

Abstract
Here we show that tyrosine phosphorylation of caveolin-2 (Cav-2) negatively regulates the anti-proliferative function of transforming growth factor beta (TGF-beta) in endothelial cells. In contrast to wild-type-Cav-2, retroviral re-expression of Y19/27F-Cav-2 in Cav-2 knockout endothelial cells did not affect anti-proliferative effect of TGF-beta compared to empty vector. Conversely, although less effective than wild-type, re-expression of S23/36A-Cav-2 reduced the effect of TGF-beta compared to empty vector. This differential effect of tyrosine and serine phosphorylation mutants of Cav-2 correlated with TGF-beta-induced Smad3 phosphorylation and transcriptional activation of plasminogen activator inhibitor-1. Thus tyrosine-phosphorylated Cav-2 counteracts anti-proliferative effect of TGF-beta in endothelial cells.
Keywords: Caveolin-2, N-terminal tyrosine and serine, phosphorylation, TGF-beta, Smad3, PAI-1, Endothelial cell proliferation
1. Introduction
Caveolins are key components of detergent resistant cholesterol lipid rich membranes including lipid rafts and caveolae. Three members were identified within the caveolin protein family: Caveolin-1 (Cav-1), Cav-2, and Cav-3. Cav-1 and -2 are ubiquitously co-expressed, while Cav-3 is muscle specific [1-5]. Caveolins play numerous important roles. In addition to being key structural proteins that organize caveolae, caveolin proteins are important in regulating endocytosis and various aspects of cellular signaling [6] including endothelial cells [7]. Although relative to Cav-1, the functional role of Cav-2 is less well defined, recent studies have started to present a growing body of evidence suggesting tissue/cell specific role for Cav-2 [8]. For example, initial observations involving Cav-2 knockout (KO) mice revealed hyperplasia in the lung [9], suggesting a role for Cav-2 in regulating lung cell proliferation and/or differentiation.
Transforming growth factor-β (TGF-β) is a multifunctional dimeric polypeptide growth factor capable of regulating proliferation, differentiation, migration, extracellular matrix production and survival of various cell types. Cell responses to TGF-β are mediated through specific transmembrane type I and type II Ser/Thr kinase receptors [10,11]. The signaling pathway is initiated by TGF-β binding to the TGF-β type II receptor (TβR-II). Upon ligand binding, TβR-II recruits and phosphorylates TβR-I, also known as activin receptor-like kinase (Alk), which transduces the signal to the nucleus through members of the Smad family [12,13]. Most cell types express a form of TβR-I known as Alk5. ECs also co-express an additional TβR-I known as Alk1. Activated Alk5 induces the phosphorylation of Smad2 and Smad3 and activation of Alk5/Smad2/3 pathway leads to inhibition of cell proliferation and is associated with a mature endothelium with increased expression of genes such as plasminogen activator inhibitor-1 (PAI-1), collagen type I (Col-1).
Recently, we have determined that Cav-2 is the negative regulator of anti-proliferative effect of TGF-β/Alk-5/Smad2/3 pathway in ECs [14]. The purpose of the present study is to investigate the molecular mechanisms of Cav-2-dependent inhibition of TGF-β/Alk5 pathway in ECs. Specifically, we have examined the relative contribution of N-terminal serine and tyrosine phosphorylation of Cav-2 in regulating anti-proliferative function of TGF-β/Alk5/Smad3/PAI-1 pathway in ECs. Our data indicates that tyrosine but not serine phosphorylation is essential for the negative regulation of anti-proliferative function and signaling of TGF-β in ECs.
2. Materials and methods
The detailed Materials and methods can be found in Supplementary Data.
2.1. Antibodies
Antibodies against total Cav-2, PY27-Cav-2, Cav-1 and Hsp-90 were from BD Transduction Labs; Antibodies to phospho-Smad3 and Lamin A/C were from Cell Signaling Biotech; Antibody to phospho-histone 3 was from Santa Cruz Biotech; Antibodies to PAI-1 and PY19-Cav-2 were from Abcam.
2.2. Endothelial cells (MLECs)
Mouse lung endothelial cells (MLECs) were isolated from 2 to 3-week-old wild type (WT) and Cav-2 KO mice as originally described [15] (For details see Supplementary Data). Human lung microvascular endothelial cells (HULECs) were obtained from the Centers for Disease Control and Prevention.
2.3. Retroviral re-expression of WT as well as serine and tyrosine phosphorylation-deficient mutants of Cav-2 in Cav-2 KO MLECs
Cav-2 KO MLECs stably expressing empty vector pBABE-puro or pBABE-puro plus WT-Cav-2, S23/36A-Cav-2, and Y19/27F-Cav-2 were generated as previously described [14] (For details see Supplementary Data).
2.4. MTT colorimetric proliferation assay
MLECs were seeded onto 24-well plates at 4 × 103/well in complete medium. Next day, medium was replaced with fresh medium without or with human TGF-β1 (Peprotech). After 6 h (day 0) or 5 days of incubation in the absence or presence of TGF-β (1 ng/ml), MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Sigma) stock solution was added and the samples were processed and measured as described in Supplementary Data.
2.5. Cell counts based proliferation assay
MLECs were plated onto six well plates at 104 cells per well for 24 h, followed by a medium change and further incubation in complete medium in the absence or presence of TGF-β for 6 days. At the end of the experiment, cells were trypsinized and viable cells counted with an automatic cell counter Countess (Invitrogen corporation). Cell number was expressed as mean ± S.D. (n = 3) from one representative out of a total three experiments.
2.6. Immunofluorescence microscopy
Cells were fixed with 3% paraformaldehyde in Dulbecco’s phosphate-buffered saline, pH 7.4 (DPBS), for 30 min, and washed three times with DPBS. Cells were then incubated sequentially with 0.1% Triton X-100 (v/v) in DPBS for 10 min, DPBS plus 5% goat serum for 30 min, and thereafter with anti-Cav-2 antibody (1:100) in 0.2% BSA for 2 h, washed three times, and incubated with Alexa Fluor 488 nm-labeled secondary antibody (Invitrogen Corp.) diluted 1:500, followed by staining with DAPI (Sigma). Slides were mounted with Slowfade (Molecular Probes, Inc., Eugene, OR), and cells were observed and images captured with 20× objective using an Olympus IX70 epifluorescence microscope.
2.7. Immunoblotting
Cells were lysed in Laemmli SDS loading buffer, followed by boiling for 5 min. An equal protein amount was loaded on SDS–PAGE, and proteins were electro-transferred onto nitrocellulose membranes. The membranes were washed in Tris-buffered saline with 0.1% Tween, blocked in 5% milk, and incubated with the appropriate primary antibodies diluted 1:1000–1:20000 at 4 °C overnight, followed by incubation with horseradish peroxidase labeled secondary antibodies diluted 1:10000, and developed by enhanced chemiluminescence.
2.8. Triton-100 insolubility assay
MLECs were lysed with 0.1% Triton X-100 in MBS (pH 6.5), lysates were incubated for 10 min on ice, and centrifuged at 48000×g at 4 °C for 30 min. The supernatant was collected and considered as Triton X-100 soluble fraction (+), while the Triton X-100 insoluble (−) pellet was solubilized with an equal volume of SDS–PAGE loading buffer, equal volumes of both samples were loaded on SDS–PAGE gel and immunoblotted. % Distribution for each detected protein in TX-100 soluble versus insoluble fraction was calculated based on the densitometric values obtained using Image J (NIH) and expressed as Mean ± S.D. (n = 3).
2.9. Nuclear/Cytosol fractionation
The nuclear and cytosolic fractions were isolated using the Nuclear/Cytosol Extraction Kit (BioVision) according to the manufacturer’s protocol, followed by standard immunoblotting of nuclear and cytosolic fractions. In addition, the densitometric ratios of p-Smad3/Lamin A/C were assessed for nuclear fractions using Image J (NIH) and expressed as Mean ± S.D. (n = 3).
2.10. RNA isolation and quantification of specific gene expression by real-time PCR
Total RNA isolation and RT-PCR of control and TGF-β-treated MLECs were performed as previously described [14] (For details see Supplementary Data).
3. Results
3.1. Expression levels and subcellular targeting of retrovirally re-expressed serine and tyrosine phosphorylation-deficient mutants are comparable to WT Cav-2
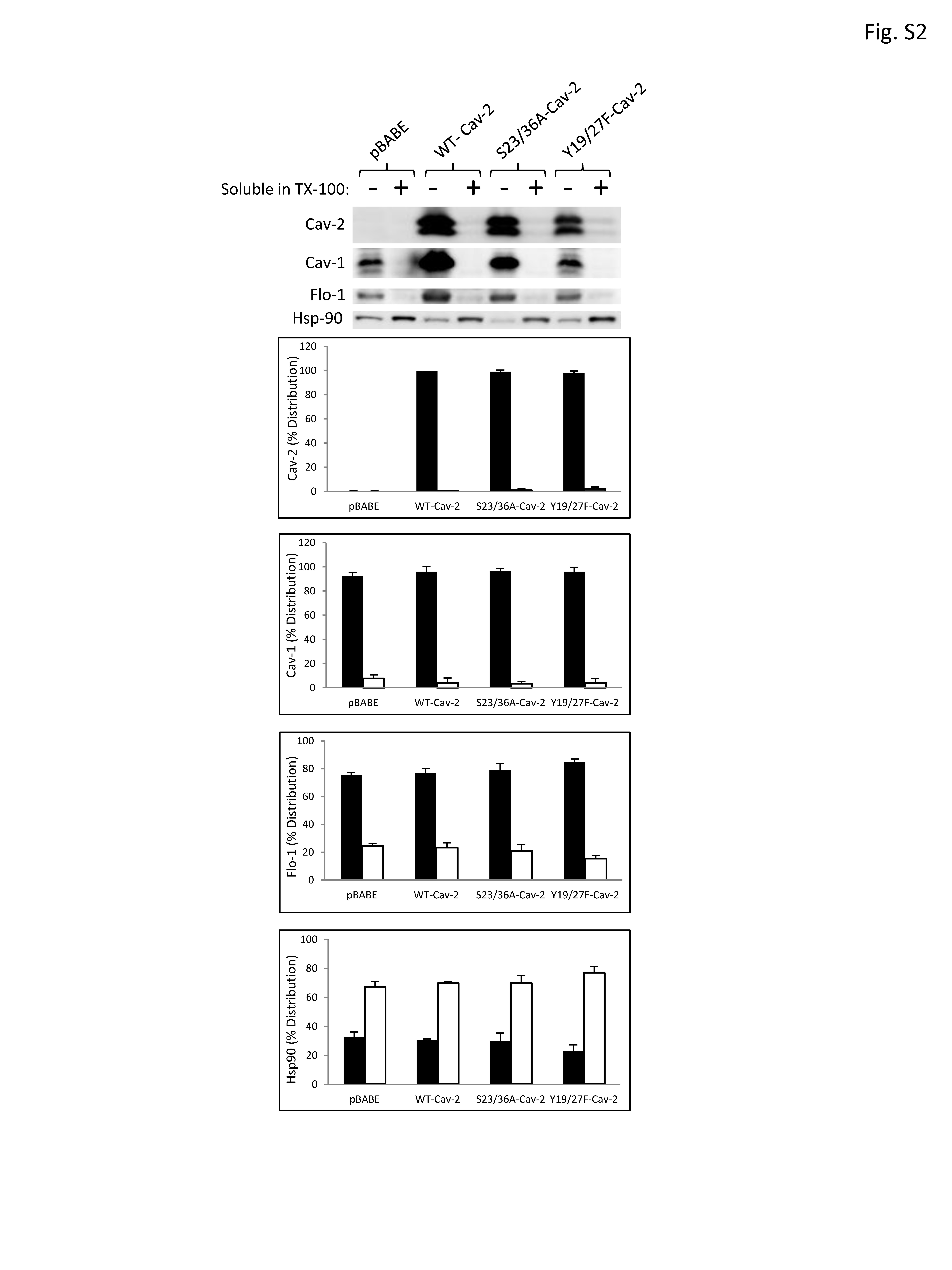
Previously, we have determined that Cav-2 KO MLECs were more sensitive than WT MLECs to anti-proliferative effect of TGF-β and that retroviral re-expression of WT Cav-2 in Cav-2 KO MLECs resulted in a similar response to TGF-β as in WT MLECs [14]. However, the detailed molecular mechanisms of this inhibitory role of Cav-2 in anti-proliferative effect of TGF-β in ECs have not been examined. Because Cav-2 has been previously shown to be phosphorylated at serine residues 23 and 36 [16,17] as well as tyrosine residues 19 [18] and 27 [19], in the current study we have examined the role of N-terminal serine and tyrosine phosphorylation of Cav-2 in negating the anti-proliferative effect and signaling of TGF-β in ECs. Specifically, we have re-expressed WT-Cav-2, serine residues 23 and 36 phosphorylation-deficient mutant (S23/36A-Cav-2) as well as tyrosine residues 19 and 27 phosphorylation-deficient mutant (Y19/27F-Cav-2) in Cav-2 KO MLECs. Using standard immunoblotting technique we have determined comparable expression levels of Cav-2 in Cav-2 KO MLECs re-expressing WT-Cav-2 as well as S23/36A-Cav-2 and Y19/27F (For details see Supplementary Results and Fig. S1A). Next, using immunofluorescence labeling with Cav-2 antibody, we have also determined that similar to WT-Cav-2, the re-expressed S23/36A-Cav-2 and Y19/27F-Cav-2 targeted to perinuclear and plasma membrane regions (For details see Supplementary Results and Fig. S1B). Finally, using TX-100 insolubility assay, we have also determined that just as retrovirally-expressed WT-Cav-2, both S23/36A-Cav-2 and Y19/27F-Cav-2 were TX-100-insoluble and co-fractionated with Cav-1 and flotillin-1 (Flo-1), protein markers of caveolae and lipid rafts, respectively but not with the cytosolic marker, Hsp-90 (For details see Supplementary Results and Fig. S2).
3.2. N-terminal tyrosine phosphorylation is more critical than serine phosphorylation of Cav-2 in negating anti-proliferative effect of TGF-β in MLECs
Here, we have used previously (Fig. S1) characterized pBABE, WT-Cav-2, S23/36A-Cav-2 and Y19/27F-Cav-2 MLECs and compared the inhibitory effect of TGF-β on proliferation using the two independent proliferation assays: cell count and MTT as well as immunoblotting with anti-phospho-histone 3 (mitotic marker) antibody.
In a cell count proliferation assay (Fig. 1A) treatment with TGF-β (1 ng/ml) reduced cell number by c.a. 52.4 ± 3.8% in pBABE cells and this inhibitory effect of TGF-β was reduced to 5.5 ± 3% by re-expression of WT-Cav-2. Re-expression of S23/36A-Cav-2 still reduced the inhibitory effect of TGF-β to 15.2 ± 5.8%. In contrast to S23/36A, re-expression of Y19/27F-Cav-2 only marginally reduced inhibitory effect of TGF-β to 45.2 ± 4.8%.
Fig. 1.
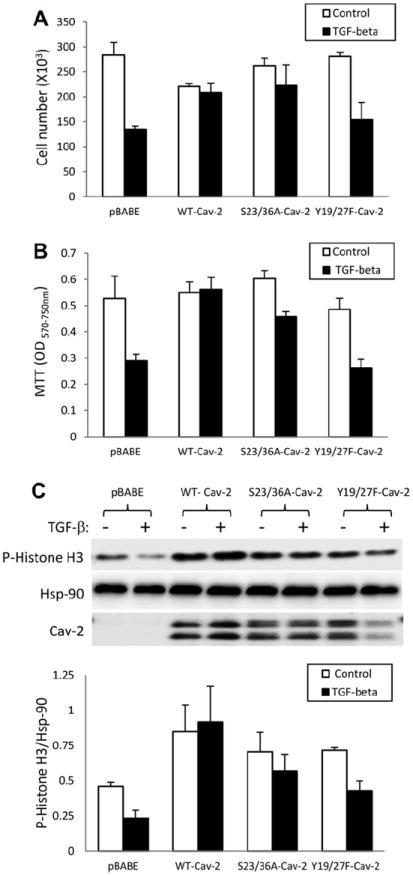
Effect of TGF-β on proliferation of Cav-2 KO MLECs re-expressing WT and phosphorylation-deficient mutants of Cav-2. (A) Effect of TGF-β on MLECs proliferation determined by cell count. pBABE, WT-Cav-2, S23/36A-Cav-2, and Y19/27F-Cav-2 MLECs were plated at 104 cells per well of six well plates and 24 h later incubated without or with TGF-β (1 ng/ml) for 5 days. Cell numbers were determined using Cell Countess and expressed as Mean ± S.D. of replicate samples (n = 3) from one representative out of a total three experiments. (B) Effect of TGF-β on MLECs proliferation determined by MTT proliferation assay. pBABE, WT-Cav-2, S23/36A-Cav-2, and Y19/27F-Cav-2 MLECs were plated at 2 × 103 cells per well of 24 well plates and 24 h later incubated without or with TGF-β (1 ng/ml) for 5 days, followed by processing for MTT proliferation assay. Optical density (OD) was measured using a plate reader with a test wavelength of 570 nm and a reference wavelength of 750 nm (570–750 nm) to obtain a sample signal, and expressed as Mean ± S.D. of replicate samples (n = 3). Values are from one representative out of a total four experiments. (C) Effect of TGF-β on MLECs proliferation determined by immunoblotting with anti-mitotic marker protein, phospho-histone H3 (P-Histone H3) antibody. WT-Cav-2, S23/36A-Cav-2, and Y19/27F-Cav-2 MLECs were plated at 3 × 105 cells onto 150 mm dishes, 24 h later incubated without or with TGF-β (1 ng/ml) for additional 48 h, lysed and processed for SDS–PAGE and immunoblotting with indicated antibodies as described in Section 2. Bottom graph: Densitometric ratios of P-Histone H3/Hsp-90 from control (blank bars) and treated with TGF-β (solid bars) samples quantified based on the above immunoblots and expressed as Means ± S.D. (n = 3).
In an MTT proliferation assay (Fig. 1B), treatment with TGF-β reduced OD values by 45.1 ± 5.7% in pBABE and re-expression of WT-Cav-2 completely reversed this inhibition. Re-expression of S23/36A-Cav-2 still reduced the inhibitory effect of TGF-β to 24.0 ± 1.1%, In contrast to S23/36A-Cav-2, re-expression of Y19/27F-Cav-2 did not affect the inhibitory effect of TGF-β (45 ± 4.9%) compared to pBABE.
Similar to cell count and MTT proliferation assays, TGF-β also reduced the relative levels of histone H3 phosphorylation by 49.5 ± 11.9% in pBABE and re-expression of WT-Cav-2 completely reversed the inhibitory effect of TGF-β (Fig. 1C). Re-expression of S23/36A-Cav-2 still reduced the inhibitory effect of TGF-β to 19.2 ± 3.9%, while re-expression of Y19/27F-Cav-2 had only marginal effect (c.a. 40.0 ± 6.3% inhibition by TGF-β). Taken together, the data obtained with the two independent proliferation assays and immunoblotting with anti-mitotic marker antibody suggest that N-terminal tyrosine phosphorylation of Cav-2 plays a critical role in inhibiting anti-proliferative effect of TGF-β in ECs. In addition, N-terminal serine phosphorylation is only modestly involved in negating anti-proliferative effect of TGF-β by Cav-2.
3.3. N-terminal tyrosine phosphorylation is more critical than serine phosphorylation of Cav-2 in negating TGF-β-stimulated Smad3 phosphorylation in MLECs
Smad3 is a key downstream mediator of TGF-β-induced anti-proliferative effect in various cell types, including ECs. Thus we have examined the role of N-terminal phosphorylation of Cav-2 in inhibiting TGF-β-stimulated Smad3 phosphorylation in MLECs. Upon stimulation with TGF-β, Smad3 is phosphorylated by Alk5 and translocates to the nucleus. To confirm that the same is true for MLECs, we isolated nuclear and cytosolic fractions from control and treated with TGF-β (1 ng/ml; 1 h) pBABE, WT-Cav-2, S23/36A-Cav-2 as well as Y19/27F-Cav-2 MLECs and determined the levels of Smad3 phosphorylation by immunoblotting with P-Smad3-specific antibody. As seen in Fig. 2A, the P-Smad3-specific signal was enriched in nuclear (N) but not cytosolic (C) fractions. Therefore, we have further determined the ratios of P-Smad3/Lamin A/C (nuclear membrane marker protein) in nuclear fractions (Fig. 2B and C). Treatment with TGF-β increased the P-Smad3/Lamin A/C ratio by c.a. twofold in pBABE MLECs, while re-expression of WT-Cav-2 blocked this increase by TGF-β. Re-expression of S23/36A-Cav-2 reduced the stimulating effect of TGF-β to only c.a. 1.14-fold. In contrast to S23/36A-Cav-2, re-expression of Y19/27F-Cav-2 did not reduce TGF-β-induced increase in Smad3 phosphorylation compared to pBABE MLECs. These data suggest that N-terminal tyrosine phosphorylation is essential molecular event allowing Cav-2 to inhibit TGF-β-induced stimulation of Smad3 phosphorylation.
Fig. 2.
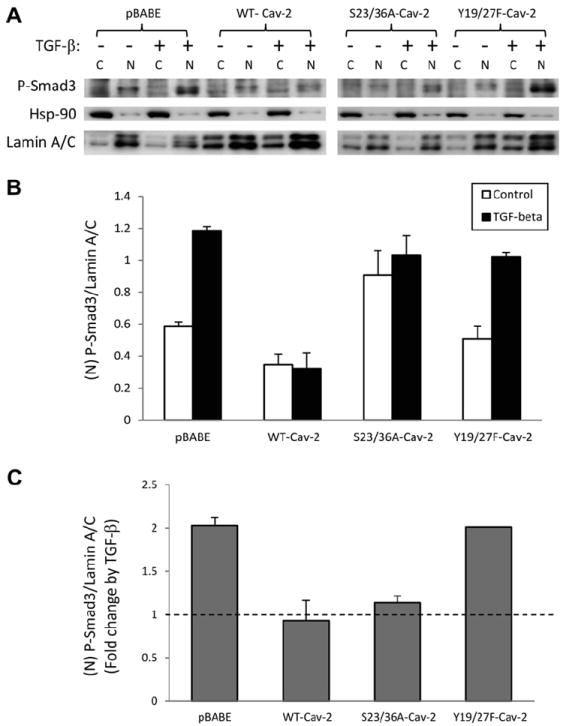
Effect of TGF-β on Smad3 phosphorylation in Cav-2 KO MLECs re-expressing WT and phosphorylation-deficient mutants of Cav-2. WT-Cav-2, S23/36A-Cav-2, and Y19/27F-Cav-2 MLECs were plated at 5 × 105 cells onto 60 mm dishes, 24 h later incubated without or with TGF-β (1 ng/ml) for 1 h, followed by cytosol (C) and nuclear (N) fractionation (Biovision). Both C and N fractions were then processed for SDS–PAGE and immunoblotting with indicated antibodies. Top graph: Densitometric ratios of phospho-Smad3 (P-Smad3)/Lamin A/C (nuclear protein marker) in N fractions from control (blank bars) and treated with TGF-β (solid bars) samples quantified based on the above immunoblots and expressed as Means ± S.D. (n = 3). Bottom graph: Fold-change by TGF-β relative to respective control samples, quantified based on N fraction and expressed as Means ± S.D. (n = 3).
3.4. N-terminal tyrosine phosphorylation is more critical than serine phosphorylation of Cav-2 in negating TGF-β-stimulated PAI-1 and Col-1 gene expression in MLECs
To gain further insights into how serine and tyrosine deficient mutants affect TGF-β/Alk5/Smad3 pathway downstream of Smad3, we have performed RT-PCR to determine mRNA levels of the two target genes of Smad3, namely PAI-1 and Col-1 in control and treated with TGF-β (1 ng/ml; 48 h) pBABE, WT-Cav-2, S23/36A-Cav-2, and Y19/27F-Cav-2 MLECs.
As seen in Fig. 3A, treatment with TGF-β increased PAI-1 mRNA expression by c.a. 10.1-fold in pBABE MLECs, while re-expression of WT-Cav-2 reduced this increase by TGF-β to c.a. 2.9-fold. Re-expression of S23/36A-Cav-2 still reduced the stimulating effect of TGF-β to c.a. 3.9-fold. In contrast to S23/36A-Cav-2, re-expression of Y19/27F-Cav-2 failed to reduce TGF-β-induced increase in PAI-1 gene expression compared to pBABE. These data suggest that N-terminal tyrosine phosphorylation of Cav-2 plays a key role in diminishing TGF-β-induced PAI-1 gene expression in MLECs, while serine phosphorylation is of marginal importance.
Fig. 3.
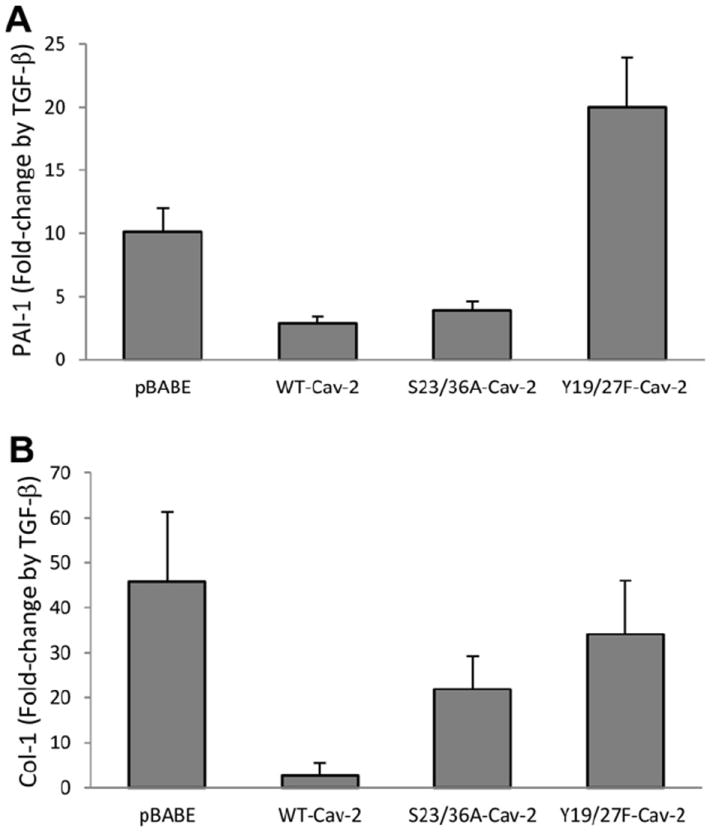
Effect of TGF-β on PAI-1 and Col 1 gene expression in Cav-2 KO MLECs re-expressing WT and phosphorylation-deficient mutants of Cav-2. WT-Cav-2, S23/36A-Cav-2, and Y19/27F-Cav-2 MLECs were plated at 3 × 105 per 150 mm dish for 24 h, incubated without or with TGF-β (1 ng/ml) for additional 48 h, followed by total RNA isolation and real-time PCR as described in Section 2. Data are expressed as fold change by TGF-β relative to control samples, calculated based on the relative amount of target mRNA normalized to the endogenous reference GAPDH mRNA and represented as Mean ± S.D of three replications done in duplicates.
Next, we have also examined the role of Cav-2 phosphorylation in TGF-β-stimulated Col-1 gene expression. As seen in Fig. 3B, treatment with TGF-β increased Col-1 mRNA expression by c.a. 45.7-fold in pBABE MLECs, while re-expression of WT-Cav-2 reduced this increase by TGF-β to only c.a. 2.6-fold. Re-expression of S23/36A-Cav-2 still reduced the stimulating effect of TGF-β to c.a. 21.9-fold. Re-expression of Y19/27F-Cav-2 only marginally reduced increase in Col-1 mRNA level by TGF-β to 34.1-fold. These data suggest that N-terminal tyrosine phosphorylation of Cav-2 is more critical than serine phosphorylation of Cav-2 in reducing TGF-β-induced Col-1 gene expression in MLECs.
3.5. N-terminal tyrosine phosphorylation is more critical than serine phosphorylation of Cav-2 in suppressing TGF-β-stimulated PAI-1 protein expression in MLECs
To further confirm if N-terminal tyrosine phosphorylation of Cav-2 in addition to PAI-1 and Col-1 transcript expression levels also inhibits TGF-β-induced PAI-1 and Col-1 protein expression levels, we have performed immunoblotting with anti-PAI-1 and Col-1 specific antibodies. As seen in Fig. 4, treatment with TGF-β: (1 ng/ml; 72 h) increased PAI-1 protein expression level by c.a. 5.3-fold in pBABE MLECs, while re-expression of WT-Cav-2 reduced this increase by TGF-β to c.a. 1.7-fold. Similar to WT-Cav-2, re-expression of S23/36A-Cav-2 still reduced the stimulating effect of TGF-β to c.a. 2.4-fold. In contrast to S23/36A-Cav-2, re-expression of Y19/27F-Cav-2 did not reduce TGF-β-induced increase in PAI-1 protein expression level compared to pBABE. These data suggest that N-terminal tyrosine phosphorylation of Cav-2 plays a key role in suppressing TGF-β-induced PAI-1 protein expression levels in MLECs, while serine phosphorylation is of marginal importance. We have also tried to determine if N-terminal phosphorylation of Cav-2 affects Col-1 protein expression levels by immunoblotting with the two independent Col-1-specific antibodies (Millipore and Santa Cruz Biotech) but were unable to detect specific signal (data not shown).
Fig. 4.
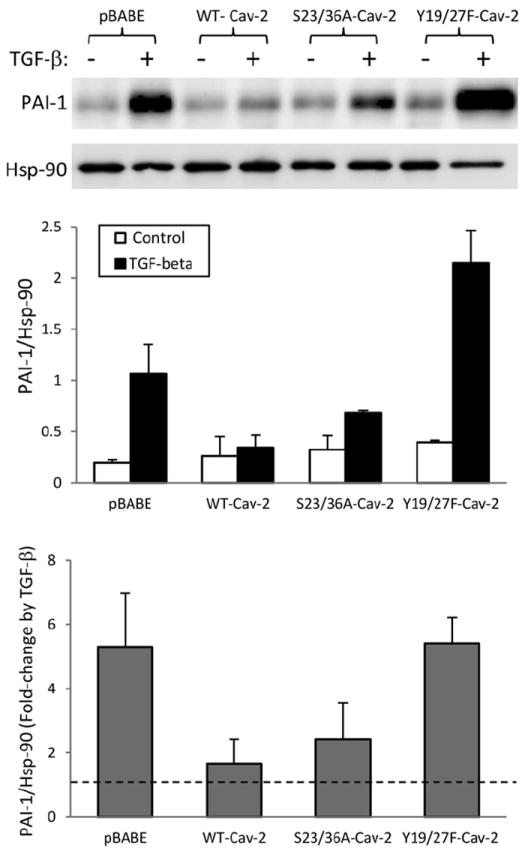
Effect of TGF-β on PAI-1 protein expression in Cav-2 KO MLECs re-expressing WT and phosphorylation-deficient mutants of Cav-2. WT-Cav-2, S23/36A-Cav-2, and Y19/27F-Cav-2 MLECs were plated at 3 × 105 cells onto 150 mm dishes, 24 h later incubated without or with TGF-β (1 ng/ml) for additional 48 h, lysed and processed for SDS–PAGE and immunoblotting with indicated antibodies as described in Section 2. Top graph: Densitometric ratios of PAI-1/Hsp-90 from control (blank bars) and treated with TGF-β (solid bars) samples quantified based on the above immunoblots and expressed as Means ± S.D. (n = 3). Bottom graph: Fold-change by TGF-β relative to respective control samples expressed as Means ± S.D. (n = 3).
3.6. N-terminal tyrosine phosphorylation of Cav-2 is stimulated by inhibition of tyrosine phosphatases in mouse and human ECs and suppressed by pharmacological inhibition of Src and Abl kinases in human ECs
Because of newly described here functional significance of tyrosine phosphorylation in regulating TGF-β signaling and function in ECs, next we have examined if tyrosine 19 and/or 27 phosphorylation of Cav-2 is regulated in ECs. Specifically, we have treated WT MLECs with vascular endothelial growth factor (VEGF; 100 ng/ml), H2O2 (0.5 mM), and sodium pervanadate (PV; 50 μM) for 30 min and analyzed phosphorylation of Cav-2 using tyrosine 19 (PY19-Cav-2) and tyrosine 27 (PY27-Cav-2) specific antibodies. As seen in Fig. 5A, treatment with PV yielded an obvious increase in PY19-Cav-2 within the examined time frame. Unlike PY19-Cav-2, no increase in PY27-Cav-2 was observed in PV-treated MLECs, presumably due to lack of specificity of PY27-Cav-2 antibody to mouse Cav-2. Thus next we have examined potential regulation of tyrosine phosphorylation of Cav-2 in human lung microvascular ECs (HULECs). Specifically, we have confirmed that just as in MLECs, PV treatment also resulted in stimulation of PY19-Cav-2 (Fig. 5B; top immunoblot; lane #2 vs. #1 and Fig. 5C). In contrast to MLECs, we have also demonstrated specific increase in PY27-Cav-2 by PV (Fig. 5B; 2nd immunoblot from the top; lane #2 vs. #1 and Fig. 5D). We have also tested a possible regulation of tyrosine phosphorylation of Cav-2 by TGF-β. 24 h pre-treatment with TGF-β had only a modest inhibitory effect on PV-stimulated PY19-Cav-2 and PY27-Cav-2 (lane # 3 vs. #2). In contrast, pre-treatment with Src inhibitor, PP2 resulted in ca. 6.8-fold and 6.6-fold reduction of PY19-Cav-2/Cav-2 and PY27-Cav-2/Cav-2 densitometric ratios, respectively. Moreover, pre-treatment with Abl inhibitor, imatinib resulted in ca. 2.3-fold reduction of both PY19-Cav-2 and PY27-Cav-2. Our data suggest that N-terminal tyrosine phosphorylation of Cav-2 can be stimulated by tyrosine phosphatase inhibition in mouse and human ECs and may be mediated by Src family kinase and possibly by Abl kinase in human ECs.
Fig. 5.
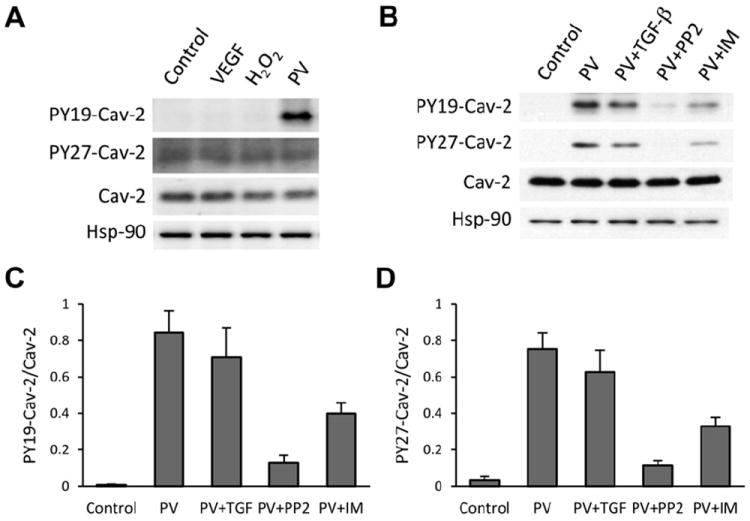
Effect of selected pathophysiological activators and tyrosine kinase inhibitors on phosphorylation of Cav-2 at tyrosine residues 19 and 27. (A) MLECs were treated without (control) or with VEGF (100 ng/ml), H2O2 (0.5 mM), or sodium pervanadate (PV; 50 μM) for 30 min, followed by lysis and immunoblotting with indicated antibodies. (B) Human lung microvascular endothelial cells (HULECs) were treated without (control; lane #1) or with PV (lane #2) for 30 min, followed by lysis and immunoblotting with indicated antibodies. In addition, cells were also pretreated before PV with 1 ng/ml of TGF-β for 24 h (PV + TGF-β; lane #3); 100 μM of Src family kinase inhibitor, PP2 for 30 min (PV + PP2; lane #4); and 20 μM of Abl kinase inhibitor, imatinib (PV + IM; lane #5). (C) Graphical representation of densitometric ratio of PY19-Cav-2/Cav-2 quantified based on immunoblot in B and expressed as Means ± S.D. (n = 3). (D) Graphical representation of densitometric ratio of PY27-Cav-2/Cav-2 quantified based on immunoblot in B and expressed as Means ± S.D. (n = 3).
4. Discussion
Here by re-expressing WT and mutated forms of Cav-2 in Cav-2 KO MLECs, we have evaluated the relative contribution of N-terminal serine and tyrosine phosphorylation sites to the inhibitory role of Cav-2 in preventing the anti-proliferative function and signaling of TGF-β in ECs. Specifically, we show that N-terminal tyrosine phosphorylation of Cav-2 plays a critical role in inhibiting anti-proliferative effect of TGF-β in ECs. In contrast to tyrosine, serine phosphorylation is only modestly involved in negating anti-proliferative effect of TGF-β by Cav-2. Although Cav-2 has previously been shown to be phosphorylated at N-terminal tyrosine residues 19 and 27 [18,19] and serine residues 23 and 36 [16,17], to our best knowledge, no concurrent and comparative analysis of the relative contribution of tyrosine and serine phosphorylation of Cav-2 to any functional effect of Cav-2 has been reported to date. Thus the current study is the first one to simultaneously examine the involvement of both N-terminal tyrosine and serine phosphorylation of Cav-2 in regulating any cell function. In addition, in the present study we provide first evidence suggesting that tyrosine 19 and 27 phosphorylation of Cav-2 may be mediated by Src family kinases and that Abl kinase could possibly contribute to this process in human ECs. Given the significance of Src family kinases in numerous EC specific processes, further studies examining if tyrosine phosphorylation of Cav-2 could affect these processes in ECs are clearly warranted.
The full dependence of anti- TGF-β-induced function and signaling of Cav-2 on N-terminal tyrosine phosphorylation and only partial dependence on N-terminal serine phosphorylation of Cav-2 could be observed using the two independent proliferation assays: cell count and MTT, and correlated with the respective effect on the levels of a mitotic marker protein, phospho-histone H3 in MLECs. We have recently reported that SB-505124, a compound which inhibits Alk5 without affecting Alk1 [20], blocked anti-proliferative effect of TGF-β on Cav-2 KO MLECs as well as Smad2 hyper-activation [14]. This data suggests that Alk5/Smad2/3 pathway is responsible for anti-proliferative effect of TGF-β in MLECs, consistent with previous reports in other ECs [21,22]. Importantly, in the current study, we show that N-terminal tyrosine phosphorylation of Cav-2 is essential for the inhibitory effect of Cav-2 on TGF-β-induced stimulation of Smad3 phosphorylation. Moreover, by analyzing the transcriptional activation of the two known transcription target genes of phosphorylated Smad2/3, we show that tyrosine phosphorylation is more critical than serine phosphorylation for the suppressive effect of Cav-2 on PAI-1 and to a lesser extent Col-1 gene induction by TGF-β. In particular, the inability of tyrosine phosphorylation-deficient mutant of Cav-2 to suppress TGF-β-induced PAI-1 mRNA and protein expression levels correlates well with the data obtained using proliferation assays. Interestingly, PAI-1 has previously been shown to be a specific target of Alk5 which was implicated in inhibiting EC proliferation [22]. Thus, it is possible that tyrosine phosphorylation of Cav-2 inhibits anti-proliferative effect of TGF-β, at least partially via suppressing PAI-1 transcriptional activation and subsequent protein expression.
What is the mechanism via which tyrosine phosphorylation of Cav-2 regulates TGF-β function and signaling in ECs? Based on the fact that specific inhibitor of Alk5 reversed the hyper-inhibitory effect of TGF-β on Cav-2 KO MLECs [14] and that re-expression of WT Cav-2 inhibits Smad3 phosphorylation, the direct interaction between tyrosine phosphorylated Cav-2 and Alk5 seems to be plausible. However, we were unable to detect direct interactions between Cav-2 and Alk5 in MLEC or primary human EC lysates by co-immunoprecipitation, presumably due to low expression levels of Alk5 in examined ECs (not shown). In addition to direct interaction, we have previously examined the possibility that Cav-2 could modify Alk5 or TGRII as well as Smad2/3 targeting to caveolar/lipid raft domains. However, we could not see any significant differences in caveolar/lipid raft targeting of previously mentioned protein components of the TGF-β signaling pathway [14]. Alternatively, tyrosine-phosphorylated Cav-2 could possibly affect the anti-proliferative signaling and function of TGF-β in ECs indirectly, perhaps via interacting with one or more of Src homology 2 domain (SH2) domain containing proteins which in turn could suppress Alk5/Smad2/3 pathway. Of note, tyrosine phosphorylation-dependent interaction of Cav-2 with such SH2 domain containing proteins as c-Src, Nck, or Ras-GAP was previously reported using Gst-fused Cav-2 approach and c-Src-over-expressing 3T3 cells [18,19].
In summary, our data indicate that N-terminal tyrosine phosphorylated Cav-2 negatively regulates anti-proliferative function and Alk5/Smad3-dependent signaling of TGF-β in MLECs, while N-terminal serine phosphorylation plays only a minor role.
Further studies elucidating the detailed mechanisms responsible for inhibitory regulation of TGF-β-induced signaling and function in ECs by tyrosine-phosphorylated Cav-2 will be necessary. In particular, investigation into the specific contribution of tyrosine 19 versus 27, identifying possibly different interacting protein partner(s) for WT Cav-2 versus tyrosine phosphorylation-deficient mutant(s) of Cav-2 might provide further mechanistic insights into the observed inhibitory effect of tyrosine phosphorylated Cav-2 on anti-proliferative signaling and function of TGF-β.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We would like to thank Drs. Michael Lisanti and Phillippe Frank for enabling us to isolate MLECs from Cav-2 KO mice, Dr. William C. Sessa for EcoPack2 cells stably producing retrovirus expressing polyoma middle T antigen. This work was supported by the grant from the National Institute of Health (1R01HL081860 to G.S.).
Footnotes
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.febslet.2012.07.008.
References
- 1.Glenney JR, Jr, Soppet D. Sequence and expression of caveolin, a protein component of caveolae plasma membrane domains phosphorylated on tyrosine in Rous sarcoma virus-transformed fibroblasts. Proc Natl Acad Sci U S A. 1992;89:10517–10521. doi: 10.1073/pnas.89.21.10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kurzchalia TV, Dupree P, Parton RG, Kellner R, Virta H, Lehnert M, Simons K. VIP21, a 21-kD membrane protein is an integral component of trans-Golgi-network-derived transport vesicles. J Cell Biol. 1992;118:1003–1014. doi: 10.1083/jcb.118.5.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scherer PE, Okamoto T, Chun M, Nishimoto I, Lodish HF, Lisanti MP. Identification, sequence, and expression of caveolin-2 defines a caveolin gene family. Proc Natl Acad Sci U S A. 1996;93:131–135. doi: 10.1073/pnas.93.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang Z, et al. Molecular cloning of caveolin-3, a novel member of the caveolin gene family expressed predominantly in muscle. J Biol Chem. 1996;271:2255–2261. doi: 10.1074/jbc.271.4.2255. [DOI] [PubMed] [Google Scholar]
- 5.Way M, Parton RG. M-caveolin, a muscle-specific caveolin-related protein. FEBS Lett. 1995;376:108–112. doi: 10.1016/0014-5793(95)01256-7. Corrected and republished in FEBS Lett. 1996, 378(1), 108–12. [DOI] [PubMed] [Google Scholar]
- 6.Parat MO. The biology of caveolae: achievements and perspectives. Int Rev Cell Mol Biol. 2009;273:117–162. doi: 10.1016/S1937-6448(08)01804-2. [DOI] [PubMed] [Google Scholar]
- 7.Sowa G. Caveolae, caveolins, cavins, and endothelial cell function: new insights. Front Physiol. 2012;2:120. doi: 10.3389/fphys.2011.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sowa G. Novel insights into the role of caveolin-2 in cell- and tissue-specific signaling and function. Biochem Res Int. 2011;2011:809259. doi: 10.1155/2011/809259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Razani B, et al. Caveolin-2-deficient mice show evidence of severe pulmonary dysfunction without disruption of caveolae. Mol Cell Biol. 2002;22:2329–2344. doi: 10.1128/MCB.22.7.2329-2344.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19:1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29:265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 12.Itoh S, Itoh F, Goumans MJ, Ten Dijke P. Signaling of transforming growth factor-beta family members through Smad proteins. Eur J Biochem. 2000;267:6954–6967. doi: 10.1046/j.1432-1327.2000.01828.x. [DOI] [PubMed] [Google Scholar]
- 13.Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-beta signal transduction. J Cell Sci. 2001;114:4359–4369. doi: 10.1242/jcs.114.24.4359. [DOI] [PubMed] [Google Scholar]
- 14.Xie L, Vo-Ransdell C, Abel B, Willoughby C, Jang S, Sowa G. Caveolin-2 is a negative regulator of anti-proliferative function and signaling of transforming growth factor-beta in endothelial cells. Am J Physiol Cell Physiol. 2011;301:C1161–C1174. doi: 10.1152/ajpcell.00486.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie L, Frank PG, Lisanti MP, Sowa G. Endothelial cells isolated from caveolin-2 knockout mice display higher proliferation rate and cell cycle progression relative to their wild-type counterparts. Am J Physiol Cell Physiol. 2010;298:C693–C701. doi: 10.1152/ajpcell.00401.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sowa G, Pypaert M, Fulton D, Sessa WC. The phosphorylation of caveolin-2 on serines 23 and 36 modulates caveolin-1-dependent caveolae formation. Proc Natl Acad Sci U S A. 2003;100:6511–6516. doi: 10.1073/pnas.1031672100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sowa G, Xie L, Xu L, Sessa WC. Serine 23 and 36 phosphorylation of caveolin-2 is differentially regulated by targeting to lipid raft/caveolae and in mitotic endothelial cells. Biochemistry. 2008;47:101–111. doi: 10.1021/bi701709s. [DOI] [PubMed] [Google Scholar]
- 18.Lee H, Park DS, Wang XB, Scherer PE, Schwartz PE, Lisanti MP. Src-induced phosphorylation of caveolin-2 on tyrosine 19. Phosphocaveolin-2 (Tyr(P)19 is localized near focal adhesions, remains associated with lipid rafts/caveolae, but no longer forms a high molecular mass heterooligomer with caveolin-1. J Biol Chem. 2002;277:34556–34567. doi: 10.1074/jbc.M204367200. [DOI] [PubMed] [Google Scholar]
- 19.Wang XB, Lee H, Capozza F, Marmon S, Sotgia F, Brooks JW, Campos-Gonzalez R, Lisanti MP. Tyrosine phosphorylation of caveolin-2 at residue 27: differences in the spatial and temporal behavior of phospho-Cav-2 (pY19 and pY27) Biochemistry. 2004;43:13694–13706. doi: 10.1021/bi049295+. [DOI] [PubMed] [Google Scholar]
- 20.Harrison CA, Gray PC, Vale WW, Robertson DM. Antagonists of activin signaling: mechanisms and potential biological applications. Trends Endocrinol Metab. 2005;16:73–78. doi: 10.1016/j.tem.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 21.Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, ten Dijke P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell. 2003;12:817–828. doi: 10.1016/s1097-2765(03)00386-1. [DOI] [PubMed] [Google Scholar]
- 22.Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-beta type I receptors. EMBO J. 2002;21:1743–1753. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.