

Abstract
Osteosarcoma (OS) is the most common primary malignancy of bone. Mortality is determined by the presence of metastatic disease, but little is known regarding the biochemical events that drive metastases. Two murine OS cell lines, K7M2 and K12, are related but differ significantly in their metastatic potentials: K7M2 is highly metastatic whereas K12 displays much less metastatic potential. Using this experimental system, the mammalian target of rapamycin (mTOR) pathway has been implicated in OS metastasis. We also discovered that aldehyde dehydrogenase (ALDH, a stem cell marker) activity is higher in K7M2 cells than K12 cells. Rapamycin treatment reduces the expression and enzymatic activity of ALDH in K7M2 cells. ALDH inhibition renders these cells more susceptible to apoptotic death when exposed to oxidative stress. Furthermore, rapamycin treatment reduces bone morphogenetic protein-2 (BMP2) and vascular endothelial growth factor (VEGF) gene expression and inhibits K7M2 proliferation, migration, and invasion in vitro. Inhibition of ALDH with disulfiram correlated with decreased mTOR expression and activity. In conclusion, we provide evidence for interaction between mTOR activity, ALDH activity, and metastatic potential in murine OS cells. Our work suggests that mTOR and ALDH are therapeutic targets for the treatment and prevention of OS metastasis.
1. Introduction
Osteosarcoma (OS), the most common primary malignancy of bone, usually occurs in the long bones during childhood and adolescence at sites of rapid bone turnover [1–3]. Despite pre- and postoperative chemotherapy and wide surgical resection of the tumor, overall survival for patients without radiographically detectable metastases is only 65–70% [1, 2, 4, 5, 8]. The prognosis for patients with detectable metastases at the time of diagnosis is particularly poor, ranging from 15 to 30% [1, 7, 8]. It is thus the presence of pulmonary metastatic disease that ultimately determines OS mortality [9]. However, little is known about the biochemical signaling pathways that drive the progression of metastases and the molecular biology of OS remains poorly understood. As a result, we have yet to develop therapeutic strategies that specifically target metastatic disease.
The mammalian target of rapamycin (mTOR) is a highly conserved serine/threonine kinase, and as its name implies, mTOR activity is specifically inhibited by the drug rapamycin [10–13]. Rapamycin is an antimicrobial agent produced by Streptomyces hygroscopius that also exhibits potent immunosuppressive and antitumor properties, likely due to its ability to arrest the cell cycle in G1-phase [14]. mTOR signaling regulates a number of critical cellular processes including cellular growth, metabolism, and aging via an extraordinarily complex intercellular signaling network [15, 16]. Dysregulation of this mTOR signaling network can participate in a variety of human disease processes including cancer [17].
In mammals, mTOR associates with the proteins Raptor or Rictor to form mTOR complexes 1 and 2 (mTORC1 and 2), respectively. mTORC1 activity is sensitive to rapamycin, whereas mTORC2 is not [18, 19]. The best characterized substrates of mTORC1 are p70 ribosomal protein S6 kinase (S6 K1) and the eukaryotic initiation factor 4E-binding protein 1 (4E-BP1), through which mTOR activity can regulate protein synthesis and cell growth [17]. A role for rapamycin-sensitive and rapamycin-insensitive mTOR signaling in cell motility and cancer metastasis is evolving but our current understanding is limited [14]. It is, however, widely recognized that mTOR signaling plays a critical role in protein synthesis, cell proliferation, growth, and survival [10, 20–22]. Dysregulated mTOR signaling is found in a variety of human cancers including hematologic, lung, breast, liver, pancreas, renal, skin, and gastrointestinal tract neoplasms [17]. In addition, it was recently discovered that mTOR signaling is activated in human osteosarcoma and correlates with surgical stage, metastasis, and disease-free survival [23]. The primary goal of this study was to investigate the role of mTOR signaling in OS metastasis and mTOR inhibition with rapamycin.
K7M2 and K12 are related murine OS cell populations derived from the same spontaneously-occurring OS in a Balb-C mouse. K7M2 cells are highly metastatic to the lungs and were clonally derived from the much less metastatic K12 cells [24]. K7M2 and K12 cells are thus very similar genetically but differ significantly in their metastatic potentials. As such, they represent excellent tools for determining critical biochemical pathways in OS metastasis. It has been reported that mTOR signaling activity is enhanced in K7M2 cells compared to K12 cells [25]. Here we report that mTOR signaling in K7M2 cells is associated with higher aldehyde dehydrogenase (ALDH, a cancer stem cell marker) activity, increased resistance to oxidative stress, increased proliferation, migration, and invasion, and higher bone morphogenetic protein (BMP2) and vascular endothelial growth factor (VEGF) expression than in the less metastatic K12 cells. All of these metastatic phenotypes were reversed with rapamycin treatment. Interestingly, we also report that ALDH inhibition with disulfiram is correlated with decreased mTOR activity and causes morphological alterations to K7M2 cells. This study provides evidence that the mTOR pathway promotes ALDH activity and metastatic potential in OS cells. We conclude that mTOR and ALDH are potential therapeutic targets in the treatment and prevention of OS metastasis.
2. Materials and Methods
2.1. Cell Culture and Rapamycin Treatment
K7M2 cells and K12 cells were cultured with proliferation medium (PM; DMEM with 10% FBS and 5% penicillin and streptomycin). For mTORC1 inhibition of K7M2 cells, rapamycin (Sigma) was dissolved in DMSO (10 mM) and diluted 1 : 1000 in proliferation medium to a working concentration of 10 μM. K7M2 cells were seeded in 12-well plastic plates at 5,000 cells per well, and 1 mL treatment medium containing rapamycin was added to each well. 1 mL of medium containing the same amount of DMSO served as control treatment. Treatment medium was refreshed each day and cells were treated for 2 to 4 days.
2.2. Fluorescence-Activated Cell Sorting (FACS) Analysis of ALDH Activity and Sorting of Cells
The Aldelfluor Kit (STEMCELL Technologies) was used to determine ALDH enzymatic activity. Cultured K7M2 cells and K12 cells, with or without rapamycin treatment (10 μM for 48 hrs), were resuspended in Aldefluor buffer (1 × 106 cells/mL) and incubated at 37°C according to the manufacturer's instructions. Cells were washed in Aldefluor buffer and maintained in 4°C throughout the cell sorting process. ALDH activity was assessed using the FL1 channel of a BD FACSAria Cell Sorting System and FACSDiva software (version 6.1.2; Becton, Dickinson and Company, San Jose, CA). Collected cells were sorted with fluorescence-activated cell sorting (FACS), according to their fluorescence intensity, which corresponds to their ALDH activity levels, as well as low side scatter (SCClo). ALDH-high cells and ALDH-low cells were separately harvested and cultured.
2.3. Cell Proliferation Assay
K7M2 and K12 cells were plated at 1000 cells per well in a 12-well plate and cultured in PM. A time-lapsed microscopic live-cell imaging (LCI) system (Automated Cell, Inc.) was used to take images of cells per each field of view at 15 minute intervals for 4 days. The approximate population doubling time (PDT) as determined as follows: 2n = cell number at harvest time/cell number initially plated; “n” refers to the number of doublings during the period of cell culture (96 hrs), thus PDT = 96 hrs/n.
2.4. Cell Survival Assay after Exposure to Oxidative Stress
The antioxidant capacities of K7M2 (unsorted, ALDH-high, and ALDH-low fraction) and K12 cells cultured in 12-well plastic plates were compared by exposure to oxidative stress (250 μM H2O2 in PM) for 6 hrs. Also, to test the role of mTOR inhibition on the antioxidant capacity of K7M2 cells, K7M2 cells were pretreated with rapamycin (10 μM) for 48 hrs prior to exposure to oxidative stress (0, 250, or 500 μM H2O2 in PM) conditions for 6 hours. Propidium iodide (PI) was added to the medium (1 μg/mL) and apoptotic cells were identified with positive PI staining.
2.5. In Vitro Single Cell Migration Assay
An automated time-lapsed microscopy system (Biorad) was used to track the single cell migration on plastic surface. Cells were observed at 15 minute increments over 96 hours, the composite images were analyzed, the tracks of migration of 10 preselected single cells were monitored for each cell group, and cell velocities were calculated.
2.6. Cell In Vitro Invasion Assay
In vitro invasion capacity of K7M2 cells with or without rapamycin treatment, as well as ALDH-high and ALDH-low fractions of untreated K7M2 cells, was assessed using a real-time cell invasion and migration (RT-CIM) assay system (ACEA Biosciences, Inc.), with a 16-well trans-well plate (CIM-plate 16, Roche Diagnostics GmbH). The surface of the wells in the upper chamber was coated with Matrigel (BD BioSciences, Bedford, MA USA) of different concentrations (2.5%, 5%, and 10%). Serum-containing medium (10% FBS) was added to the wells of the lower chamber. Cells (4 × 104 per well) in serum-free medium were seeded in the upper chamber. The migration of the cells through the Matrigel was monitored by the system every 15 minutes for 24 hours. Data analysis was carried out using RTCA Software 1.2 supplied with the instrument.
2.7. Semiquantitative Reverse Transcription Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from the cells using the RNeasy plus mini kit (Qiagen) and cDNA was generated using the iScript cDNA Synthesis kit (Bio-Rad). The sense and antisense primers for RT-PCR and their product sizes are found in Table 1. The cycling parameters used for all reactions were as follows: 94°C for 5 minutes; 30 cycles of the following: denature for 45 seconds at 95°C, anneal for 30 seconds (53°C–56°C), and extend for 45 seconds at 72°C. RT-PCR was performed using a Bio-Rad MyiQ thermal cycler (Bio-Rad). GAPDH served as a control gene, and the expression of target genes was normalized to the expression of GAPDH. Gradient dilution (1 : 1, 1 : 2, and 1 : 4) of RNA samples from different cell groups was compared respectively to verify the quantitative difference of gene expression. RT-PCR analysis was performed using ImageJ software (version 1.32j, National Institutes of Health, Bethesda, MD) where the integrated density (product of the area and the mean gray value) of bands was calculated. All molecular bands were normalized to GAPDH.
Table 1.
Primer Sequences.
| Gene | Primer sequence | Band size (bp) |
|---|---|---|
| GAPDH | Forward: TCCATGACAACTTTGGCATTG | 103 |
| Reverse: TCACGCCACAGCTTTCCA | ||
| BMP2 | Forward: TCTTCCGGGAACAGATACAGG | 126 |
| Reverse: TGGTGTCCAATAGTCTGGTCA | ||
| BMP4 | Forward: ATTCCTGGTAACCGAATGCTG | 89 |
| Reverse: CCGGTCTCAGGTATCAAACTAGC | ||
| VEGF | Forward: GCCAGACAGGGTTGCCATAC | 108 |
| Reverse: GGAGTGGGATGGATGATGTCAG | ||
| c-Myc | Forward: TGACCTAACTCGAGGAGGAGCTGGAATC | 170 |
| Reverse: AAGTTTGAGGCAGTTAAAATTATGGCTGAAGC | ||
| ALDH | Forward: GACAGGCTTTCCAGATTGGCTC | 142 |
| Reverse: AAGACTTTCCCACCATTGAGTGC | ||
| mTOR | Forward: CAGTTCGCCAGTGGACTGAAG | 130 |
| Reverse: GCTGGTCATAGAAGCGAGTAGAC |
2.8. Actin Staining
Organization of actin in ALDH-high, ALDH-low, and disulfiram treated cells was assessed using the phalloidin conjugated with Alexa Fluor 488 (Invitrogen). Populations of ALDH-high and ALDH-low cells were plated on to 12 well plates (50,000 cells/plate) and grown overnight in proliferation medium. The following day, the plates were washed twice with PBS, fixed in 3.7% formaldehyde solution for 10 minutes at room temperature, and washed two more times with PBS. The cells were then permeabilized in 0.1% Trition X-100 for 20 minutes and washed again with PBS. For each well a staining solution of 5 μL of methanolic stock solution phalloidin with 200 μL PBS and 1% BSA was added. The staining solution was kept in the wells for 20 minutes, and then the wells were washed again with PBS. This made the actin appear green under a fluorescence microscope. The nucleus was stained by adding 300 μL of 300 nM DAPI to wells for 5 minutes and then rising twice with PBS. This made the nucleus appear blue under a fluorescence microscope.
2.9. Disulfiram Treatment
Disulfiram is an ALDH inhibitor that has been used to treat alcoholism by blocking the enzymatic conversion of ethanol to acetic acid. In this study it was used to determine the effect of blocking ALDH in K7M2 cells vis-à-vis mTOR activity and cell morphology. ALDH-high and ALDH-low cells were plated and left to grow for approximately 2 hours before adding the disulfiram. A concentration of 250 nM was found to be the high nontoxic concentration able to be used on the cells.
2.10. mTOR Immunostaining
In order to determine the relative expression of mTOR in disulfiram-treated versus untreated cells, the cells were incubated with p-4E-BP1 (a rabbit antibody to mTOR). After a period of 2 hours, the cells were rinsed and then incubated with a fluorescent anti-rabbit antibody for one hour. The cells were then treated with DAPI as described earlier in order to visualize the nuclei. Pictures were then taken with a fluorescence microscope.
2.11. Statistical Analysis
At least three samples obtained from each subject were pooled for statistical analysis of all results from this study, and the results are expressed as a mean ± SD. The differences between two means were considered to be statistically significant if P value was <0.05. A Student's t-test was used to determine statistically significant differences between two means.
3. Results
3.1. Rapamycin Inhibits BMP2, VEGF, and ALDH-1A1 Gene Expression in K7M2 Cells
K7M2 cells are known to exhibit greater mTORC1 kinase activity [25]. In order to confirm elevated mTORC1 kinase activity in our K7M2 cells we compared the amount of its phosphorylated substrate, phospho-4E-BP1 (p-4E-BP1), in K7M2 and K12 cells by immunofluorescence using a phosphospecific antibody (anti-phospho-threonine-37/46). Figure 1(a) shows a representative image of fixed K7M2 and K12 cell populations processed for p-4E-BP1 and DAPI immunofluorescence. Consistent with published results [25], we observed a greater than twofold increase in p-4E-BP1 immunofluorescence in K7M2 cultures compared to K12 cultures (Figure 1(b)), suggestive of elevated mTORC1 kinase activity in the more metastatic K7M2 cells.
Figure 1.
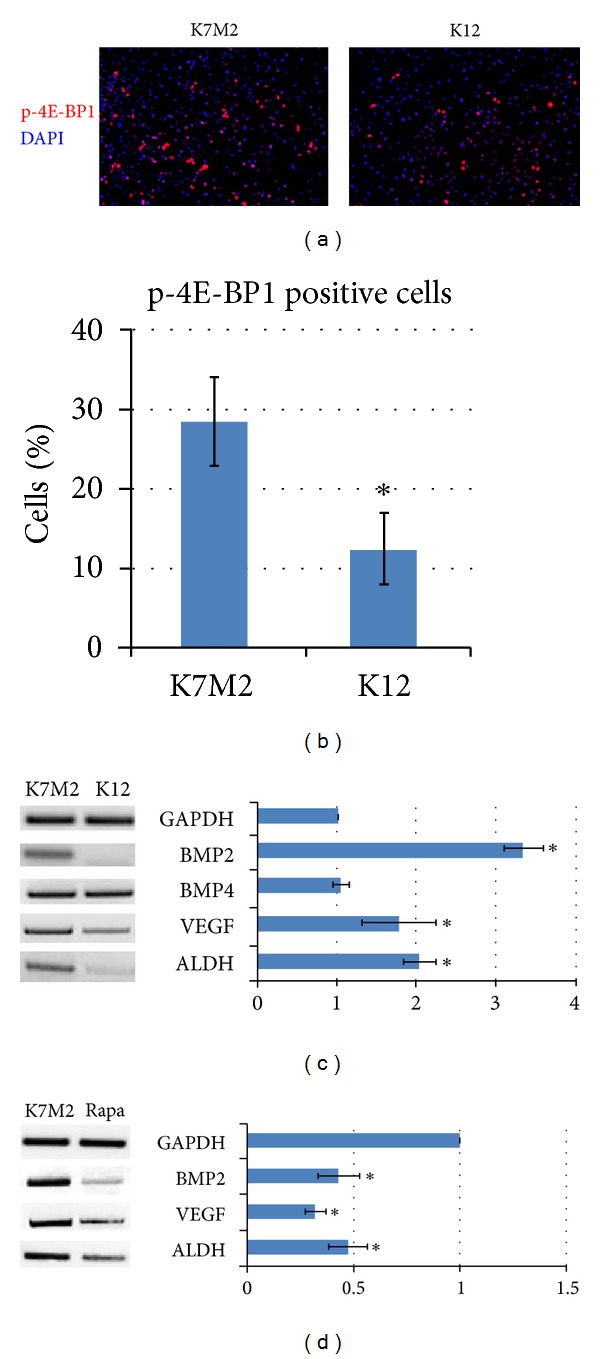
Phospho-4E-BP1 levels and rapamycin-sensitive gene expression in murine OS cells. (a) Immunofluorescence analysis of K7M2 and K12 cells depicting phospho-4E-BP1 (p-4E-BP1) counterstained with DAPI. (b) Quantitative analysis of p-4E-BP1 staining comparing the percentage of positive K7M2 and K12 cells. (c) RT-PCR was performed on cellular RNA extracted from K7M2 and K12 cells in order to quantitate the relative expression of BMP2, BMP4, VEGF, and ALDH-1A1. GAPDH serves as a loading control. (d) RT-PCR was performed on cellular RNA extracted from K7M2 cells treated with rapamycin or DMSO only (K7M2) in order to quantitate the relative expression of BMP2, VEGF, and ALDH-1A1 in the presence of mTORC1 kinase inhibition. Again, GAPDH serves as a loading control. Asterisks (∗) indicate statistically significant differences (P < 0.05).
ALDH activity, a “cancer stem cell marker,” is found in a variety of human cancers and has been associated with metastasis, drug resistance, and a poor prognosis [26–30]. In order to investigate the role of mTORC1 activity in the regulation of ALDH in K7M2 cells we first performed reverse transcription polymerase chain reaction (RT-PCR) using RNA extracted from K7M2 and K12 cells treated with rapamycin or DMSO control. We have previously shown that BMP2 and VEGF expressions are upregulated in K7M2 cells compared to K12 cells [31]. Therefore, we analyzed the expression of BMP2, BMP4, and VEGF, in addition to ALDH-1A1 expression. GAPDH served as a loading control. Figure 1(c) shows the upregulation of these genes, except BMP4, in K7M2 cells compared to K12 cells, in the absence of rapamycin. After the addition of rapamycin to the media, we observed a significant down-regulation of BMP2, VEGF, and ALDH-1A1 mRNA transcripts of K7M2 cells (Figure 1(d)).
3.2. Rapamycin Treatment Reduces ALDH Activity and Sensitizes K7M2 Cells to Oxidative Stress
In order to further investigate the effect of rapamycin on ALDH we performed fluorescence-associated cell sorting (FACS) analysis to quantitate the percentage of cells with ALDH activity. Consistent with our RT-PCR results, we observed a threefold increase in the percentage of K7M2 cells with ALDH activity (23.1 ± 3.5% versus 7.4 ± 2.7%) compared to K12 cells (Figure 2(a)). However, after rapamycin treatment, the percentage of K7M2 cells with ALDH activity reduced significantly, compared to DMSO control group, and approached a level of activity more comparable to the less metastatic K12 cells (Figure 2(b)).
Figure 2.
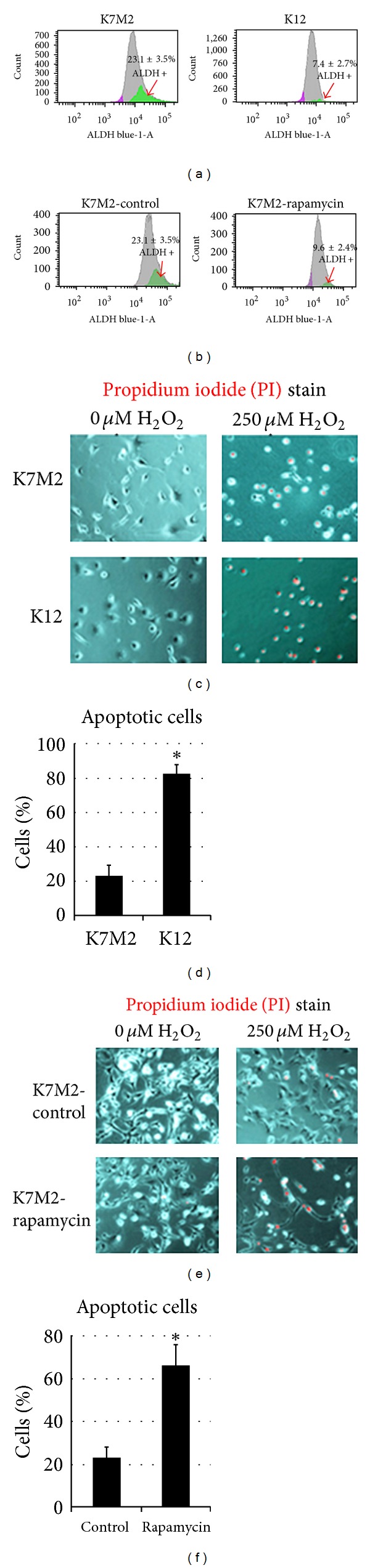
Rapamycin treatment reduces ALDH activity and sensitizes K7M2 cells to oxidative stress. (a) ALDH activity was detected in K7M2 and K12 cells using FACS analysis and the relative amount of cells positive for ALDH is shown for each cell population. (b) K7M2 cells were treated with rapamycin or DMSO only (control) and analyzed by FACS as in (a). (c) K7M2 and K12 cells were treated with or without H2O2 (250 μM) and apoptosis was detected using PI staining. (d) A quantitative analysis of (c) illustrating the percentage of apoptotic cells after H2O2 treatment compared to untreated controls. (e) K7M2 cells were treated with or without H2O2, in the presence or absence (DMSO only control) of rapamycin, and apoptotic cells were detected as in (c). (f) A quantitative analysis of (e) illustrating the percentage of apoptotic cells after H2O2 compared to untreated controls. Asterisks (∗) indicate statistically significant differences (P < 0.05).
After observing that rapamycin effectively reduced ALDH-1A1 expression and ALDH activity in K7M2 cells, we next wanted to investigate the effect of rapamycin treatment on K7M2 resistance to oxidative stress because the activity of ALDH in cancer may function to neutralize oxidative stress and provide chemoresistance [32, 33]. We tested resistance to oxidative stress using H2O2 treatment. Apoptosis was monitored by nuclear inclusion of propidium iodide (PI). Figure 2(c) contains representative images of K7M2 cells and K12 cells treated with or without H2O2. After treatment with H2O2, the large majority (>85%) of K12 cells underwent apoptosis as indicated by PI inclusion, whereas most of the K7M2 cells maintained viability (>65%) as indicated by nuclear exclusion of PI (Figure 2(d)). Therefore, K7M2 cells are more resistant to oxidative stress from H2O2 exposure than K12 cells. In order to test if this resistance to H2O2 was related to mTORC1 or ALDH activity we repeated these experiments on K7M2 cells in the presence or absence of rapamycin, and 0 μM or 250 μM of H2O2. Representative images are shown in Figure 2(e). We observed an increased sensitivity to H2O2 with rapamycin treatment as rapamycin caused a threefold increase in apoptosis (Figure 2(f)). Rapamycin alone is not proapoptotic in this assay, as rapamycin in the absence of H2O2 did not increase the frequency of PI nuclear inclusion in K7M2 cells.
3.3. Rapamycin Treatment Reduces K7M2 Cell Proliferation
We analyzed K7M2 cell proliferation with and without rapamycin treatment because rapamycin has been shown to induce a G1-arrest in many tumor cell lines including osteosarcoma [10, 34]. After four days of culture in the presence or absence of rapamycin, K7M2 cells cultured in the presence of rapamycin appeared less dense on the culture dishes (Figure 3(a)). We then determined the population doubling time (PDT) of K7M2 and K12 cells with or without rapamycin treatment, over a culture period of 4 days (Figures 3(b) and 3(c)). It showed that PDT of both K7M2 and K12 was increased by rapamycin treatment, while its effect on K7M2 cells is more profound than that of K12 cells.
Figure 3.
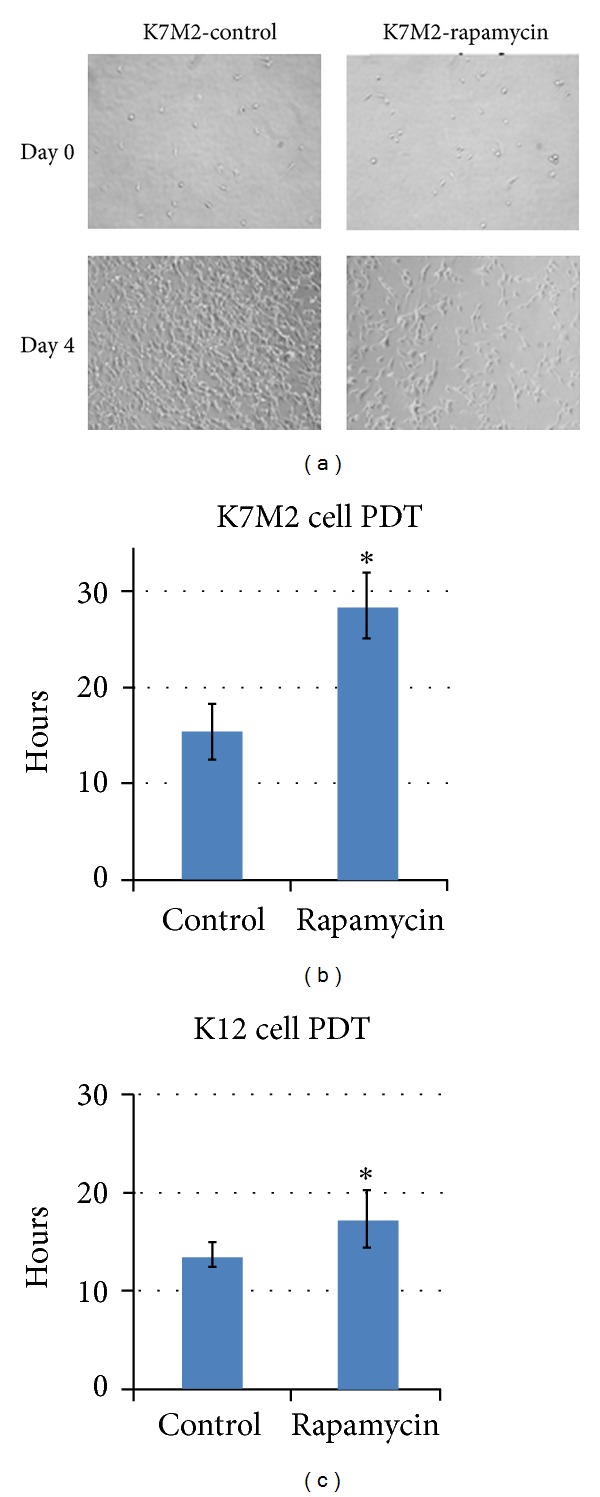
Rapamycin treatment reduces K7M2 cell proliferation. (a) K7M2 cells were cultured with media containing 10 μM rapamycin or DMSO only (control) for four days. Representative images of cell density at the beginning and end of treatment are shown. (b) A quantitative analysis of (a) to determine the population doubling time (PDT) of K7M2 cells in rapamycin-treated and control groups (DMSO only). (c) A quantitative analysis determining the population doubling time (PDT) of K12 cells in rapamycin-treated and control groups (DMSO only). Asterisks (∗) indicate statistically significant differences (P < 0.05).
3.4. Rapamycin Treatment Reduces K7M2 Cell Migration and Invasion
K7M2 cells display much greater metastatic potential in vivo compared to K12 cells [24]. Consistent with this enhanced metastatic ability, we performed an in vitro tracking cell migration assay and observed more migratory activity in K7M2 cells compared to K12 cells and both K7M2 and K12 migration was sensitive to rapamycin (Figure 4(a)). We then determined the velocity of cell migration in this assay and the results are similar and displayed in Figure 4(b). Lastly, we evaluated the invasion capacity of K7M2 and K12 cells in 2.5% Matrigel and found that K7M2 cells have much stronger invasion capacity than K12 cells (data not shown). Furthermore, it was found that rapamycin greatly reduces K7M2 cell invasion in this assay (Figure 4(c)).
Figure 4.
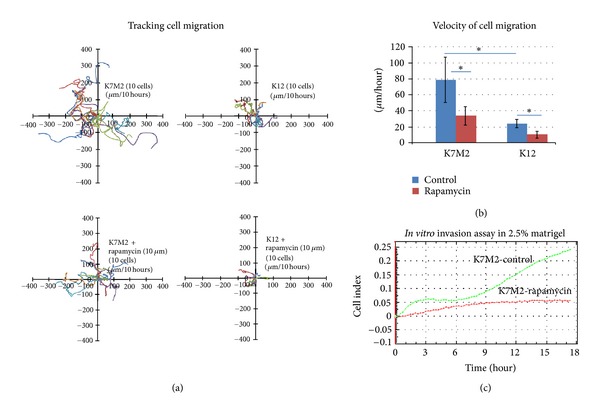
OS cell migration and invasion is sensitive to rapamycin treatment. (a) K7M2 and K12 cell migration was tracked in the presence or absence (DMSO only control) of rapamycin (10 μM) and the data obtained is displayed. (b) The velocity of cell migration was determined in each group in (a) and the results are displayed. (c) K7M2 cell invasion through a 2.5% Matrigel was monitored for 18 hours in the presence or absence (DMSO only) of 10 μM rapamycin. Asterisks (∗) indicate statistically significant differences (P < 0.05).
3.5. ALDH-High K7M2 Cells Have Greater Invasiveness, Morphologic Changes, Resistance to Oxidative Stress, and Expression of Oncogenic Factors Than ALDH-Low K7M2 Cells
K7M2 cells have been sorted according to the differential ALDH activity. The sorted populations of ALDH-high and ALDH-low K7M2 cells have been expanded in vitro (Figure 5(a)). In vitro invasion assay with 2.5% Matrigel demonstrated that ALDH-high K7M2 cells have much higher invasion capacity compared to ALDH-low K7M2 cells (Figure 5(b)). ALDH-high K7M2 cells were found to be more spread out and irregular in shape than ALDH-low K7M2 cells. They also displayed characteristics of typical motile behavior such as more organized structure of actin and the presence of filopodia (Figure 5(c)). Certain types of filopodia have been associated with increased invasiveness and metastasis rate [35]. Antioxidative stress assay showed that ALDH-high K7M2 cells are more resistant to the treatment of hydrogen peroxide, compared to ALDH-low K7M2 cells (Figure 2). RT-PCR also showed that ALDH-high cells had a higher expression of mTOR and c-Myc.
Figure 5.
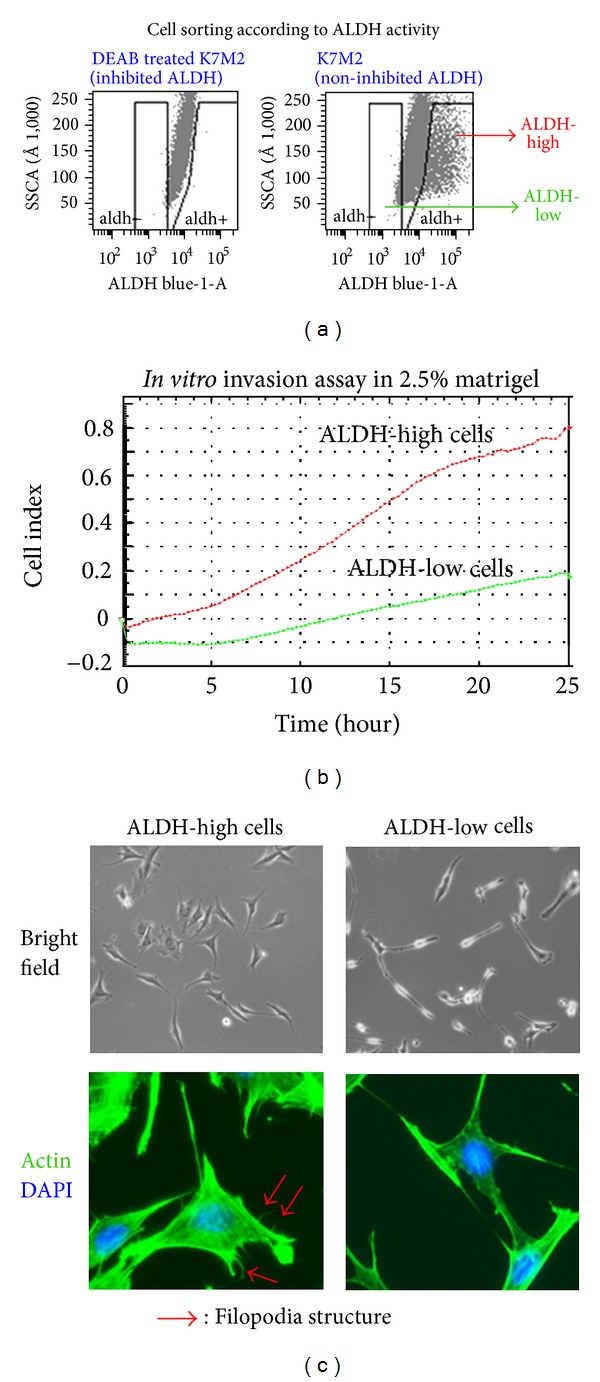
Cell sorting of K7M2 cells via ALDH activity and differential features of ALDH-high cells and ALDH-low cells. (a) K7M2 cells were suspended in Aldefluor buffer and sorted according to their fluorescence. Cells were treated with DEAB to block ALDH fluorescence, and cells were deemed ALDH-high if their fluorescence was higher than that of the DEAB-treated controls. Cells were deemed ALDH-low if their fluorescence was lower than that of the DEAB-treated controls. (b) ALDH-high and ALDH-low K7M2 cell invasion was tracked over a period of 24 hours and the data obtained is displayed. The ALDH-high cells displayed a much higher invasion potential, with more fraction of cells invading through the matrigel (2.5%). (c) Under bright field microscopy and under fluorescent microscopy (after staining for actin (green) and nucleic acids (blue)) ALDH-high K7M2 cells displayed more filopodia than ALDH-low cells. Also the ALDH-high cells were more irregularly shaped and were more pleomorphic than ALDH-low K7M2 cells.
3.6. ALDH-High K7M2 Cells Treated with an ALDH-Inhibitor (Disulfiram) Show Reduced ALDH Activity, Oncogene Expression, and Morphologic Alterations
ALDH-high cells treated with disulfiram were verified to show a much reduced ALDH activity (data not shown). ALDH-high cells treated with disulfiram were found to have a lower expression of mTOR and c-Myc than untreated ALDH-high cells as shown via RT-PCR (Figure 6(a)). Treated cells also displayed less mTOR activity as shown via immunostaining with phosphpo-4E-BP1 (Figure 6(b)). Finally, the addition of disulfiram altered the cells' morphologies: they appeared much more like ALDH-low cells than untreated ALDH-high cells. There were much fewer filapodia, and the cells appeared much more uniform in shape (Figure 6(c)).
Figure 6.
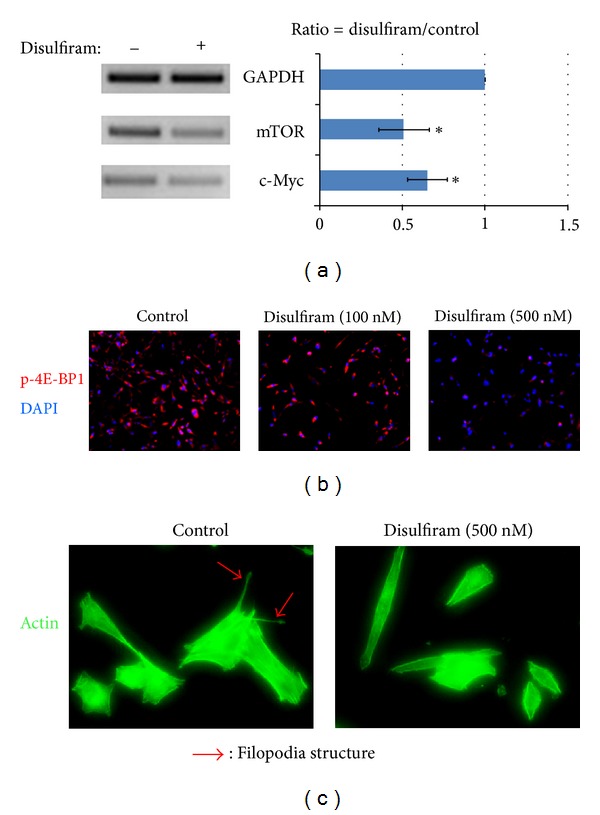
Inhibition of ALDH with disulfiram inhibits metastatic properties of ALDH-high K7M2 cells. (a) Disulfiram (250 nM) was added to ALDH-high K7M2 cells and the cells were cultured for at least 24 hours in 10% FBS growth medium. RT-PCR was used to determine gene expression differences as a result of this treatment, with GADPH used as a control. Both mTOR and c-Myc expression were reduced as a result of the treatment with disulfiram. (b) Immunostaining with phospho-4E-BP1 (a mouse anti-mTOR antibody) was done to confirm that treatment with disulfiram reduced expression of mTOR As the concentration of disulfiram was increased (0 nm, 100 nm and 500 nm) cells displayed progressively less mTOR expression. (c) Morphologic differences after treatment with disulfriam were also present, with disulfiram treated cells (stained for actin) appearing less pleomorphic and with fewer filopodia.
4. Discussion
K7M2 murine OS cells are highly metastatic to the lungs and were clonally derived from the less metastatic K12 OS cells [24]. Thus, the K7M2 and K12 cell lines are very similar genetically but differ significantly in their metastatic phenotypes and represent excellent tools for determining the critical biochemical pathways of OS metastasis. Here we report that, in comparison with K12 cells, K7M2 cells feature “rapamycin-sensitive” mTOR signaling which promotes cellular behaviors associated with metastasis, including higher ALDH activity, increased resistance to oxidative stress, proliferation, migration, invasion, and upregulation of BMP2 and VEGF expression. These results imply that mTORC1 activity may contribute to the enhanced metastatic potential of K7M2 cells.
There are several limitations to this study not the least of which is that all of our experiments were performed in vitro. Additionally, all of our experiments, and therefore our conclusions, are based entirely on the study of two murine cell lines. Although the observations reported above are interesting, care must be taken not to overstate these data. Fortunately, the metastatic phenotypes of K7M2 and K12 cells have been well-characterized in previous studies, including the effect of rapamycin treatment on K7M2 metastasis [24, 36]. We believe that our conclusions are sound, but future studies evaluating ALDH and mTOR activity in other OS cell lines and in vivo will be essential to validate these observations.
A role for mTOR signaling in osteosarcoma metastasis is slowly evolving. Human osteosarcoma tissue samples have stained positive for mTOR signaling and this has been correlated with surgical stage, metastasis pattern, and disease-free survival [23]. Our current understanding of the molecular biology remains limited. It has been shown in K7M2 cells that Ezrin (a plasma membrane to actin cytoskeleton linker protein) expression is upregulated compared to K12 cells, and that dynamic regulation of Ezrin activity via phosphorylation is required for metastasis to occur in a mouse xenograft model. It appears that Ezrin must be turned “on” and “off” in order to regulate cellular metabolism and protect K7M2 OS cells against stress after early arrival in the lungs. Furthermore, Ezrin, phosphatidylinositol-3-kinase (PI3K), and AKT are each associated with phosphorylation of S6K1 and 4E-BP1. Interestingly, Ezrin-associated phosphorylation of S6K1 and 4E-BP1 is sensitive to rapamycin, suggesting that these phosphorylation events depend on the mTORC1 kinase complex [25, 37].
mTOR signaling can regulate cell motility through mTORC1 and mTORC2-dependent kinase activity. Here we show that rapamycin can inhibit K7M2 cell migration in vitro, presumably by blocking mTORC1 activity. The regulation of cell motility by mTORC1 is most likely to be the mechanism that influences cell migration. Both mTORC1/S6K1 and mTORC1/4E-BP1 pathways are known to regulate cell motility. S6K1 participates in the phosphorylation of the focal adhesion proteins and F-actin reorganization in order to modulate cell motility but little is known about the manner in which 4E-BP1 regulates this process [14]. Thus, rapamycin treatment could block migration by interfering with mTORC1-mediated cell motility but would potentially leave mTORC2 unchecked and free to regulate cell motility via control of the actin cytoskeleton through activation of the Rho GTPases [14].
ALDH activity has been identified as a “cancer stem cell” (CSC) marker in multiple neoplasms and has been associated with metastasis and a poor prognosis, but such a role has yet to be established in osteosarcoma [26–30]. The CSC hypothesis predicts that if certain genetic alterations occur in the right context, and within a more primitive cell, then a cancer-initiating cell is born that retains all the qualities of stem cells including self-renewal, proliferation, differentiation, resistance to drugs, stress, apoptosis, and the capacity to migrate, invade, and induce angiogenesis [38]. Our results show that ALDH-high OS cells display a significantly greater metastatic phenotype than ALDH-low cells. Although the relationship between CSCs and metastasis has not been clearly elucidated, it has been demonstrated that the number of metastatic cancer colonies correlates with the frequency of CSCs in the primary tissue. Furthermore, CSC subpopulations display a higher potential for invasiveness than subpopulations of nonstem tumor cells [39].
In this study, we observed that K7M2 cells exhibit higher levels of ALDH activity and this is dependent on mTORC1 activity as indicated by treatment with rapamycin. Furthermore, we showed that K7M2 cells, in comparison to K12 cells, are more resistant to oxidative stress caused by H2O2 treatment. The activity of ALDH in cancer may function to neutralize oxidative stress and provide chemoresistance, and thus upregulation of ALDH activity by mTOR may be one mechanism that confers resistance to oxidative stress in K7M2 cells [32, 33]. Alternatively, disulfiram, an ALDH inhibitor, has been shown to reduce the invasiveness of U2OS cells and the expression of matrix metalloproteinases, raising the possibility that ALDH activity may have a more direct role in tumor invasion [28]. However, the mechanisms for disulfiram's effects on U2OS invasion remain unclear.
In terms of stem cell regulation, an integral role for mTOR signaling in the proliferation and self-renewal of embryonic stem cells (ESCs) has been implicated. Loss of mTOR activity in human ESCs impairs pluripotency, blocks proliferation, and enhances mesoderm and endoderm activities [40]. The repression of developmental genes through mTOR signaling is thought to maintain pluripotency. Differentiation induces an “anabolic switch” and an increase in protein synthesis and protein content coincide with cell fate determination. Specifically, mTOR, and 4E-BP1 may function to regulate global and selective protein synthesis during self-renewal and differentiation [41]. Therefore, mTORC1 kinase activity may serve a similar role in the regulation of osteosarcoma stem cell proliferation and self-renewal.
Indeed, the ability of mTOR to promote proliferation without differentiation has been demonstrated in the literature. Many studies have shown that rapamycin treatment induces a G1-arrest in various cell lines, including osteosarcoma [10, 34], and here we have shown that rapamycin has a similar effect on K7M2 OS cells. This G1-arrest is likely to occur through the down-regulation of cyclin D1 as was previously shown in human OS cells by Gazitt et al. [42]. mTOR activity has also been shown to block differentiation as rapamycin treatment can enhance osteoblast differentiation [43]. Although an OS cancer stem cell has not been identified to date, evidence is accumulating that suggests this discovery is merely a matter of time [44]. If an OS cancer stem cell exists, we suspect that mTOR and ALDH activity play pivotal roles in the maintenance of this subpopulation of cancer cells.
Here we also demonstrated that BMP2 and VEGF expression in K7M2 cells is reduced by rapamycin treatment. Previously we had shown that BMP2 and VEGF expression is upregulated in K7M2 cells compared to K12 cells [31]. Furthermore, treatment with the BMP antagonist Noggin caused decreased motility, altered morphology, and increased cell death in K7M2 cells. It is also known that BMP2 is activated through hypoxia/AKT/mTOR/HIFα pathway in osteoblasts [45]. VEGF, on the other hand, is a potent inducer of neovascularization whose activity is thought to be an absolute requirement for tumor growth and metastasis [46]. Consistent with the present study, rapamycin has been shown to reduce VEGF production and inhibit angiogenesis in mice in vivo [47]. Therefore, down-regulation of BMP2 and VEGF (by mTORC1 inhibition) may each contribute to alter the metastatic behavior of K7M2 cells. Both BMP2 and VEGF may prove to be therapeutic targets in the treatment of OS metastasis. In vivo studies are essential to investigate the role of these secreted proteins in OS metastatic biology.
Our results show that ALDH inhibition via disulfiram actually decreased mTOR activity. This implies that under certain circumstances, mTOR activity may be regulated by ALDH. This has profound implications as mTOR activity is important in many cancers and is downstream of important tumor suppressors and oncogenes that are frequently mutated in human cancers [17]. Our work supports an oncogenic role for mTORC1 activity in osteosarcoma and provides evidence to link mTOR signaling, ALDH activity, and metastatic behavior. To our knowledge the present study is the first to implicate mTOR in the regulation of ALDH activity. Given our data and other evidence that exists to implicate mTOR and ALDH function in CSCs, we suspect that if an OS stem cell exists, mTOR and ALDH may be important for its proliferation, self-renewal, drug resistance, cell survival, and metastasis. The search for surface markers of OS cells has been long and disappointing, but ALDH may represent great promise as an OS stem cell surface marker. At this point in time we cannot be certain of the mechanisms by which ALDH and mTOR interact. Future investigations are required to further delineate the roles and relationships of mTOR, ALDH, and OS cancer stem cells.
In conclusion, mTOR signaling is upregulated in highly metastatic K7M2 OS cells. This upregulation is associated with increased ALDH expression and activity, resistance to oxidative stress, greater proliferation, migration, and invasion, and higher BMP2 and VEGF expression. This work implicates rapamycin and its analogs as potential agents in the treatment and prevention of OS metastasis. It also suggests a relationship between mTOR and ALDH. ALDH may, in its own right, represent a possible marker and therapeutic target of OS cell metastatic potential.
Conflict of Interests
The coauthor, J. Huard, receives remuneration for consulting and royalties from Cook Myosite Inc. The corresponding author, K. Weiss, is on the scientific advisory board of Eleison Pharmaceuticals. The remaining authors have no conflict of interests to disclose.
Acknowledgments
The authors wish to acknowledge the generous support of the Pittsburgh Foundation and the Houy family in loving memory of Jon Houy. The flow cytometry work was made possible by Lynda Guzik at the McGowan Institute for Regenerative Medicine, University of Pittsburgh, Pittsburgh, PA, USA.
References
- 1.Bielack SS, Kempf-Bielack B, Delling G, et al. Prognostic factors in high-grade osteosarcoma of the extremities or trunk: an analysis of 1,702 patients treated on neoadjuvant cooperative osteosarcoma study group protocols. Journal of Clinical Oncology. 2002;20(3):776–790. doi: 10.1200/JCO.2002.20.3.776. [DOI] [PubMed] [Google Scholar]
- 2.Carrle D, Bielack SS. Current strategies of chemotherapy in osteosarcoma. International Orthopaedics. 2006;30(6):445–451. doi: 10.1007/s00264-006-0192-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayden JB, Hoang BH. Osteosarcoma: basic science and clinical implications. Orthopedic Clinics of North America. 2006;37(1):1–7. doi: 10.1016/j.ocl.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Ferrari S, Smeland S, Mercuri M, et al. Neoadjuvant chemotherapy with high-dose ifosfamide, high-dose methotrexate, cisplatin, and doxorubicin for patients with localized osteosarcoma of the extremity: a joint study by the italian and Scandinavian Sarcoma Groups. Journal of Clinical Oncology. 2005;23(34):8845–8852. doi: 10.1200/JCO.2004.00.5785. [DOI] [PubMed] [Google Scholar]
- 5.Messerschmitt PJ, Garcia RM, Abdul-Karim FW, Greenfield EM, Getty PJ. Osteosarcoma. Journal of the American Academy of Orthopaedic Surgeons. 2009;17(8):515–527. doi: 10.5435/00124635-200908000-00005. [DOI] [PubMed] [Google Scholar]
- 6.Vo K, Amarasinghe B, Washington K, Gonzalez A, Berlin J, Dang TP. Targeting notch pathway enhances rapamycin antitumor activity in pancreas cancers through PTEN phosphorylation. Molecular Cancer. 2011;10, article 138 doi: 10.1186/1476-4598-10-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meyers PA, Schwartz CL, Krailo M, et al. Osteosarcoma: a randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. Journal of Clinical Oncology. 2005;23(9):2004–2011. doi: 10.1200/JCO.2005.06.031. [DOI] [PubMed] [Google Scholar]
- 8.Meyers PA, Schwartz CL, Krailo MD, et al. Osteosarcoma: the addition of muramyl tripeptide to chemotherapy improves overall survival—a report from the children’s oncology group. Journal of Clinical Oncology. 2008;26(4):633–638. doi: 10.1200/JCO.2008.14.0095. [DOI] [PubMed] [Google Scholar]
- 9.Meyers PA, Gorlick R. Osteosarcoma. Pediatric Clinics of North America. 1997;44(4):973–989. doi: 10.1016/s0031-3955(05)70540-x. [DOI] [PubMed] [Google Scholar]
- 10.Brown EJ, Albers MW, Shin TB, et al. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature. 1994;369(6483):756–758. doi: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- 11.Chiu MI, Katz H, Berlin V. RAPT1, a mammalian homolog of yeast Tor, interacts with the FKBP12/rapamycin complex. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(26):12574–12578. doi: 10.1073/pnas.91.26.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78(1):35–43. doi: 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- 13.Sabers CJ, Martin MM, Brunn GJ, et al. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. The Journal of Biological Chemistry. 1995;270(2):815–822. doi: 10.1074/jbc.270.2.815. [DOI] [PubMed] [Google Scholar]
- 14.Zhou H, Huang S. MTOR signaling in cancer cell motility and tumor metastasis. Critical Reviews in Eukaryotic Gene Expression. 2010;20(1):1–16. doi: 10.1615/critreveukargeneexpr.v20.i1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124(3):471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 16.Zoncu R, Efeyan A, Sabatini DM. MTOR: from growth signal integration to cancer, diabetes and ageing. Nature Reviews Molecular Cell Biology. 2011;12(1):21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dazert E, Hall MN. mTOR signaling in disease. Current Opinion in Cell Biology. 2011;23:744–755. doi: 10.1016/j.ceb.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 18.Jacinto E, Loewith R, Schmidt A, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nature Cell Biology. 2004;6(11):1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 19.Loewith R, Jacinto E, Wullschleger S, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Molecular Cell. 2002;10(3):457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 20.Crouch MF. Regulation of thrombin-induced stress fibre formation in Swiss 3T3 cells by the 70-kDa S6 kinase. Biochemical and Biophysical Research Communications. 1997;233(1):193–199. doi: 10.1006/bbrc.1997.6419. [DOI] [PubMed] [Google Scholar]
- 21.Dufner A, Thomas G. Ribosomal S6 kinase signaling and the control of translation. Experimental Cell Research. 1999;253(1):100–109. doi: 10.1006/excr.1999.4683. [DOI] [PubMed] [Google Scholar]
- 22.Ferrari S, Bandi HR, Hofsteenge J, Bussian BM, Thomas G. Mitogen-activated 70K S6 kinase. Identification of in vitro 40 S ribosomal S6 phosphorylation sites. The Journal of Biological Chemistry. 1991;266(33):22770–22775. [PubMed] [Google Scholar]
- 23.Zhou Q, Deng Z, Zhu Y, Long H, Zhang S, Zhao J. mTOR/p70S6K signal transduction pathway contributes to osteosarcoma progression and patients’ prognosis. Medical Oncology. 2010;27(4):1239–1245. doi: 10.1007/s12032-009-9365-y. [DOI] [PubMed] [Google Scholar]
- 24.Khanna C, Prehn J, Yeung C, Caylor J, Tsokos M, Helman L. An orthotopic model of murine osteosarcoma with clonally related variants differing in pulmonary metastatic potential. Clinical and Experimental Metastasis. 2000;18(3):261–271. doi: 10.1023/a:1006767007547. [DOI] [PubMed] [Google Scholar]
- 25.Wan X, Mendoza A, Khanna C, Helman LJ. Rapamycin inhibits ezrin-mediated metastatic behavior in a murine model of osteosarcoma. Cancer Research. 2005;65(6):2406–2411. doi: 10.1158/0008-5472.CAN-04-3135. [DOI] [PubMed] [Google Scholar]
- 26.Charafe-Jauffret E, Ginestier C, Iovino F, et al. Aldehyde dehydrogenase 1-positive cancer stem cells mediate metastasis and poor clinical outcome in inflammatory breast cancer. Clinical Cancer Research. 2010;16(1):45–55. doi: 10.1158/1078-0432.CCR-09-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheung AMS, Wan TSK, Leung JCK, et al. Aldehyde dehydrogenase activity in leukemic blasts defines a subgroup of acute myeloid leukemia with adverse prognosis and superior NOD/SCID engrafting potential. Leukemia. 2007;21(7):1423–1430. doi: 10.1038/sj.leu.2404721. [DOI] [PubMed] [Google Scholar]
- 28.Cho HJ, Lee TS, Park JB, et al. Disulfiram suppresses invasive ability of osteosarcoma cells via the inhibition of MMP-2 and MMP-9 expression. Journal of Biochemistry and Molecular Biology. 2007;40(6):1069–1076. doi: 10.5483/bmbrep.2007.40.6.1069. [DOI] [PubMed] [Google Scholar]
- 29.Honoki K, Fujii H, Kubo A, et al. Possible involvement of stem-like populations with elevated ALDH1 in sarcomas for chemotherapeutic drug resistance. Oncology Reports. 2010;24(2):501–505. doi: 10.3892/or_00000885. [DOI] [PubMed] [Google Scholar]
- 30.Huang EH, Hynes MJ, Zhang T, et al. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Research. 2009;69(8):3382–3389. doi: 10.1158/0008-5472.CAN-08-4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weiss KR, Cooper GM, Jadlowiec JA, McGough RL, III, Huard J. VEGF and BMP expression in mouse osteosarcoma cells. Clinical Orthopaedics and Related Research. 2006;450:111–117. doi: 10.1097/01.blo.0000229333.98781.56. [DOI] [PubMed] [Google Scholar]
- 32.Magni M, Shammah S, Schiró R, Mellado W, Dalla-Favera R, Gianni AM. Induction of cyclophosphamide-resistance by aldehyde-dehydrogenase gene transfer. Blood. 1996;87(3):1097–1103. [PubMed] [Google Scholar]
- 33.Russo JE, Hilton J. Characterization of cytosolic aldehyde dehydrogenase from cyclophosphamide resistant L1210 cells. Cancer Research. 1988;48(11):2963–2968. [PubMed] [Google Scholar]
- 34.Albers MW, Williams RT, Brown EJ, Tanaka A, Hall FL, Schreiber SL. FKBP-rapamycin inhibits a cyclin-dependent kinase activity and a cyclin D1-Cdk association in early G1 of an osteosarcoma cell line. The Journal of Biological Chemistry. 1993;268(30):22825–22829. [PubMed] [Google Scholar]
- 35.Machesky LM. Lamellipodia and filopodia in metastasis and invasion. FEBS Letters. 2008;582(14):2102–2111. doi: 10.1016/j.febslet.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 36.Khanna C, Khan J, Nguyen P, et al. Metastasis-associated differences in gene expression in a murine model of osteosarcoma. Cancer Research. 2001;61(9):3750–3759. [PubMed] [Google Scholar]
- 37.Ren L, Hong SH, Chen QR, et al. Dysregulation of ezrin phosphorylation prevents metastasis and alters cellular metabolism in osteosarcoma. Cancer Research. 2012;72:1001–1012. doi: 10.1158/0008-5472.CAN-11-0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wicha MS, Liu S, Dontu G. Cancer stem cells: an old idea—a paradigm shift. Cancer Research. 2006;66(4):1883–1890. doi: 10.1158/0008-5472.CAN-05-3153. [DOI] [PubMed] [Google Scholar]
- 39.Hu YY, Zheng MH, Zhang R, Liang YM, Han H. Notch signaling pathway and cancer metastasis. Advances in Experimental Medicine and Biology. 2012;727:186–198. doi: 10.1007/978-1-4614-0899-4_14. [DOI] [PubMed] [Google Scholar]
- 40.Zhou J, Su P, Wang L, et al. mTOR supports long-term self-renewal and suppresses mesoderm and endoderm activities of human embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(19):7840–7845. doi: 10.1073/pnas.0901854106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sampath P, Pritchard DK, Pabon L, et al. A hierarchical network controls protein translation during murine rmbryonic stem cell self-renewal and differentiation. Cell Stem Cell. 2008;2(5):448–460. doi: 10.1016/j.stem.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 42.Gazitt Y, Kolaparthi V, Moncada K, Thomas C, Freeman J. Targeted therapy of human osteosarcoma with 17AAG or rapamycin: characterization of induced apoptosis and inhibition of mTOR and Akt/MAPK/Wnt pathways. International Journal of Oncology. 2009;34(2):551–561. [PubMed] [Google Scholar]
- 43.Ogawa T, Tokuda M, Tomizawa K, et al. Osteoblastic differentiation is enhanced by rapamycin in rat osteoblast-like osteosarcoma (ROS 17/2.8) cells. Biochemical and Biophysical Research Communications. 1998;249(1):226–230. doi: 10.1006/bbrc.1998.9118. [DOI] [PubMed] [Google Scholar]
- 44.Gibbs CP, Jr., Levings PP, Ghivizzani SC. Evidence for the osteosarcoma stem cell. Current Orthopaedic Practice. 2011;22:322–326. doi: 10.1097/BCO.0b013e318221aee8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tseng WP, Yang SN, Lai CH, Tang CH. Hypoxia induces BMP-2 expression via ILK, Akt, mTOR, and HIF-1 pathways in osteoblasts. Journal of Cellular Physiology. 2010;223(3):810–818. doi: 10.1002/jcp.22104. [DOI] [PubMed] [Google Scholar]
- 46.Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nature Reviews Cancer. 2008;8(8):579–591. doi: 10.1038/nrc2403. [DOI] [PubMed] [Google Scholar]
- 47.Guba M, Von Breitenbuch P, Steinbauer M, et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nature Medicine. 2002;8(2):128–135. doi: 10.1038/nm0202-128. [DOI] [PubMed] [Google Scholar]