

Abstract
The copper (Cu) exporter ATP7B mediates resistance to cisplatin (cDDP) but details of the mechanism are unknown. We explored the role of the CXXC motifs in the metal binding domains (MBDs) of ATP7B by investigating binding of cDDP to the sixth metal binding domain (MBD6) or a variant in which the CXXC motif was converted to SXXS. Platinum measurement showed that cDDP bound to wild type MBD6 but not to the SXXS variant. Wild type ATP7B rendered ovarian 2008 cells resistant to cDDP. In 2008 and in HEK293T cells, wild type ATP7B trafficked from TGN to peripheral locations in response to Cu or cDDP. A variant in which the CXXC motifs in all 6 MBDs were converted to SXXS localized correctly to the TGN but failed to traffic when exposed to either Cu or cDDP. Deletion of either the first 5 MBDs or all 6 MBDs resulted in failure to localize to the TGN. Neither the SXXS variant nor the deletion variant was able to mediate resistance to cDDP. We conclude that cDDP binds to the CXXC motifs of ATP7B and that this interaction is essential to the trafficking of ATP7B and to its ability to mediate resistance to cDDP.
Keywords: ATP7A, ATP7B, Metal binding sequences, Cisplatin, Platinum containing drugs
1. Introduction
Copper (Cu) transporters CTR1, ATP7A, ATP7B and the metallochaperone ATOX1 modulate the cellular pharmacology of the Pt containing drugs by controlling their uptake, efflux and subcellular distribution. CTR1 mediates the influx of cisplatin (cDDP) [1-4] and the exporters ATP7A (Menkes disease protein) and ATP7B (Wilson’s disease protein) sequester this and other Pt drugs into secretory vesicles and mediate their eventual efflux [5-7]. ATP7A and ATP7B are P1B-type ATPases that reside in the trans-Golgi network; they receive Cu1+ from ATOX1 that interacts with their N-terminal cytoplasmic domains and transport it across the vesicle membrane by utilizing ATP [8]. Each of the six N-terminal metal binding domains (MBD) of ATP7A and ATP7B contains a core CXXC motif to which Cu1+ becomes chelated. The structure of MBD is conserved in evolution with a ferredoxin βαββαβ fold [9,10] which is also found in ATOX1 and is the site to which cDDP binds [11,12]. NMR studies of ATOX1 [13], ATP7A [14] and ATP7B [15] show that MBD binds Cu1+ to the two surface-exposed cysteines in the metal binding loop and undergo structural changes that are similar in all of these proteins [10,16-20]. The CXXC motifs in the single MBD of ATOX1 and the six MBD units in the N-terminal cytosolic regions of ATP7A and ATP7B are required for the efflux of Cu. Among the 6 MBD units of ATP7A and ATP7B, mutational studies have identified a particularly important role for the sixth MBD which is the closest to the trans-membrane pore. The sixth MBD is likely to receive Cu from the fourth MBD and propel it toward the central pore [20-23]. In the case of ATP7B, the importance of the sixth MBD has been documented by the fact that missense mutations in this region, of which the G591D in turn 2 is the best known, cause Wilson’s disease [24]. This mutation is likely to alter the stability of MBD6 and renders it insoluble [20,24].
The finding that the cysteines that bind Cu in ATOX1 also chelate cDDP [11,12] raises the possibility that the MBDs of ATP7A and ATP7B could potentially chelate this Pt-containing drug. cDDP has been reported to form complexes with several other proteins through cysteine and methionine residues [25-27] and thus can be expected to interact with ATP7A and ATP7B. Our previous study showed that cDDP can be transported by ATP7B into subcellular vesicles in Sf9 cells thus indicating a direct interaction between cDDP and ATP7B but left unanswered the question of whether the interaction was mediated by the CXXC motifs.
To understand how ATP7A and ATP7B modulate the cellular pharmacology of cDDP, this study examined the role of cysteine residues in the CXXC motifs of ATP7B. We show here that the cDDP binds to the CXXC motif in the sixth MBD of ATP7B and causes its multimerization. Truncation of most of the MBDs of ATP7B, or conversion of the CXXC motifs of all 6 MBDs to SXXS, eliminated the ability of ATP7B to confer resistance to cDDP, to regulate the accumulation of this drug and to undergo Cu- and cDDP-induced trafficking.
2. Experimental section
2.1. Reagents
The lentiviral vector, pLVX-mCherry-C1, In-Fusion cloning kit, packaging kit and anti-mCherry antibodies were from Clonetech, Palo Alto, CA. pMAL/c2x vector, restriction enzymes, maltose binding resin, and antibodies to maltose binding protein were from New England Biolabs, Ipswich, MA. Secondary antibodies, fluorescent reagents, Superscript III, PCR kits, Genetailor mutagenesis kit, PCR2.1- TOPO vector, media and sera were from Invitrogen, Carlsbad, CA. P230 monoclonal antibody was from BD Biosciences, San Diego, CA. Cisplatin was from Teva Parental Inc., Irvine, CA. Antibodies to tubulin were from Sigma, St. Louis, MO. Complete protease inhibitor was from Roche, Nutley NJ. Electrophoresis gels and Bradford reagent were from Bio Rad, Richmond, CA. Other chemicals were from Sigma, St. Louis, MO.
2.2. DNA constructs
All constructs of ATP7B were generated from a 2008 ovarian carcinoma cell cDNA library generated by Superscript III and amplified by PCR using oligonucleotides listed in Table 1. The MBD6 sequence spanning from amino acids 561–633 was cloned into pCR2.1-TOPO from which it was excised by EcoR1 and cloned into pMAL/c2x. After transformation into Escherichia coli BL21 (DE3), and sequence verification, a bacterial clone was selected for the expression of MBD6 fused to the maltose binding protein which will be referred to herein as MBD6. Conversion of both cysteines in MBD6 to serines was accomplished with the Genetailor kit using the wild type pMAL-MBD6 as template. The wild type ATP7B, ATP7B Δ1–5 (in which MBDs 1–5 spanning from amino acids 1–539 were truncated), and ATP7B Δ1–6 (in which MBDs 1–6 spanning from amino acids 1–599 were truncated) were PCR amplified from a 2008 cDNA library. The ATP7B variant in which all the CXXC motifs were converted to SXXS was PCR amplified from the plasmid vector 0CMB398 that was generously provided by Dr. S. La Fontaine and Dr. J.F. Mercer (University of Melbourne, Melbourne Australia) [22]. All ATP7B variants were cloned into pLVX-mCherry-C1 vector using the In-Fusion cloning kit.
Table 1.
Oligonucleotides used for cloning ATP7B variants.
| MBD6 | Fa TCCGATGGCAACATTGAGCT |
| Ra TTACTGGGCCAGGGAAGCATGAA | |
| SXXS | F GACAATCACAGGGATGACCTCTGCGTCCTCTGTCCACAAC |
| R GTCATCCCTGTGATTGTCAGC | |
| ATP7B | F ATGCCTGAGCAGGAGAGACAG |
| R TCAGATGTACTGCTCCTCAT | |
| ATP7B Δ1–6 | F GCCACCAGCAAAGCCCTTGTTAAG |
| R TCAGATGTACTGCTCCTCATC | |
| ATP7B Δ1–5 | F CTCGAGATAGCTCAGTTCATC |
| R TCAGATGTACTGCTCCTCATC | |
| ATP7B SXXS | F ATGCCTGAGCAGGAGAGACAG |
| R TCAGATGTACTGCTCCTCAT |
F, forward; R, reverse.
2.3. Cell culture and expression of lentiviral constructs of ATP7B
Human ovarian carcinoma 2008 cells were maintained in RPMI medium containing 10% fetal calf serum; HEK293T cells were cultured in high glucose DMEM with 1 nM sodium pyruvate, and 1 nM essential amino acids. Cells were incubated at 37 °C, 5% CO2. Lentiviral stocks of ATP7B variants were produced in HEK293T cells and used to transduce 2008 ovarian carcinoma cells or HEK293T cells [28]. Selection was made with 10 μg/mL puromycin. A pool of cells expressing high levels of the fluorescent mCherry tag was obtained by three rounds of FACS sorting.
2.4. Production and purification of recombinant MBD6
Plasmids expressing either maltose binding protein alone or maltose binding protein (MBP) fused to MBD6 were transformed into competent E. coli BL21 (DE3) and grown in LB containing 100 μg/mL ampicillin. For protein purification, cultures were grown in minimal medium M9 containing 3% LB medium and incubated at 37 °C at 260 rpm until OD600 reached ~0.6 after which the temperature was reduced to 30 °C and 0.3 μM IPTG (isopropyl-β-d-thiogalactoside) was added. The bacteria were harvested at 4 °C by centrifugation at 12,000×g for 45 min. The pellets were resuspended in 20 mL lysis buffer (10 mM HEPES, pH 7.6, 150 mM NaCl, 1% DMSO, 1 μg/mL DNAse 1, 0.25 mg/mL lysozyme, Complete protease inhibitor), incubated at room temperature for 30 min, sonicated on ice for 6 min and following centrifugation at 4 °C and 16,000×g for 30 min, incubated for 1 h with 200 mM of Cu chelators tetrathiomolybdate or bathocuproine sulfate and 0.5 mM of the reducing agent Tris-(hydroxypropyl)phosphine at 4 °C. The lysate was loaded onto amylose columns that were pre-equilibrated with 10 column volumes of binding buffer (100 mM NaCl, 10 mM HEPES, pH 7.5, 1 mM NaN3, 20mM β-mercaptoethanol (BME)). After 4 washes with 5–10 column volumes of the same buffer, MBP or MBP-MBD6 was eluted with binding buffer containing 10mM maltose. For some experiments, MBD6 was excised fromthemaltose binding protein using Factor Xa in a buffer containing 100 mM NaCl, 50mM HEPES, pH 7.5, and 20 mM BME. The cut protein was injected into an FPLC system (BIO-RAD, Richmond CA) and purified on a Superdex75 column (GE, Piscataway, NJ) and concentrated with an Amicon Ultra Cell filtration unit (Millipore, Billerica, MA). All samples were kept in the presence of 20mM BME until use. BME was removed by washing the samples under anaerobic conditions with binding buffer in a Millipore filtration unit.
2.5. Analysis of the interaction of cDDP with MBD6 with UV spectrometry
The absorbance at 280 nm reflecting the formation of Pt–sulfur bonds and disulfides was measured as a function of time using a single beam spectrophotometer (Beckman model DU530). Triplicate samples of 1.0 mL each containing 50 μM protein in the binding buffer were used. The spectrophotometer was immediately zeroed after cDDP addition and the reaction progress was measured in a 1-cm path length CVD-UV disposable Cuvettes (Ocean Optics, Dunedin, FL) for 2 h at room temperature. The control sample contained the same reaction mixture without cDDP.
2.6. Analysis of the interaction of cDDP with MBD6 and MBD6-SXXS using gel filtration, ICP-MS or ICP-OES
250 μL aliquots of a reaction mixture containing 0.2 mg/mL protein in binding buffer were incubated with either native cDDP (4:1 molar ratio cDDP to protein) or with 1% volatile reducing agent triethylphosphene for 24 h at 37 °C. The reaction mixtures were placed in a Millipore filtration unit and washed with 100 volumes of binding buffer (without BME) under anaerobic conditions. The Pt levels were measured by ICP-MS. Control levels of Pt were obtained from the analysis of Pt binding to MBP which was subtracted from values obtained for MBP-MBD6 and the mutant MBD6-SXXS. Some experiments were performed by interacting a twofold molar excess cDDP with the proteins while they were bound to the amylose resin in columns and incubating them for 5, 30, or 60 min. The columns were then washed thoroughly with 10 column volumes of binding buffer (without BME) and then assayed for Pt by ICP-OES. Protein levels were determined by Bradford assay (BioRad, Richmond, CA).
2.7. Chemical modification of cysteine residues in MBD6
All manipulations were conducted in an anaerobic chamber. The apo form of the MBD6 was first washed thoroughly with binding buffer to remove the reducing agent BME and then incubated for 3 h with 40 mM of N-ethyl maleimide (NEM) at 37 °C. NEM was removed by 4 washes with binding buffer using a Millipore filtration unit, followed by dilution with the binding buffer and protein determination by Bradford assay. After this step 14 μg aliquots of the sample were incubated with 100 fold molar excess of cDDP (3 mM) for 1 h at 37 °C and then resolved on non-denaturing polyacrylamide gels. Gel electrophoresis was carried as previously reported [29].
2.8. Assay of cytotoxicity and cellular accumulation of cDDP
Sensitivity to cDDP was determined using the Cell Count Kit 8 (CCK-8) colorimetric assay (Dojindo, Gaithersburg, MD) in which highly water-soluble tetrazolium salt, WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt] is reduced by dehydrogenase activities in cells to give a yellow-color formazan dye. The amount of yellow formazan formed during a 4 h exposure to the tetrazolium salt is an indicator of the number of live cells in the culture at the time the tetrazolium is added. We used a 1 h exposure to cDDP and a 5 day period of growth prior to addition of the tetrazolium salt to the culture. The data is expressed as percent survival which is calculated as the number of cells in the cDDP treated cultures relative to that in the untreated control cultures. Pt content was measured by ICP-MS as previously reported [6].
2.9. Fluorescent microscopy
Cells were cultured on coverslips coated with 5 μg/mL gelatin. They were allowed to attach overnight and then they were exposed for 15 min to either 30 μM cDDP or 100 μM CuCl2 at 37 °C. Cells were quickly rinsed with cold PBS and fixed with 3.7% formaldehyde in PBS for 5 min, permeabilized with 0.3% Triton X 100 in PBS for another 5 min, blocked with 5% BSA in PBS for 1 h, incubated successively with primary and secondary antibodies for 1 h each, for 30 min with Hoechst 33342 and FITC-tagged phalloidin, washed for 15 min three more times and then mounted on slides with Gelvatol. Imaging was performed using a DeltaVision deconvolution microscope system (Applied Precision, Inc., Issaquah, WA) at the UCSD Cancer Center’s Digital Imaging Shared Resource Facility.
3. Results
3.1. cDDP binds to MBD6 via the CXXC motif
The wild type form of the sixth MBD of ATP7B was produced in E. coli as a recombinant protein fused to the C-terminal end of maltose binding protein. We also produced a mutant form of the sixth MBD in which both cysteines in the CXXC motif were converted to serines. Maltose binding protein without the MBD was produced as a control. The proteins were captured on amylose resin and reacted with a two-fold molar excess of cDDP for 5, 30, or 60 min following which the resin was washed, the proteins eluted and the amount of bound Pt measured by ICP-MS. As shown in Fig. 1A significant amounts of Pt were found bound to the wild type MBD6 at all 3 time points. The mutant MBD6 in which the CXXC motif was converted to SXXS failed to bind any more Pt than the maltose binding protein by itself. By the end of the 1 h incubation, the wild type MBD6 had bound 1.75±0.19 mol Pt/mol, the SXXS mutant 0.42±0.013 mol Pt/mol (p=0.0023) and the maltose binding protein 0.57±0.012 mol Pt/mol (p=0.0057). The wild type MBD6 accumulated cDDP at a rate of 0.08 mol Pt/min/mol, the SXXS mutant at 0.005±0.0001 and the maltose binding protein alone at 0.007±0.001Pt/min/mol.
Fig. 1.
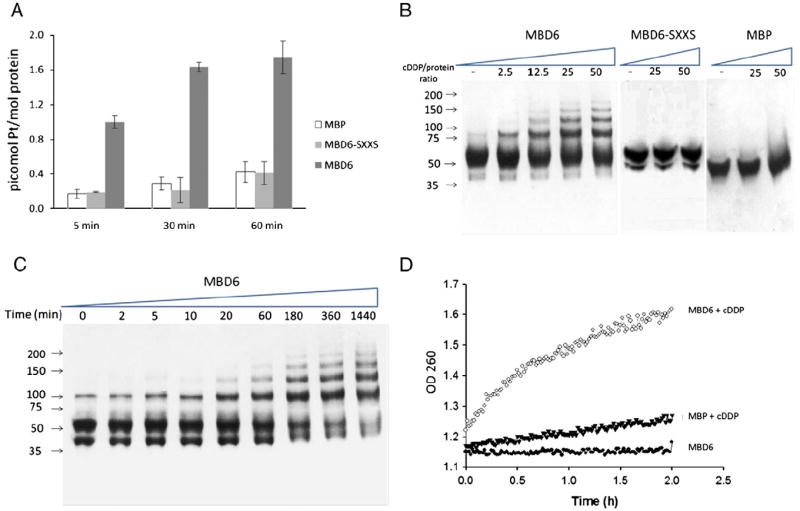
cDDP binds and multimerizes MBD6. (A) ICP-MS analysis of cDDP binding when a two-fold molar excess was incubated with maltose binding protein (MBP), MBD6-SXXS and MBD6 for 5, 30 or 60 min. (B) Interaction of MBD6, MBD-SXXS and MBP with cDDP analyzed by gel electrophoresis following incubation for 1 h of the protein samples with increasing levels of cDDP as indicated. (C) Time course of interaction of MBD6 with a 25 molar excess cDDP analyzed by gel electrophoresis. (D) UV spectrometric analysis of the interaction of cDDP with MBD6. Absorption was measured at 280 nm at 60 s intervals for 2 h. (●), MBD6 in the absence of cDDP; (▼) MBP and (○) MBD6 in the presence of a 25 molar excess cDDP. Each data point in (A) is the average of 3 independent experiments.
3.2. cDDP causes CXXC-dependent multimerization of MBD6
PAGE and UV spectrometric analysis demonstrated that incubation of MBD6 with various molar ratios of cDDP, or with a fixed concentration of cDDP for varying periods of time, resulted in a progressive multimerization of MBS6 (Fig. 1B–D). Non-denaturing PAGE analysis showed that incubation of wild type MBS6 for 1 h with increasing molar excess of cDDP (0 to 50-fold) caused multimerization to produce forms whose molecular weights were consistent with dimers, trimers, tetramers and pentamers (Fig. 1B). This was dependent on the CXXC motif, and specific to MBD6, was shown by the fact that the mutant MBD6 in which the CXXC motif had been converted to SXXS, and the maltose binding protein itself, failed to form multimers. Multimerization of wild type MBD6 was progressive with time as shown in Fig. 1C, and exposure of MBD6 to a 25 molar excess of cDDP for long time periods resulted in the formation of high molecular weight complexes. DDP-induced multimerization of MBD6 was rapid as complexes with molecular weights consistent with dimers and trimers were detected as early as 10 min; at the end of the 24 h incubation very little MBD monomer was left but high levels of dimers, trimers, tetramers and pentamers were visible. Multimerization of wild type MBD6 was induced by cDDP but not by comparable concentrations of Cu or incubation in atmospheric oxygen (data not shown).
The binding of cDDP to the wild type MBD6 was confirmed using a UV280 spectrophotometric assay which permitted sequential measurements over a period of 2 h. Wild type MBD6 and the maltose binding protein were exposed to a 20-fold molar excess of cDDP and the UV280 absorption was recorded at 1 min intervals. In the absence of cDDP there was no change in the absorbance of either protein during the 2 h incubation at 37 °C under aerobic conditions. However, as shown in Fig. 1D, in the presence of cDDP there was a large and progressive increase in the UV280 absorbance of MBD6; a much more muted change was observed when maltose binding protein alone was incubated with cDDP.
DTT is a strong reducing agent that reacts readily with DDP. The addition of 20 mM DTT to the reaction blocked the ability of 1 mM DDP to induce multimerization. However, when MBD6 was incubated with a 20-fold molar excess of cDDP for 1 h, washed extensively and then reacted with 20 mM DTT, it took 24 h to reverse the multimerization to visible degrees as shown in Fig. 2A indicating that the binding of cDDP to MBD was extremely strong.
Fig. 2.
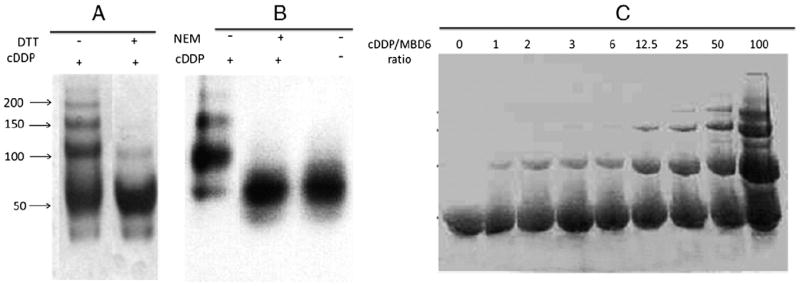
Effect of DTT, NEM and Cu on the interaction of cDDP with MBD6. (A) MBD6 was treated for 1 h with a 25 molar excess cDDP (first lane), then washed and incubated with 40 mM DTT. Visible reversal of MBD6 oligomerization was only detected after 24 h of incubation with 40 mM DTT. (B) Effect of blocking cysteine residues in cDDP-induced multimerization of MBD6 was assayed by pre-incubating MBD6 with 1000-fold molar excess NEM for 3 h. First lane, MBD incubated with 100 fold molar excess cDDP; second lane, MBD pre-treated with NEM for 3 h and then washed and incubated with 100 fold molar excess cDDP; third lane is MBD treated without NEM or cDDP.
To provide further evidence that the cysteine residues in the wild type MBD6 were responsible for the binding of DDP and its multimerization, we first blocked the cysteine residues of MBD6 by exposing the purified protein for 3 h to 1000-fold molar excess N-ethyl maleimide (NEM), a reagent that specifically binds to exposed cysteines on proteins. Following removal of the NEM by excessive washing, the wild type MBD6 was exposed to a 100-fold molar excess of cDDP for 1 h. Fig. 2B shows that pre-treatment with NEM blocked the ability of cDDP to trigger multimerization suggesting that the cysteine residues of the wild type MBD6 are necessary for this reaction.
3.3. Pretreatment with Cu does not affect cDDP binding to MBD6
Since the cysteine residues are involved in the binding of MBD6 with Cu, we sought to determine whether cDDP is capable of replacing Cu in MBD6. MBD6 was incubated for 1 h with 1 mM Cu1+ under anaerobic conditions following which excess Cu was removed by filtration and then 40 μg protein aliquots were interacted with increasing concentrations of cDDP. As shown in Fig. 2C, cDDP caused multimerization of MBS6 in all of the samples consistent with the conclusion that cDDP dislodges Cu from its binding site.
3.4. CXXC motifs of ATP7B MBDs are required for resistance to cDDP
To determine the extent to which the MBDs, and specifically their CXXC motifs, are required for ATP7B to mediate resistance to cDDP, a series of vectors was constructed that was capable of expressing wild type or variant forms of ATP7B fused to the C-terminal end of the red fluorescent protein mCherry (mC). For this purpose we used the pLVX-mCherry C1 lentiviral vector and expressed the various forms of ATP7B in human ovarian carcinoma 2008 cells. The variants tested included: mC-ATP7B, encoding the full length wild type ATP7B; mCATP7B Δ1–5, in which MBDs 1–5 were deleted; mC-ATP7B Δ1–6 that lacked all six MBDs; and, mC-ATP7B SXXS in which the cysteine residues in the CXXC motifs in all of the six MBDs were converted to serines. Fig. 3 shows both flow cytometric and western blot analyses documenting the expression of each of these variants in 2008 cells along with the expression of wild type mCherry alone (2008-mC) that served as a control. With the exception of the 2008-mC that expressed very high levels of the native mCherry protein, the level of expression of each of the mC-tagged ATP7B variants was similar.
Fig. 3.
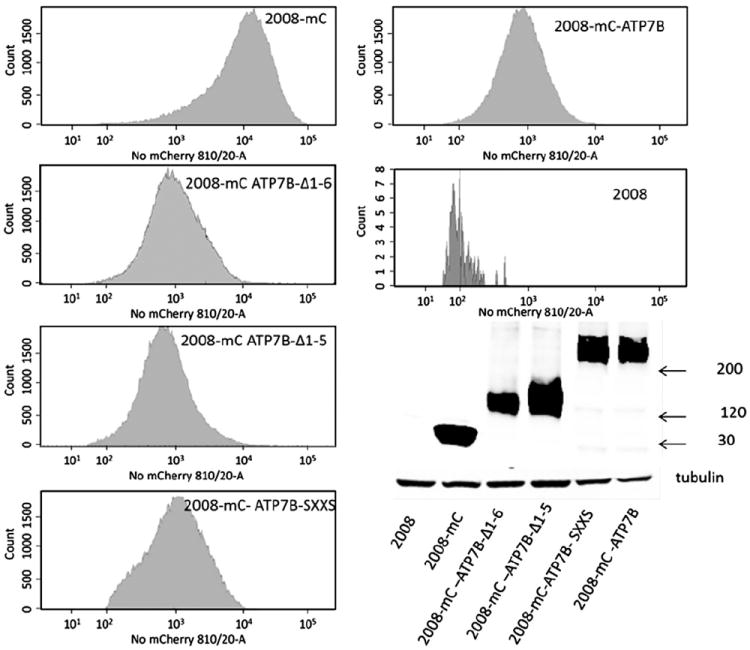
Characterization of 2008 cells expressing mCherry alone or mCherry-tagged ATP7B variants by flow cytometry and Western blotting.
The sensitivity of each 2008 subline to the growth inhibiting effect of cDDP was determined by exposing the cells for 1 h to increasing concentrations of cDDP and determining survival 5 days later with the CCK-8 assay. The IC50 values are presented in Table 2. The 2008 cells expressing wild type ATP7B (mC-ATP7B) were 5-fold resistant to cDDP relative to the 2008-mC cells expressing just mCherry alone. Confirming the results obtained with the recombinant proteins, the IC50 value for the cells expressing the mC-ATP7B SXXS variant was similar to that of the 2008-mC cells indicating that loss of the CXXC motifs disabled the ability of ATP7B to mediate cDDP resistance. Truncation of the N-terminal region of ATP7B containing all six MBDs also eliminated the ability of ATP7B to produce cDDP resistance. The same effect was observed for the mC-ATP7B Δ1–5 cells in which only the sixth MBD remained, indicating that this MBD alone was not sufficient to mediate cDDP resistance.
Table 2.
IC50 values of 2008 cells expressing mCherry or mCherry-tagged ATP7B variants.
| 2008 | 2008-mC | 2008-mC ATP7B | 2008-mC ATP7B SXXS | 2008-mC ATP7B Δ1–5 | 2008-mC ATP7B Δ1–6 | |
|---|---|---|---|---|---|---|
| IC50 | (μM) | 15.17 | ± 0.25 | 12.7±3.1 | 70.5 ±1.3 | 12.7±1.9 |
| 7.9 | ±1.2 | 10.4 ± 1.1 | ||||
| p | 3.27 ×10−12 | 0.19 | 0.20 | 0.47 |
3.5. CXXC motifs of ATP7B MBDs are required for control of cDDP accumulation
Previous studies have shown that 2008 cells that over-express ATP7B accumulate lower levels of cDDP due to increased efflux [30]. To determine whether the effect of ATP7B on the accumulation of cDDP required the MBDs, and specifically their CXXC motifs, Pt accumulation levels were assayed following incubation with 30 μM cDDP for 1 h. As shown in Fig. 4E, expression of wild type mC-ATP7B significantly reduced cDDP uptake. At the end of 1 h, cells expressing mC-ATP7B had accumulated only 1.85±0.17 pmol Pt/ng sulfur which was significantly less than the accumulation of 3.84±0.26 pmol Pt/ng sulfur in the 2008-mC cells (p=0.0001). Expression of mC-ATP7B SXXS failed to reduce uptake at all, whereas expression of mC ATP7B Δ1–5 and mC ATP7B Δ1–6 slightly but significantly reduced cDDP accumulation relative to that in the control 2008-mC cells (2.98±0.22 and 2.78±0.08, respectively; p=0.008 and 0.02, respectively).
Fig. 4.
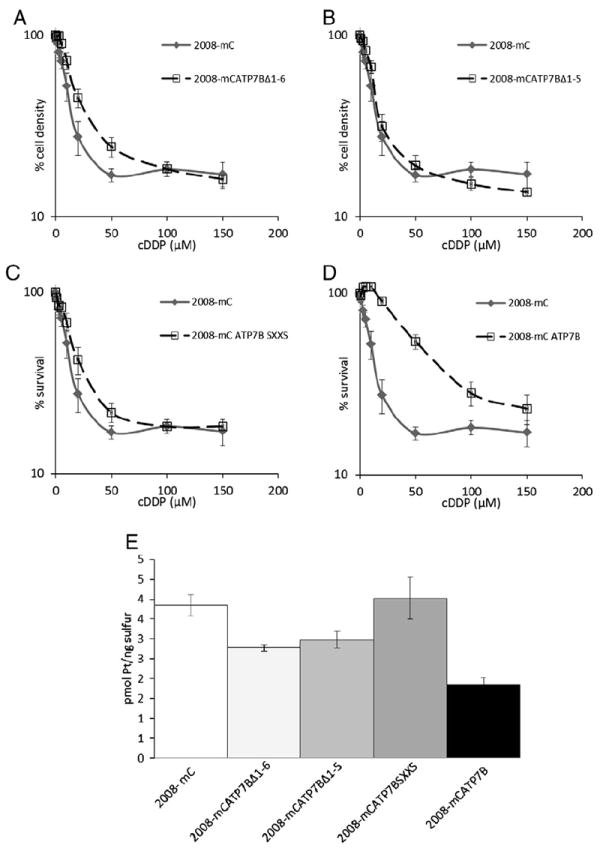
Cytotoxicity and accumulation of cDDP. (A–D) Assay of cell survival with CCK-8 method 5 days after exposure of cells for 1 to increasing concentrations of cDDP. (E) Pt accumulation measured by ICP-MS following exposure of cells for 1 h to 30 μM cDDP. Each data point represents the mean of at least 3 independent experiments performed with triplicate cultures; vertical bars, ±SEM.
3.6. CXXC motifs of ATP7B MBDs are required for Cu- and cDDP-induced trafficking of ATP7B
The distribution of mCherry-tagged AT7B and its variants in 2008 and HEK293T cells was studied before and following exposure for 15 min to 100 μM CuCl2 or 30 μM cDDP. Deconvoluting fluorescence microscopy showed that, in the absence of drug exposure, the distribution of wild type ATP7B was different from that of its variants and from that of native mCherry. Fig. 5 shows that wild type ATP7B was found almost exclusively in the perinuclear region where it colocalized with the trans-Golgi marker p230. The mC-ATP7B SXXS variant was similarly distributed. However, both the mC ATP7B Δ1–5 and the mC ATP7B Δ1–6 exhibited a much more diffuse distribution to peripheral vesicular structures with only a minority of the protein residing in the perinuclear region suggesting retention in the ER due to loss of signal for trans-Golgi localization. To confirm this observation, HEK293T cells, which are larger and spread out more than the 2008 cells, were infected with the lentiviral vectors expressing wild type ATP7B and its variants. As shown in Fig. 7, in the absence of drug exposure, the same alterations in ATP7B distribution were observed in the HEK293T cells indicating that the changes were not cell type specific.
Fig. 5.
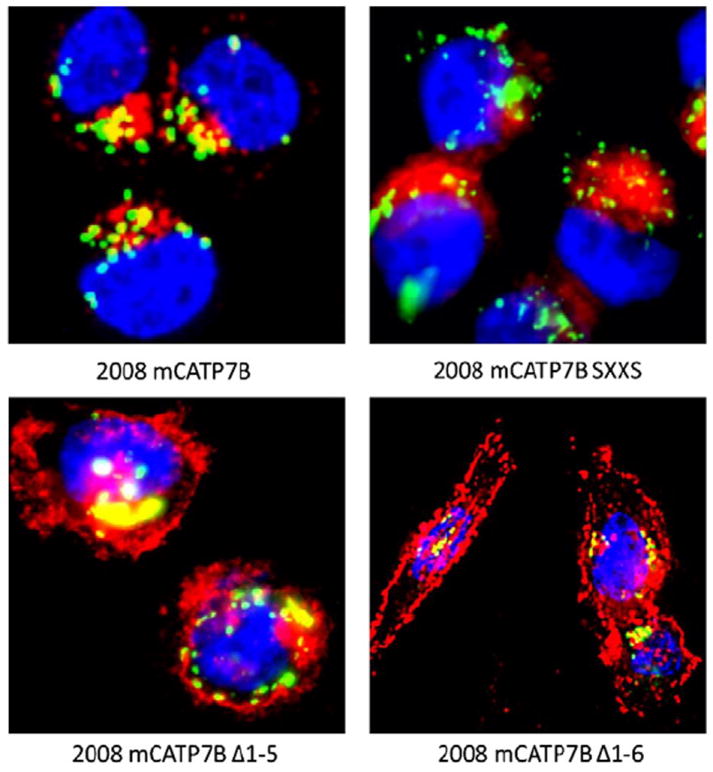
Subcellular localization of mCherry-tagged ATP7B variants in 2008 cells as detected by deconvoluting fluorescence microscopy. An Alexafluor 488-tagged antimouse antibody was used for detection of p230 (green); nuclei are stained with Hoechst 33342 (blue); mCherry protein is red.
Fig. 7.
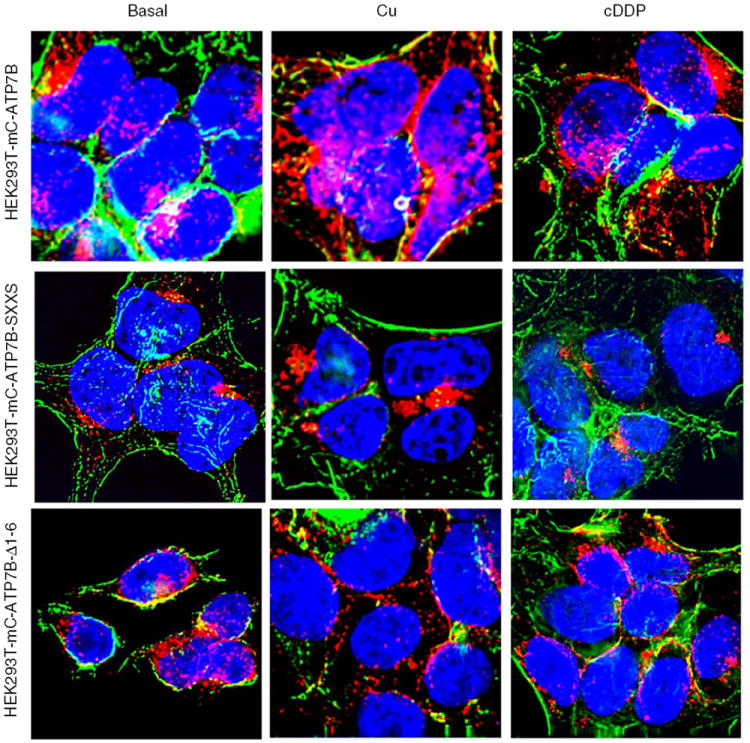
Deconvoluting fluorescent microscopic analysis of the subcellular distribution in HEK293T cells of mCherry tagged ATP7B and variants mC-ATP7B SXXS, and mC-ATP7B Δ1–6, in basal medium and following exposure for 15 min to 100 μM CuCl2 or 30 μM cDDP. Green is FITC-phalloidin; nuclei are stained blue with Hoechst 33342; mCherry protein is red.
Exposure of cells to 30 μM cDDP or 100 μM Cu for 15 min produced different patterns of trafficking for the wild type and variant ATP7Bs. In response to either Cu or cDDP the wild type mC-ATP7B that was localized to the perinuclear region became dispersed to vesicles throughout the cell in both the 2008 cells (Fig. 6) and the HEK293T cells (Fig. 7). In contrast, neither Cu nor cDDP triggered redistribution of the mC-ATP7B SXXS in either the 2008 (Fig. 6) or HEK293T cells (Fig. 7), indicating that the CXXC motifs of the MBDs are required for trafficking in response to both agents. Neither Cu nor cDDP produced a perceptible change in the already diffuse distribution of the mC-ATP7B Δ1–5 or the mC-ATP7B Δ1–6 variants.
Fig. 6.
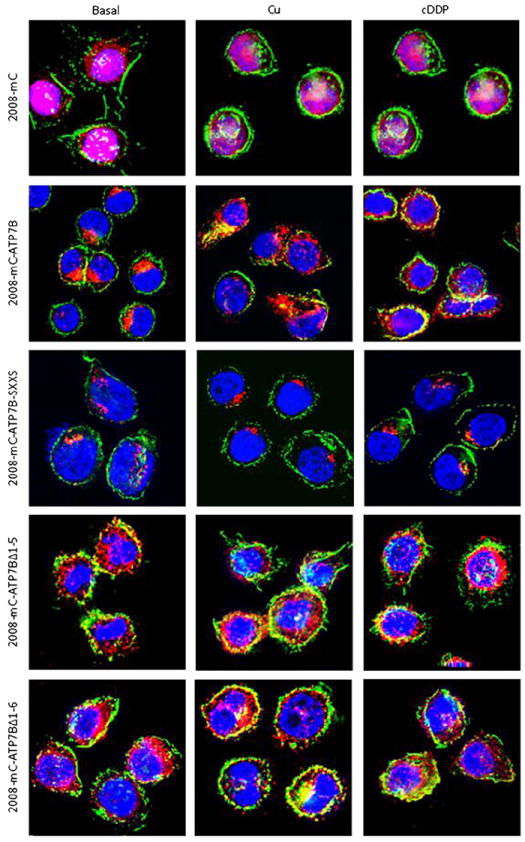
Deconvoluting fluorescence microscopic analysis of the subcellular distribution in 2008 cells of mCherry (mC) and mCherry tagged ATP7B and variants mC-ATP7B SXXS, mC-ATP7B Δ1–5 and mC-ATP7B Δ1–6, in basal medium and following exposure for 15 min to 100 μM CuCl2 or 30 μM cDDP. Green is FITC-phalloidin; nuclei are stained blue with Hoechst 33342; mCherry protein is red.
4. Discussion
This study demonstrates that cDDP binds to MBD6 and that this interaction occurs via its CXXC motif. The results also establish that in whole cells CXXC motifs in the MDB of ATP7B are required for the interaction with cDDP, and are essential to the ability of ATP7B to control the uptake of cDDP, and thus its ability to mediate resistance to this drug. Using recombinant MBD6 we provide three lines of evidence that this domain of ATP7B, which was previously shown to have an essential role in the transport of Cu [22], is one of the sites at which ATP7B interacts with cDDP. First, purified recombinant MBD6 was found to bind MBD6 in a CXXC-dependent manner. Second, cDDP caused oligomerization of MBD6 in both concentration and time-dependent manners, and this was dependent on an intact CXXC motif. Third, cDDP caused CXXC-dependent structural changes in the recombinant MBD6 in a time-dependent manner as evidenced by UV260 spectrometry. Given that each of the 6 MBD in ATP7B shares the same ferredoxin βαββαβ fold and contains similarly positioned CXXC motifs, it seems likely that cDDP can bind to each of the MBDs independently. Like Cu, cDDP triggers the acyl phosphorylation of ATP7B and its relocalization from the TGN to peripheral vesicles [6,7,31]. Since cDDP is behaving in a manner similar to Cu in binding to the CXXC motifs in the MBDs, it may be achieving these effects through the same mechanism by which Cu is working.
The fact that blocking the cysteines on MBD6 by prior exposure to NEM, or conversion of these cysteines to serines, prevented the formation of multimers when the protein was subsequently exposed to cDDP indicates that the cysteine residues of the MBD6 are essential for the interaction of this domain with cDDP. This conclusion is consistent with the observation of Dolgova et al. [32]. The finding that conversion of the CXXC motifs to SXXS blocked the binding of cDDP to MBD6 suggests that it is the CXXC motif itself that is the site of binding. Further credence is given to the concept that cDDP binds directly to the CXXC motif by the fact that cDDP has been shown in NMR structural studies to bind to the CXXC motif in ATOX1, a protein that shares the ferredoxin βαββαβ fold with the MBDs of ATP7B [11].
Previous reports have established that cDDP can crosslink cysteines in adjacent proteins [25]. A cysteine-based mechanism of multimerization is consistent with the observation that high concentrations of DTT were able to reverse the cDDP-induced multimerization. The finding that reversal required high concentrations of DTT and a long period of incubation suggests that the binding of cDDP is very strong. This is consistent with the finding that cDDP becomes very tightly bound to the CXXC motif in ATOX1 [11] and that it causes eventual denaturation of the protein [12]. cDDP can cause multimerization of proteins [33] due to the facile formation of a thiolate bridge between cDDP and cysteines. In many cases the polymer products are of unknown composition and size [34-37]. In the case of the interaction of cDDP with cysteine and methionine, formation of strong dative bonds between the Pt and S atoms can be expected according to the Hard–Soft-Acid–Base principle [38]. Since both the sulfur and platinum atoms are soft atoms with a relatively high polarizability, interactions between such atoms are generally considered to be strong. While it will require further mutagenesis studies to determine which amino acids are essential to the interaction of cDDP with other parts of ATP7B, the results obtained with MBD6, and reports by others, are consistent with the idea that the cysteines rather than methionines are most likely to be involved [27]. In binding Cu, the methionine residue in the MXCXXC metal binding domain is proposed only to stabilize the structure of the loop and not to play a direct role in metal binding [39].
One question that arises from the demonstration that cDDP triggers multimerization of ATP7B is whether this occurs in intact cells, and whether such aggregates contribute to the cytotoxic effect of cDDP. The observation that multimerization can be reversed with a thiol suggests that thioredoxin and glutaredoxin may assist in the removal of the Pt residues from ATP7B in whole cells. Support for this idea is provided by the fact that the N-terminal domain of ATP7B physically associates with the reducing enzyme glutaredoxin [40], and cDDP resistant cells over-express glutaredoxin and thioredoxin [41-43]. GSH has been shown to remove cDDP from proteins very efficiently [33]. The discovery that inhibitors of thioredoxin reductase, such as 4-mercaptopyridine and 2-mercaptopyridine, increase cDDP efficacy in experimental animals provides additional support to this concept [42,44-47].
When expressed in 2008 cells, the ATP7B variant in which all of the CXXC motifs had been converted to SXXS (mC-ATP7B SXXS) was unable to reduce the cellular accumulation of cDDP or confer resistance to this agent. The mC-ATP7B SXXS protein, like wild type ATP7B, localized to the perinuclear region and co-localized with the trans-Golgi marker p230 indicating that the failure of mC-ATP7B SXXS variant to mediate resistance was not due to misdistribution under basal conditions. However, the mC-ATP7B SXXS variant failed to traffic to peripheral vesicles upon exposure to either Cu or cDDP. This is consistent with prior observations made in CHO cells that this variant does not traffic in response to Cu [22]. The failure of this construct to confer resistance to cDDP leaves open the question of whether the inability to mediate resistance is due to failure to bind cDDP or to relocate to vesicles that might be essential to the efflux process.
The results reported here confirm and extend our prior discovery that cDDP triggers the relocalization of ATP7B from the TGN even in cells that lack the polarization of benign epithelial cells [7]. We found that deletion of either just the first 5 MBDs (amino acids 1–539), or all 6 MBDs (amino acids 1–599) resulted in forms of ATP7B that were not entirely localized to the TGN under basal conditions in both the 2008 and HEK293T cells. Neither of these proteins showed an alteration in subcellular distribution in response to Cu or cDDP. This is consistent with the discovery that amino acids 37–45 constitute a signal sequence that is required for both the complete TGN localization of ATP7B under basal conditions and its relocalization in response to Cu [48]. It remains to be seen whether addition of this signal sequence to the mC-ATP7B Δ1–5 or mC-ATP7B Δ1–6 variants would restore TGN localization or ability to control cDDP accumulation or cytotoxicity.
Whether cDDP is transferred to the interior of the secretory vesicles or just becomes bound to the CXXC motifs of the MDB that reside on the cytoplasmic surface of the vesicles is a subject of speculation at present. One possibility is that, although cDDP binds to ATP7B, the drug is stripped off by GSH and the GSH–cDDP complex is then transported into vesicles by another transporter such as MRP2. It is also possible that microenvironmental factors such as very low pH [6] and high levels of reducing molecules would enable the ATP7B to function as an ATPase pump in transporting cDDP across the vesicular membrane.
Acknowledgments
We are deeply grateful to Drs. Sharon La Fontaine and Julian Mercer for providing a vector containing the ATP7B-SXXS variant. We thank Angela Robles for her generous assistance and operational support, Dennis Young for assistance with flow cytometric analysis, and Kersi Pestonjumasp for assistance with fluorescent microscopy.
Abbreviations
- BME
β-mercaptoethanol
- DTT
dithiothreitol
- GSH
glutathione
- IPTG
isopropyl-β-d-thiogalactoside
- MDB
metal binding domain
- MBP
maltose binding protein
- NEM
N-ethyl maleimide
Footnotes
This work was supported by the National Institutes of Health [CA152185 and CA095298] and the Department of Defense [W81XWH-08-1-0135]. Additional support for core laboratories was from grant P30 NS047101 for the UCSD Neuroscience Microscopy Shared Facility and the UCSD Cancer Center Specialized Support grant P30 CA23100.
Nomenclature
ATP7B ATPase, Cu++ transporting, beta polypeptide
GenBank ID ENSG00000123191
PDB ID P35670
EC 3.6
References
- 1.Larson CA, Blair BG, Safaei R, Howell SB. Mol Pharmacol. 2008;75:324–330. doi: 10.1124/mol.108.052381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holzer AK, Samimi G, Katano K, Naerdemann W, Lin X, Safaei R, Howell SB. Mol Pharmacol. 2004;66:817–823. doi: 10.1124/mol.104.001198. [DOI] [PubMed] [Google Scholar]
- 3.Holzer AK, Manorek GH, Howell SB. Mol Pharmacol. 2006;70:1390–1394. doi: 10.1124/mol.106.022624. [DOI] [PubMed] [Google Scholar]
- 4.Song I, Savaraj N, Siddik Z, Liu P, Wei Y, Wu C, Kuo M. Mol Cancer Ther. 2004;3:1543–1549. [PubMed] [Google Scholar]
- 5.Samimi G, Katano K, Holzer AK, Safaei R, Howell SB. Mol Pharmacol. 2004;66:25–32. doi: 10.1124/mol.66.1.25. [DOI] [PubMed] [Google Scholar]
- 6.Safaei R, Larson BJ, Otani S, Rasmussen ML, Howell SB. Mol Pharmacol. 2008;73:461–468. doi: 10.1124/mol.107.040980. [DOI] [PubMed] [Google Scholar]
- 7.Katano K, Safaei R, Samimi G, Holzer A, Tomioka M, Goodman M, Howell SB. Clin Cancer Res. 2004;10:4578–4588. doi: 10.1158/1078-0432.CCR-03-0689. [DOI] [PubMed] [Google Scholar]
- 8.Solioz M, Vulpe C. Trends Biochem Sci. 1996;21:237–241. [PubMed] [Google Scholar]
- 9.Marchler-Bauer A, Anderson JB, Derbyshire MK, DeWeese-Scott C, Gonzales NR, Gwadz M, Hao L, He S, Hurwitz DI, Jackson JD, Ke Z, Krylov D, Lanczycki CJ, Liebert CA, Liu C, Lu F, Lu S, Marchler GH, Mullokandov M, Song JS, Thanki N, Yamashita RA, Yin JJ, Zhang D, Bryant SH. Nucleic Acids Res. 2007;35:D237–D240. doi: 10.1093/nar/gkl951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arnesano F, Banci L, Bertini I, Ciofi-Baffoni S, Molteni E, Huffman DL, O’Halloran TV. Genome Res. 2002;12:255–271. doi: 10.1101/gr.196802. [DOI] [PubMed] [Google Scholar]
- 11.Arnesano F, Banci L, Bertini I, Felli IC, Losacco M, Natile G. J Am Chem Soc. 2011;133:18361–18369. doi: 10.1021/ja207346p. [DOI] [PubMed] [Google Scholar]
- 12.Palm ME, Weise CF, Lundin C, Wingsle G, Nygren Y, Bjorn E, Naredi P, Wolf-Watz M, Wittung-Stafshede P. Proc Natl Acad Sci U S A. 2011;108:6951–6956. doi: 10.1073/pnas.1012899108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arnesano F, Banci L, Bertini I, Huffman DL, O’Halloran TV. Biochemistry. 2001;40:1528–1539. doi: 10.1021/bi0014711. [DOI] [PubMed] [Google Scholar]
- 14.Gitschier J, Moffat B, Reilly D, Wood WI, Fairbrother WJ. Nat Struct Biol. 1998;5:47–54. doi: 10.1038/nsb0198-47. [DOI] [PubMed] [Google Scholar]
- 15.Banci L, Bertini I, Cantini F, Rosenzweig AC, Yatsunyk LA. Biochemistry. 2008;47:7423–7429. doi: 10.1021/bi8004736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeSilva TM, Veglia G, Porcelli F, Prantner AM, Opella SJ. Biopolymers. 2002;64:189–197. doi: 10.1002/bip.10149. [DOI] [PubMed] [Google Scholar]
- 17.Banci L, Bertini I, Ciofi-Baffoni S, Huffman DL, O’Halloran TV. J Biol Chem. 2001;276:8415–8426. doi: 10.1074/jbc.M008389200. [DOI] [PubMed] [Google Scholar]
- 18.Pufahl RA, Singer CP, Peariso KL, Lin SJ, Schmidt PJ, Fahrni CJ, Culotta VC, Penner-Hahn JE, O’Halloran TV. Science. 1997;278:853–856. doi: 10.1126/science.278.5339.853. [DOI] [PubMed] [Google Scholar]
- 19.Rosenzweig AC, Huffman DL, Hou MY, Wernimont AK, Pufahl RA, O’Halloran TV. Struct Fold Des. 1999;7:605–617. doi: 10.1016/s0969-2126(99)80082-3. [DOI] [PubMed] [Google Scholar]
- 20.Achila D, Banci L, Bertini I, Bunce J, Ciofi-Baffoni S, Huffman DL. Proc Natl Acad Sci U S A. 2006;103:5729–5734. doi: 10.1073/pnas.0504472103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Forbes JR, Hsi G, Cox DW. J Biol Chem. 1999;274:12408–12413. doi: 10.1074/jbc.274.18.12408. [DOI] [PubMed] [Google Scholar]
- 22.Cater MA, Forbes J, La Fontaine S, Cox D, Mercer JF. Biochem J. 2004;380:805–813. doi: 10.1042/BJ20031804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iida M, Terada K, Sambongi Y, Wakabayashi T, Miura N, Koyama K, Futai M, Sugiyama T. FEBS Lett. 1998;428:281–285. doi: 10.1016/s0014-5793(98)00546-8. [DOI] [PubMed] [Google Scholar]
- 24.Loudianos G, Dessi V, Lovicu M, Angius A, Nurchi A, Sturniolo GC, Marcellini M, Zancan L, Bragetti P, Akar N, Yagci R, Vegnente A, Cao A, Pirastu M. Hum Mutat. 1998;12:89–94. doi: 10.1002/(SICI)1098-1004(1998)12:2<89::AID-HUMU3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 25.Zimmermann T, Zeizinger M, Burda JV. J Inorg Biochem. 2005;99:2184–2196. doi: 10.1016/j.jinorgbio.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 26.Mandal R, Kalke R, Li XF. Rapid Commun Mass Spectrom. 2003;17:2748–2754. doi: 10.1002/rcm.1259. [DOI] [PubMed] [Google Scholar]
- 27.Soldatovic T, Bugarcic ZD. J Inorg Biochem. 2005;99:1472–1479. doi: 10.1016/j.jinorgbio.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 28.Larson CA, Adams PL, Blair BG, Safaei R, Howell S. Mol Pharmacol. 2010;78:333–339. doi: 10.1124/mol.110.064766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Safaei R, Holzer AK, Katano K, Samimi G, Howell SB. J Inorg Biochem. 2004;98:1607–1613. doi: 10.1016/j.jinorgbio.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 30.Katano K, Safaei R, Samimi G, Holzer A, Rochdi M, Howell SB. Mol Pharmacol. 2003;64:466–473. doi: 10.1124/mol.64.2.466. [DOI] [PubMed] [Google Scholar]
- 31.Huster D, Lutsenko S. J Biol Chem. 2003;278:32212–32218. doi: 10.1074/jbc.M305408200. [DOI] [PubMed] [Google Scholar]
- 32.Dolgova NV, Olson D, Lutsenko S, Dmitriev OY. Biochem J. 2009;419:51–56. doi: 10.1042/BJ20081359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peleg-Shulman T, Gibson D. J Am Chem Soc. 2001;123:3171–3172. doi: 10.1021/ja005854y. [DOI] [PubMed] [Google Scholar]
- 34.el-Khateeb M, Appleton TG, Gahan LR, Charles BG, Berners-Price SJ, Bolton AM. J Inorg Biochem. 1999;77:13–21. doi: 10.1016/s0162-0134(99)00146-4. [DOI] [PubMed] [Google Scholar]
- 35.Berners-Price SJ, Kuchel PW. J Inorg Biochem. 1990;38:327–345. doi: 10.1016/0162-0134(90)80006-j. [DOI] [PubMed] [Google Scholar]
- 36.Dedon PC, Borch RF. Biochem Pharmacol. 1987;36:1955–1964. doi: 10.1016/0006-2952(87)90494-1. [DOI] [PubMed] [Google Scholar]
- 37.Heudi O, Mercier-Jobard S, Cailleux A, Allain P. Biopharm Drug Dispos. 1999;20:107–116. doi: 10.1002/(sici)1099-081x(199903)20:2<107::aid-bdd161>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 38.Chattaraj PK, Maiti B. J Am Chem Soc. 2003;125:2705–2710. doi: 10.1021/ja0276063. [DOI] [PubMed] [Google Scholar]
- 39.Odermatt A, Solioz M. J Biol Chem. 1995;270:4349–4354. doi: 10.1074/jbc.270.9.4349. [DOI] [PubMed] [Google Scholar]
- 40.Lim CM, Cater MA, Mercer JF, La Fontaine S. Biochem Biophys Res Commun. 2006;348:428–436. doi: 10.1016/j.bbrc.2006.07.067. [DOI] [PubMed] [Google Scholar]
- 41.Zunino F, Pratesi G, Micheloni A, Cavalletti E, Sala F, Tofanetti O. Chem Biol Interact. 1989;70:89–101. doi: 10.1016/0009-2797(89)90065-3. [DOI] [PubMed] [Google Scholar]
- 42.Richardson ME, Siemann DW. Int J Radiat Oncol Biol Phys. 1994;29:387–392. doi: 10.1016/0360-3016(94)90295-x. [DOI] [PubMed] [Google Scholar]
- 43.Marverti G, Giuseppina Monti M, Pegg AE, McCloskey DE, Bettuzzi S, Ligabue A, Caporali A, D’Arca D, Moruzzi MS. Carcinogenesis. 2005;26:1677–1686. doi: 10.1093/carcin/bgi129. [DOI] [PubMed] [Google Scholar]
- 44.Ahmadi R, Urig S, Hartmann M, Helmke BM, Koncarevic S, Allenberger B, Kienhoefer C, Neher M, Steiner HH, Unterberg A, Herold-Mende C, Becker K. Free Radic Biol Med. 2006;40:763–778. doi: 10.1016/j.freeradbiomed.2005.09.031. [DOI] [PubMed] [Google Scholar]
- 45.Suzuki Y, Kondo Y, Himeno S, Nemoto K, Akimoto M, Imura N. Prostate. 2000;43:144–149. doi: 10.1002/(sici)1097-0045(20000501)43:2<144::aid-pros9>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 46.Sasada T, Nakamura H, Ueda S, Sato N, Kitaoka Y, Gon Y, Takabayashi A, Spyrou G, Holmgren A, Yodoi J. Free Radic Biol Med. 1999;27:504–514. doi: 10.1016/s0891-5849(99)00101-x. [DOI] [PubMed] [Google Scholar]
- 47.Biaglow JE, Miller RA. Cancer Biol Ther. 2005;4:6–13. doi: 10.4161/cbt.4.1.1434. [DOI] [PubMed] [Google Scholar]
- 48.Braiterman L, Nyasae L, Guo Y, Bustos R, Lutsenko S, Hubbard A. Am J Physiol Gastrointest Liver Physiol. 2009;296:G433–G444. doi: 10.1152/ajpgi.90489.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]