proteins used to make stable cell lines. Evc2ΔW lacks the W-peptide (blue) at the C terminus.
(D) Evc2-YFP and Evc2ΔW-YFP were expressed at similar levels in stable cell lines (top, anti-YFP blot), and Evc2-YFP and endogenous Evc2 were present at similar levels (bottom, anti-Evc2 blot). Anti-Evc2 recognized Evc2ΔW-YFP poorly.
(E) SAG (12 hr) induced Gli1 protein in Evc2-YFP cells but not in Evc2ΔW-YFP cells.
(F) Localization of endogenous Evc2 (anti-Evc2, green) at cilia (red) marked by anti-acetylated tubulin (acTub) in NIH/3T3 cells.
(G) Mean (± 95% confidence interval [CI], n = 24) Evc2 fluorescence at cilia from such images.
(H) Localization of stably expressed Evc2-YFP (anti-YFP, green) relative to markers of various ciliary compartments. Plots below the images indicate normalized fluorescent intensities for each of the channels along the cilium from base (left) to tip (right).
(I) Images of Evc2 localization captured by STED microscopy. (J) Localization of Evc2-YFP (green) and endogenous Evc (red) at cilia (white) in cells exposed to SAG (2 hr). Scale bars: 2.5 μm and 0.5 μm.
See also Figure S1.
In contrast to EvC, Weyers is a dominant syndrome caused by specific mutations that delete the last 43 amino acids (aa) at the C terminus of Evc2, generating a dominant-negative protein that can inhibit Hh signaling (Valencia et al., 2009). Hereafter, we refer to this truncated protein as Evc2ΔW, and to the missing fragment in the protein as the Weyers peptide (W-peptide). We constructed two stable NIH/3T3 cell lines expressing full-length Evc2 or Evc2ΔW, each tagged with a YFP-FLAG tag (Figure 1C). Evc2-YFP, Evc2ΔW-YFP, and endogenous Evc2 were all expressed at equivalent levels in these cells (Figure 1D).
As predicted by the genetics of Weyers syndrome, the expression of Evc2ΔW produced a near-total block of Hh signaling stimulated by the direct Smo agonist SAG (Figure 1E). To ensure that the large YFP-FLAG tag did not alter function, we confirmed the effect of Evc2ΔW in stable cell lines expressing Evc2 or Evc2ΔW tagged with a single FLAG epitope (Figure S1F). Given their identical behavior, we hereafter refer to both sets of stable cell lines as Evc2 and Evc2ΔW cells. Taken together, both siRNA-based loss-of-function and dominant-negative expression confirm the critical role of Evc2 in Hh signaling and establish the Evc2ΔW cell line as a relevant model for dissecting the mechanistic role of Evc2.
Importantly, neither the frequency of ciliation nor the length distribution of cilia was affected when Evc2 was disabled by siRNA transfection or Evc2ΔW expression (Figures S1G–S1J). In addition, three proteins commonly used to label cilia—IFT88, Arl13b, and Inv (Pazour et al., 2000; Watanabe et al., 2003; Caspary et al., 2007)—all showed normal ciliary localization in Evc2ΔW cells (Figures S1K–S1M). The lack of a general requirement for Evc2 in cilia structure and function, along with the strong effects of Evc2 on Hh signaling, provided a unique opportunity to understand how a ciliopathy protein can regulate Hh signaling in the context of otherwise normal cilia.
EvC and Weyers syndromes have been classified as ciliopathies on the basis of a report that both Evc2 and Evc localize to basal bodies of primary cilia (Blair et al., 2011). Both endogenous Evc2 (Figures 1F and 1G) and stably expressed Evc2-YFP (Figure 1H) localized in a punctum near the base of the ciliary axoneme regardless of Hh pathway activity (Figure 1J). We defined the subciliary localization of Evc2-YFP relative to a series of markers for known ciliary compartments. In contrast to the previous reports, Evc2 was not localized at the basal body (Figure 1H), and instead was found just distal to the transition zone (TZ). The TZ was recently shown to form a barrier between the ciliary membrane and the plasma membrane (Craige et al., 2010; Chih et al., 2012; Garcia-Gonzalo et al., 2011; Williams et al., 2011). Analogously to some TZ proteins, Evc2 formed a ring-like collar around the cilium (Figure 1I). Evc, the second protein that is mutated in EvC syndrome, precisely colocalized with Evc2, consistent with the observation that these proteins form a complex (Figure 1J) (Blair et al., 2011).
In summary, Evc and Evc2 can traverse the TZ barrier and are colocalized in a narrow region, hereafter called the EvC zone, that sits between the TZ and the Inv compartment. This compartment is likely a general feature of primary cilia, since endogenous Evc and Evc2 display a similar localization pattern at the cilia base in both epithelial and mesenchymal cell lines and primary tissues (Figures S1N–S1P).
Identification of the Step in Hedgehog Signaling that Requires Evc2
Heretofore, the step in Hh signaling that is impaired in EvC and Weyers was unknown. We found that the expression of Evc2ΔW did not affect the levels of endogenous Evc2 or several key Hh pathway proteins (Figure S2A). We tested the requirement for Evc2 when Hh signaling was activated at different levels in the pathway. Evc2 and Evc2ΔW cells were treated with Shh, which activates the pathway at the level of Ptc1, or with SAG or 20-hydroxycholesterol, both of which activate Smo (Chen et al., 2002; Nachtergaele et al., 2012). In all three cases, Evc2ΔW blocked signaling (Figure 2A and Figures S2B and S2C). Strikingly, Evc2 was also required for Hh signaling initiated by a constitutively active oncogenic mutant of Smo, SmoM2 (Figure 2B) (Xie et al., 1998).
Figure 2. Evc2 Is Required for Hh Signaling at a Step between Smo and PKA.
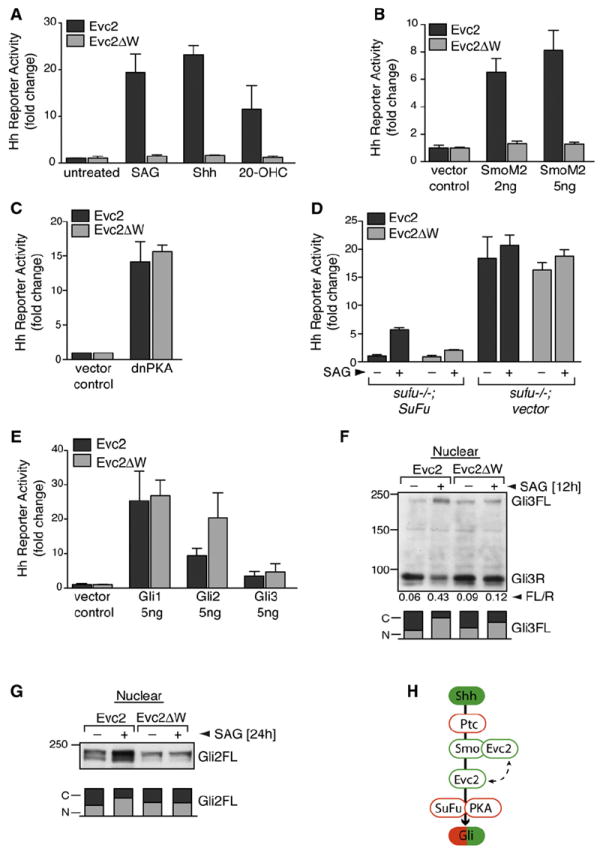
(A) Hh reporter activity in response to SAG, Shh, or 20-OHC (10 μM) in cells stably expressing Evc2 or Evc2ΔW. Effects on the induction of endogenous Gli1 are shown in Figure S2B.
(B and C) Hh reporter activity measured in Evc2 or Evc2ΔW cells transiently transfected with constitutively active SmoM2 (B) or dnPKA (C).
(D) Effect of Evc2ΔW on Hh signaling (36 hr) in sufu−/− MEFs or sufu−/− MEFs rescued by the stable expression of SuFu.
(E) Hh reporter activity measured in Evc2 or Evc2ΔW cells transiently transfected with the indicated Gli constructs.
(F and G) Levels of full-length (Gli3FL) and repressor (Gli3R) forms of Gli3 (F) or full-length Gli2 (G) in the nucleus of Evc2 and Evc2ΔW cells left untreated or treated with SAG. The number below each lane in (F) denotes the Gli3FL/R ratio, and the bar below each lane denotes the relative fraction of Gli2/3 in the nucleus [N] and the cytoplasm [C].
(H) Evc2 functions at the level of Smo or at a step between Smo and SuFu/PKA. Positive and negative regulators are colored in green and red, respectively. For (A)–(E), the mean values (± SD) are plotted.
See also Figure S2.
Although the precise signaling events downstream of Smo remain poorly understood, it is known that they depend on the negative regulators PKA and SuFu. PKA is a powerful suppressor of Hh signaling, and blockage of PKA activity is sufficient to fully activate Hh target gene transcription even in the absence of Shh (Lepage et al., 1995; Hammerschmidt et al., 1996; Tuson et al., 2011). Expression of dominant-negative PKA (dnPKA) induced Hh target gene transcription to equal degrees in both Evc2 and Evc2ΔW cells (Figure 2C). Mouse embryonic fibroblasts (MEFs) lacking SuFu (sufu−/− cells) also showed high levels of Hh target gene transcription consistent with activated signaling (Svärd et al., 2006). Hh reporter gene activity in sufu−/− cells was unaffected by the expression of either Evc2 or Evc2ΔW, and reintroduction of SuFu to these cells restored sensitivity to Evc2ΔW (Figure 2D). Finally, Evc2ΔW did not inhibit Hh reporter activity that was directly stimulated by coexpression of the three Hh transcription factors, Gli1, Gli2, and Gli3 (Figure 2E).
Smo activation regulates the Gli transcription factors in two distinct ways. When the Hh signal is received, Gli3R production is turned off, and instead Gli2FL and Gli3FL are converted to transcriptional activator proteins (GliA) that translocate into the nucleus to activate target genes. In the absence of signaling, both Evc2 and Evc2ΔW cells had equal amounts of Gli3R in the nucleus, showing that Evc2 does not regulate the conversion of Gli3FL to Gli3R (Figure 2F). However, Hh signaling was unable to extinguish Gli3R production in Evc2ΔW cells (Figure 2F). In addition, all aspects of SAG-induced GliA formation were blocked in Evc2ΔW cells. Gli3FL failed to dissociate from SuFu (Figure S2F), and both Gli3FL and Gli2FL proteins failed to translocate from the cytoplasm to the nucleus (Figures 2F and 2G; Figures S2D and S2E).
To summarize the above epistasis analysis, Evc2 is required in the Hh signaling pathway at the level of or immediately downstream of Smo but upstream of PKA and SuFu (Figure 2H). Evc2 may be required for Smo to adopt its fully active conformation in cilia, or it may relay the signal from Smo to downstream steps in signaling.
The Role of Evc2 in Trafficking Hh Pathway Proteins to Primary Cilia
Given that Evc2 is critical for all aspects of Smo signaling, we expected it to play a role in the trafficking of Smo to primary cilia. Surprisingly, we found that Smo accumulated in cilia normally in response to SAG in both Evc2 and Evc2ΔW cells and in NIH/3T3 cells depleted of endogenous Evc2 by siRNA (Figures 3A and 3B; Figures S3A and S3B). Smo activation and accumulation in cilia are correlated with an increase in the levels of Gli2, Gli3, and SuFu at the tips of cilia, a step that is thought be critical for the nuclear translocation of the Gli proteins (Kim et al., 2009; Tukachinsky et al., 2010). SAG-induced recruitment of endogenous Gli2, Gli3, and SuFu to the tips of cilia was blunted in Evc2ΔW cells and in cells depleted of Evc2 by siRNA (Figures 3C–3H; Figures S3C–S3E). Thus, Evc2 is required to relay Smo activation to the recruitment of SuFu and Gli proteins to the tips of primary cilia.
Figure 3. Evc2 Is Required for the Accumulation of Gli2, Gli3, and SuFu in Cilia.
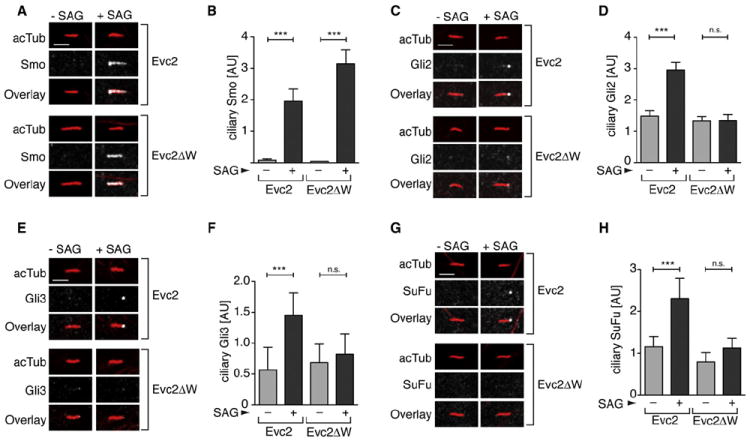
Confocal images show levels of endogenous Smo (A), Gli2 (C), Gli3 (E), and SuFu (G) (all white) at representative cilia (red) from Evc2 and Evc2ΔW cells left untreated or treated with SAG. Multiple cilia from such images were used to quantitate the mean (± 95% CI) fluorescence of Smo (n = 49) (B), Gli2 (n = 25) (D), Gli3 (n = 22) (F), and SuFu (n = 22) (H) at cilia. Treatment times were 4 hr for (B), (D), and (F), and 12 hr for (H). Statistical test: one-way ANOVA with Tukey posttest; p < 0.001 (***) and p > 0.05 (ns). Scale bars: 2.5 μm.
See also Figure S3.
A Localization Signal for the EvC Zone
To shed light on the mechanism of Evc2 function, we focused on Evc2ΔW, a dominant inhibitor of Hh signaling. Because Evc2ΔW was inserted into the membrane appropriately and transported along the secretory pathway normally (Figures S4A–S4C), we asked whether it localized in cilia like the wild-type (WT) protein. Evc2ΔW-YFP was efficiently targeted to cilia; however, its pattern of localization within the cilium was markedly different from that of Evc2-YFP. Instead of being concentrated in a punctum at the EvC zone, Evc2ΔW-YFP was distributed along the entire length of the ciliary membrane (Figures 4A–4C).
Figure 4. The W-Peptide Is a Localization Signal for the EvC Zone.

(A) Evc2ΔW-YFP (green) is localized along the entire length of cilia (red) in Evc2ΔW cells regardless of Hh signaling (2 hr).
(B) STED image of Evc2ΔW-YFP distribution at cilia.
(C) Evc2ΔW-YFP localization relative to various ciliary compartments (compare with Figure 1H).
(D) Mean (± SD) SAG-induced Hh reporter activity in cells transfected with the indicated deletion mutants of Evc2.
(E) Protein sequence of the region between residues 1177 and 1198 of Evc2 that was analyzed by alanine-scanning mutagenesis. For each of the ten mutants (Evc2A1–A10), the indicated stretch of four aa was mutated to alanines (A). The FV motif, which is critical for suppression of Hh signaling, is circled in red.
(F) Effects of the Evc2 mutants from (E) on Hh signaling induced by SAG (mean ± SD). The red line marks a 50% reduction in reporter activity compared with WT Evc2.
(G) T-Coffee multiple sequence alignment of the W-peptide in mouse, human, chicken, and fugu Evc2. Boxes encircle positions where Ala mutations produce (red) or fail to produce (green) a dominant-negative effect (see Figures S4D and S4E). The red line underlines the region subjected to Ala-scanning mutagenesis in (E). Identical (*), conserved (:) and semiconserved (●) residues are indicated.
(H) YFP-SmoW was constructed by transferring the W-peptide (blue) from Evc2 to the C terminus of YFP-Smo. Yellow star: YFP tag.
(I) Localization of YFP-Smo and YFP-SmoW (green) at cilia (red) after transient expression in NIH/3T3 cells. Figure S4F shows that YFP-SmoW is localized to the EvC zone.
(J) Localization of YFP-Smo and YFP-SmoW at the EvC zone (orange) or the whole cilium (blue) in individual cells (circles) expressing varying amounts of the two proteins after transient transfection. Whole-cell YFP fluorescence is plotted for each cell; the black bar marks the mean fluorescence (or expression level) for each construct (n = 30).
(K) A mutant W-peptide carrying the A5 mutation that overlaps the FV motif (YFP-SmoWA5) (E) and inhibits Hh signaling (F) cannot target Smo to the EvC zone. Scale bars: 2.5 μm and 0.5 μm in (B).
See also Figure S4.
The ability of Evc2ΔW to potently block Hh signaling provided a facile assay to uncover important sequence elements within the W-peptide. Using a combination of deletion analysis, alanine-scanning mutagenesis, and directed mutagenesis, we identified two contiguous residues within the W-peptide (F1185 and V1186) that have strong effects on the dominant-negative phenotype (Figures 4D–4G). Alanine (Ala) mutants overlapping these residues, but not several other Ala mutants of the W-peptide, blocked Hh signaling (Figures 4F and 4G; Figures S4D and S4E), demonstrating the selective importance of this FV motif.
Given that the W-peptide was required for EvC zone localization, we tested whether it could target a different ciliary membrane protein to this compartment. YFP-tagged Smo (YFP-Smo) is known to localize along the entire length of the ciliary membrane when overexpressed in cells (Rohatgi et al., 2009). Remarkably, the W-peptide was sufficient to restrict Smo to the EvC zone when fused to the C terminus of YFP-Smo (Figures 4H–4J and Figure S4F). The FV motif was again critical because mutation of this motif in the W-peptide abrogated its ability to target Smo to the EvC zone (Figure 4K). The W-peptide did not induce ciliary or EvC zone localization when fused to a Smo truncation mutant (SmoΔC) that was unable to localize to cilia (Figure S4G). Thus, the W-peptide deleted from Evc2 in patients with Weyers is not a cilia-targeting signal; rather, it is a portable sequence that is both necessary and sufficient to mediate the localization of ciliary membrane proteins to the EvC zone.
The Localization of Evc2 Mutants Predicts Their Phenotype in Hh Signaling
Our results suggested the intriguing idea that Evc2ΔW is a dominant-negative protein in Weyers patients because it is dispersed throughout the ciliary membrane rather than being restricted to the EvC zone. To investigate whether the ciliary localization of Evc2ΔW was necessary for its dominant-negative phenotype, we generated C-terminally truncated Evc2 proteins modeled on recessive disease alleles found in EvC patients (Ruiz-Perez and Goodship, 2009; Valencia et al., 2009). In comparison with Evc2ΔW (which lacks the C-terminal 43 aa), Evc2Δ1157-1220 and Evc2Δ1133-1220 lack slightly longer fragments spanning the C-terminal 64 and 88 aa, and Evc2Δ1157-1176 carries an internal deletion in this region (Figure 5A). Among these mutants, only Evc2Δ1157-1220 blocked Hh signaling (Figure 5A). Analogously to Evc2ΔW, Evc2Δ1157-1220 was targeted to cilia but distributed along the entire ciliary membrane (Figure 5A). In contrast, neither Evc2Δ1133-1220 nor Evc2Δ1157-1176 strongly blocked Hh signaling. The former was not targeted to cilia at all, and the latter was appropriately restricted to the EvC zone (Figure 5A).
Figure 5. The Targeting of Evc2 to the EvC Zone Is Critical for Its Function.

(A) A series of Evc2-YFP deletion mutants (ii) were characterized for their effects on SAG-stimulated Hh reporter activity (mean ± SD) (i) and for their localization in cilia (iii and iv). The localization of the Evc2 variants (green, anti-YFP) at cilia (red) was consistent across a range of expression levels in transiently transfected cells (graph in iv is drawn as in Figure 4J).
(B) Ciliary localization of selected Evc2 mutants (green, anti-YFP) described in Figure 4 at cilia (red) of transiently transfected cells (n ~ 20 for each construct). Scale bars, 2.5 μm.
To further test the correlation between mislocalization along the entire ciliary membrane and the dominant-negative phenotype, we examined a series of Evc2 mutants with varying effects on Hh signaling. In each case, mutants restricted to the EvC zone did not block Hh signaling, whereas those that were dispersed along the entire cilium were dominant inhibitors of the pathway (Figures 4F and 5B). Ala mutations (Evc2A4 and Evc2A5) overlapping the FV motif of the W-peptide that blocked Hh signaling also mislocalized Evc2 throughout the ciliary membrane (Figure 5B). We propose that this FV motif is required for a protein interaction that restricts Evc2 to the EvC zone and facilitates Smo signaling. More generally, the above correlations provide functional evidence that both targeting to the cilium and restriction to the EvC zone are critical for the proper function of Evc2 in Hh signaling. It is important to emphasize that prior to the work presented here, the only evidence that Evc2 functioned in cilia (or that EvC and Weyers are indeed ciliopathies) was the observation that it was localized in cilia (Blair et al., 2011).
Evc2 Associates with Smo in Response to Hh Signaling
The requirement for Evc2 at a specific step in Hh signaling led us to consider whether it associated with Hh pathway components that function at this step. We tested this association in Evc2-YFP cells, where both the Hh components and Evc2-YFP are expressed at endogenous levels (Figure 1D). To stabilize protein interactions during the detergent-solubilization step required to extract Evc2 from membranes, we treated cells with the membrane-permeable, reversible crosslinker dithiobis(succinimidyl propionate) (DSP). DSP was added to intact cells and then inactivated prior to cell lysis to circumvent artifactual crosslinking during lysis and extraction. In this binding assay, Evc2-YFP showed an interaction with Evc and Smo, but not with Gli2, SuFu, or PKA (Figure 6A). Although the Evc2–Evc interaction was constitutive, the interaction between Evc2-YFP and Smo was only detectable when Hh signaling was activated by Shh or SAG (Figures 6A and 6B).
Figure 6. Hh Signaling Promotes the Association of Smo with Evc2.
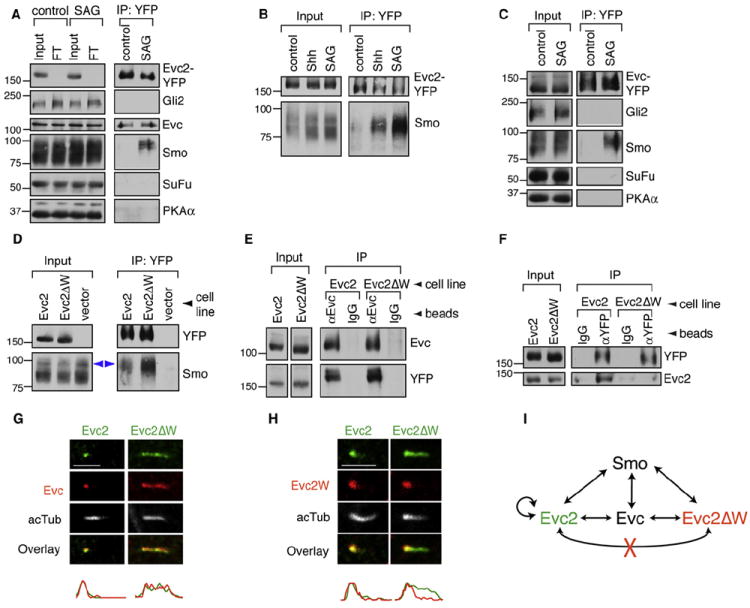
(A) Immunoblotting was used to assess the presence of Gli2, Evc, Smo, SuFu, and PKA-Cα in anti-YFP immunoprecipitates from Evc2-YFP cells left untreated (control) or treated with SAG (2 hr). Input and flow-through (FT) samples show the amount of each protein in the whole extract before and after the IP, respectively. In this panel and in (B)–(D), IP was performed after whole-cell crosslinking (DSP, 0.1 mM).
(B) Immunoblots showing the amount of endogenous Smo that coprecipitated with Evc2-YFP from cells left untreated or treated with SAG or Shh for 2 hr.
(C) Immunoblots showing the amounts of Gli2, Smo, SuFu, and PKA-Cα that coprecipitated with Evc-YFP from SAG-treated (2 hr) cells stably expressing the protein.
(D) Immunoblots showing the amount of Smo that coprecipitated with Evc2-YFP or Evc2ΔW-YFP from SAG-treated (2 hr) stable cell lines. A control IP was performed with cells lacking a YFP-tagged Evc2 bait protein (vector control). Blue arrows denote the higher molecular weight fraction of Smo that associated with Evc2.
(E) Immunoblots showing the amount of Evc2-YFP and Evc2ΔW-YFP that coprecipitated with endogenous Evc from SAG-treated (2 hr) cells on anti-Evc beads or control IgG beads.
(F) Immunoblots showing amounts of endogenous Evc2 that coprecipitated with Evc2-YFP or Evc2ΔW-YFP using anti-YFP beads or control IgG beads from cells treated with SAG (2 hr).
(G) Localization of endogenous Evc (red) at cilia (white) in Evc2 and Evc2ΔW cells (anti-YFP, green).
(H) An Evc2 antibody directed exclusively against the W-peptide (anti-Evc2W; Figure S5A) was used to assess the ciliary localization of Evc2 (red) in Evc2-YFP and Evc2ΔW-YFP cells (anti-YFP, green).
(I) Summary of the interactions between Evc2, Evc2ΔW, Evc, and Smo. Evc2 can self-associate, but Evc2ΔW cannot associate with Evc2. Scale bars: 2.5 μm.
See also Figure S5.
To demonstrate the specificity of the Smo-Evc2 interaction, we used a panel of different antibodies (summarized in Figure S5A) to assay the interaction between YFP-Smo and Evc2 in extracts from smo−/−;YFP-Smo MEFs (Rohatgi et al., 2009) without the use of a protein crosslinker. The interaction between Evc2 and YFP-Smo could be readily detected when endogenous Evc2 was isolated using an anti-Evc2 antibody or, in reciprocal immunoprecipitations (IPs), when YFP-Smo was isolated using three different antibodies (Figures S5B–S5D). Crosslinking with DSP substantially stabilized the interaction between Evc2 and YFP-Smo in a dose-dependent fashion (Figure S5E). Taken together, these IP experiments using five antibodies against independent epitopes in two different cell lines confirmed the Hh-stimulated interaction between Smo and Evc2. Consistent with the observation that Evc and Evc2 form a complex (Blair et al., 2011), we also detected an interaction between Smo and Evc (Figure 6C).
Having established these interaction assays, we sought to understand the molecular basis for the dominant-negative activity of Evc2ΔW. We started by testing whether Evc2ΔW could still bind to Smo and Evc. Compared with Evc2-YFP, Evc2ΔW-YFP showed a substantially stronger interaction with Smo (Figure 6D and Figure S5F). Thus, the interaction of Smo and Evc2 does not require the W-peptide. Moreover, the W-peptide was not required for the binding of Evc2 to Evc (Figure 6E). Next, we tested the ability of Evc2-YFP and Evc2ΔW-YFP to form complexes with endogenous Evc2. Evc2-YFP was able to bind endogenous Evc2, suggesting that Evc2 can form homo-oligomers in cells (Figure 6F). This ability to multimerize was compromised by loss of the W-peptide, since Evc2ΔW-YFP failed to bind endogenous Evc2 (Figure 6F). Consistent with these biochemical data, we found that Evc2ΔW expression could displace endogenous Evc (Figure 6G), but not endogenous Evc2 (Figure 6H), from the EvC zone and cause it to spread along the entire length of the cilium. Taken together, we propose that the critical biochemical defect in Evc2ΔW is its inability to multimerize with endogenous Evc2 (Figure 6I).
Spatial Restriction of the Smo-Evc2 Interaction to Primary Cilia
During our interaction analyses, we found that Evc2 selectively bound the fraction of total Smo that demonstrated slower mobility on SDS-PAGE (Figure 6D and Figure S5F), consistent with a post-Golgi glycosylation pattern (Figure S4C). Indeed, Smo that coprecipitated with Evc2 and Evc2ΔW was resistant to digestion with EndoH and sensitive to digestion with PNGaseF (Figure S6A), formally showing that it carried the glycosylation hallmark of transit through the Golgi.
Thus, the Smo-Evc2 interaction was both induced by Hh signaling and spatially restricted to a post-Golgi compartment. The primary cilium was the most likely location for this interaction because Evc2 and Smo were colocalized in the EvC zone after Hh pathway activation (by both conventional and superresolution microscopy), with little colocalization throughout the rest of the cell (Figure 7A; Figures S6B and S6C). Consistent with our IP data, Evc2ΔW, which was distributed throughout the cilium, showed a greater degree of overlap with Smo in the cilium (Figure 7A). An in situ proximity ligation assay (Duolink) demonstrated that Evc2 and Smo were located within 40 nm of each other at the cilium (Figures S6D–S6F).
Figure 7. Association of Evc2 and Smo at Primary Cilia.
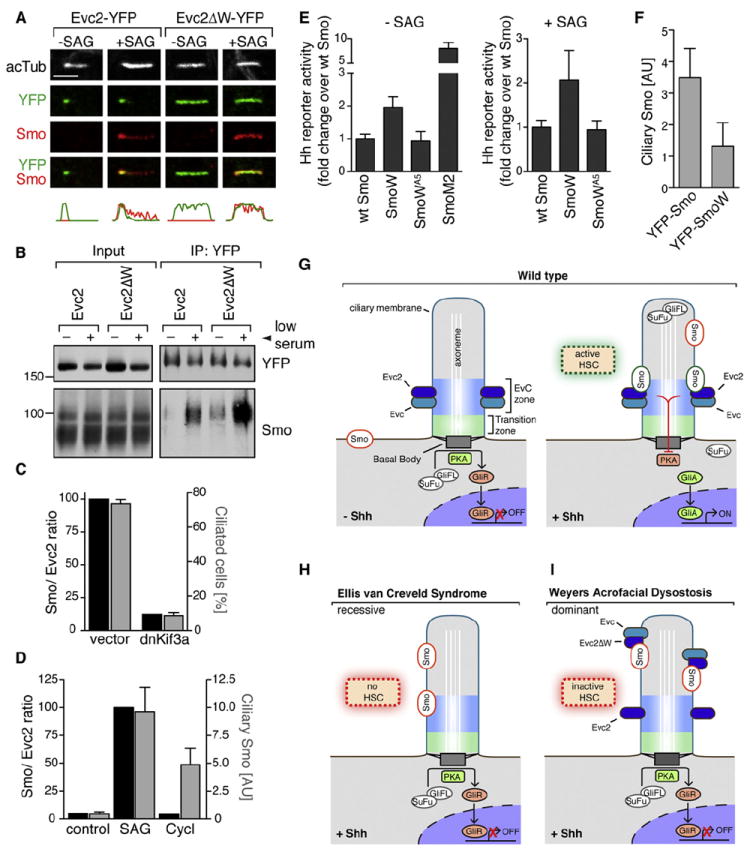
(A) Colocalization of Smo (red) and Evc2-YFP or Evc2ΔW-YFP (anti-YFP, green) at cilia (white) of stable cell lines left untreated or treated with SAG (4 hr). Scale bar: 2.5 μm.
(B) Immunoblots showing the amount of endogenous Smo that coprecipitated with Evc2-YFP and Evc2ΔW-YFP on anti-YFP beads from cells grown in high serum or low serum (to efficiently induce ciliation) and exposed to SAG (2 hr).
(C) Association of endogenous Smo with Evc2-YFP in cells expressing dnKif3a (to ablate cilia) or cells expressing an empty control vector. In dnKif3a or control cells, Smo-Evc2 association (left axis), expressed as the ratio of Smo pulled down per unit of Evc2, is compared with the percentage of cells that form cilia (right axis, mean ± SD). Primary data are shown in Figures S7A and S7B.
(D) Association between Smo and Evc2-YFP in cells left untreated or treated with SAG or cyclopamine (Cycl, 5 μM) for 2 hr. In control-, cyclopamine-, or SAG-treated cells, Smo-Evc2 association (left axis) is compared with the intensity of Smo fluorescence (mean ± 95% CI) at cilia (right axis). Primary data are shown in Figures S7G and S7H.
(E) Mean (± SD) Hh reporter activity in cells transiently expressing YFP-Smo, YFP-SmoW, or YFP-SmoWA5 and left untreated or treated with SAG. In each case, data are shown relative to WT YFP-Smo.
(F) Bar graph showing the mean (± 95% CI; n = 19) fluorescence of YFP-Smo or YFP-SmoW at cilia.
(G–I) Models for the function of Evc2 in Hh signaling. In all three models, the localization of WT Evc2 and Evc is restricted to the EvC zone, located just distal to the transition zone in cilia. (G) In the absence of Shh (left), PKA channels Gli proteins toward the formation of transcriptional repressors (Gli3R) and away from the formation of Gli activators (GliA). Shh (right) promotes the association of Smo with Evc/Evc2 in the EvC zone, leading to an active Hh signaling complex (HSC) that relieves the negative effect of PKA on the Gli proteins. The Gli-SuFu complex translocates to the tip of the cilium, ultimately leading to the dissociation of SuFu, the nuclear translocation of Glis, and the transcription of target genes. Only the population of Smo at the EvC zone participates in an active HSC. (H) In the recessive EvC syndrome, mutant Evc2 proteins fail to localize in cilia, preventing the formation of an active HSC. (I) In the dominant Weyers syndrome, the mutant Evc2ΔW protein is not confined to the EvC zone, thus sequestering Smo and Evc in inactive complexes and preventing assembly of a functional HSC. In all panels, active Smo is outlined in green and inactive Smo is outlined in red.
See also Figures S6 and S7.
Three independent experiments suggested that cilia were required for the interaction of Smo and Evc2. First, growth of cells in the presence of high serum, which is known to block ciliogenesis (Tucker et al., 1979), substantially inhibited the interaction between Smo and Evc2 (Figure 7B). Similar results were obtained with cells that stably expressed a dominant-negative fragment of the anterograde motor Kif3a (dnKif3a), which led to the loss of cilia in a majority of cells (Figure 7C; Figures S7A and S7B). Finally, the interaction between Smo and Evc2 was abrogated by independent mutations in Smo and in Evc2 that prevented the ciliary localization of either protein (Figures S7C–S7F).
Smo can adopt either an active or an inactive conformation in cilia (Rohatgi et al., 2009). Both SAG, a Smo agonist, and cyclopamine, a Smo antagonist, can induce the accumulation of Smo in cilia. However, the association between Smo and Evc2 was only detected in cells treated with SAG (Figure 7D; Figures S7G and S7H). Thus, the Smo-Evc2 interaction appears to be sensitive to both the integrity of the primary cilia and the active conformation of Smo.
Within the cilium, the EvC zone is the most likely place for an association between Smo and Evc2. However, because Smo is often found throughout the ciliary membrane, this raises an important question: precisely where in the cilium is Smo activity required to transduce the Hh signal? We tested the signaling activity of a Smo protein that was tethered to the EvC zone by fusion of the W-peptide (YFP-SmoW; Figure 4H). If Smo activity is required at the tips of cilia, as proposed by some models (Goetz et al., 2009), YFP-SmoW should show less signaling activity compared with the WT YFP-Smo protein, which localizes throughout the cilium. Instead, both with and without SAG, YFP-SmoW was ~2-fold more active than YFP-Smo, even though the former was present in cilia at ~2.6-fold lower levels (Figures 7E and 7F). Notably, SmoW is not a fully activated protein, because its activity is dwarfed by that of the constitutively active SmoM2 mutant and its activity can be suppressed by cyclopamine (Figure 7E and Figure S7I). Taken together, these results are consistent with the notion that Smo signaling at the cilium is compartmentalized in the EvC zone and facilitated by association with Evc2.
DISCUSSION
The primary cilium has been called a “Hedgehog signal transduction machine” (Goetz et al., 2009). The input into this machine is the accumulation of Smo in the ciliary membrane, and the output is activated Gli transcription factors that can transit to the nucleus and initiate the Hh transcriptional program. Our discovery of a Smo-Evc2 signaling complex sheds light on the signal transduction step between Smo and PKA/SuFu, a step that is poorly understood in mammalian Hh signaling. The association of Smo with Evc2 in a distinct compartment in primary cilia, the EvC zone, is required for the two outputs of Smo, the inhibition of Gli3 repressor formation and the induction of Gli2 and Gli3 activator formation (Figure 7G).
The EvC zone may represent an important signaling compartment in primary cilia, with implications for understanding ciliopathies and skeletal dysplasia syndromes. Interestingly, the EvC zone is located immediately adjacent to the TZ, which forms a gate for ciliary protein entry and is also populated by several ciliopathy proteins (Czarnecki and Shah, 2012). Crosstalk between these adjacent zones would provide a mechanism by which signaling events at the EvC zone could impact trafficking decisions made at the TZ. Targeting of Evc2 to the EvC zone is accomplished by two hierarchical but separable steps: (1) trafficking to the cilium and (2) restriction of Evc2 distribution to the Evc zone. This two-step mechanism for Evc2 localization provides a parsimonious explanation for the opposite modes of inheritance seen in recessive EvC and dominant Weyers disease. The mutant Evc2 proteins found in EvC are nonfunctional because they fail to localize to cilia (Figure 7H), whereas the mutants in Weyers interfere with Hh signaling because they are mislocalized throughout the ciliary membrane (Figure 7I).
Although Smo is absolutely required for Hh signaling in all tissues, Evc2 and Evc likely play more tissue-specific roles. In humans, loss-of-function mutations in EVC or EVC2 lead to clear Hh phenotypes in the skeleton, heart, and orofacial tissues, but few phenotypes in other Hh-patterned tissues, such as the nervous system and the gut (Ruiz-Perez and Goodship, 2009). At least in humans, this tissue specificity cannot be explained by a redundancy between EVC and EVC2, because those rare patients who carry a deletion that eliminates both genes do not show more severe phenotypes (Temtamy et al., 2008). We favor the model in which a different protein or mechanism appropriates the function of Evc2 in tissues such as the nervous system, because, at least in the mouse, neither EVC nor EVC2 is expressed there during development (Takeda et al., 2002; Ruiz-Perez et al., 2007). Given the importance of Smo as a drug target and the importance of Hh signaling in diverse tissues, this observation suggests that we may be able to interrupt Smo signaling in a tissue-selective manner by targeting accessory proteins, such as Evc2, rather than Smo itself.
EXPERIMENTAL PROCEDURES
Constructs and Reagents
Mouse full-length Evc2 (NM_145920.3) and Evc2ΔW (aa 1–1176) were tagged with either a single FLAG tag or a dual YFP-FLAG tag at the C terminus. Mouse full-length Evc (NM_021292.2) was tagged with a dual YFP-FLAG tag at the C terminus. Mutations were introduced by inverse PCR, the QuikChange method (Agilent), or gene synthesis (DNA2.0). We obtained SAG from Enzo Life Sciences, 20-OHC from Steraloids, cyclopamine from Toronto Research Chemicals, cycloheximide and puromycin from Sigma, PNGaseF and EndoH from New England Biolabs, DSP from Thermo Scientific, and n-dodecyl-β-d-maltopyranoside and cholesteryl-hemisuccinate from Affymetrix.
Cell Lines
NIH/3T3, C3H10T1/2, and IMCD3 cells were obtained from ATCC. The stable cell lines expressing tagged Evc, Evc2, and Evc2 mutants were produced by site-specific recombination into a single site in the genome of 3T3 cells using the Flp-In system (Invitrogen). The Evc2-YFP;dnKif3a and Evc2-YFP;vector cell lines were generated by infecting Evc2-YFP cells with a retrovirus carrying dominant-negative Kif3a cloned into pRetroX-pTuner or a retrovirus carrying the empty pRetroX-pTuner vector. Stable transductants were selected in 2 μg/ml puromycin.
Antibodies
Polyclonal antibodies against the intracellular domain of mouse Evc2 (aa 232–1220) or Evc (aa 461–1005) were generated in rabbits (Cocalico Biologicals) and affinity purified prior to use (Figure S5A). Anti-Evc2W, the antibody that selectively recognizes full-length Evc2 but not Evc2ΔW, was generated by affinity purification of the antisera against the W-peptide alone. Commercially available antibodies or polyclonal antisera that were previously described are listed in Supplemental Experimental Procedures.
Assays for Hedgehog Pathway Activation
Fibroblasts were grown to confluence in medium (high-glucose Dulbecco’s modified Eagle’s medium (DMEM), 0.05 mg/ml penicillin, 0.05 mg/ml streptomycin, 2 mM GlutaMAX, 1 mM sodium pyruvate, 0.1 mM MEM nonessential amino acid supplement) containing 10% fetal bovine serum (FBS; HyClone) and then switched to medium containing 0.5% FBS with pathway agonists for 24 hr (unless indicated otherwise). In all cases, SAG was used at 100 nM and Shh conditioned medium at a 1/4 dilution. Whole-cell lysates were prepared in 50 mM Tris pH 7.4, 150 mM NaCl, 2% [v/v] NP-40, 0.25% [w/v] deoxycholate, 1 mM dithiothreitol (DTT), and 1X Sigma Fast EDTA-free protease inhibitor cocktail. For Hedgehog reporter assays, cells were transiently transfected using Fugene 6 (NIH/3T3 cells) with a mixture (4:1) of a firefly luciferase reporter driven by a Gli-responsive promoter and a constitutive Renilla luciferase reporter (Sasaki et al., 1997) together with the construct of interest. Cells were grown to confluence and switched to medium containing 0.5% FBS with pathway agonists or antagonists for 24 hr. In the graphs, each data point represents the mean of triplicate wells, with error bars representing the standard deviation (SD).
Alkaline Phosphatase Assay
For the alkaline phosphatase expression assay (Hyman et al., 2009), C3H10T1/2 cells were transfected with an Evc2 or NT control siRNA, grown to confluency, and treated with SAG (100 nM) for 36 hr in 0.5% FBS containing DMEM. After lysis (100 mM Tris pH 9.4, 250 mM NaCl, 25 mM MgCl2, 1% [v/v] Triton X-100), alkaline phosphatase activity was measured using the CDP-Star Chemiluminescence reagent (Perkin Elmer).
IP and DSP Crosslinking
Whole-cell extracts or membrane extracts were incubated with antibodies covalently coupled to Protein A-coated magnetic beads or GFP-binding protein (GBP) covalently coupled to M270 carboxylic acid beads (both Dynal). To crosslink proteins prior to IP, cells were washed with D-PBS (PBS, 1 mM CaCl2, 1 mM MgCl2) and incubated with the crosslinker DSP for 30 min on ice. Following DSP inactivation (100 mM Tris pH 7.4, 30 min), cells were harvested and lysed in a DTT-free buffer (50 mM Tris pH 7.4, 150 mM NaCl, 2% [v/v] NP-40, 0.25% [w/v] deoxycholate, 10 μg/ml LPC, and 1X Sigma Fast EDTA-free protease inhibitor cocktail). After this extract was incubated with antibody-coupled beads, proteins were eluted and crosslinks were reversed with a reducing SDS sample buffer containing 50 mM tris-2-carboxyethylphosphine hydrochloride (TCEP).
siRNA and Endoribonuclease-Prepared siRNA Transfections
siRNA and endoribonuclease-prepared siRNA (esiRNA) reagents for Evc2 (siRNA: SASI_Mm_01_00106977; esiRNA: EMU014101), Smo (siRNA: SASI_ Mm_02_00346929, esiRNA: EMU047471), and nontargeting controls were obtained from Sigma. NIH/3T3 cells were reverse transfected with 38 nM of siRNA or esiRNA using Lipofectamine RNAiMax (Invitrogen) according to the manufacturer’s instructions.
Immunofluorescence and Microscopy
Cultured cells were fixed in 4% [w/v] paraformaldehyde (PFA) in PBS for 10 min at room temperature (RT) and washed in PBS (3 × 5 min). Fixed cells were incubated with blocking buffer (1% [v/v] normal donkey serum, 10 mg/ml [w/v] bovine serum albumin, 0.1% [v/v] Triton X-100 in PBS) for 30 min at RT. Primary antibodies were diluted in blocking buffer and used to stain cells for 1 hr at RT or overnight at 4°C. After washing (3 × 5 min in PBS containing 0.1% [v/v] Triton X-100) was completed, secondary antibodies were added in blocking buffer and incubated for 1 hr at RT. Stained cells were washed as above and mounted with DAPI containing anti-fade medium (Mowiol).
Microscopy was performed on an inverted Leica SP2 or SP5 laser scanning confocal microscope or an inverted Leica DMIRE2 epifluorescence microscope. Epifluorescence images, taken as z-stacks, were deconvolved (Auto-Quant). In all figures, cilia are oriented with their base to the left and their tip to the right. Although markers of the cilia base are only shown in selected images, ciliary orientation was explicitly established using a basal body marker (gamma tubulin) in all cases. For superresolution microscopy, cells were seeded on #1.5H coverslips (Zeiss) and processed for IF using secondary antibodies (Atto425 and Alexa488) compatible with two-color stimulated emission depletion (STED). Images were taken as z-stacks on a Leica TCS STED CW system and deconvolved using the built-in Leica STED-2D-deconvolution software.
Data Analysis
Films were scanned with an Epson Perfection V700 photoscanner as grayscale TIFF files (600 dpi). Quantitative analysis of band intensities was performed with ImageJ, and the data were transferred to GraphPad Prism for analysis. To calculate the fraction of GliFL in the nucleus or cytoplasm, we calculated the ratios N/(N+C) and C/(N+C), where N and C are the intensities of the GliFL band in nuclear and cytoplasmic fractions, respectively.
For the quantitative analysis of fluorescence intensities, all images were obtained with identical gain, offset, and laser power settings. To compare the mean Smo, Gli2, Gli3, and SuFu fluorescence levels at the primary cilium, we used a one-way ANOVA with a Tukey posttest to assess statistical significance. In addition, a Kruskal-Wallis test was used to compare the median fluorescence data to avoid assumptions about the normality of the underlying distribution. There was no difference between the two tests.
For the single-cell analysis of the ciliary localization of YFP-tagged Smo or Evc2 mutants (Figures 4E, 5B, and 5D), we measured whole-cell YFP immunofluorescence intensity (taken as a measure of protein expression) by outlining the entire cell and using the integrated density function of ImageJ. In the same cell, the localization of Smo or Evc2 was classified as absent, present in the EvC zone, or present throughout the whole cilium. The mean fluorescence derived from measurements of multiple cells reflects the average protein expression in the cell population.
Supplementary Material
Acknowledgments
We thank S. Peyrot for first drawing our attention to Evc2. We also thank the following for their contributions to this work: R. Toftgard (sufu−/− cells), P. Beachy (smo−/− cells and Gli constructs), B. Wang (Gli constructs), W. Benson (Evc2 clone), B. Yoder (anti-IFT88), T. Yokoyama (anti-Inv), E. Nigg (anti-CEP164), H. Khanna (anti-CEP290), J. Eggenschwiler (anti-Gli2), R. Moon (dnPKA construct), M. Scott (use of a confocal microscope and guidance), L. Milenkovic (help with IF of mouse embryonic tissues), K. Yeung and T. Desai (help with IF of mouse tissues), A. Seki (Arl13b antibody), P. Niewiadomski and S. Khan (dnKif3a construct), A. Lebensohn (GBP), S. Nachtergaele (Smo mutagenesis), and J. Kong and B. Novitch (chicken neural tube analysis). We thank U. Rüther, E.W. Humke, A. Guse, S. Nachtergaele, P. Niewiadomski, and F. Willmund for discussions and comments on the manuscript. K.V.D. and R.R. conceived and designed the experiments. K.V.D. performed the experiments and analyzed the data. C.E.H. contributed plasmids. K.V.D. and R.R. wrote the paper. This work was supported by the March of Dimes Foundation, the National Cancer Institute (CA129174), the Pew Foundation, and the Sontag Foundation.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.devcel.2012.07.004.
References
- Blair HJ, Tompson S, Liu Y-N, Campbell J, MacArthur K, Ponting CP, Ruiz-Perez VL, Goodship JA. Evc2 is a positive modulator of Hedgehog signalling that interacts with Evc at the cilia membrane and is also found in the nucleus. BMC Biol. 2011;9:14. doi: 10.1186/1741-7007-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspary T, Larkins CE, Anderson KV. The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell. 2007;12:767–778. doi: 10.1016/j.devcel.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of Smoothened activity. Proc Natl Acad Sci USA. 2002;99:14071–14076. doi: 10.1073/pnas.182542899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, Sandoval W, Peterson AS. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol. 2012;14:61–72. doi: 10.1038/ncb2410. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DYR, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- Craige B, Tsao CC, Diener DR, Hou Y, Lechtreck KF, Rosenbaum JL, Witman GB. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J Cell Biol. 2010;190:927–940. doi: 10.1083/jcb.201006105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarnecki PG, Shah JV. The ciliary transition zone: from morphology and molecules to medicine. Trends Cell Biol. 2012;22:201–210. doi: 10.1016/j.tcb.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdzicka M, Patnala S, Hirshman MG, Cai J-F, Nitowsky H, Egeland JA, Ginns EI. A new gene, EVC2, is mutated in Ellis-van Creveld syndrome. Mol Genet Metab. 2002;77:291–295. doi: 10.1016/s1096-7192(02)00178-6. [DOI] [PubMed] [Google Scholar]
- Garcia-Gonzalo FR, Corbit KC, Sirerol-Piquer MS, Ramaswami G, Otto EA, Noriega TR, Seol AD, Robinson JF, Bennett CL, Josifova DJ, et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat Genet. 2011;43:776–784. doi: 10.1038/ng.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz SC, Ocbina PJR, Anderson KV. The primary cilium as a Hedgehog signal transduction machine. Methods Cell Biol. 2009;94:199–222. doi: 10.1016/S0091-679X(08)94010-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerschmidt M, Bitgood MJ, McMahon AP. Protein kinase A is a common negative regulator of Hedgehog signaling in the vertebrate embryo. Genes Dev. 1996;10:647–658. doi: 10.1101/gad.10.6.647. [DOI] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Humke EW, Dorn KV, Milenkovic L, Scott MP, Rohatgi R. The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev. 2010;24:670–682. doi: 10.1101/gad.1902910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman JM, Firestone AJ, Heine VM, Zhao Y, Ocasio CA, Han K, Sun M, Rack PG, Sinha S, Wu JJ, et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc Natl Acad Sci USA. 2009;106:14132–14137. doi: 10.1073/pnas.0907134106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kato M, Beachy PA. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc Natl Acad Sci USA. 2009;106:21666–21671. doi: 10.1073/pnas.0912180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepage T, Cohen SM, Diaz-Benjumea FJ, Parkhurst SM. Signal transduction by cAMP-dependent protein kinase A in Drosophila limb patterning. Nature. 1995;373:711–715. doi: 10.1038/373711a0. [DOI] [PubMed] [Google Scholar]
- Nachtergaele S, Mydock LK, Krishnan K, Rammohan J, Schlesinger PH, Covey DF, Rohatgi R. Oxysterols are allosteric activators of the oncoprotein Smoothened. Nat Chem Biol. 2012;8:211–220. doi: 10.1038/nchembio.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novarino G, Akizu N, Gleeson JG. Modeling human disease in humans: the ciliopathies. Cell. 2011;147:70–79. doi: 10.1016/j.cell.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, Cole DG. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg 737, are required for assembly of cilia and flagella. J Cell Biol. 2000;151:709–718. doi: 10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317:372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Corcoran RB, Scott MP. Hedgehog signal transduction by Smoothened: pharmacologic evidence for a 2-step activation process. Proc Natl Acad Sci USA. 2009;106:3196–3201. doi: 10.1073/pnas.0813373106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Perez VL, Goodship JA. Ellis-van Creveld syndrome and Weyers acrodental dysostosis are caused by cilia-mediated diminished response to hedgehog ligands. Am J Med Genet C Semin Med Genet. 2009;151C:341–351. doi: 10.1002/ajmg.c.30226. [DOI] [PubMed] [Google Scholar]
- Ruiz-Perez VL, Ide SE, Strom TM, Lorenz B, Wilson D, Woods K, King L, Francomano C, Freisinger P, Spranger S, et al. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat Genet. 2000;24:283–286. doi: 10.1038/73508. [DOI] [PubMed] [Google Scholar]
- Ruiz-Perez VL, Tompson SWJ, Blair HJ, Espinoza-Valdez C, Lapunzina P, Silva EO, Hamel B, Gibbs JL, Young ID, Wright MJ, Goodship JA. Mutations in two nonhomologous genes in a head-to-head configuration cause Ellis-van Creveld syndrome. Am J Hum Genet. 2003;72:728–732. doi: 10.1086/368063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Perez VL, Blair HJ, Rodriguez-Andres ME, Blanco MJ, Wilson A, Liu Y-N, Miles C, Peters H, Goodship JA. Evc is a positive mediator of Ihh-regulated bone growth that localises at the base of chondrocyte cilia. Development. 2007;134:2903–2912. doi: 10.1242/dev.007542. [DOI] [PubMed] [Google Scholar]
- Sasaki H, Hui C, Nakafuku M, Kondoh H. A binding site for Gli proteins is essential for HNF-3beta floor plate enhancer activity in transgenics and can respond to Shh in vitro. Development. 1997;124:1313–1322. doi: 10.1242/dev.124.7.1313. [DOI] [PubMed] [Google Scholar]
- Svärd J, Heby-Henricson K, Persson-Lek M, Rozell B, Lauth M, Bergström Å, Ericson J, Toftgård R, Teglund S. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev Cell. 2006;10:187–197. doi: 10.1016/j.devcel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Takeda H, Takami M, Oguni T, Tsuji T, Yoneda K, Sato H, Ihara N, Itoh T, Kata SR, Mishina Y, et al. Positional cloning of the gene LIMBIN responsible for bovine chondrodysplastic dwarfism. Proc Natl Acad Sci USA. 2002;99:10549–10554. doi: 10.1073/pnas.152337899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temtamy SA, Aglan MS, Valencia M, Cocchi G, Pacheco M, Ashour AM, Amr KS, Helmy SMH, El-Gammal MA, Wright M, et al. Long interspersed nuclear element-1 (LINE1)-mediated deletion of EVC, EVC 2, C4orf 6, and STK32B in Ellis-van Creveld syndrome with borderline intelligence. Hum Mutat. 2008;29:931–938. doi: 10.1002/humu.20778. [DOI] [PubMed] [Google Scholar]
- Tucker RW, Pardee AB, Fujiwara K. Centriole ciliation is related to quiescence and DNA synthesis in 3T3 cells. Cell. 1979;17:527–535. doi: 10.1016/0092-8674(79)90261-7. [DOI] [PubMed] [Google Scholar]
- Tukachinsky H, Lopez LV, Salic A. A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J Cell Biol. 2010;191:415–428. doi: 10.1083/jcb.201004108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuson M, He M, Anderson KV. Protein kinase A acts at the basal body of the primary cilium to prevent Gli2 activation and ventralization of the mouse neural tube. Development. 2011;138:4921–4930. doi: 10.1242/dev.070805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia M, Lapunzina P, Lim D, Zannolli R, Bartholdi D, Wollnik B, Al-Ajlouni O, Eid SS, Cox H, Buoni S, et al. Widening the mutation spectrum of EVC and EVC2: ectopic expression of Weyer variants in NIH 3T3 fibroblasts disrupts Hedgehog signaling. Hum Mutat. 2009;30:1667–1675. doi: 10.1002/humu.21117. [DOI] [PubMed] [Google Scholar]
- Watanabe D, Saijoh Y, Nonaka S, Sasaki G, Ikawa Y, Yokoyama T, Hamada H. The left-right determinant Inversin is a component of node monocilia and other 9+0 cilia. Development. 2003;130:1725–1734. doi: 10.1242/dev.00407. [DOI] [PubMed] [Google Scholar]
- Williams CL, Li C, Kida K, Inglis PN, Mohan S, Semenec L, Bialas NJ, Stupay RM, Chen N, Blacque OE, et al. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J Cell Biol. 2011;192:1023–1041. doi: 10.1083/jcb.201012116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, Bonifas JM, Lam CW, Hynes M, Goddard A, et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391:90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.