

Abstract
Recent studies have highlighted large genomic regions prone to undergo epigenetic changes in cancers. In this issue of Cancer Cell, Bert and colleagues describe novel genomic domains with aberrant epigenetic changes involving concordant activation of neighboring genes. These domains involve aberrant CpG-island hypermethylation similar to that observed in gene silencing.
One of the most exciting areas of biology in the last decade is the explosion of knowledge about the organization and regulatory features of human normal (Dunham et al., 2012) and cancer (Baylin and Jones, 2011) epigenomes. Increasingly, it has become evident that epigenetic changes in cancer constitute driver events in tumorigenesis, and exome sequencing studies have revealed recurrent mutations in key chromatin modifiers in multiple tumor types (You and Jones, 2012). Most studies concerning epigenetic deregulation in cancer have concentrated on abnormal transcriptional silencing (Baylin and Jones, 2011). In this issue of Cancer Cell, Bert et al. (2013) describe a phenomenon of epigenetic deregulation involving exactly the opposite: abnormal upregulation of gene expression that occurs in clusters of genes, hence termed “long-range epigenetic activation” (LREA) domains (Figure 1A). These findings extend a growing theme in cancer epigenetics: abnormal epigenetic deregulation of adjacent genes within large chromosomal regions, a concept with important ramifications for how chromatin boundaries exist normally and abnormally and how they go awry in cancer. We consider the current paper in the context of topological changes in the cancer genome.
Figure 1. Patterns of Long-Range Epigenetic Control Regions in Normal and Cancer Cells.
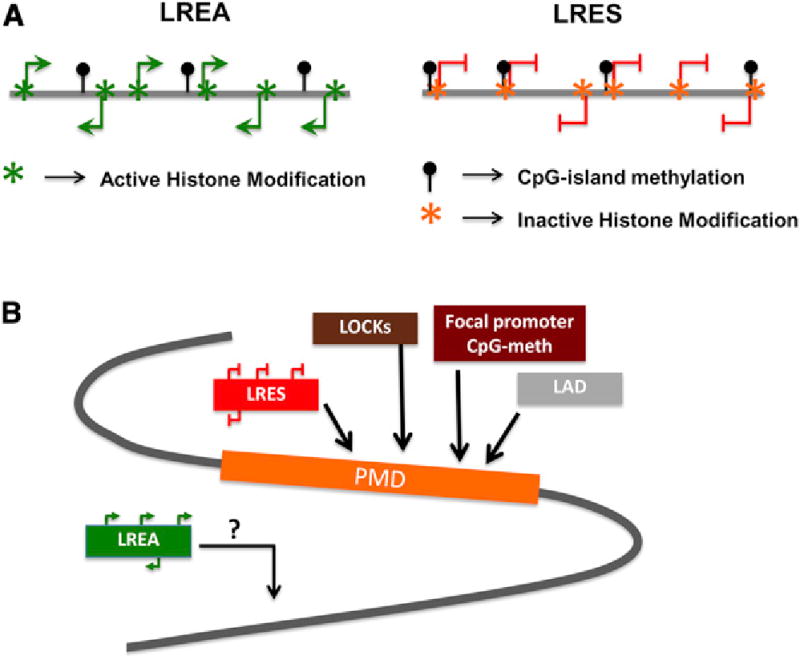
(A) Long-range epigenetic activation (LREA) domains and long-range epigenetic silenced (LRES) domains harbor gains in CpG-island methylation. The CpG-island methylation gains, more usually associated with abnormal gene silencing, may, in LREAs, result in gene activation if associated with the use of alternate TSS, inhibition of repressor binding factors, or association with activating histone modifications. LRES domains harbor inactivating histone modifications along with promoter CpG-methylation at gene TSS.
(B) Shows the association of the multiple long-range chromatin domains with epigenomic alterations in cancers. Cancer-specific promoter CpG-island methylation and LRES genes often occur within PMDs, which are also associated with other key epigenetic domains like the lamin associated domains (LADs) and large organized chromatin-lysine-(K) modifications (LOCKs). LREAs may constitute an additional type of abnormality in cancer.
Cancer-specific abnormal expression changes of genes that have wild-type DNA sequences are attributable to at least two mechanisms: gains and losses in DNA methylation as well as switches in histone modifications (Baylin and Jones, 2011). The most highly explored epigenetic change in cancer is abnormal DNA methylation in promoter CpG-islands, it is attendant gene silencing (Baylin and Jones, 2011). These methylation changes involve the participation of inactivating histone modifications, silencing protein complexes, and nucleosome occupancy surrounding gene transcription start sites (TSS) (Jones, 2012). As introduced above, such epigenetic changes can concordantly occur in adjacent genes spanning large regions involving inactive histone modifications and DNA-methylation in a process called long-range epigenetic silencing (LRES) (Coolen et al., 2010) (Figure 1A).
In the context of epigenetic gene silencing, there are new revelations regarding CpG-island hypermethylation, including LRES, and these are by no means restricted to or even best understood from focusing solely on abnormal scenarios like cancer. Deep sequencing efforts for genome-wide DNA methylation and chromatin mapping have revealed broad domains of dense DNA methylation harboring developmental genes in embryonic stem cells (ESCs), which mosaically lose their DNA-methylation in normal adult cells to form “partially methylated domains” or PMDs (Figure 1B). Importantly, these also seem to be regions where reestablishment of ESC-like CpG methylation patterns is incomplete during cellular reprogramming and cancer-like abnormalities can evolve (Lister et al., 2011). Moreover, during tumorigenesis, very significant epigenetic changes are targeted to these regions. For example, in colon cancer, PMDs are shown to lose even more DNA methylation (20%–60% methylated-CpGs) than normal colon cells (Berman et al., 2012). Perhaps most surprisingly, significantly large numbers of the genes with cancer-specific promoter CpG-island hypermethylation appear as focal changes within the PMDs, including LRES-genes.
The widespread and focal shifts in chromatin and DNA methylation in PMDs suggest that in cancers there is a loss of boundaries that regulate chromatin domains. Excitingly, the field is moving forward in identifying some of the molecular underpinnings of PMDs and their alterations in cancer. This understanding is pivotal to the entire concept of how epigenomes are organized. First, although there is global CpG-methylation loss within these PMDs, there is a concomitant increase in the inactive histone modification H3K9me2 observed as an overlap of the PMDs with large organized chromatin-lysine-(K)-modifications (LOCKs) (Hansen et al., 2011). In general, genes within these PMDs and LOCKs have low expression. Second, PMDs in normal cells largely encompass late-replicating lamin-associated domains in the nuclear periphery (Berman et al., 2012). These attributes of PMDs have important functional ramifications. In normal settings, DNA methylation maintenance is less efficient at late-replicating heterochromatic DNA in the PMDs, and this may underlie progressive loss of DNA methylation in these regions during normal differentiation (Hansen et al., 2010). This replication-timing relationship may be accentuated in cancers, possibly leading to further losses of DNA methylation in PMDs. Intriguingly, PMDs harbor significant numbers of developmental genes, which in ESCs are silenced by polycomb protein complex (PcG) occupancy, but not with DNA methylation in the promoter CpG-islands. Often, this promoter chromatin is in a context where PcG and the active histone modification, H3K4me3, co-reside to yield “bivalent” chromatin, which holds genes in a poised/low expression state until activation or repression during lineage commitment (Baylin and Jones, 2011). Studies from our lab suggest that about 70% of the promoter CpG-island hypermethylated genes in various cancers are the above PcG-related genes (Easwaran et al., 2012). These relationships suggest that late-replicating PMD regions are vulnerable to abnormal silencing with the accrual of promoter DNA-methylation of developmental genes in cancer, which cumulatively could help maintain subgroups of cells with abnormal self-renewal and/or blocks in differentiation capacity.
So where do the LREA domains fit in this concept of large epigenomic domains in cancer cells? As stated earlier, multiple genes in LREA are abnormally upregulated in prostate cancer cells compared to normal prostate epithelial cells or ESCs. Not surprisingly then, as opposing phenomena, LRES and LREA generally show opposing promoter chromatin modifications: the former harboring inactive histone modifications whereas the latter active histone modifications (Figure 1A). Intriguingly, however, the LREA domains have the same frequency of focal CpG island hypermethylation gain as in the rest of the cancer genome. Moreover, even though the authors observe that LREAs are separated from PMDs, the genes involved with CpG island hypermethylation are, as for PMDs, often those with a history of PcG-regulation in development (Bert et al., 2013). This juxtaposition of promoter CpG island hypermethylation with upregulated genes would seem to contradict its usual association with gene silencing. One explanation offered by Bert et al. (2013) is that gene activation may occur via use of alternate TSS. For other genes, activation might involve methylation-mediated inhibition of repressor binding to promoter regulatory elements. These proposals merit further investigation.
An important take-home message from the aforementioned observations is that focal promoter CpG island hypermethylation within broad regions of losses of DNA methylation is a key epigenetic abnormality in cancer. The study by Bert et al. (2013) adds to this and raises additional questions about these domain changes. The focal promoter DNA hypermethylation, while mostly associated with gene silencing, can, in a context-dependent manner, be involved in the activation of a minority of genes as in the LREA domains. Further studies in multiple cancer types are required to define the LREA domains, their differences from the PMDs, and importantly, which genes or groups of genes in these regions are important to tumorigenesis. For the last question, we need to determine how often epigenetic abnormalities in cancer involve genes residing within long-range chromatin abnormalities. The precise molecular alterations underlying long-range boundary shifts and altered gene regulation are being revealed, but much more remains to be clarified. The role of nuclear structure and chromatin configurations in this phenomenon remains to be elucidated. Further, unbiased determination of DNA methylation, chromatin modification patterns, and nucleosome localization using next-generation sequencing across these domains will help understand the molecular basis of these long-range chromosome changes. One fascinating question is how cancer mutations in chromatin-regulating genes relate to these various domains. Are these long-range regions and corresponding gene expression changes a common downstream readout for many of these genetic changes? If so, will therapeutic targeting of epigenetic modifiers provide unified approaches for treating patients according to chromatin-related gene mutation patterns in their tumors? An exciting period of research surely is upon us for learning more about the biology and translational ramifications of LRES, LREA, and PMDs in cancers.
Acknowledgments
Portions of the authors work cited in this article were supported by ES011858 from the National Institutes of Environmental Health Sciences (NIEHS), CA043318 from the National Cancer Institute (NCI), and the Hodson Junior Scholarship Award.
References
- Baylin SB, Jones PA. Nat Rev Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y, Noushmehr H, Lange CP, van Dijk CM, Tollenaar RA, et al. Nat Genet. 2012;44:40–46. doi: 10.1038/ng.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bert SA, Robinson MD, Strbenac D, Statham AL, Song JZ, Hulf T, Sutherland RL, Coolen MW, Stirzaker C, Clark SL. Cancer Cell. 2013;23:9–22. doi: 10.1016/j.ccr.2012.11.006. this issue. [DOI] [PubMed] [Google Scholar]
- Coolen MW, Stirzaker C, Song JZ, Statham AL, Kassir Z, Moreno CS, Young AN, Varma V, Speed TP, Cowley M, et al. Nat Cell Biol. 2010;12:235–246. doi: 10.1038/ncb2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R, et al. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easwaran H, Johnstone SE, Van Neste L, Ohm J, Mosbruger T, Wang Q, Aryee MJ, Joyce P, Ahuja N, Weisenberger D, et al. Genome Res. 2012;22:837–849. doi: 10.1101/gr.131169.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, et al. Nat Genet. 2011;43:768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, Stamatoyannopoulos JA. Proc Natl Acad Sci USA. 2010;107:139–144. doi: 10.1073/pnas.0912402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O’Malley R, Castanon R, Klugman S, et al. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You JS, Jones PA. Cancer Cell. 2012;22:9–20. doi: 10.1016/j.ccr.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]