

Abstract
Akt kinase plays a central role in cell growth, metabolism and tumorigenesis. Although TRAF6 E3 ligase orchestrates IGF-1-mediated Akt ubiquitination and activation, it is unclear whether TRAF6 is involved in Akt activation by other growth factor receptors as well. Here we show that Akt ubiquitination is also induced by activation of ErbB receptors; unexpectedly, Skp2 SCF complex, but not TRAF6, is a critical E3 ligase for ErbB receptor-mediated Akt ubiquitination and membrane recruitment. Interestingly, Skp2 deficiency impairs Akt activation, Glut1 expression, glucose uptake and glycolysis, and breast cancer progression in various tumor models. Moreover, Skp2 overexpression correlates with Akt activation, breast cancer metastasis, and serves as a marker for poor prognosis in Her2-positive patients. Finally, we showed that Skp2 silencing sensitizes Her2-overexpressing tumors to Herceptin treatment. Our study suggests that distinct E3 ligases are utilized by diverse growth factors for Akt ubiquitination and activation.
Introduction
Akt kinase is a key factor that conveys growth factor signals from outside the cell to inside the cell. It serves as a central node for the regulation of cell proliferation, cell survival, metabolism and tumorigenesis (Brazil et al., 2002; Liu et al., 2009; Manning and Cantley, 2007; Yang et al., 2010a). The recruitment of Akt kinase to the plasma membrane is a critical step for Akt phosphorylation and activation by growth-factor stimuli. Although the PIP3 formation-induced by PI3K activation is essential for Akt membrane recruitment, our recent study reveals that K63-linked ubiquitination of Akt is also required for this process. TRAF6 is found to be an ubiquitin ligase (E3) for Akt and plays a crucial role in Akt ubiquitination, membrane translocation, and phosphorylation upon stimulation with insulin-like growth factor-1 (IGF-1) (Yang et al., 2009; Yang et al., 2010b). Thus, Akt ubiquitination and PIP3 binding are two important events required for Akt membrane recruitment and activation in response to IGF-1. However, it remains largely unclear whether Akt ubiquitination is universally engaged in Akt membrane translocation and activation triggered by other growth factor receptors, such as ErbB family.
Under normoxic condition, differentiated cells primarily utilize mitochondria oxidative phosphorylation to generate adenosine 5′-triphosphate (ATP) for biogenesis and cellular processes (Aragones et al., 2009; Vander Heiden et al., 2009). However, under hypoxia these cells switch their metabolism from aerobic oxidative phosphorylation to anaerobic glycolysis. Notably, tumor cells utilize aerobic glycolysis regardless of the oxygen levels, known as the Warburg effect. The elevated aerobic glycolysis seen in tumor cells rapidly generates ATP in order to meet their increased need for energy and biosynthesis to sustain tumor growth (Birnbaum, 2004; Plas and Thompson, 2005; Robey and Hay, 2009). Akt kinase is frequently activated in various tumor types and represents one of the main drivers for the Warburg effect (Elstrom et al., 2004). Akt increases glucose uptake by enhancing transcription and membrane translocation of glucose transporters. It promotes glycolytic flux through increasing hexokinase and phosphofructokinase activity (Robey and Hay, 2009). Accumulating evidence shows that the activation of the Akt pathway causes increased dependency on aerobic glycolysis (Elstrom et al., 2004; Wieman et al., 2007), suggesting that therapeutic strategies that target the Akt pathway can block glucose metabolism and consequently result in tumor regression. Although numerous downstream players involved in Akt-mediated glycolysis have been proposed, current knowledge regarding the upstream regulators of Akt-dependent glycolytic pathway remains limited.
In this study, we unexpectedly discover that Skp2, rather than TRAF6, is critically involved in ErbB family-induced Akt ubiquitination, aerobic glycolysis and tumorigenesis. Importantly, targeting glycolysis by Skp2 deficiency sensitizes Her2-positive tumors to Herceptin treatment, highlighting the clinical value of Skp2 targeting in breast cancer therapy.
Results
Skp2 is responsible for EGF-mediated Akt ubiquitination
To determine whether Akt ubiquitination is a common event induced by growth factors, we examined whether Akt ubiquitination is induced by activation of epidermal growth factor (EGF) receptor, a member of the ErbB receptor family. Indeed, in vivo ubiquitination assay revealed that endogenous Akt ubiquitination is also induced upon EGF treatment (Figures 1A and 1E and Figure S1F, upper panel), suggesting that Akt ubiquitination is a general event triggered by growth factors. As TRAF6 is important for IGF-1-mediated Akt ubiquitination and activation (Yang et al., 2009), we determined whether EGF-mediated Akt ubiquitination and activation depend on TRAF6. To our surprise, EGF-induced ubiquitination of Akt and phosphorylation of Akt and Foxo1 were comparable between WT and Traf6-/- MEFs (Figure S1A and S1B), suggesting that TRAF6 is dispensable for EGF-induced Akt activation.
Figure 1. Skp2 is required for EGF-mediated Akt ubiquitination.
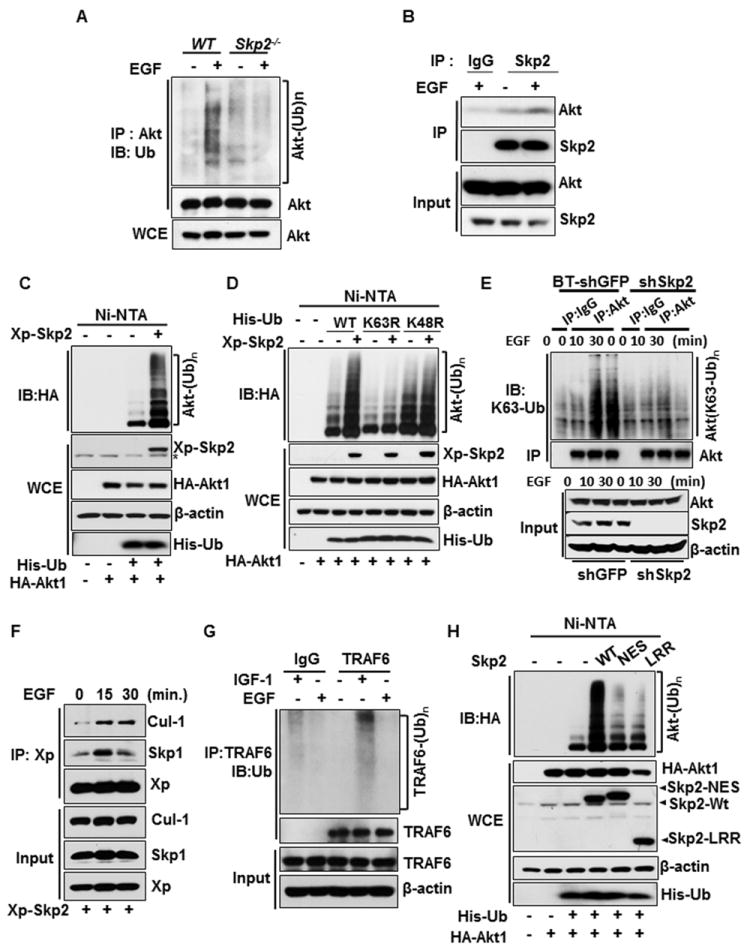
(A) WT and Skp2-/- MEFs were serum-starved, treated with or without EGF, and harvested for in vivo ubiquitination assay. (B) 293T cells were serum-starved, treated with or without EGF and harvested for Co-immunoprecipitation (IP) assay followed by immunoblot (IB) analysis. (C) In vivo ubiquitination assay in 293T cells transfected with hemagglutinin (HA)-Akt1 and His-ubiquitin (His-Ub), along with Xpress-Skp2 (Xp-Skp2). Ni–nitrilotriacetic acid (NTA) indicates nickel bead precipitate; WCE indicates whole-cell extracts. * indicates non-specific signal. (D) In vivo ubiquitination assay in 293T cells transfected with various constructs. (E) Control- and Skp2-knockdown BT-474 cells were serum-starved, treated with or without EGF and harvested for in vivo ubiquitination assay. (F) 293T cells were transfected with Xp-Skp2, serum-starved, treated with or without EGF, and harvested for Co-IP assay followed by IB analysis. (G) Cos1 cells were serum-starved, treated with or without IGF-1 or EGF and harvested for in vivo ubiquitination assay. (H) In vivo ubiquitination assay in 293T cells transfected with HA-Akt1, His-Ub, along with WT Skp2 and E3-ligase dead mutants (Skp2-NES and Skp2-LRR). See also Figure S1.
Skp2 is an F-Box protein that forms a Skp2 SCF complex with Skp1, Cullin-1 (Cul-1), and RBx1 to constitute an E3 ligase activity that triggers protein ubiquitination and degradation (Chan et al., 2010b; Nakayama and Nakayama, 2006). Skp2 displays oncogenic activities by regulating cell cycle progression, senescence, and metastasis (Chan et al.; Chan et al., 2010a; Lin et al., 2010; Lin et al., 2009). As we have previously demonstrated that Akt physically interacts with Skp2 in vivo in response to IGF-1 (Lin et al., 2009), we determined whether Skp2 is engaged in EGF-mediated Akt ubiquitination. while Akt ubiquitination upon EGF stimulation was induced in WT MEFs or control-knockdown cells, it was impaired in Skp2-/- MEFs and Skp2 knockdown cells (Figures 1A and 1E and Figure S1F, upper panel), indicating that Skp2 is required for EGF-induced Akt ubiquitination. Moreover, the interaction between endogenous Skp2 and Akt was also enhanced by EGF (Figure 1B and Figure S1C), which occurred both in cytosol and nucleus (Figure S1D). Notably, Skp2 overexpression induced Akt ubiquitination in the absence of proteosome inhibitor, which did not cause Akt degradation (Figure 1C). Skp2 overexpression promoted lysine (K) 63-linked, rather than K48-linked, ubiquitination of Akt, which was diminished upon Skp2 deficiency (Figure 1D). We further demonstrated that EGF markedly promoted endogenous K63-linked, rather than K48-linked, ubiquitination of Akt, which was diminished upon Skp2 deficiency (Figure 1E and Figure S1E, S1F). While TRAF6 knockdown reduced the basal Akt ubiquitination and IGF-1-induced Akt phosphorylation and activation, Skp2 overexpression was still able to efficiently induce Akt ubiquitination (Figure S1G), suggesting that Skp2 promotes Akt ubiquitination independent of TRAF6.
Diverse growth factors utilize distinct E3 ligase for Akt ubiquitination and activation
While TRAF6 plays a critical role in IGF-1-mediated Akt ubiquitination and activation (Yang et al., 2009), this current study shows that EGF selectively utilizes the Skp2 SCF complex to ubiquitinate Akt. To gain further mechanistic insights into how ErbB and IGF-1 selectivity utilize distinct E3 ligases for Akt activation, we investigated the possibility that EGF and IGF-1 may do so by regulating their E3 ligase activities. Skp2, but not TRAF6, interacted with EGFR and its E3 ligase activity was enhanced upon EGF treatment, as judged by the formation of the Skp2 SCF complex (Figure 1F and Figure S1H). In contrast, TRAF6 associated with IGF-1Rβ and its E3 ligase activity, as determined by TRAF6 auto-ubiquitination, was induced by IGF-1, but not by EGF stimulation (Figure 1G and Figure S1I). Moreover, while IGF-1 readily activated TRAF6 within 15 min, it could not promote the formation of Skp2-SCF complex at this early time point, although it indeed does so after 1h (Figure S1J), consistent with our previous observation (Lin et al., 2009). Additionally, the endogenous expression of Skp2 was not affected by IGF-1 stimulation within 1h (Figure S1K). Thus, these results demonstrated that Skp2 is not activated at early time points, but it is activated at longer time points. Since IGF-1 induces the association of TRAF6 with Akt (Yang et al., 2009) and EGF stimulates the interaction between Skp2 and Akt (Figure 1B and Figure S1C), we postulated that a potential mechanism responsible for this distinctive dependence on TRAF6 and Skp2 may be due to their different binding affinity for Akt. Indeed, we found that EGF selectively induced the interaction of Akt with Skp2, but not with TRAF6 (Figure S1L), providing an explanation of why Skp2, but not TRAF6, is involved in ErbB family-induced Akt ubiquitination.
Given that phosphorylation is a well-characterized posttranslational modification that regulates protein-protein interaction, we hypothesized that IGF-1 and EGF may regulate the interaction of Akt with Skp2 or TRAF6 by inducing their phosphorylation. In support of this notion, TRAF6 was tyrosine-, but not serine/threonine-, phosphorylated upon IGF-1 treatment (Figure S1M, upper and middle panels). In contrast, EGF failed to induce TRAF6 tyrosine phosphorylation (Figure S1M, lower panel). To further understand whether IGF-1-mediated TRAF6 phosphorylation may regulate TRAF6 and Akt interaction, we treated the TRAF6 immunocomplex with phosphatase to induce TRAF6 dephosphorylation and found that IGF-1-induced TRAF6 and Akt interaction was diminished (Figure S1N). While Skp2 underwent tyrosine and serine/threonine phosphorylation upon EGF stimulation, the phosphatase treatment resulted in Skp2 dephosphorylation, accompanied by Skp2 and Akt dissociation (Figures S1O and S1P). Collectively, these results suggest that various receptor tyrosine kinases (RTKs) can recruit, activate distinct E3 ligases and regulate the interaction between Akt and distinct E3 ligases, thereby contributing to RTK-mediated Akt ubiquitination.
The Skp2-SCF complex is required for Akt ubiquitination and activation triggered by ErbB family signaling
Whereas Skp2 overexpression promoted in vivo Akt ubiquitination, Skp2-NES and Skp2-LRR mutants, both of which have defects in Skp2 SCF E3 ligase activity (Chan et al., 2010a; Kim et al., 2003; Lin et al., 2009), compromised this effect (Figure 1H). Of note, the impairment of Skp2-NES mutant in promoting Akt ubiquitination was not due to its defect in Akt binding (Figure S1Q). Likewise, Skp2-mediatd in vivo Akt ubiquitination was profoundly compromised upon Cul-1 or Skp1 knockdown (Figures 2A and 2B). Interestingly, silencing both UbcH5c and Ubc13, two ubiquitin-conjugating enzymes (E2s) critical for K63-linked ubiquitination (Xia et al., 2009; Zeng et al., 2010)., attenuated Skp2-mediated Akt ubiquitination (Figure 2C). We found that Skp2, but not Skp1 and Cul-1, could directly interact with Akt (Figure S2) and that Skp2 SCF complex readily triggered in vitro Akt ubiquitination, in a manner similar to that of TRAF6 (Figure 2D). Thus, Skp2 SCF complex is a direct E3 ligase for Akt.
Figure 2. Skp2 SCF complex is a direct E3 ligase for Akt.
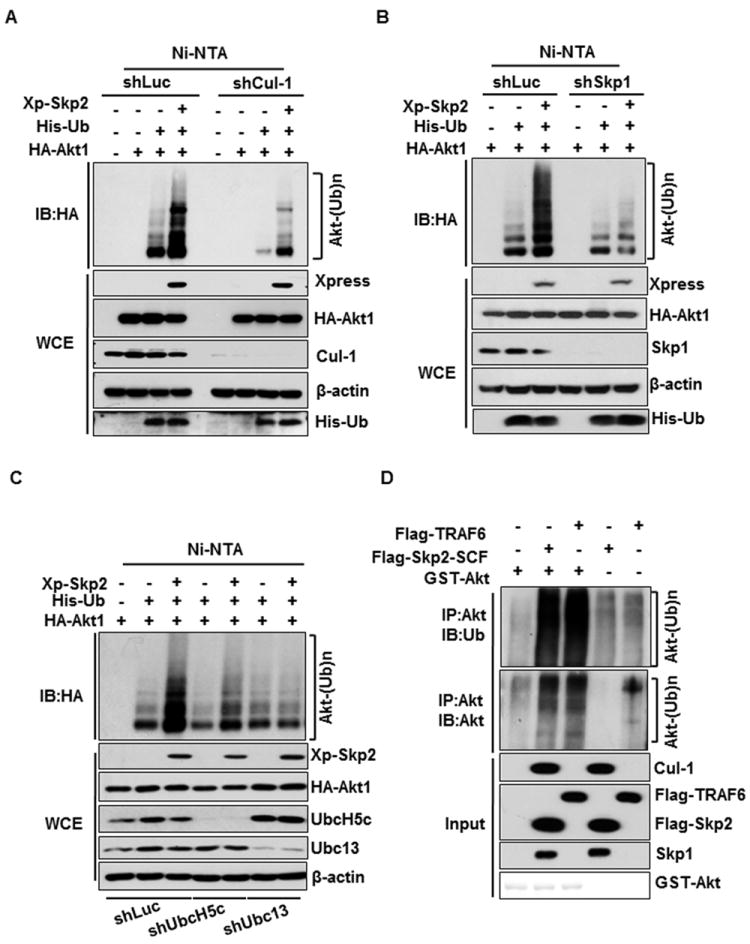
(A) Luciferase or Cul-1-silenced 293 cells transfected with various constructs were harvested for in vivo ubiquitination assay. (B) In vivo ubiquitination assay in Luciferase or Skp1-silenced 293 cells transfected with various constructs. (C) In vivo ubiquitination assay in various 293 knockdown cells transfected with different plasmids. (D) GST-Akt proteins were incubated with adenosine triphosphate, E1, and E2 (both UbcH5c and Ubc13/Uev1) along with or without Flag-Skp2-SCF or Flag-TRAF6 for in vitro Akt ubiquitination assay. See also Figure S2.
We next determined whether Skp2 SCF complex is required for EGF-mediated Akt activation. Strikingly, EGF-induced phosphorylation of Akt and Foxo1 were markedly reduced in Skp2-/- MEFs compared to that in WT MEFs (Figure 3A). We observed a similar impairment in EGF-mediated Akt phosphorylation upon Skp2, Cul-1, Skp1, UbcH5c, or Ubc13 knockdown (Figure 3B and Figure S3A-S3C). Accordingly, these results indicate that the Skp2 SCF complex is critical for EGF-triggered Akt phosphorylation and activation. To determine whether Skp2 is also involved in Akt signaling activation by other ErbB family proteins, BT-474 breast cancer cells were treated with Heregulin (HRG), which is known to activate ErbB2 and ErbB3 (Agus et al., 2002; Lee-Hoeflich et al., 2008). Notably, Skp2 knockdown also impaired Akt phosphorylation and activation in BT-474 cells upon HRG treatment (Figure 3C). These results suggest that Skp2 is generally involved in Akt activation in response to ErbB receptor signaling.
Figure 3. Skp2 SCF complex is required for Akt activation and membrane recruitment.
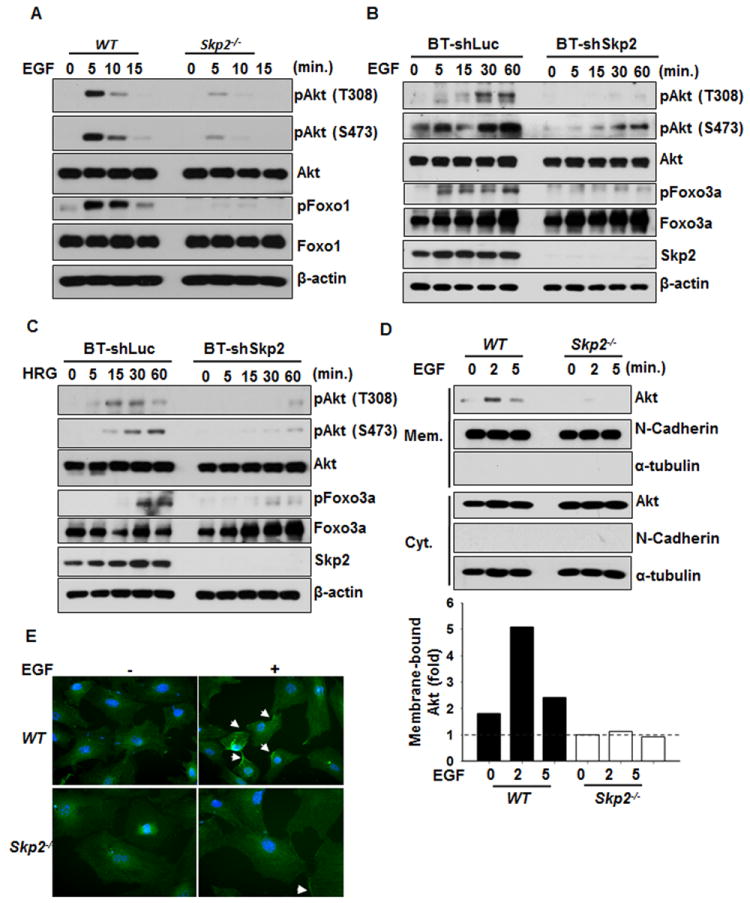
(A) WT and Skp2-/- MEFs were serum-starved, treated with EGF for various time points and harvested for IB analysis. (B, C) BT-474 cells with control- and Skp2-silenced were serum-starved, treated with EGF (B) or HRG (C) for various time points and harvested for IB analysis. (D) WT and Skp2-/- MEFs were serum-starved, treated with EGF for various time points, and the membrane (mem) and cytosolic (cyt) fractions were isolated for IB analysis. The relative intensity of was quantified with ImageQuant software and normalized with the Akt levels in WT MEFs without EGF treatment. (E) WT and Skp2-/- MEFs were serum-starved, treated with EGF for 5 min and fixed for immunofluorescence assay. The arrow indicates the membrane localization of Akt. The quantification results were shown in Figure S3E. See also Figure S3.
We next examined whether Skp2 regulates the ubiquitination of other Akt isoforms. Notably, we found ubiquitination of both Akt1 and Akt2, but not Akt3 were induced by Skp2 overexpression, although basal ubiquitination of Akt2 was lower than that of Akt1 (Figure S3D). Furthermore, Skp2 deficiency attenuated EGF-mediated Akt1 and Akt2 phosphorylation (Figure S3E), indicating that Skp2 is required for EGF-promoted activation of both Akt1 and Akt2.
As TRAF6-mediated Akt ubiquitination at K8 and K14 residues is critical for its membrane recruitment and activation (Yang et al., 2009), we then validated whether these two sites are also being utilized by Skp2 SCF complex. Indeed, we found that K8 and K14 residues are essential sites for Skp2-mediated Akt ubiquitination. Biochemical fractionation assay and immunofluorescence assay revealed that EGF-induced Akt membrane recruitment was dramatically impaired in Skp2-/- MEFs compared to that in WT MEFs (Figures 3D and 3E and Figure S3G).
Although the binding of Akt to PIP3 is an essential step for Akt membrane recruitment and activation, our recent report reveals that K63-linked ubiquitination of Akt plays a dispensable role in Akt and PIP3 binding (Yang et al., 2009). We further excluded the possibility that Skp2 regulates Akt membrane recruitment and activation by affecting Akt and PIP3 binding (Figure S3H). While Akt dimerization is also proposed to be important for Akt activation (Datta et al., 1995; Kunstle et al., 2002; Noguchi et al., 2007), ubiquitination-dead mutant of Akt displayed similar capability to form Akt dimer as WT Akt did (Figure S3I), suggesting that Akt ubiquitination does not regulate Akt dimerization.
Skp2 regulates glycolysis through inducing Akt ubiquitination and activation
Cancer cells evolve to develop a mechanism that increases glucose uptake and glycolysis to generate higher ATP levels, a phenomenon called Warburg effect (Warburg, 1956). The Warburg effect is critically regulated by Akt, which is highly activated in human cancers (Elstrom et al., 2004; Manning and Cantley, 2007; Plas and Thompson, 2005; Robey and Hay, 2009). As Skp2 orchestrates Akt ubiquitination and activation, it is conceivable that Skp2 may regulate glucose uptake and glycolysis. Indeed, we found that Skp2 knockdown suppressed glucose uptake and glycolysis in breast cancer cells upon EGF or HRG stimulation, as determined by lactate production and glucose incorporation assays (Figures 4A and 4B and Figures S4A-S4C and S4E), in a manner similar to that of PI3K/Akt inhibition (Figures 4B and 4C and Figures S4D and S4F). To further investigate whether Skp2 regulates in vivo glycolysis and breast cancer development, we monitored the impact of Skp2 expression on in vivo glucose uptake and subsequent tumor growth in a xenograft model bearing Her2-overexpressing breast tumors. While in vivo glucose uptake, was enriched in control-silenced breast tumors, it was profoundly reduced upon Skp2 knockdown, which correlated with tumor suppression upon Skp2 silencing (Figures 4D-4G). Accordingly, Skp2 promotes in vivo glycolysis and breast cancer development.
Figure 4. Skp2 regulates glucose uptake and glycolysis in vitro and in vivo.
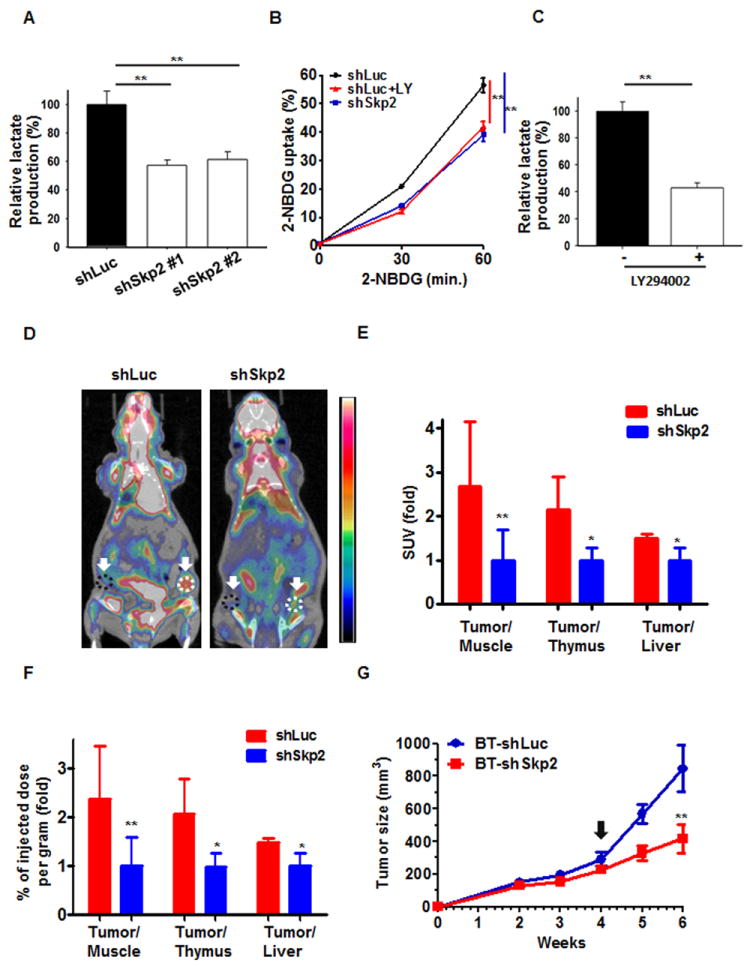
(A) Lactate production was measured in BT-474 cells with Luciferase and Skp2 knockdown. (B) Glucose uptake was measured in BT-474 cells with Luciferase and Skp2 knockdown. Cells treated with or without LY294002 were grown in the presence of the fluorescent analog NBDG for various time points, and glucose uptake was quantified using FACS analysis. (C) Lactate production was measured in BT-474 cells treated with or without LY294002. (D) Representative PET/CT images in nude mice bearing breast tumors with Luciferase- or Skp2-knockdown. White dotted lines indicate area of the breast tumors and black dotted lines indicate area of muscle tissues that were analyzed for in vivo glucose uptake. (E, F) Glucose uptake was expressed as Standard Uptake Value (SUV) ratio (E) or as percent of injected dose per gram (F) of labeled [18F] FDG-glucose incorporation in mice bearing Luciferase- or Skp2-silenced breast tumors. Glucose uptake in breast tumors was normalized with muscle, thymus or liver tissues in each mouse. The quantified results are presented as means ± s.d. (n = 5). (G) Breast tumor development in nude mice bearing breast tumors with Luciferase- or Skp2-knockdown (n=5). The arrow indicates the time point for in vivo glucose uptake analysis. **, p<0.01. See also Figure S4.
Intriguingly, introduction of Myr-Akt, a constitutively active form of Akt, rescued the defect in lactate production of Skp2-knockdown cells in the presence of EGF or HRG (Figure 5A and Figures S5A and S5B), suggesting that Skp2 regulates glycolysis in cancer cells through modulating Akt activation. Since Akt regulates glucose uptake by inducing gene expression and membrane translocation of Glut1 (glucose transporter type 1), a predominant glucose transporter expressed in most cell types (Barthel et al., 1999; Wieman et al., 2007), we examined whether Glut1 is a downstream target of Skp2 in regulating glycolytic phenotype. As expected, Skp2 deficiency reduced Glu1 transcription and protein expression in breast cancer cells, recapitulating the phenotype driven by Akt inactivation (Figures 5B, 5C and 5F and Figures S4E-S4G). Importantly, we found that Glut1 protein expression in both membrane fraction and total cell extracts was markedly induced by EGF in control-knockdown cells; however, this induction was impaired in Skp2-deficient cells (Figures 5D and 5E and Figure S5C). The inhibition of Glut1 protein expression in Skp2-knockdown cancer cells was also rescued by the introduction of Myr-Akt (Figure 5F), underscoring that Skp2 regulates Glut1 expression through Akt activation.
Figure 5. Skp2 regulates glycolysis through promoting Akt ubiquitination and activation.
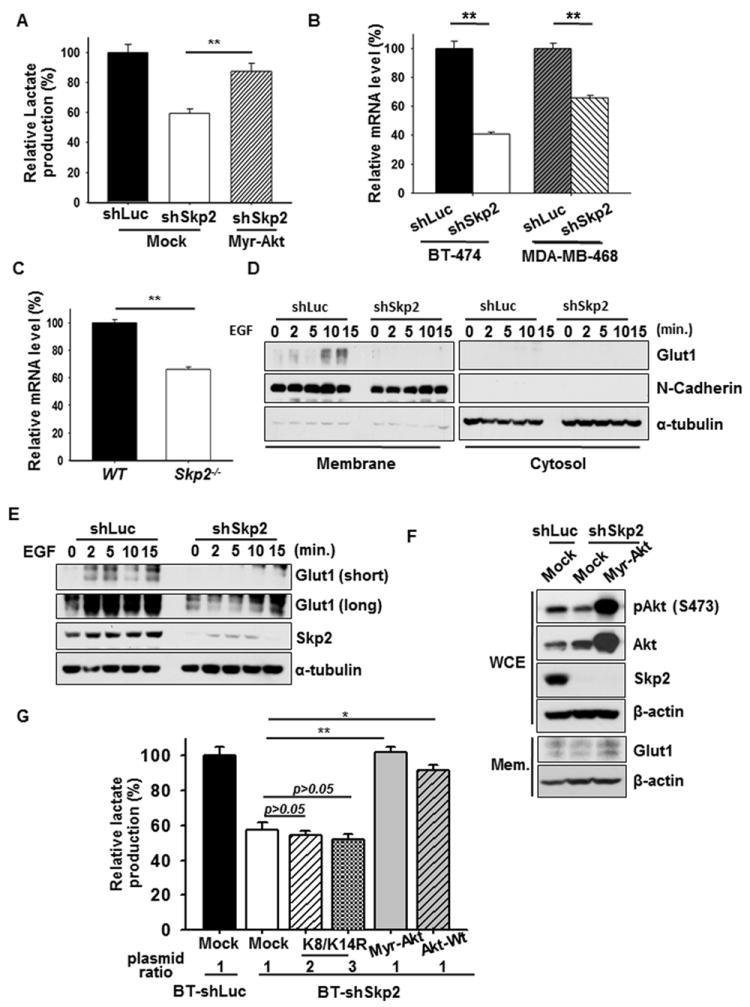
(A) Lactate production was determined in BT-474 cells with Luciferase, Skp2 knockdown, or Skp2 knockdown plus Myr-Akt overexpression upon EGF stimulation. (B) Real-Time PCR analysis of Glut1 mRNA levels in BT-474 cells or MDA-MB-468 cells with Luciferase or Skp2 knockdown. (C) Real-Time PCR analysis of Glut1 mRNA levels in WT and Skp2-/- MEFs. (D) Cos1 cells with Luciferase or Skp2 knockdown were serum-starved, treated with EGF for various time points and harvested for the isolation of membrane and cytosolic fractions, followed by IB analysis. (E) Cos1 cells with Luciferase or Skp2 knockdown were serum-starved, treated with EGF for various time points and harvested for IB analysis. (F) IB analysis of Glut1 protein expressions in whole cell extracts (WCE) or membrane fractions (Mem) of BT-474 cells with Luciferase, Skp2 knockdown or Skp2 knockdown plus Myr-Akt overexpression. (G) Lactate production was measured in Luciferase- and Skp2-silenced BT-474 cells transfected with various constructs as indicated upon EGF stimulation. See also Figure S5.
To further support our notion that Skp2 regulates glycolysis through Akt ubiquitination, we introduced Akt-Wt, Myr-Akt and Akt-K8R/K14R in Skp2-deficient cells to investigate their effects on glucose metabolism. Notably, while the introduction of Akt-Wt or Myr-Akt rescued the defect in lactate production of Skp2-knockdown cells in the presence of EGF or HRG, Akt-K8R/K14R mutant failed to do so (Figure 5G and Figures S5A and S5B). These results indicate that Skp2 controls glycolysis in cancer cells through regulating Akt ubiquitination and activation.
Skp2 loss attenuates Akt activity, Glut1 expression and mammary tumor development in MMTV-Neu mice
As Skp2 is critical for ErbB receptor signaling, we examined the role of Skp2 loss in Akt activation, Glut1 expression, and tumorigenesis in MMTV-Neu transgenic mice, which develop primary and metastatic breast cancer (Oshima et al., 2004). Kaplan-Meier survival analysis revealed that Skp2 deficiency significantly prolonged the survival of MMTV-Neu mice (Figure 6A). Notably, Skp2 deficiency markedly delayed breast cancer development and reduced breast tumor volume in MMTV-Neu mice, thereby restricting breast cancer metastasizing to the lungs (Figures 6B-6E). Skp2 loss also inhibited in vivo Akt activation and Glut1 expression in MMTV-Neu mice, correlated with the reduced Ki-67 expression of MMTV-Neu mice (Figure 6F). Collectively, Skp2 deficiency inhibits Akt activation and Glut1 expression in vitro and in vivo, in turn repressing breast cancer development.
Figure 6. Skp2 deficiency restricts in vivo Akt activation and mammary tumor development upon Neu overexpression.
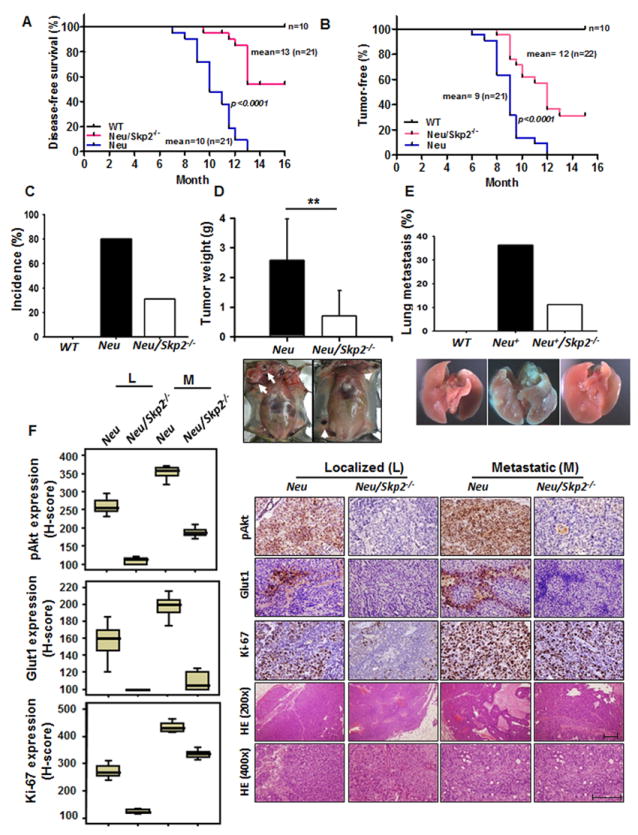
(A) Kaplan–Meier plot analysis of cumulative disease-free survival of WT, MMTV-Neu and MMTV-Neu/Skp2-/- mice. (B) Kaplan–Meier plot analysis of tumor-free incidence of WT, MMTV-Neu and MMTV-Neu/Skp2-/- mice. (C) The percentage of mice that develop mammary tumor was analyzed from a cohort of WT, MMTV-Neu and MMTV-Neu/Skp2-/- mice at age around 10 months (WT, n=10; MMTV-Neu, n=13; MMTV-Neu/Skp2-/-, n=10). (D) Mammary tumors were obtained and weighed from MMTV-Neu and MMTV-Neu/Skp2-/- mice at the age around 10 months (MMTV-Neu, n=11; MMTV-Neu/Skp2-/-, n=10). Arrows indicate mammary tumors. **, p<0.01. (E) The percentage of mice that develop lung metastasis was analyzed from a cohort of WT, MMTV-Neu and MMTV-Neu/Skp2-/- mice at age around 12 months (WT, n=10; MMTV-Neu, n=11; MMTV-Neu/Skp2-/-, n=9). (F) Histological and quantification analysis of pAkt, Glut1 and Ki-67 protein expression in MMTV-Neu and MMTV-Neu/Skp2-/- mice. Scale bar indicates 200 μm. All p-value < 0.001 by using Mann-Whitney U Test, except the p-value for Glut1 expression between localized and metastatic MMTV-Neu/Skp2-/- (p=0.105).
Skp2 serves as a marker for poor prognosis in Her2-positive breast cancer patients
Since Skp2 regulates Akt activation and tumorigenesis in MMTV-Neu mouse model, we next investigated whether Skp2 correlates with Akt activation and serves as a marker for poor prognosis in Her2-positive breast cancer patients. For this purpose, we have retrospectively identified 213 consecutively treated breast cancer patients that received modified radical mastectomy with curative intent and without neither pre- nor post-operative adjuvant chemotherapy and/or radiation therapy. Among them, 80 cases were Her2-positve (defined as 3+), while 132 were Her2-negative (defined as 0 to 2+). In these 80 cases of Her2-positive breast carcinomas, Skp2 overexpression was significantly correlated with numerous adverse clinicopathological factors, including increments of primary tumor status (pT, p=0.05), nodal metastasis (pN, p=0.012), and stage (p=0.026) (Table S1). Skp2 expression also significantly correlated with the upregulation of pAkt (S473) (p<0.001) (Figure 7A and Table S1). We next asked the question whether Skp2 expression also correlates with both Akt1 and Akt2 phosphorylation in breast tumors. Interestingly, phosphorylation of both Akt isoforms was higher in Skp2-high tumors than that in Skp2-low tumors, and Skp2 expression nicely correlated with Her2 levels in breast tumors (Figure 7B). Our results suggest that Skp2 upregulation in Her2-positve breast tumors may contribute to activation of Akt1 and Akt2, thereby promoting breast cancer progression in Her2-positive breast cancer patients.
Figure 7. Skp2 deficiency prolongs survival of Her2-positve patients and confers Herceptin sensitivity in Her2-positive cells and tumors.
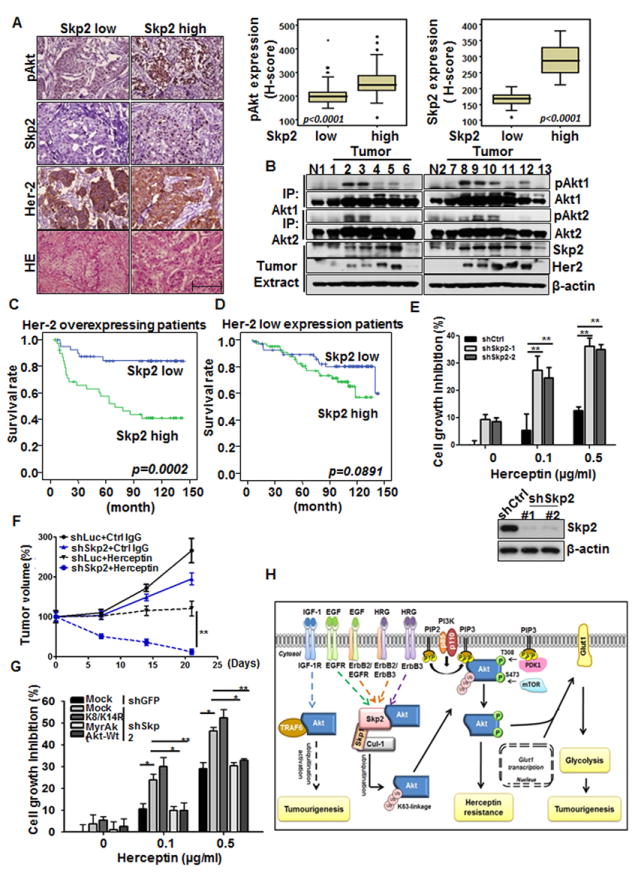
(A) Histological and quantification analysis of pAkt expressions in Her2-positive patients with low or high expression of Skp2. Scale bar indicates 200 μm. All p-value < 0.001 by using Mann-Whitney U Test. (B) Breast tumors and normal breast tissues were extracted and subjected for IP assay, followed by IB analysis. (C) Kaplan–Meier plot analysis of metastasis-free survival of 80 cases of Her2-positive patients with low or high expression of Skp2. (D) Kaplan–Meier plot analysis of metastasis-free survival of 132 cases of Her2 low-expressing patients with low or high expression of Skp2. (E) Cell growth inhibition assay and IB analysis in BT-474 cells with Luciferase or Skp2 knockdown. BT-474 cells were treated with various doses of Herceptin for 6 days and cell numbers were counted using hemocytometer. (F) Tumor volume of Her2-positive tumors with or without Skp2 silencing upon treatment with IgG or Herceptin. Tumor volume at various time points of treatment is presented as percentage of original tumor size (~200 mm3) at day zero of treatment. (G) BT-474 cells with Luciferase or Skp2 knockdown were transfected with various plasmids, treated with various doses of Herceptin and viable cell numbers were counted using hemocytometer. (H) The working model of Skp2 in ErbB family-regulated Akt activation, glycolysis and tumorigenesis. See also Figure S6 and Tables S1-S4.
In the univariate survival analysis, Skp2 (p=0.0048 by continuous scoring) and pAkt (S473) (p=0.0134) overexpression, together with pT status (p<0.0001), pN status (p=0.0016), and stage (p<0.0001) effectively predicted inferior metastasis-free survival (p=0.0002 by binary cut-offs) (Figure 7C and Table S2). In the multivariate analysis, while Skp2 overexpression remained prognostically significant for metastasis-free survival in Her2-positive breast cancer patients (p=0.0343 by continuous scoring in multivariate analysis, p=0.0048 by continuous scoring in univariate analysis) (Table S2), this prognostic effect was not significant in our 132 in-house Her2-negative breast cancers (p=0.1065 by continuous scoring in univariate analysis and p=0.0891 by binary cut-offs) (Figure 7D and Table S3). Moreover, Skp2 level did not significantly correlate with pAkt (S473) level in Her2-negative breast cancer samples (p=0.672, Table S4). To further strengthen our notion that Skp2 serves as a biomarker for poor prognosis, we have analyzed the correlation between Skp2 and Her2 mRNA levels in NKI dataset (van de Vijver et al., 2002) and found that high Skp2 mRNA level significantly predicted inferior distal metastasis-free survival (p=0.0018) in Her2 overexpressing breast cancer, but not in Her2 low expressing breast cancer (p=0.1364) (Figures S6A and S6B). Our results collectively suggest that the status of Skp2 expression can predict survival outcome of Her2-positive breast cancer patients.
Skp2 deficiency enhances Herceptin sensitivity in Her2-positive cancer cells and tumors
Herceptin is the standard treatment for Her2-positive breast tumors which substantially improves the clinical outcomes of these patients. However, there are still a number of patients that display intrinsic or acquired resistance to this treatment. The discordant Herceptin responses among Her2-positive patients are possibly due to the heterogeneous nature of tumors. As Skp2 deficiency or knockdown inhibits glycolysis in mouse tumor models (Figures 4D-4F and 6F), we examined whether Skp2 targeting sensitizes Herceptin response in Her2-positive cancer cells and tumors. Indeed, the Herceptin sensitivity was significantly enhanced in Skp2-silenced cells (Figures 7E and Figure S6C). In preclinical mouse model, although Herceptin by itself inhibited tumor growth, it did not cause tumor shrinkage (Figure 7F). Strikingly, Skp2 silencing in conjunction with Herceptin treatment resulted in substantial tumor regression (Figure 7F), highlighting that Skp2 is an appealing therapeutic target to combine with Herceptin for anti-cancer treatment.
To support the notion that Skp2 regulates Herceptin resistance through promoting Akt ubiquitination and activation, we introduced Akt-K8R/K14R, Akt-WT and Myr-Akt in Skp2-silenced cells to examine their effects on Herceptin sensitivity. Notaby, overexpression of WT Akt or Myr-Akt prevented heightened Herceptin sensitivity in Skp2-silecned cells, whereas Akt-K8/K14R overexpression failed to do so (Figure 7G), elucidating that Skp2 regulates Herceptin resistance through promoting Akt ubiquitination.
Discussion
Our results reveal several unexpected findings with important clinical implications. We identify that the Skp2 SCF complex is a crucial E3 ligase for promoting Akt ubiquitination and activation, thereby resulting in elevated glycolysis and tumorigenesis in response to ErbB receptor signaling (Figure 7H). Importantly, we provide pre-clinical evidence demonstrating that targeting glycolysis drastically benefits therapeutic outcomes for current anti-cancer treatments.
Different growth factors utilize distinct E3 ligases for Akt ubiquitination and activation
Our current and previous findings establish that K63-linked ubiquitination of Akt is a general event triggered by growth factors and cytokines, such as IGF-1, EGF and interleukin-1, and plays a critical role in Akt membrane recruitment and activation. While TRAF6 is required for IGF-mediated Akt ubiquitination and activation, our current study reveals that Skp2, but not TRAF6, is selectively engaged in Akt ubiquitination and activation driven by activation of ErbB family receptors. Given that other growth factors like PDGF and FGF are known to activate Akt signaling and we also revealed that Skp2 increased association with Akt upon stimulation of PDGF, but not FGF (Figure S7A), it remains to be determined whether Skp2 is involved in PDGF-mediated Akt ubiquitination and activation.
Recent studies reveal that Akt-mediated Skp2 S72 phosphorylation stabilizes Skp2 expression and enhances its E3 ligase activity (Gao et al., 2009; Lin et al., 2009). Consistent with this notion, we found that the Skp2 S72A mutant defective in Akt-mediated phosphorylation reduced its ability to promote Akt ubiquitination compared to WT Skp2 (Figure S7B), indicating that Akt-mediated phosphorylation may serve as a positive feedback loop for Akt ubiquitination. Moreover, we found that the Skp2-S72A mutant was defective in promoting Akt phosphorylation and activation in breast cancer cells (Figure S7C). Interestingly, the level of Skp2 S72 phosphorylation was upregulated and correlated with pAkt levels in multiple breast cancer cell lines (Figure S7D), suggesting that Skp2 phosphorylation is upregulated in breast cancer cells and correlates with Akt activation. Moreover, Skp2 S72A mutant compromised its ability to promote breast cancer cell migration and invasion compared to that of WT Skp2 (Figures S7E and S7F), correlated with its defect in promoting Akt ubiquitination and activation (Figures S7B and S7C). Future work would be required to further understand whether Skp2 phosphorylation orchestrates breast cancer metastasis.
Skp2 SCF complex regulates non-proteolytic K63-linked ubiquitination
Skp2 SCF complex is known to play a critical role in cell cycle regulation and tumorigenesis by promoting ubiquitination and degradation of p27 and p21 (Chan et al., 2010b; Lin et al., 2010; Nakayama and Nakayama, 2006). We identify that Akt is a Skp2 SCF substrate, whose ubiquitination does not lead to degradation. Thus, Akt is a substrate of Skp2 SCF complex that undergoes K63-linked ubiquitination. As Skp2 and other F-box proteins are long thought to promote ubiquitination and degradation of their substrates, our current study challenges this dogma by identifying Akt as a non-proteolytic substrate for Skp2. In light of this unexpected finding, we speculate that there may be more Skp2 protein substrates undergoing non-proteolytic K63-linked ubiquitination.
Skp2 regulates aerobic glycolysis, the Warburg effect
Elevated aerobic glycolysis is a hallmark in various tumor origins, as evidenced by the wide application of PET scan in clinical diagnosis for detecting cancer cells with actively glucose uptake (Zhu et al., 2011). Thus, identifying factors that control cancer cell glycolysis not only can advance current knowledge of how tumor cells seize glucose metabolism to acquire survival advantage, but also provide innovative approaches for cancer therapy. Our present study uncovers an unrecognized function of Skp2 in glucose metabolism. We identify Skp2 as a critical regulator for glycolysis by regulating Akt ubiquitination and activation.
Hypoxia-inducible factor-1α (HIF-1α) can activate transcription of genes encoding Glut1, Hexokinase II, Lactate dehydrogenase A (LDH-A) as well as pyruvate dehydrogenase kinase 1 (PDK1). As such, HIF-1α accumulation stimulates glucose metabolism by increasing both glucose consumption and lactate production as PDK1 inhibits conversion of pyruvate to acetyl-CoA and suppresses oxidative phosphorylation. Since activation of Akt/mTOR is known to enhance HIF-1α translation (Jiang et al., 2001; Majumder et al., 2004), it is likely that Skp2 may also upregulate HIF-1α translation and induces HIF-1-dependent glycolysis through the Akt/mTOR pathway.
In addition to the Skp2-Akt axis, we cannot rule out the possibility that Skp2 may also cooperate with other key regulators to participate in glucose metabolism or other metabolic pathways. For instance, Myc overexpression increases transcription of many metabolic enzymes, including glycolytic enzymes (Glut1 and Hexokinase II), LDH-A, and several enzymes required for nucleotide biosynthesis (DeBerardinis et al., 2008; Osthus et al., 2000; Shim et al., 1997). Since Skp2 cooperates with Myc to activate various Myc target genes involved in cell cycle transition or cell migration (Chan et al., 2010a; Kim et al., 2003; von der Lehr et al., 2003), it implies that Skp2 may participate in Myc-dependent metabolic processes through inducing the transcription of metabolic target genes. Future study will be needed to explore this possibility.
Skp2 is a marker for poor survival outcomes and a potential therapeutic target for Her2-positive breast cancer
The PI3K/Akt is one of the most important oncogenic pathways activated downstream of Her2/ErB2 in cancers (Hynes and MacDonald, 2009). Our study reveals that Skp2 overexpression correlates with Akt activation and poor survival outcomes in Her2-posivtive breast cancer patients, but not in Her2-negative ones, supporting the in vivo relevance of Skp2 in ErbB family receptor-induced Akt activation and breast cancer progression. Our findings demonstrating the pronounced defect of Skp2 silencing in glucose uptake of Her2-overexpressing tumor suggests that Skp2 targeting is a promising strategy for inhibiting glycolysis and cancer (Figures 4D-4G). Since Skp2 also recognizes and promotes degradation of p27 and p21 (Bornstein et al., 2003; Nakayama et al., 2000; Nakayama and Nakayama, 2006), it is possible that downregulation of p27 or p21 may also contribute to tumor progression or poor survival outcome driven by Skp2 overexpression.
In addition to Her/Neu overexpression, Skp2 loss also impacts tumorigenesis driven by Pten, pRB, and p19ARF inactivation in various transgenic mouse models (Lin et al., 2010; Wang et al., 2010). It is possible that Skp2 silencing may globally inhibit glycolysis driven by various oncogenic insults through regulating Akt activation or other mechanisms. Our findings along with these recent reports thereby suggest that targeting glycolysis pathways can be an important therapeutic approach for cancer treatment.
Multiple genetic or epigenetic alterations have been revealed in various cancer types. Therefore, designing a strategy that universally targets fundamental features of cancer cells, such as glycolysis, may serve as a valuable anti-cancer approach. Several small molecules that pharmacologically inactivate glycolysis have emerged and shown promising anticancer activities as single agent or in combination with other therapeutic modalities (Pelicano et al., 2006; Vander Heiden et al., 2009). In light of this notion, our discovery reveals that Skp2 silencing as a therapeutic strategy to inactivate glycolysis can sensitize Her2-positive cells/tumors response to Herceptin (Figures 7E-7G and Figure S6C). As such, our study provides a proof-of-principle that targeting glycolysis is a compelling therapeutic strategy as a single or a combinatory therapy for cancer treatment.
Experimental Procedures
Mice, cell culture, and reagents
MMTV-Neu and Skp2-/- mice were described (Lin et al., 2010; Nakayama et al., 2000; Oshima et al., 2004). Mouse embryonic fibroblasts (MEFs) from wild-type and Skp2-/- mice were prepared as previously described (Lin et al., 2004; Lin et al., 2010). 293T, Cos1, BT-474 and MDA-MB-231 cells were cultured in DMEM containing 10% fetal bovine serum (FBS). (His)6-ubiquitin, (His)6-ubiquitin-K48R and (His)6-ubiquitin-K63R, GST-Akt1, HA-Akt1 constructs was described previously (Yang et al., 2009). Skp2-LRR construct was from W. Tansey. Herceptin was a gift from Dr. M. H. Lee.
In vivo and in vitro ubiquitination assay
In vivo and in vitro ubiquitination assays were performed as described (Lin et al., 2009; Yang et al., 2009). For in vivo ubiquitination assay, 293T cells were transfected with the indicated plasmids for 48 h and lysed by the denatured buffer (6M guanidine-HCl, 0.1M Na2HPO4/NaH2PO4, 10 mM imidazole). The cell extracts were then incubated with nickel beads for 3 h, washed, and subjected to immunoblotting analysis. For in vitro ubiquitination assays, recombinant GST and GST-Akt proteins were purified from the bacterial lysates of BL21 competent cells. Flag-Skp2 SCF complex and Flag-TRAF6 were expressed in 293T cells, immunoprecipitated by anti-Flag antibody, and eluted from Protein A/G beads using Flag peptides according to manufacturers’ standard procedures. Purified GST, GST-Akt, Flag-SCF and Flag-TRAF6 proteins were incubated for 3 h at 37°C in 20 μl of reaction buffer [20 mM Hepes (pH 7.4), 10 mM MgCl2, 1 mM DTT, 59 μM ubiquitin, 50 nM E1, 850 nM of Ubc13/Uev1a, 1 mM ATP, 30 μM creatine phosphate, and 1 U of creatine kinase]. After incubation, protein mixtures were diluted in RIPA Buffer and the supernatant fluid was precleared with Protein A/G beads for 1 h, and immunoprecipitated overnight with anti-Flag antibody, after which Protein A/G beads were added for an additional 1 h. Beads were washed 4 times with E1A Buffer. Proteins were eluted in SDS-sample buffer and subjected to immunoblotting analysis.
Glucose uptake assay
Cells were seeded in 60 mm plates. 24 h later, cells were refreshed with serum-starved (0.1% FBS) and glucose-free DMEM. 16 h later, cells treated with or without 10 μM LY294002 were grown in the presence of 50 μM 2-NBDG for 30 min. and 60 min., respectively, and glucose uptake was quantified using FACS analysis
Lactate production assay
Cells were plated in 24-well plate and cultured overnight. After pretreated with or without 10 μM LY294002, cells were treated with EGF (50 ng/ml) for 8 hrs. Culture medium was removed from cells and lactate concentration was determined using lactate test strips and Accutrend Lactate analyzer (Accutrend Lactate, Roche). Next, cells were harvested, stained with trypan blue and viable cell numbers were counted directly under the microscope using hemocytometer. Last, the rate of lactate production were determined (lactate production rate=lactate concentration/cells/time) and normalized with the rate detected in control group.
In vivo tumorigenesis assay and in vivo glucose uptake assay
For in vivo tumorigenesis assays, 5 × 106 of BT-474-M1 cells were injected into mammary fat pad of age-matched athymic female nude mice (5 mice for each group). Tumor size was measured weekly with a caliper, and tumor volume was determined with the standard formula: L × W2 × 0.52, where L is the longest diameter and W the shortest diameter. While developed tumors have reach to the volume around 200 mm3, mice were imaged and analyzed with [18F]FDG for in vivo glucose uptake. [18F]FDG were administered via a single tail-vein injection and PET/CT images were scanned and collected on Inveon CT/PET system (Siemens). Mice were awake during the uptake period and maintained on a heating pad. Images then were reconstructed Images were reconstructed using two dimensional ordered subsets expectation maximization (OSEM) algorithm. PET and CT image fusion and image analysis were performed using software ASIPro 5.2.4.0 (Siemens). For Herceptin treatment, 8 × 106 of BT-474-M1 cells were injected into mammary fat pad of age-matched athymic female nude mice (5 mice for each group). While developed tumors have reach to the volume around 200 mm3, mice were injected with 5 mg/kg of Herceptin or vehicle control (IgG) intraperitoneally once per week, and tumor size was measured weekly with a caliper.
Cell growth assay
For Herceptin administration, 8 × 103 of BT-474 cells with control, Skp2 knockdown were seeded in 12 wells in triplicate, 24 h later, treated with Herceptin (refreshed every two days). Six days later, cells were harvested, stained with trypan blue, and viable cells were counted directly under the microscope using hemocytometer.
Immunohistochemistry and scoring
The procedures of immunohistochemical studies were performed as previously described(Huang et al., 2006). In brief, sections were cut onto an adhesive-coated glass slides at 3-μm thickness. For stainings in human samples, the slides were incubated with primary antibodies targeting Her-2 (Thermo Scientific, Clone SP3, 1:100), Skp2 (Invitrogen, clone 2C8D9, 1:100), and pAkt(Ser473) (Cell Signaling, clone D9E, 1:25). The slides from mouse samples were incubated with primary antibodies for pAkt(Ser473) (Cell Signaling, clone D9E, 1:25), Ki-67 (Abcam, polyclonal, 1:200), and Glut1 (Abcam, clone SPM498, 1:200), respectively. Primary antibodies were detected using the ChemMate DAKO EnVision kit (DAKO, K5001). The slides were incubated with the secondary antibody for 30 minutes and developed with 3,3-diaminobenzidine for 5 minutes. Incubation without the primary antibody was used as a negative control. Immunoexpression was scored by two pathologists (C.F.L & H.Y.H) using a multiheaded microscope to reach a consensus for each case. The staining was evaluated based on a combination of both the percentage and intensity of positively stained tumor cells to generate an H-score, which was calculated using the following equation: H-score = ΣPi (i + 1), where i is the intensity of the stained tumor cells (0 to 4 +), and Pi is the percentage of stained tumor cells for each intensity.
Statistical analysis
Statistical analyses were performed using the SPSS 14 software package. For human breast samples, the Mann-Whitney U test was used to assess the differential expression level of Skp2 and pAkt(S473) expression in relation to Her-2 expression status. The Spearman’s rank correlation coefficient was used to clarify the association between Skp2 expression to clinicopathological variables and pAkt(Ser473) expression levels. The endpoint analyzed was distal metastasis-free survival, calculated from the starting date of surgery to the date of event. The median period of follow-up was 103 months (range, 6 to 143). Survival analysis was performed using the Cox proportional hazards model. Survival curves were plotted using the Kaplan-Meier method, and log-rank tests were performed to evaluate prognostic differences between groups for categorical variables. For mouse samples, the expression levels of pAkt, Ki-67 and Glut1 between various groups were assessed by using Mann-Whitney U test. For all analyses, two-sided tests of significance were used with p=< 0.05 considered significant.
Supplementary Material
Highlights.
Distinct E3 ligases regulate Akt ubiquitination, activation and cancer development.
Skp2 regulates Akt-mediated cancer glycolysis and tumorigenesis.
Skp2 serves as a marker for poor prognosis in Her2-positive patients.
Targeting glycolysis sensitizes Her2-positive tumors to Herceptin treatment.
Acknowledgments
We thank Mrs. J. Delacerda and C. V. Kingsley at small animal imaging facility of MD Anderson Cancer Center for their assistance in CT/PET imaging. We also thank the members from Lin’s laboratory for their valuable comments and suggestions. This work was supported by the MD Anderson Cancer Center Trust Scholar Award, NIH grants, CPRIT grant, DOD prostate cancer New Investigator Award to H.K.L., the MD Anderson Cancer Center Breast SPORE Career Development Award to C. H. C. and the grant from the Department of Health in Taiwan to C.F.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agus DB, Akita RW, Fox WD, Lewis GD, Higgins B, Pisacane PI, Lofgren JA, Tindell C, Evans DP, Maiese K, et al. Targeting ligand-activated ErbB2 signaling inhibits breast and prostate tumor growth. Cancer Cell. 2002;2:127–137. doi: 10.1016/s1535-6108(02)00097-1. [DOI] [PubMed] [Google Scholar]
- Aragones J, Fraisl P, Baes M, Carmeliet P. Oxygen sensors at the crossroad of metabolism. Cell Metab. 2009;9:11–22. doi: 10.1016/j.cmet.2008.10.001. [DOI] [PubMed] [Google Scholar]
- Barthel A, Okino ST, Liao J, Nakatani K, Li J, Whitlock JP, Jr, Roth RA. Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem. 1999;274:20281–20286. doi: 10.1074/jbc.274.29.20281. [DOI] [PubMed] [Google Scholar]
- Birnbaum MJ. On the InterAktion between hexokinase and the mitochondrion. Dev Cell. 2004;7:781–782. doi: 10.1016/j.devcel.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Bornstein G, Bloom J, Sitry-Shevah D, Nakayama K, Pagano M, Hershko A. Role of the SCFSkp2 ubiquitin ligase in the degradation of p21Cip1 in S phase. J Biol Chem. 2003;278:25752–25757. doi: 10.1074/jbc.M301774200. [DOI] [PubMed] [Google Scholar]
- Brazil DP, Park J, Hemmings BA. PKB binding proteins. Getting in on the Akt. Cell. 2002;111:293–303. doi: 10.1016/s0092-8674(02)01083-8. [DOI] [PubMed] [Google Scholar]
- Chan CH, Gao Y, Moten A, Lin HK. Novel ARF/p53-independent senescence pathways in cancer repression. J Mol Med. doi: 10.1007/s00109-011-0766-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CH, Lee SW, Li CF, Wang J, Yang WL, Wu CY, Wu J, Nakayama KI, Kang HY, Huang HY, et al. Deciphering the transcriptional complex critical for RhoA gene expression and cancer metastasis. Nat Cell Biol. 2010a;12:457–467. doi: 10.1038/ncb2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CH, Lee SW, Wang J, Lin HK. Regulation of Skp2 expression and activity and its role in cancer progression. ScientificWorldJournal. 2010b;10:1001–1015. doi: 10.1100/tsw.2010.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta K, Franke TF, Chan TO, Makris A, Yang SI, Kaplan DR, Morrison DK, Golemis EA, Tsichlis PN. AH/PH domain-mediated interaction between Akt molecules and its potential role in Akt regulation. Mol Cell Biol. 1995;15:2304–2310. doi: 10.1128/mcb.15.4.2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- Gao D, Inuzuka H, Tseng A, Chin RY, Toker A, Wei W. Phosphorylation by Akt1 promotes cytoplasmic localization of Skp2 and impairs APCCdh1-mediated Skp2 destruction. Nat Cell Biol. 2009;11:397–408. doi: 10.1038/ncb1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HY, Kang HY, Li CF, Eng HL, Chou SC, Lin CN, Hsiung CY. Skp2 overexpression is highly representative of intrinsic biological aggressiveness and independently associated with poor prognosis in primary localized myxofibrosarcomas. Clin Cancer Res. 2006;12:487–498. doi: 10.1158/1078-0432.CCR-05-1497. [DOI] [PubMed] [Google Scholar]
- Jiang BH, Jiang G, Zheng JZ, Lu Z, Hunter T, Vogt PK. Phosphatidylinositol 3-kinase signaling controls levels of hypoxia-inducible factor 1. Cell Growth Differ. 2001;12:363–369. [PubMed] [Google Scholar]
- Kim SY, Herbst A, Tworkowski KA, Salghetti SE, Tansey WP. Skp2 regulates Myc protein stability and activity. Mol Cell. 2003;11:1177–1188. doi: 10.1016/s1097-2765(03)00173-4. [DOI] [PubMed] [Google Scholar]
- Kunstle G, Laine J, Pierron G, Kagami Si S, Nakajima H, Hoh F, Roumestand C, Stern MH, Noguchi M. Identification of Akt association and oligomerization domains of the Akt kinase coactivator TCL1. Mol Cell Biol. 2002;22:1513–1525. doi: 10.1128/mcb.22.5.1513-1525.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Hoeflich ST, Crocker L, Yao E, Pham T, Munroe X, Hoeflich KP, Sliwkowski MX, Stern HM. A central role for HER3 in HER2-amplified breast cancer: implications for targeted therapy. Cancer Res. 2008;68:5878–5887. doi: 10.1158/0008-5472.CAN-08-0380. [DOI] [PubMed] [Google Scholar]
- Lin HK, Bergmann S, Pandolfi PP. Cytoplasmic PML function in TGF-beta signalling. Nature. 2004;431:205–211. doi: 10.1038/nature02783. [DOI] [PubMed] [Google Scholar]
- Lin HK, Chen Z, Wang G, Nardella C, Lee SW, Chan CH, Yang WL, Wang J, Egia A, Nakayama KI, et al. Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature. 2010;464:374–379. doi: 10.1038/nature08815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HK, Wang G, Chen Z, Teruya-Feldstein J, Liu Y, Chan CH, Yang WL, Erdjument-Bromage H, Nakayama KI, Nimer S, et al. Phosphorylation-dependent regulation of cytosolic localization and oncogenic function of Skp2 by Akt/PKB. Nat Cell Biol. 2009;11:420–432. doi: 10.1038/ncb1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder PK, Febbo PG, Bikoff R, Berger R, Xue Q, McMahon LM, Manola J, Brugarolas J, McDonnell TJ, Golub TR, et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat Med. 2004;10:594–601. doi: 10.1038/nm1052. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, Shirane M, Tsunematsu R, Tsukiyama T, Ishida N, et al. Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. Embo J. 2000;19:2069–2081. doi: 10.1093/emboj/19.9.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- Noguchi M, Ropars V, Roumestand C, Suizu F. Proto-oncogene TCL1: more than just a coactivator for Akt. FASEB J. 2007;21:2273–2284. doi: 10.1096/fj.06-7684com. [DOI] [PubMed] [Google Scholar]
- Oshima RG, Lesperance J, Munoz V, Hebbard L, Ranscht B, Sharan N, Muller WJ, Hauser CA, Cardiff RD. Angiogenic acceleration of Neu induced mammary tumor progression and metastasis. Cancer Res. 2004;64:169–179. doi: 10.1158/0008-5472.can-03-1944. [DOI] [PubMed] [Google Scholar]
- Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA, Dang CV. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275:21797–21800. doi: 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25:4633–4646. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- Plas DR, Thompson CB. Akt-dependent transformation: there is more to growth than just surviving. Oncogene. 2005;24:7435–7442. doi: 10.1038/sj.onc.1209097. [DOI] [PubMed] [Google Scholar]
- Robey RB, Hay N. Is Akt the “Warburg kinase”?-Akt-energy metabolism interactions and oncogenesis. Semin Cancer Biol. 2009;19:25–31. doi: 10.1016/j.semcancer.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R, Dang CV. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc Natl Acad Sci U S A. 1997;94:6658–6663. doi: 10.1073/pnas.94.13.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Lehr N, Johansson S, Wu S, Bahram F, Castell A, Cetinkaya C, Hydbring P, Weidung I, Nakayama K, Nakayama KI, et al. The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol Cell. 2003;11:1189–1200. doi: 10.1016/s1097-2765(03)00193-x. [DOI] [PubMed] [Google Scholar]
- Wang H, Bauzon F, Ji P, Xu X, Sun D, Locker J, Sellers RS, Nakayama K, Nakayama KI, Cobrinik D, et al. Skp2 is required for survival of aberrantly proliferating Rb1-deficient cells and for tumorigenesis in Rb1+/- mice. Nat Genet. 2010;42:83–88. doi: 10.1038/ng.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Wieman HL, Wofford JA, Rathmell JC. Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell. 2007;18:1437–1446. doi: 10.1091/mbc.E06-07-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia ZP, Sun L, Chen X, Pineda G, Jiang X, Adhikari A, Zeng W, Chen ZJ. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature. 2009;461:114–119. doi: 10.1038/nature08247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WL, Wang J, Chan CH, Lee SW, Campos AD, Lamothe B, Hur L, Grabiner BC, Lin X, Darnay BG, et al. The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science. 2009;325:1134–1138. doi: 10.1126/science.1175065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WL, Wu CY, Wu J, Lin HK. Regulation of Akt signaling activation by ubiquitination. Cell Cycle. 2010a;9:486–497. doi: 10.4161/cc.9.3.10508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WL, Zhang X, Lin HK. Emerging role of Lys-63 ubiquitination in protein kinase and phosphatase activation and cancer development. Oncogene. 2010b;29:4493–4503. doi: 10.1038/onc.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, Xu M, Chen ZJ. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell. 2010;141:315–330. doi: 10.1016/j.cell.2010.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu A, Lee D, Shim H. Metabolic positron emission tomography imaging in cancer detection and therapy response. Semin Oncol. 2011;38:55–69. doi: 10.1053/j.seminoncol.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.