

Abstract
The finding that arsenic trioxide is an effective treatment for acute promyelocytic leukemia has renewed interest in the pharmacological uses of inorganic and organic arsenicals. Here we synthesize and characterize the reactivity of an arsenical-maleimide (As-Mal) that can be efficiently conjugated to exposed cysteine residues in peptides and proteins with the ultimate goal of directing these As(III) species to vicinal thiols in susceptible targets within cells and tissues. As-Mal conjugated to a surface cysteine in thioredoxin provides a more potent inhibitor for Escherichia coli thioredoxin reductase than comparable simple inorganic or organic arsenicals. As-Mal can be coupled to all of the eight cysteine residues of reduced unfolded ribonuclease A, or to site-specific locations using appropriate cysteine mutations. We demonstrate particularly strong binding to the two CxxC motifs of protein disulfide isomerase using a mutant RNase in which As-Mal is specifically incorporated at residues 26 and 110. As-Mal will provide a facile reagent for the incorporation of As(III) species into a wide range of thiol-containing proteins, biomaterials and surfaces.
Inorganic arsenicals, including the sulfur-containing minerals realgar and orpiment and the oxide, As2O3, have been used for millennia to treat a wide range of medical conditions including leukemia, skin cancers and solid tumors.1,2 In the early 1900’s Ehrlich and Hata surveyed a range of organoarsenicals as anti-syphilitics and introduced arsphenamine as the first rational chemotherapeutic. The related melarsen oxide (Figure 1, 1) and its derivatives have been widely used as antitrypanosomals.1,2 While the medical applications of arsenicals declined in the 1940’s with the development of antibiotics and other modern therapies, they are now the subject of renewed interest following the demonstration that As2O3 is remarkably effective as a treatment for acute promyelocytic leukemia.3,4 In addition to inorganic arsenicals, several organoarsenicals are now in clinical trials for the treatment of leukemias and solid tumors.2,5
Figure 1.
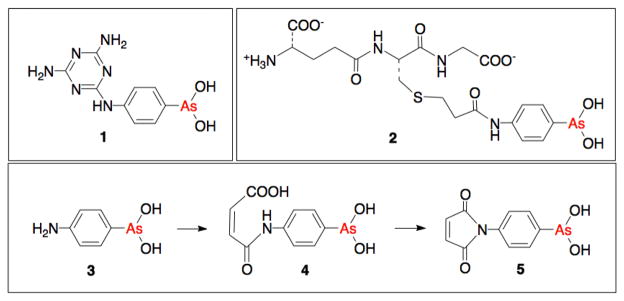
Structures of melarsen oxide 1 and 4-(N-(S-glutathionylacetyl)amino) phenylarsonous acid 2. The bottom panel shows the synthesis of the arsenical-maleimide 5 (4-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-
There is general agreement that the biological effects of As(III) species largely reflect their coordination to vicinal thiols.1,6 Hence a continuing challenge is the efficient targeting of arsenicals, while minimizing extraneous labeling and the toxicity associated with the many off-target thiol-containing proteins. For example, compound 2 (Figure 1) uses the tripeptide glutathione (GSH) as a vehicle for arsenical delivery via conjugation with 4-(2-bromoacetylamino)phenylarsonous acid.2,7 However, the bromoacetyl function reacts rather slowly under conditions typically used in thiol-bioconjugation reactions. Hence we sought a simple arsenical reagent that could be introduced rapidly and stoichiometrically at cysteine residues on peptides and proteins. We chose maleimides because they exhibit high specificity for thiols, they react several orders of magnitude more rapidly than bromoacetamide derivatives,8,9 and they have been widely used to conjugate cytotoxic agents to cysteine residues on monoclonal antibodies.10–12 Here we show selective and efficient incorporation of arsenicals into peptides and proteins, and demonstrate that these protein-arsenical conjugates are more effective inhibitors than simple arsenical derivatives. This new reagent will also allow the facile preparation of designed arsenical materials, including resins for thiol-based chromatography.
Figure 1 shows the synthesis of the arsenical-maleimide (As-Mal, 5) in overall 76% yield via the cyclization of maleamic acid 4 (Supporting Information; Figure S1). As-Mal shows two modes of reaction with thiols: one via Michael addition at the maleimide, and the other through coordination at the As(III) center. A titration of As-Mal with GSH showed a decline in 320 nm absorbance (with a sharp endpoint at 1.0 GSH/As-Mal; Figure S2). These data are consistent with comparable experiments with N-phenylmaleimide (not shown), and reflect a rapid conjugation of thiol with maleimide without undue interference from reversible binding to the arsenical moiety. The reaction between 100 μM As-Mal and equimolar GSH at pH 7.5, 25 °C, is essentially complete in 40 s (half-complete in 2.2 s, and is comparable to the behavior of N-phenylmaleimide under these second-order conditions; Figure S3).
These experiments suggest that As-Mal should react rapidly with protein thiols providing that they are solvent-accessible. We first demonstrated placement of a single As-Mal group at the surface of a folded protein. Here, Escherichia coli thioredoxin (an oxidoreductase containing a redox-active pair of cysteine residues, C32 and C35) was mutated to leave the surface-accessible C32 available for conjugation. Treatment of this C35S mutant with one equivalent of As-Mal rapidly generated a monolabeled derivative (at 100 μM concentrations the reaction was half-complete in <5 s; Figure S4). Monoalkylation was confirmed by mass spectrum (Figure S5), by the loss of the single DTNB reactive thiol group, and by titration of the conjugated arsenical reagent with dithiothreitol (Figures S6 and S7).
Arsenical-peptide/protein conjugates might be expected to provide more specificity towards their targets than the parent arsenical reagent alone. Figure 2 provides the first confirmation of this with a thioredoxin reductase from E. coli.13 The flow of reducing equivalents between NADPH and thioredoxin is schematically depicted in Figure 2. After the CxxC motif on the reductase has received a pair of reducing equivalents from the FAD moiety, a conformational change leads to formation of a mixed disulfide with a thioredoxin docked against the reductase, and to the eventual release of reduced thioredoxin.13,14 Figure 2 shows that 1 μM of the simple arsenicals sodium arsenite, monomethylarsenous acid (MMA) and p-succinylamidophenylarsenoxide (PSAO) provide only modest inhibition of this bacterial thioredoxin reductase. However the same concentration of the As-Mal Trx conjugate is considerably more potent, with a rapid and progressive loss of enzymatic activity.
Figure 2.

Inhibition of thioredoxin reductase by arsenicals. Panel A shows the reduction of Trx by NADPH catalyzed by thioredoxin reductase, together with the arsenicals tested at 1 μM. Reoxidation of reduced Trx by 5,5′-dithiobis(2-nitrobenzoate) (DTNB) is followed in the assay shown in the panel B (see Supporting Information)
We next wanted to test arsenic protein conjugates as potential inhibitors of protein disulfide isomerase (PDI). PDI has recently been suggested as a target for antiviral,15 anti-thrombotic,16 and anti-cancer therapies.17 Human PDI is comprised of four thioredoxin domains with the a and a′ domains carrying CxxC motifs that are responsible for the varied oxidoreductase and isomerase activities of the enzyme.18,19 PDI proteins have multiple intracellular roles in eukaryotes. However they are also found at the cell surface, where they can modulate extracellular redox poise 20–22 and are involved in platelet activation,20,23,24 enhancing metastasis,25,26 and viral fusion 22,27 and where they might be targeted by protein-based inhibitors and antibodies. Typically, the substrates of PDI family members are proteins that retain disordered or conformationally-mobile regions.18,19 A widely-employed substrate of PDI is reduced, or disulfide-mispaired, pancreatic ribonuclease A (RNase), and hence we used this protein as an initial vehicle for arsenic conjugation. Reduction of the four native disulfides of RNase leads to a tractable unfolded protein that can be labeled with eight As-Mal moieties (Figure S8). However here we prepared a limited series of site-directed mutants, including those in which only cysteines 1 and 4, or 4 and 8, or 1 and 8 were retained in the sequence. These were subsequently labeled with As-Mal (Figure 3, panel A; Supporting Information and Figures S9 and S10).
Figure 3.

RNase-based arsenicals. Panel A depicts cysteine residues along the 124 residue native chain of reduced RNase (at positions 26, 40, 58, 65, 72, 84, 95 and 110; labeled, for clarity, as cysteines 1-8). Sextuple cysteine to serine mutants of RNase are shown below, allowing arsenicals to be installed with a range of spacings (here 1-4, 4-8, and 1-8). Panel B depicts the domain structure of reduced PDI with CxxC motifs in a and a′ domains and the 1,8 bis-As-Mal RNase used in the titrations in panel C. Panel C is representative of four titrations. Panel D shows comparison between mono-, bis- and tris-As-Mal RNase derivatives in the insulin reductase assay using the same colors as shown in the bar diagram in Panel E. The concentration of inhibitors was chosen to reflect a total of 10 μM arsenic in the assay.
The widely-used insulin reductase assay was chosen to assess these inhibitors.28,29 Here PDI catalyzes the reduction of insulin disulfides driven by the water-soluble phosphine, TCEP, and the accumulation of the isolated B chain is followed by light scattering. In the absence of the As-Mal RNase derivatives the onset of turbidity occurs at about 230 s whereas it is strongly suppressed with the 1,8 bis-As-Mal RNase and to a lesser extent with the 1,4 and 4,8 bis-As-Mal RNase (Figure 3, panel E). For perspective, the monoarsenical C4 As-Mal RNase is considerably less effective than 1,8 bis-As-Mal RNase, and the 1,4,8 tris-As-Mal RNase is roughly comparable to the 1,8 derivative (Figure 3). To confirm that inhibition reflected the conjugation of arsenicals to RNase we evaluated the effect of the parent unconjugated cysteine RNase mutant proteins and found no significant inhibition of insulin reduction (Figure S11). Further, the simple monoarsenical PSAO fails to significantly delay the onset of turbidity at a concentration of 10 μM (Figure 3, panel E). Thus the placement and spacing of arsenicals along a protein chain can be exploited to modulate inhibitory potency.
A spectrophotometric titration of the 1,8 bis-As-Mal RNase with reduced PDI following the increase in absorbance at 300 nm accompanying thiol coordination to hydrated As(III) species is shown in Figure 3 (panel C). The data are fit to a Kd of 22 ± 7 nM with a stoichiometry of 1.21 ± 0.16 RNase per PDI. These data are consistent with the complexation of both the a and a′ CxxC motifs of reduced PDI with the 1,8 bis-As-Mal RNase (Figure 3, panel B). For comparison, a direct assessment of binding of PSAO to reduced PDI showed much weaker binding with a Kd of 1.1 μM29 consistent with failure to significantly impact the insulin reductase assay. Finally, since the levels of GSH in the extracellular matrix reach about 10 μM,30,31 we included this concentration in the assay and found that it did not significantly attenuate the inhibition observed with the 1,8 bis-As-Mal RNase. In contrast intracellular concentrations of GSH are in the 0.5 – 10 mM range32 and we have found that inclusion of 5 mM GSH provides insignificant inhibition of PDI by these arsenicals (not shown).
In summary, this work provides a simple way to conjugate arsenicals to cysteine-bearing peptides and proteins and demonstrates that the resulting adducts are more potent inhibitors than simple monoarsenical derivatives. Arsenicals might be delivered intracellularly via cell penetrating peptides33 or thiol-bearing dendrimers34 for targeting species that bind arsenical avidly in competition with GSH. While maleimide-thiol conjugates can be subject to reverse Michael reactions over extended times in physiological media, this is not necessarily a disadvantage because it provides the potential for targeted, controlled, release of cytotoxic species.35 Finally, the facility of the conjugation chemistry described here could allow multiple peptidic and protein scaffolds to be screened prior to manipulation of linker stability.11,36
Supplementary Material
Acknowledgments
Funding Sources
The work was supported by NIH grant GM26643.
We thank Jennifer Codding and Benjamin Israel for gifts of materials and Drs. Melissa Blackman, Joseph Fox, Danny Ramadan, Ronak Rughani, and Sharon Rozovsky for helpful discussions.
Footnotes
The authors declare no competing financial interest.
Supporting Information. Materials and Methods, synthesis of As-Mal, and characterization of As-Mal derivatized proteins. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nidhubhghaill OM, Sadler PJ. Struct Bond. 1991;78:129. [Google Scholar]
- 2.Dilda PJ, Hogg PJ. Cancer Treat Rev. 2007;33:542. doi: 10.1016/j.ctrv.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Beauchamp EM, Uren A. Vitamins and Hormones. 2012;88:333. doi: 10.1016/B978-0-12-394622-5.00015-8. [DOI] [PubMed] [Google Scholar]
- 4.Emadi A, Gore SD. Blood Rev. 2010;24:191. doi: 10.1016/j.blre.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu JX, Zhou GB, Chen SJ, Chen Z. Curr Op Chem Biol. 2012;16:92. doi: 10.1016/j.cbpa.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 6.Spuches AM, Kruszyna HG, Rich AM, Wilcox DE. Inorg Chem. 2005;44:2964. doi: 10.1021/ic048694q. [DOI] [PubMed] [Google Scholar]
- 7.Dilda PJ, Decollogne S, Weerakoon L, Norris MD, Haber M, Allen JD, Hogg PJ. J Med Chem. 2009;52:6209. doi: 10.1021/jm9008339. [DOI] [PubMed] [Google Scholar]
- 8.Gilbert HF. Methods Enzymol. 1995;251:8. doi: 10.1016/0076-6879(95)51107-5. [DOI] [PubMed] [Google Scholar]
- 9.Hansen RE, Winther JR. Anal Biochem. 2009;394:147. doi: 10.1016/j.ab.2009.07.051. [DOI] [PubMed] [Google Scholar]
- 10.Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, Lu Y, Meng YG, Ng C, Yang J, Lee CC, Duenas E, Gorrell J, Katta V, Kim A, McDorman K, Flagella K, Venook R, Ross S, Spencer SD, Lee Wong W, Lowman HB, Vandlen R, Sliwkowski MX, Scheller RH, Polakis P, Mallet W. Nature Biotech. 2008;26:925. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- 11.Shen BQ, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu SF, Mai E, Li D, Tibbitts J, Baudys J, Saad OM, Scales SJ, McDonald PJ, Hass PE, Eigenbrot C, Nguyen T, Solis WA, Fuji RN, Flagella KM, Patel D, Spencer SD, Khawli LA, Ebens A, Wong WL, Vandlen R, Kaur S, Sliwkowski MX, Scheller RH, Polakis P, Junutula JR. Nature Biotech. 2012;30:184. doi: 10.1038/nbt.2108. [DOI] [PubMed] [Google Scholar]
- 12.Lyon RP, Meyer DL, Setter JR, Senter PD. Methods Enzymol. 2012;502:123. doi: 10.1016/B978-0-12-416039-2.00006-9. [DOI] [PubMed] [Google Scholar]
- 13.Williams CH, Arscott LD, Muller S, Lennon BW, Ludwig ML, Wang PF, Veine DM, Becker K, Schirmer RH. Eur J Biochem. 2000;267:6110. doi: 10.1046/j.1432-1327.2000.01702.x. [DOI] [PubMed] [Google Scholar]
- 14.Lennon BW, Williams CH, Jr, Ludwig ML. Science. 2000;289:1190. doi: 10.1126/science.289.5482.1190. [DOI] [PubMed] [Google Scholar]
- 15.Khan MM, Simizu S, Lai NS, Kawatani M, Shimizu T, Osada H. ACS Chem Biol. 2011;6:245. doi: 10.1021/cb100387r. [DOI] [PubMed] [Google Scholar]
- 16.Jasuja R, Passam FH, Kennedy DR, Kim SH, van Hessem L, Lin L, Bowley SR, Joshi SS, Dilks JR, Furie B, Furie BC, Flaumenhaft R. J Clin Invest. 2012;122:2104. doi: 10.1172/JCI61228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu S, Butkevich AN, Yamada R, Zhou Y, Debnath B, Duncan R, Zandi E, Petasis NA, Neamati N. P Natl Acad Sci USA. 2012;109:16348. doi: 10.1073/pnas.1205226109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Appenzeller-Herzog C, Ellgaard L. Biochim Biophys Acta. 2008;1783:535. doi: 10.1016/j.bbamcr.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 19.Hatahet F, Ruddock LW. Antioxid Redox Signal. 2009;11:2807. doi: 10.1089/ars.2009.2466. [DOI] [PubMed] [Google Scholar]
- 20.Manickam N, Sun X, Li M, Gazitt Y, Essex DW. Br J Haematol. 2008;140:223. doi: 10.1111/j.1365-2141.2007.06898.x. [DOI] [PubMed] [Google Scholar]
- 21.Jordan PA, Gibbins JM. Antioxid Redox Signal. 2006;8:312. doi: 10.1089/ars.2006.8.312. [DOI] [PubMed] [Google Scholar]
- 22.Bi S, Hong PW, Lee B, Baum LG. Proc Natl Acad Sci USA. 2011;108:10650. doi: 10.1073/pnas.1017954108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holbrook LM, Watkins NA, Simmonds AD, Jones CI, Ouwehand WH, Gibbins JM. Br J Haematol. 2009;148:627. doi: 10.1111/j.1365-2141.2009.07994.x. [DOI] [PubMed] [Google Scholar]
- 24.Jurk K, Lahav J, Van Aken H, Brodde MF, Nofer JR, Kehrel BE. J Thromb Haemostasis. 2011;9:2278. doi: 10.1111/j.1538-7836.2011.04509.x. [DOI] [PubMed] [Google Scholar]
- 25.Kaiser BK, Yim D, Chow IT, Gonzalez S, Dai Z, Mann HH, Strong RK, Groh V, Spies T. Nature. 2007;447:482. doi: 10.1038/nature05768. [DOI] [PubMed] [Google Scholar]
- 26.Goplen D, Wang J, Enger PO, Tysnes BB, Terzis AJ, Laerum OD, Bjerkvig R. Cancer Res. 2006;66:9895. doi: 10.1158/0008-5472.CAN-05-4589. [DOI] [PubMed] [Google Scholar]
- 27.Jain S, McGinnes LW, Morrison TG. J Virol. 2007;81:2328. doi: 10.1128/JVI.01940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holmgren A. J Biol Chem. 1979;254:9627. [PubMed] [Google Scholar]
- 29.Ramadan D, Rancy PC, Nagarkar RP, Schneider JP, Thorpe C. Biochemistry. 2009;48:424. doi: 10.1021/bi801988x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith CV, Jones DP, Guenthner TM, Lash LH, Lauterburg BH. Toxicol Appl Pharm. 1996;140:1. doi: 10.1006/taap.1996.0191. [DOI] [PubMed] [Google Scholar]
- 31.Saito G, Swanson JA, Lee KD. Adv Drug Deliv Rev. 2003;55:199. doi: 10.1016/s0169-409x(02)00179-5. [DOI] [PubMed] [Google Scholar]
- 32.Meister A, Anderson ME. Ann Rev Biochem. 1983;52:711. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- 33.Koren E, Torchilin VP. Trends Mol Med. 2012;18:385. doi: 10.1016/j.molmed.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 34.Hermandson G. Bioconjugate Techniques. Academic Press; Boston, MA: 2008. [Google Scholar]
- 35.Baldwin AD, Kiick KL. Bioconj Chem. 2011;22:1946. doi: 10.1021/bc200148v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palanki MS, Bhat A, Lappe RW, Liu B, Oates B, Rizzo J, Stankovic N, Bradshaw C. Bioorg Med Chem Lett. 2012;22:4249. doi: 10.1016/j.bmcl.2012.05.040. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.