

Abstract
Gonadotropin-releasing hormone-1 (GnRH-1) neurons migrate from the nasal placode to the forebrain where they control gonadal function via the hypothalamic–pituitary–gonadal axis. The birth of GnRH-1-expressing neurons is one of the first neurogenic events in the developing nasal placode. By gene expression screening on single GnRH-1 neurons, amyloid precursor binding protein-1 (FE65) was identified in migratory GnRH-1 neurons. FE65 has been shown to modulate β1-integrin dynamics, actin cytoskeleton, cell motility, and FE65/amyloid precursor protein signaling has been described in neuro/glial cell fate determination as well as in modulating neurogenesis. Analysis of two mouse lines, one deficient for the 97 kDa FE65 isoform and a second deficient for the 97 and 60 kDa forms of FE65, showed overlapping phenotypes. In both lines, no migratory defects of the GnRH-1 neurons were observed, but a 25% increase in GnRH-1 neuronal number during embryonic development was found. Bromodeoxyuridine birth tracing and spatiotemporal tracking of GnRH-1 cell precursors demonstrated that the lack of the N-terminal portion of FE65, which includes part of the functional nuclear translocation/gene transcription domain of FE65 (WW domain), extends the timing of GnRH-1 neurogenesis in the developing nasal placode without affecting proliferation of GnRH-1 neuronal progenitors or cell death. The observed changes in the dynamics of GnRH-1 neurogenesis highlight a unique role for the 97 kDa isoform of FE65 and suggest that GnRH-1 cells, which have a short neurogenic window, originate from multipotent progenitors able to generate distinct cell types as GnRH-1 neurogenesis declines in response to environmental changes.
Introduction
Gonadotropin releasing hormone-1 (GnRH-1) is essential for vertebrate reproduction. Secreted by forebrain GnRH-1 neurons, GnRH-1 binds to receptors on pituitary gonadotropes to regulate reproduction via the hypothalamic–pituitary–gonadal axis. In mammals, GnRH-1 neurons are first detected in the vomeronasal organ (VNO) anlage and thereafter migrate along vomeronasal/olfactory axons into the forebrain during prenatal development (Wray, 2002). Alterations in normal GnRH-1 development can lead to reproductive dysfunction, e.g., Kallman patients exhibiting hypogonadotropic hypogonadism (Kallmann, 1944).
Gene expression screenings of GnRH-1 neurons (Kramer and Wray, 2001) identified the amyloid precursor protein (APP) binding-protein-1 (FE65) gene. This multimodular adaptor protein is expressed in neurons in two isoforms: 97 and 60 kDa (Wang et al., 2004). The 97 kDa FE65 contains a tryptophan, tryptophan (WW) domain, and two phospho-tyrosine binding (PTB) domains. The WW domain is necessary for FE65 nuclear translocation and mediated gene transcription (Telese et al., 2005). The 60 kDa protein, which originates from an alternative start codon, has a truncated WW domain and cannot activate gene expression (Wang et al., 2004). FE65 can modulate gene expression alone (Wiley et al., 2007), as a FE65/Tip60/APP intracellular domain (AICD) transcriptional complex (Cao and Südhof, 2001) or in association with the Notch intracellular domain (NICD), (Fischer et al., 2005). In addition, FE65 modulates APP trafficking, processing, and proteolysis (King and Scott Turner, 2004). FE65, in association with APP, can regulate β1-integrin dynamics, actin cytoskeleton, and cell motility (Sabo et al., 2001, 2003; Standen et al., 2003). A role for FE65/APP signaling has been described in neuro/glial cell fate determination (Chen and Dong, 2009), and increased neurogenesis has been reported in cortical neuronal progenitor cell cultures from APP−/−, TAG1−/− (a ligand of APP), and FE65−/− 97 kDa isoform-specific knock-out (KO) mice (Ma et al., 2008b). To date, no corroborating in vivo observations have been reported.
FE65 is regulated during embryonic development in neural structures (Simeone et al., 1994; Kesavapany et al., 2002). In mouse, FE65 mRNA is expressed, at approximately mid-gestation, in the neural tube basal plate, neural crest, acoustic ganglion, sympathetic ganglion, and nasal placode (Simeone et al., 1994). The latter region is where GnRH-1 neurons are first detected. The GnRH-1 system in mice comprises a small number of neurons whose migration into the CNS has been well characterized. Thus, the role of FE65 in GnRH-1 neuronal development in both neurogenesis and migration was examined.
Analysis of two mouse lines, one deficient for the 97 kDa isoform that retains a truncated FE65 60 kDa protein and the other deficient for both FE65 isoforms, showed no changes in GnRH-1 neuronal migration. However, a 25% increase in total GnRH-1 cell number during embryonic development was found. Analysis of early events in development of GnRH-1 neurons indicated that neurogenesis of specific progenitor cells in the VNO anlage increased in the absence of the fully functional WW domain of FE65. These data highlight a unique role for the 97 kDa isoform in controlling GnRH-1 neurogenesis that is not redundant with the 60 kDa isoform of FE65.
Materials and Methods
Animals
FE65 mutant mouse strains.
p97FE65 (C57BL/6) and p97/60FE65 (backcrossed four times into C57BL/6 background) were provided by Drs. G. M. Martin (University of Washington, Seattle, WA) and S. Guénette, (Massachusetts General Institute for Neurodegenerative Disease, Boston, MA), respectively. p97FE65 and p97/60FE65 null and controls were generated by time-mated heterozygous crosses. Because no differences for the described phenotype have been observed between WT and heterozygous mice, heterozygous mice have been included in control groups when needed. Mice were harvested from embryonic day (E) 11.5 (plug day, E0.5) to adult. All mice were killed in accordance with the National Institutes of Health (NIH)/National Institute of Neurological Stroke and Disorders (NINDS) guidelines.
Bromodeoxyuridine treatment.
Time-mated pregnant females (NIH Swiss or P97FE65) were injected intraperitoneally with bromodeoxyuridine (BrdU) (Sigma-Aldrich) at 50 g/kg in saline solution (0.9% NaCl2 in sterile H2O). Single or multiple injections were performed depending on the experimental plan, and embryos were harvested between 24 and 96 h after injection. All procedures were approved by the NINDS Animal Care and Use Committee and performed in accordance with NIH guidelines.
Tissues.
Whole embryos (E12.5–E14.5), dissected head [E17.5 and postnatal day 0 (P0)], or brain (adult) were immediately frozen on dry ice and stored at −80°C until sectioning. E11.5 mice were fixed in 4% formaldehyde for 3 h, washed in PBS, cryoprotected in 30% sucrose/PBS overnight, transferred to Tissue-Tek OCT compound (Sakura Finetek), frozen, and stored at −80°C until sectioning (see below).
PCR on single GnRH-1 cells from nasal explants
Nasal explants were cultured as described previously (Fueshko and Wray, 1994). Briefly, embryos were obtained from timed-pregnant NIH Swiss mice in accordance with NIH guidelines. Bilateral olfactory pits were dissected, trimmed, and adhered onto coverslips by a plasma (Cocalico Biologicals)/thrombin (Sigma-Aldrich) clot. Explants were maintained in defined serum-free medium (SFM) (Fueshko and Wray, 1994) at 37°C with 5% CO2. On culture day 3, fresh media containing fluorodeoxyuridine (8 × 10−5 m; Sigma) was given to inhibit proliferation of dividing olfactory neurons and non-neuronal explant tissue. On culture day 6, the media was changed with fresh SFM. cDNA was extracted and PCR amplified at 3.5, 4.5, 6, and 7 d in vitro (DIV) (five single GnRH-1 cells/DIV) (Kramer and Wray, 2000; Sharifi et al., 2002). All cDNA pools were initially screened by PCR for GnRH-1 (to ensure the correct cell phenotype) and β-tubulin and L19, two housekeeping genes (microtubule and ribosomal, respectively; see primer sequences in the study by Giacobini et al., 2004). Only GnRH-1 cells positive for all three transcripts were used here. FE65 primers were designed from published GenBank sequences and screened using BLAST (basic local alignment search tool) to ensure specificity of binding. The FE65 primers used were as follows: forward primer, 5′-GCTCCCAGACCTCCACCTCC-3′; reverse primer, 5′-GGAAGAACATCCACAGCCCC-3′. These primers were pretested on brain cDNA to determine optimal concentration and temperature conditions. For each reaction, 30.5 μl of nuclease free H2O, 5 μl of 10× PCR GOLD buffer (Applied Biosystems), 4 μl of 25 mm MgCl2, 5 μl of dNTP mix (2.5 mm), and 0.5 μl of Amplitaq Gold (Applied Biosystems) were mixed. Primers (125 nm) and template cDNA (1 μl) were added to the mixture. The PCR program was as follows: 10 min at 94°C pre-run, 30 s at 94°C, 30 s at 64°C, 2 min at 72°C for 40 cycles, and 10 min at 72°C post-run. Amplified products were run on a 1.5% agarose gel.
Reverse transcription-PCR of nasal region
Total RNA extraction of nasal tissue from E11.5–E17.5 mice, whole embryo at E17.5, and adult brain samples was conducted using RNA STAT-60 (Tel-Test) following the protocol of the manufacturer. Briefly, tissue was homogenized (1 ml of RNA STAT-60 per 50–100 mg of tissue), chloroform was added (0.2 ml/ml homogenate), and the mixture was spun. Isopropanol (0.5 ml) was added to the aqueous layer to precipitate RNA. RNA pellet was washed (75% ethanol), air dried, and resuspended in DEPC-treated water. One nanogram of each sample was used for reverse transcription (RT)-PCR reaction using the Access-Quick RT-PCR System (Promega). The reaction was performed at 48°C for 45 min, 94°C for 2 min, 40 cycles of 94°C for 30 s, 64°C for 1 min, 68°C for 2 min, and final extension at 68°C for 7 min. One round of PCR was conducted, and the PCR products were resolved on a 1.5% agarose gel by electrophoresis.
FE65 RT-PCR of laser capture microdissected tissue
Laser microdissection.
Fresh frozen embryos at E11.8 were cryosectioned (12 μm) and placed alternately on standard subbed glass slides or on 0.17 mm, 1.35 μm naphthalate polyethylene slides (PEN; P.A.L.M. Microcur Technologies). Laser capture microdissection (LCM) was performed at 20× magnification using a PALM LCM system (Carl Zeiss).
RT-PCR.
The LCM tissues were popped into a sterile Microfuge cap containing 10 μl of 0.1% Triton X-100 and DEPC H2O. After centrifugation, RNase inhibitor (RNaseOut; Invitrogen) was added. First strand was prepared as described previously (Giacobini et al., 2004). Transcript analysis was performed by PCR on two samples per genotype (40 cycles) using the following sequences: FE65 exon 2 forward primer, 5′-CTCGGCCCACAATGCAGCCA-3′; FE65 exon 2 reverse primer, 5′-CCGGGGAGCCATAGGAGGGG-3′; FE65 exon 7 forward primer, 5′-CCCCAGGACGCAGCAGTGTG-3′; FE65 exon 10 reverse primer, 5′-GCGAGCATTGCGCCGTTCAG-3′; FE65 exon 13 forward primer, 5′-GCTCCCAGACCTCCACCTCC-3′; FE65 exon 14 reverse primer, 5′-GGAAGAACATCCACAGCCCC-3′; glyceraldehyde-3-phosphate dehydrogenase (GAPDH) forward primer, 5′ GTCATCATCTCCGCCCCTTC-3′; GAPDH reverse primer, 5′-ATCCACGACGACGGACACATTGG-3′. PCR products were resolved on a 1.5% agarose gel by electrophoresis.
Western blot
Nasal tissue was removed from E12.5, E14.5, E17.5, and P1 mice and olfactory bulbs from adult brain, homogenized in SDS buffer (2% in H2O) supplemented with protease inhibitor cocktail tablets (Roche Molecular Biochemicals, Indianapolis, IN), mixed, and heated at 95°C for 5 min. The lysate was then centrifuged, and total protein concentration of the supernatants was determined using BCA protein assay kit (Pierce) and analyzed using a spectrophotometer (SmartSpec 3000; Bio-Rad). Conditions for SDS-PAGE and Western blotting were as described previously (Kramer and Wray, 2000). The membrane was blocked with 10% dried milk diluted in TTBS (20 mm Tris-HCl, 150 mm NaCl, and 0.2% Tween 20) at pH 7.4 for 1 h. The polyclonal antibody against FE65 (kindly donated by Dr. N. Zambrano, University Federico II, Naples, Italy) was added and incubated at 4°C overnight. The membrane was washed in TTBS, and an HRP-goat anti-rabbit secondary antibody (1:5000; Bio-Rad) was added. After incubation (2 h) and washing at room temperature, the complex was reacted using the Western blot chemiluminescence reagent plus kit (PerkinElmer Life and Analytical Sciences, DuPont) according to the directions by the manufacturer and exposed to film.
Immunocytochemistry
Fresh-frozen samples were cut in serial sections (16 μm) using a Leica CM 3050S cryostat and maintained at −80°C until processing. Three serial series were generated for E12.5 and E13.5, four series for E14.5 embryos, five series for E15.5 embryos, and six series for E17.5 and postnatal animals. Fixed embryos were cut at 14 μm into two or three serial series, depending on the experimental needs.
Primary antibodies.
GnRH-1 [SW-1; 1:3000 (Wray et al., 1988)], Rb polyclonal anti-FE65 (1:1000; Sigma-Aldrich), Rb polyclonal anti-FE65 1 (1:4000; provided by Dr. N. Zambrano), Rb polyclonal anti-peripherin (peripheral intermediate filament marker; 1:2000; Millipore Bioscience Research Reagents), Rb anti-Ki67 (marker of mitotic activity; 1:2000; Novo Castra Laboratories), Rb anti-caspase-3 active form (1:3000; BD Biosciences), rat anti-BrdU (1:1000; Accurate Chemical & Scientific), mouse monoclonal biotinylated anti-HuC-D (1:100; Invitrogen), goat polyclonal anti-Sox2 (1:800; Santa Cruz Biotechnology), Rb polyclonal anti-Sox2 (1:500; Millipore Bioscience Research Reagents), goat polyclonal anti-Notch-3 (1:400; R & D Systems), Rb polyclonal anti-APP G-369 (kind gift from Dr. C. Thomas, Jefferson University, Philadelphia, PA), anti-Nestin Rb polyconal (1:3000; kind gift from Dr. R. McKay, National Institutes of Health, Bethesda, MD), and Rb polyclonal anti-phospho-histone H3 (1:300; Millipore Bioscience Research Reagents).
Chromogen-based reactions.
Tissue was stained as described previously (Fueshko and Wray, 1994). Briefly, after fixation (4% formaldehyde), sections were washed in PBS, blocked in 10% normal horse or goat serum depending on experimental needs/0.3% Triton X-100, and then washed in PBS. Sections were incubated in primary antibody (4°C, overnight). The next day, tissues were washed (PBS) and incubated for 1 h with 1:500 biotinylated secondary antibody in PBS and 0.3% Triton X-100. The following secondary antibodies were used: goat anti-rabbit biotinylated (Vector Laboratories), goat anti-mouse biotinylated (Millipore Bioscience Research Reagents), and goat anti-rat Sp-biotinylated (Jackson ImmunoResearch). After PBS washes, the tissue was processed using a standard avidin–biotin–horseradish peroxidase/3′,3-diaminobenzidine (DAB) protocol. For most double labeling, DAB (a brown product) was used as the first chromogen and SG substrate (a blue product; Vector Laboratories). For GnRH-1/BrdU immunostaining, sections were first immunolabeled for GnRH-1 as described above, and after visualization with DAB, sections were fixed in 4% formaldehyde (15 min), incubated in 2N HCl/PBS (35 min at 37°C) (Forni et al., 2006), washed in PBS, and then incubated with anti-BrdU rat antibody (2 h at room temperature). After biotinylated secondary antibody incubation, followed by avidin–biotin–horseradish peroxidase incubation, nickel-intensified DAB (black) was used as the second chromogen. Stained sections were counterstained with methyl green, air dried, and coverslipped with Permount (Thermo Fisher Scientific). For Ki67 immunostaining, sections underwent microwave antigen retrieval treatment in citrate buffer (Forni et al., 2006). Pictures were taken on an Eclipse E800 microscope (Nikon) with a Retiga SRV camera (QImaging) using QCapture software (QImaging).
Immunofluorescence labeling.
Sections were processed as described above. Depending on the primary antibody used, sections were incubated for 45 min with the following secondary antibodies: goat anti-rabbit 488 (1:1000), goat anti-rabbit 555 (1:1000), donkey anti-rabbit 488 (1:1000), donkey anti-goat 568 (1:1000), goat anti-mouse 488 (1:1000), goat anti-mouse 555 (1:1000), and goat anti-rat cyanine 3 (1:1000) (all from Invitrogen). Sections were counterstained with 4′,6′-diamidino-2-phenylindole (1:3000; Sigma-Aldrich), rinsed in deionized water, and coverslipped with Fluoro Gel (Electron Microscopy Services). Confocal pictures were taken on a spinning disk confocal system CSU10 (Yocogawa) mounted on an Eclipse TSE200 microscope (Nikon) using an EMCCD ImageM digital camera (Hamamatsu) with I-Vision software (Biovision). Nonconfocal fluorescent pictures were taken on a Nikon Eclipse E800 with a Retiga SRV camera using QCapture software. Images were further analyzed using NIH ImageJ software (Wayne Rasband, http://rsbweb.nih.gov/ij/).
Cell counts
Single GnRH-1 cells or double-labeled GnRH-1/BrdU cells.
Cells were counted at 20× on a bright-field microscopy after immunolabeling. Analysis of GnRH-1 neuronal distribution was performed in the (1) nasal region (VNO, axonal tracks surrounding the olfactory pits, forebrain junction) and (2) brain (all the cells that accessed the olfactory bulb and were distributed within the forebrain). Number of cells per region and total cell number were calculated for each animal as the average of the counted cells per series multiplied by the number of series cut per animal (on average, at least half of the series were stained and counted per labeling).
Other counts.
Hu, BrdU, phospho-histone H3, and cleaved caspase-3-positive cell counts were performed on three serial sections per side for each VNO at E11.5. The total number of cells per animal (anlage per animal) was calculated as the average of the cells counted in the sections of the two pits multiplied by 2 (right and left pit), by the number of sections (three), and by the number of series. Counts of migratory GnRH-1 and migratory Hu cells were performed analyzing 9900 μm2 fields on four sections for right and left pit per animal. Counts were manually performed on digital images (20×) using NIH ImageJ 1.42b software (Wayne Rasband, http://rsbweb.nih.gov/ij) cell count plugin (http://rsbweb.nih.gov/ij/plugins/cell-counter.html). Means ± SEs were calculated on at least three animals per genotype. A Student's t test or ANOVA was used to assess differences among and between groups.
Results
FE65 is expressed by migrating GnRH-1 cells
GnRH-1 neurons maintained in nasal explants exhibit many characteristics displayed by GnRH-1 neurons in vivo (Wray, 2002). In this model system, GnRH-1 neurons migrate from the nasal pit into the periphery of the explant (Fig. 1A,B) and can be identified in situ (Kusano et al., 1995). Identification of GnRH-1 neurons in vitro has allowed single GnRH-1 neurons to be removed from explants and cDNA pools generated and then screened for GnRH-1 (correct cell phenotype), β-tubulin, and L19 by PCR (Kramer and Wray, 2000; Sharifi et al., 2002). Individual GnRH-1 cells were examined for FE65 transcript (Fig. 1C). A band of the correct size for FE65 (226 bp; arrow) was found in all tested cells. Double-labeled immunocytochemistry on explants demonstrated coexpression of FE65 in GnRH-1 neurons (Fig. 1D). FE65 expression was then evaluated in nasal regions at different developmental stages in vivo. Between E12.5 and E14.5, GnRH-1 cells form a continuum in the nasal region as they migrate along axonal bundles into the forebrain (Fig. 1E–G) with the majority of GnRH-1 cells in the brain by E16.5 (Wray et al., 1989b). A specific product of the correct size for FE65 mRNA (226 bp; arrow) was found in nasal regions from E11.5 to E17.5 (Fig. 1H). FE65 protein expression in the nasal region from E12.5 to E17.5 was confirmed by both Western blot analysis (Fig. 1I) and immunocytochemistry (Fig. 1J,K). At E12.5, FE65 was detected in cells of the developing VNO (Fig. 1J) and in migratory cells (Fig. 1K). Double immunofluorescence verified FE65 expression in GnRH-1 cells crossing the nasal region (Fig. 1L). Based on the expression of FE65 in migratory GnRH-1 cells in vitro and in vivo, we hypothesized that FE65 might play a role in the movement of GnRH-1 neurons into the brain.
Figure 1.
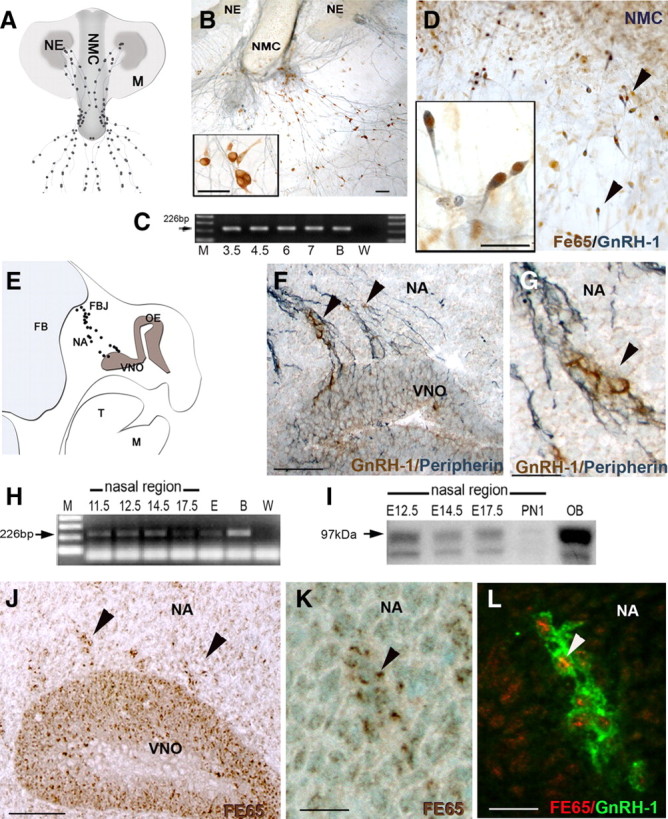
GnRH-1 neurons express FE65. A, Schematic of a nasal explant (NE, nasal pit epithelium; NMC, nasal midline cartilage; M, surrounding mesenchyme). GnRH-1 neurons (black dots) emerge from nasal pit epithelium and follow olfactory axons to the midline and then into the periphery of the explant. B, Photomicrograph of nasal explant immunocytochemically stained for GnRH-1 (brown) and peripherin (blue). C, Representative gel of FE65 PCR products from single GnRH-1 cell cDNAs at 3.5–7 DIV. FE65 transcript was detected in all cells (M, marker; B, brain; W, water). D, GnRH-1 cells (blue) express FE65 (brown) in nasal explants. E, Schematic of an E12.5 parasagittal section showing location of GnRH-1 neurons (black dots) extending from the VNO to the nasal forebrain junction (NFJ) (OE, olfactory epithelium; FB, forebrain; T, tongue; M, mandible; NA, nasal area). F, G, Photomicrograph of GnRH-1 neurons (brown) migrating out of the VNO along peripherin (blue)-positive axon bundles in vivo (E12.5). H, A band of expected size for FE65 transcript (226 bp) was detected in total RNA isolated from E11.5–E17.5 noses (E, 17.5 whole embryo; B, brain; W, water). I, Western blot analysis of nasal areas at E12.5, E14.5, E17.5, and P1 revealed a 97 kDa specific band (arrow), with a strong band detected at E12.5, which thereafter decreased as a function of age (OB, olfactory bulb; lower band degradation product). J, K, FE65 (brown) is expressed in cells in the VNO (J) and in cells (arrowheads) emerging from the VNO. L, GnRH-1 neurons (green) express FE65 (red) while migrating in the nasal area. Scale bars: B, F, J, 100 μm; insets in B, D, 25 μm; G, K, L, 25 μm.
GnRH-1 neuronal development in p97FE65 hypomorphic mice
To test the hypothesis that FE65 has a role in GnRH-1 neuronal migration, a line of FE65 hypomorph mice missing the 97 kDa form of FE65 (p97FE65 KO) was analyzed. This line was chosen because reports indicated that the 97 kDa protein was the main isoform expressed during neuronal development (Hu et al., 1999; Wang et al., 2004; Ma et al., 2008b). Sections from p97FE65 KO and control littermates, at multiple stages (E12.5 to P0), were immunocytochemically stained for GnRH-1 (Fig. 2A–D). To determine whether changes in movement of the GnRH-1 cells occurred in the absence of p97FE65, a spatiotemporal analysis was performed with GnRH-1 cells counted as a function of location (nose vs forebrain) over embryonic development.
Figure 2.

p97FE65 KOs have an increased number of GnRH-1 cells. A–D, Comparable serial sections of E14.5 WT (A, C) and FE65 mutants (B, D) immunostained for GnRH-1 (brown). The majority of GnRH-1 neurons are distributed between nasal area (NA) and forebrain junction (FBJ), with a smaller group of cells detectable in the preoptic area (POA). In FE65 mutants (B, D), larger groups of GnRH-1 cells were detected in the NA and FBJ. E, Brain region. Similar number of GnRH-1 cells migrated into the brain per day. No significant differences were detected in the number of GnRH-1 neurons accessing the forebrain between genotypes at any examined developmental stage. F, Nasal area. After E14.5, the number of GnRH-1 neurons in the p97FE65 KO was significantly higher than in control animals. Between E13.5 and E15.5, the number of GnRH-1 cells in the nasal area decreased less in KOs compared with controls, despite comparable number of cells that migrated in the brain area (E). G, Total number of GnRH-1 cells increased in FE65 mutants. Total GnRH-1 cell number was determined in serial sections of control and FE65 KO animals at the indicated developmental stages. Between E14.5 and E17.5, the number of GnRH-1 cells was significantly higher in KOs compared with control animals. In E–G, number in bars = animals, mean values ± SE; *p < 0.05, t test. Scale bars: A (for A, B), C (for C, D), 100 μm.
In both p97FE65 KO and controls, only a few GnRH-1 cells were detected in the brain at E12.5 (Fig. 2E). From E13.5 to E17.5, the number of GnRH-1 cells in the brain increased in both genotypes. A decrease in GnRH-1 cell number in the brain, associated with birth, was also detected in both genotypes. A simple regression plot indicated that similar changes occurred in the wild type (WT) and KO during development (r2 = 0.994), consistent with appropriate cell movement into the developing forebrain. In control mice, as expected, the number of GnRH-1 cells in nasal regions decreased as a function of age (Fig. 2F). In contrast, the KO showed no consistent reduction in the number of GnRH-1 cells in the nasal region between E12.5 and E14.5 (Fig. 2F). After E14.5, the changes detected in GnRH-1 cells, both the gain in brain areas and decrease in nasal areas, were similar in WT and KO mice. However, throughout the period examined, the number of GnRH-1 cells in nasal areas was significantly greater in KO versus WT mice (p < 0.05). The increase found in nasal regions correlated with the age of the mice (E12.5, 16%; E13.5, 19%; E14.5, 39%; E15.5, 44%; E17.5, 104%; P0, 70%). From E14.5 to E17.5, there were >200 more GnRH-1 cells in the nasal area of KO mice compared with controls.
Increase in total number of GnRH-1 cells during embryogenesis
The number of GnRH-1-expressing cells in C57BL/6 (background of the analyzed mouse lines) undergoes a small increase between E13.5 and E14.5 (∼90 cells; p = 0.3) (Fig. 2G) as the full complement of GnRH-1 cells express detectable levels of protein after the end of neurogenesis. As described by others (Krüger et al., 2004; Miller et al., 2009) from E14.5 to E17.5, a plateau in the total GnRH-1 cell number was reached (1050 ± 50). In the KO mice, the total number of GnRH-1 cells detected between E12.5 and 17.5 was significantly greater than WT (two-way ANOVA, p < 0.01). At E13.5, the total number of GnRH-1 neurons was ∼21% higher in the KO (Fig. 2G) (WT, 958 ± 92; KO, 1159 ± 84). From E14.5 to P0, the difference in GnRH-1 cell number between FE65 mutants and controls reached 30% (Fig. 2G) (E14.5: WT, 1046 ± 41 and KO, 1385 ± 39; E15.5: WT, 1015 ± 101 and KO, 1307 ± 27; E17.5: WT, 1109 ± 87 and KO, 1446 ± 59; P0: WT, 739 ± 32 and KO, 1013 ± 143). In contrast to the GnRH-1 cell number in WT, the GnRH-1 cell number in KOs significantly increased between E13.5 and E14.5 (Fig. 2G) (p = 0.02). As in the controls, a plateau in GnRH-1 cell number occurred from E14.5 to E17.5. However, the total GnRH-1 cell number was higher in KOs, stabilizing around 1385 ± 28. These data indicate that, between E13.5 and E14.5, while GnRH-1 cells are migrating into the brain, a higher number of GnRH-1 cells matured in the nasal regions of the KO, accounting for the increase in total GnRH-1 cell number (Fig. 2F).
P97FE65 mutants express alternative FE65 isoform in the developing VNO and in migratory neurons
Ablation of the second exon, which contains the start codon for p97FE65, in p97FE65 KOs can lead to compensatory expression of a shorter isoform of FE65 (Wang et al., 2004). Characterization of this isoform revealed that it originates from an alternative start codon in the third exon of the FE65 gene and codes for a smaller FE65 protein (p60FE65) that retains a truncated WW domain but functional PTB domains (Wang et al., 2004). To test whether compensatory expression of p60FE65 could occur at early developmental stages, RT-PCR was performed on brain and VNOs removed from E11.8 WT and p97FE65 mutant mice using laser capture microdissection (Fig. 3A–C), (Giacobini et al., 2004). Using primers for exon 2, exons 7–10 and exons 13–14, products from the N- to C-terminal portion of p97FE65, were amplified (Fig. 3A). All p97FE65 PCR products were found in cDNA from control tissues (Fig. 3D). In the p97FE65 KO samples, amplification products were found for exons 7–10 and 13–14 (Fig. 3E), indicating that these mice express the shorter isoforms (Guénette et al., 2006) of FE65 in brain and VNO. Immunostaining with a FE65 antibody that recognizes amino acids 352–369, a portion of protein conserved in p97 and p60FE65 (Wang et al., 2004), confirmed protein expression of p60FE65 in the developing VNO and in migratory neurons exiting the VNO of p97FE65 mutants (Fig. 3F,G). In contrast, only background staining was detected in sections from p97/p60FE65 Kos, a mouse line missing both 97 and 60 kDa isoforms of FE65 (Fig. 3H) (Wang et al., 2004; Guénette et al., 2006).
Figure 3.
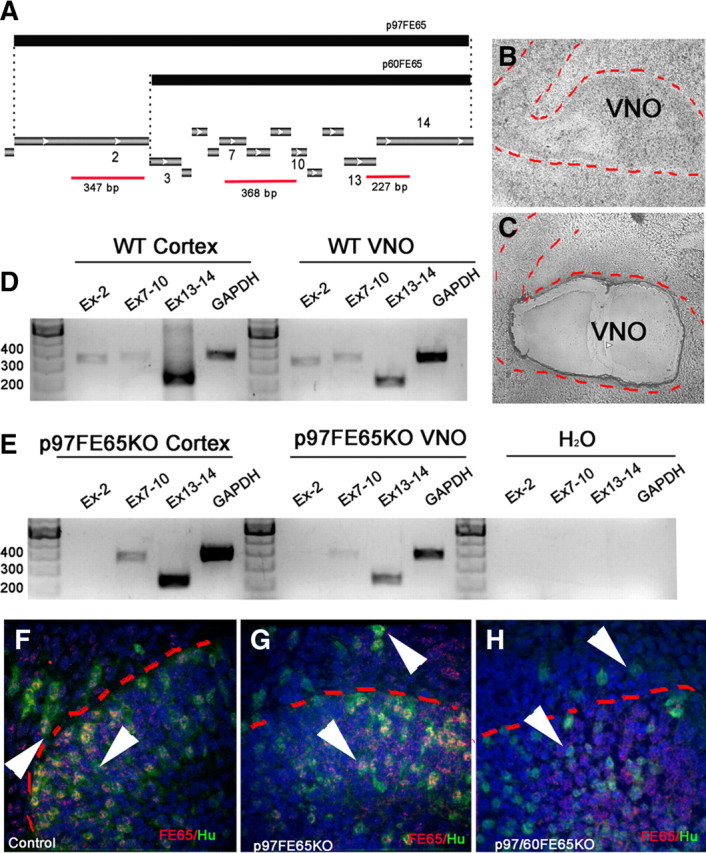
p97FE65 mice express alternative isoform of FE65 in the developing pit. A, Schematic illustrating products used to differentiate between p97 and p60 isoforms of FE65. The p97 isoform of FE65 originates from a start codon in exon 2. The truncated p60FE65 form originates from an alternative start codon in exon 3. Different regions of FE65 analyzed by RT-PCR are represented in red (EX-2, EX7–10, EX13–14) together with the expected length. B, C, Phase-contrast picture of VNO (indicated with red dashed line) before (B) and after (C) laser capture microdissection. D, E, PCR products obtained from cDNA of laser captured cortex and VNO from WT (D) and p97FE65 embryos. In the mutant material, no amplification of exon 2 was found, but bands were detected for exons 7–10 and 13–14, indicating expression of the alternative forms of FE65. F–H, Double immunofluorescence for FE65 (red) and pan-neuronal marker Hu (green) in WT (F), p97FE65 KO (G), and p97/60FE65 KO (H) E14.5 sections through the VNO. FE65 expression was found in newly formed neurons (arrowheads) in WT (F) and p97FE65 KO (G), whereas only background staining was observed in p97/60FE65 KO (H).
The p97/60FE65 KO displays an overlapping phenotype to the p97FE65 hypomorph
To determine whether the increase in GnRH-1 cell number found in the absence of the 97 kDa FE65 isoform could be exacerbated by the absence of both p97 and p60FE65 isoforms and to exclude an effect attributable to variation of expression of the 60 kDa isoform, GnRH-1 cells were counted in E14.5 p97/60FE65 KO mice, a line missing expression of both isoforms (Guénette et al., 2006). GnRH-1 cell counts revealed no additional effects in the absence of both FE65 isoforms. In fact, as observed in the p97FE65 KO at E14.5, a significant increase in the number of cells was detected in the nasal area (Fig. 4) (WT, 710 ± 70; KO, 953 ± 53), whereas no differences were detected in the number of GnRH-1 cells in the brain (Fig. 4) (WT, 285 ± 67; KO, 254 ± 54). Although the total GnRH-1 cell number in the second mouse line (background C57BL/6 N5) was lower for both controls and KO (WT, 994 ± 37; KO, 1207 ± 76) than that observed in the 97 kDa hypomorphs (background C57BL/6), the increase in cell number in the nasal area in both FE65 mutant lines was similar (∼39% in p97FE65 KO vs ∼34% in p97/60FE65 KO mice).
Figure 4.

p97/60FE65 KOs display a similar increase in GnRH-1 cell number and cell distribution as p97FE65 KOs. A, p97/60FE65 KO. At E14.5, no significant difference was found in the number of GnRH-1 neurons in brain, as observed in p97FE65 hypomorphs (B). However, more GnRH-1 cells were detected in p97/60FE65 KOs between the nasal area and forebrain junction (NA/FBJ) compared with controls (A), with a similar increase to that observed in p97FE65 mutants at the same age (B). n = animals, mean values ± SE. *p < 0.05, t test.
The data from the FE65 KO lines indicated that (1) GnRH-1 cells show spatiotemporal migration into the forebrain similar to controls, (2) the number of GnRH-1 cells able to enter the brain is limited (at the level of the forebrain junction) to a specific range per day, and (3) changes in early developmental events occur in the absence of fully functional FE65, leading to an increase in GnRH-1 cell number.
Prolonged GnRH-1 neurogenesis in the absence of p97FE65
Previous experiments showed that the GnRH-1 neurogenic peak occurs between E9.5 and E10.5, when ∼70% of the GnRH-1 neurons in a postnatal brain form (Wray et al., 1989a; Jasoni et al., 2009). BrdU incorporation at different embryonic stages was performed to evaluate whether the increased number of GnRH-1 neurons detected in the FE65 KO after E14.5 was the result of a higher number of initial progenitors or a delayed exit from the proliferative/neurogenic program (Fig. 5). One group of pregnant females received three BrdU pulses (50 μg/g) every 12 h around the peak of GnRH-1 neurogenesis (E9.5, E10, and E10.5). A second group was injected during the declining phase of GnRH-1 neurogenesis (BrdU injection at E11, E11.5, and E12). Controls and KO littermates were harvested and analyzed 3.5 d after the last BrdU injection.
Figure 5.

Increased GnRH-1 neurogenesis in FE65 mutants. A–D, Representative embryonic sections after BrdU injections and staining for GnRH-1 (brown) and BrdU (black). Double-labeled cells are indicated by arrowheads. A, B, E14.5 section of nasal area (NA)/forebrain junction (FBJ) after BrdU incorporation (50 μg/g) at E9.5, E10, and E10.5. C, D, E15.5 section after BrdU incorporations at E11, E11.5, and E12. E, Mean GnRH-1-positive/BrdU-positive cell number in control and FE65 mutants after BrdU incorporation at the indicated developmental stages; values ± SE. *p ≤ 0.05, t test. Scale bars: A, 100 μm; B, 20 μm.
In the first group (BrdU injections at E9.5–E10.5), independent of genotype, ∼40% of the total GnRH-1 neuronal population detected at E14.5 was labeled. Thus, in littermate KO mice, a significant increase in the number of BrdU-positive GnRH-1 cells was found (+25%), consistent with the increase in total cell number found in KOs at E13.5 and E14.5 (Fig. 5). In the second group (BrdU injections at E11–E12), ∼18% of the total GnRH-1 neuronal population at E15.5 was labeled. Counts of double-labeled GnRH-1/BrdU cells revealed ∼40% higher number of newly formed GnRH-1 neurons in FE65 KOs compared with control littermates. The increase (40%) in GnRH-1/BrdU-labeled cells observed in mice injected in the terminal phases of GnRH-1 neurogenesis (E11–E12) compared with the increase (25%) observed in the early phases (E 9.5–E10) suggests an active role for FE65 in controlling the exit from the neurogenic program.
To confirm that the increased number of GnRH-1 neurons detected in the FE65 KO was the result of a delayed exit from the proliferative/neurogenic program, several in vivo parameters were needed to examine early phases of GnRH-1 development in FE65 KO mice. These parameters included (1) finding an early marker for GnRH-1 neurons, (2) characterizing the relation between FE65 expression and GnRH-1 neuronal development, (3) identifying the proliferative cells in the VNO, and (4) establishing that GnRH-1 neurons arose from these proliferative VNO cells.
Hu marks early GnRH-1 cells in the developing VNO
The relation between FE65 expression and GnRH-1 neuronal development needed to be analyzed at early stages in vivo. In mice, E11.5 coincides with neurogenesis of GnRH-1 cells (Wray et al., 1989a). At this stage, few (∼150) GnRH-1 cells were detected (Fig. 6A). However, neuronal precursors positive for the early pan-neuronal RNA binding protein Hu were present (Fig. 6B) (Lendahl et al., 1990; Zimmerman et al., 1994; Graham et al., 2003). Hu-positive cells were found in the anlage of the VNO as well as in a continuum toward the brain, appearing in the route taken by GnRH-1 neurons (Fig. 6C). Double-label immunocytochemistry at E11.5 revealed that the very first cells with detectable levels of GnRH-1 protein were positive for Hu and were found both within the VNO (Fig. 6D–G) as well as along the migration path (Fig. 6H). Hu expression in GnRH-1 cells varied as the cells matured/migrated toward the forebrain as described previously for GnRH-1 neurons in chicken (Fornaro et al., 2003) and for other neuronal cell types (Okano and Darnell, 1997; Deschênes-Furry et al., 2006). Analysis at E12.5, the stage when the number of GnRH-1-expressing cells increases (∼800 cells), revealed large groups of Hu-positive cells and Hu/GnRH-1-positive cells distributed between the VNO and the forebrain (Fig. 6I,J). These observations indicated that Hu is expressed, among other migratory neurons (Fig. 6H,J), in GnRH-1 precursors in the VNO and in maturing GnRH-1-positive neurons.
Figure 6.
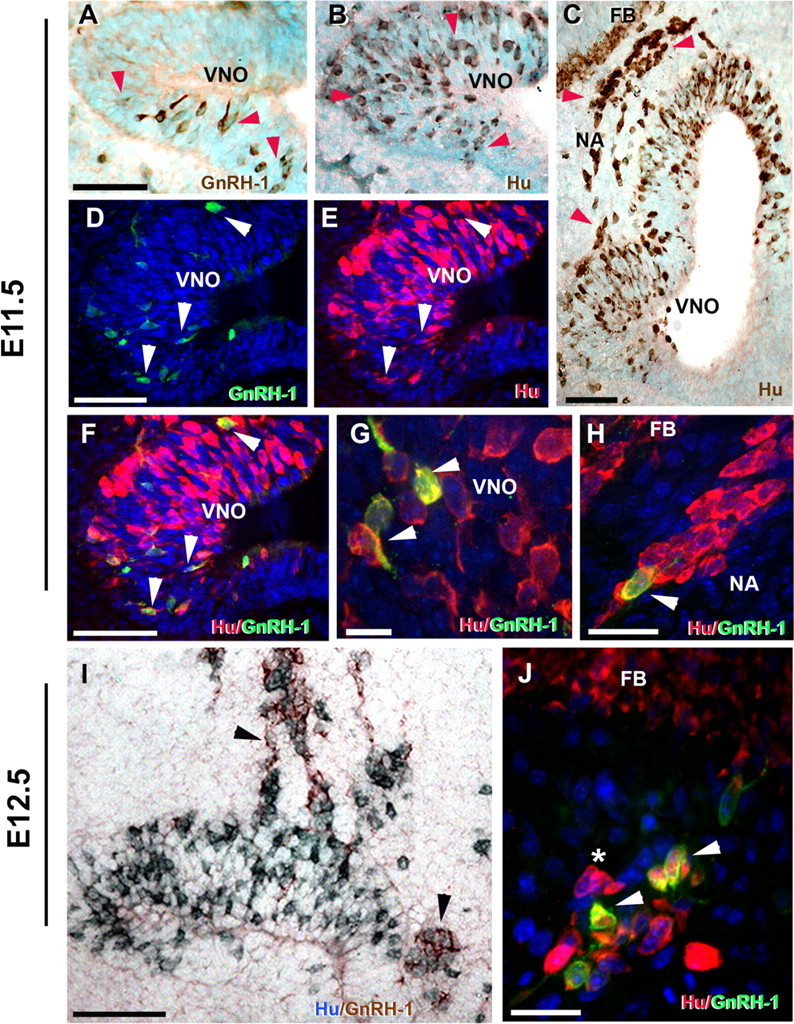
GnRH-1 cells emerge from the developing vomeronasal region as Hu-positive cells and increase GnRH-1 levels while migrating toward the forebrain junction (FBJ). Immunohistochemistry for GnRH-1 (A) and RNA binding protein Hu (B, C) on sections of the developing VNO at E11.5 show few GnRH-1-expressing neurons detectable in the VNO (A, red arrowheads). In contrast, large numbers of neuronal precursors (Hu positive) were present both within the VNO (B, red arrowhead) and in the nasal region (NA) extending from the VNO toward the forebrain (FB) (C). D–H, Double immunofluorescence for GnRH-1 (green) and Hu (red). D–F, In the developing VNO at E11.5, a few Hu-positive neuronal precursors start to express GnRH-1 (arrowheads in D, E; merge in F). G, High magnification (100×) within the VNO anlage showing Hu-positive/GnRH-1-positive GnRH-1 neuron precursors (arrowheads). H, Hu-positive cells (red) migrating toward the forebrain (see C for localization), Hu/GnRH-1 double-positive maturing GnRH-1 neuron (arrowhead). I, Double immunocytochemistry for Hu (blue) and GnRH-1 (brown) at E12.5 showed large groups of Hu-positive and maturating Hu-positive/GnRH-1-positive neurons (arrowheads) migrating out of the VNO. J, Double immunofluorescence for Hu (red) and GnRH-1 (green) at E12.5 highlighted maturing Hu/GnRH-1 neurons (arrowhead) in proximity of the forebrain as well as Hu single-labeled migratory neurons (*). Scale bars: A–F, I, 50 μm; H, J, 20 μm; G, 10 μm.
FE65 expression in proliferative neuronal progenitors
At E11.5, the apical cells directly adjacent to the lumen of the VNO were proliferative cells (Ki67 positive) (Fig. 7A). Hu-positive cells and Hu/GnRH-1-positive cells were first detected in proximity to this apical proliferative compartment (Fig. 7B,C). Although FE65 expression was detected in cells in the anlage of the VNO (Fig. 7D), double immunofluorescent staining for Ki67 and FE65 (Fig. 7E) indicated that the proliferative precursors (Ki67 positive) were negative or expressed FE65 at low levels. Double immunostaining for Sox2 (a marker of undifferentiated cells) and the neuronal marker Nestin (Zimmerman et al., 1994) confirmed that the proliferative apical cells were neuronal progenitors (Fig. 7F). Two potential partners of FE65, Notch-3 and APP, were expressed in the proliferative niche (Fig. 7G) and in the proliferative and newly formed neurons (Fig. 7H,I), respectively.
Figure 7.
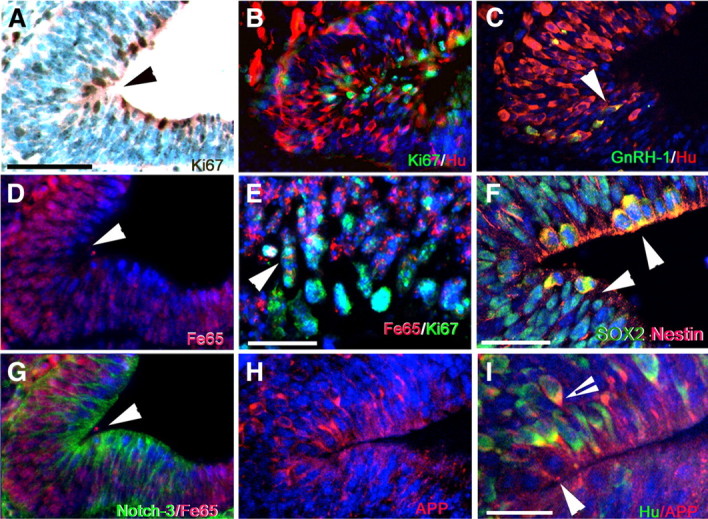
GnRH-1 neurons originate from progenitor cells facing the lumen of the developing VNO. A, Ki67 immunostaining (brown) highlighted the proliferative cells in the apical part of the anlage of the VNO. B, Ki67 (green)/Hu (red) double immunofluorescence: Hu-positive cells originated from the neurogenic proliferative compartment in the apical part of the VNO. C, Double Hu (red)/GnRH-1 (green) immunostaining highlights the first differentiating Hu/GnRH-1 neurons in the VNO (arrowhead), in proximity of the proliferating area (see A, B). D, FE65 immunofluorescence: the apical cells facing the lumen (arrowhead) expressed FE65 (red) at very low levels. E, Ki67 (green) and FE65 (red) double immunofluorescence: the neurogenic proliferative compartment of the VNO expressed FE65 at low or undetectable levels. F, Apical cells of the anlage of the VNO expressed the neurogenic progenitor markers Sox2 (green) and Nestin (red). G, Notch-3 (green) was mainly expressed in the proliferative niche, and FE65 (red) increased in the postmitotic cells. H, I, APP (red) was expressed in the apical neurogenic niche with a pattern similar to Notch-3 (G). I, Enlargement of the apical neurogenic area. APP (red) was expressed by the Hu-negative apical cells (arrowhead) and in newly formed Hu-positive (green) neuronal precursors (arrowhead with black line). Scale bars: A–D, G, H, 50 μm; E, I, 20 μm; F, 25 μm.
To verify that the proliferative neuronal progenitor cells identified in the lumen of the developing placode gave rise to the Hu/GnRH-1 migratory neurons, BrdU tracing was performed (Fig. 8). BrdU incorporation at approximately E10.5 (Fig. 8A–E) or E11 (data not shown) followed by analysis after 24, 48, and 72 h showed that the majority of postmitotic neurons (BrdU/Hu positive) originated from the proliferating neuronal precursor cells of the developing VNO and migrated out from this structure toward the brain. BrdU/Hu-positive cells were in fact detected emerging from the VNO 24 h after BrdU injection (Fig. 8B). Forty-eight and 72 h after injection, BrdU/Hu-positive cells were distributed between the VNO and forebrain junction, with few Hu-positive BrdU heavy labeled cells found within the VNO (Fig. 8C,D). In contrast to BrdU/Hu-positive cells, BrdU/GnRH-1-positive cells were only detected 48 and 72 h after injection, along the migratory route (Fig. 8E). These experiments established, both spatially and temporally, that the GnRH-1 cells arise from the proliferative neuronal progenitor cells in the lumen of the VNO, and a population of migrating Hu-positive cells start to express the GnRH-1 peptide at immunodetectable levels, 2–3 d after the cells become postmitotic.
Figure 8.
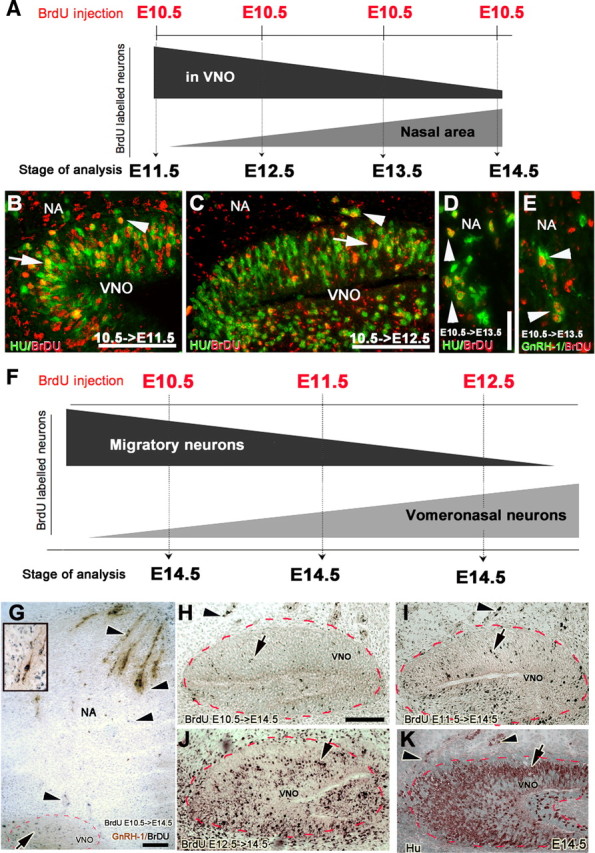
Migratory neurons in nasal region are born before VNO sensory neurons. A, Experimental paradigm: pregnant mice were injected with a single BrdU injection (50 μg/g) at E10.5, and the distribution of BrdU-positive neurons in the VNO anlage and in the nasal area was analyzed 24 h (E11.5) to 96 h (E14.5) after injection. The distribution of BrdU-labeled neurons between VNO and nasal area is schematically represented. B, E11.5 Hu (green)/BrdU (red) double labeling, 24 h after BrdU injection. Hu/BrdU double-positive neurons were distributed within (arrow) and migrating out of (arrowhead) the VNO. C, E12.5 Hu (green)/BrdU (red) double labeling 48 h after BrdU injection. Few Hu-positive and BrdU heavy labeled cells (arrow) were found within the VNO but were detected emerging from the VNO (arrowhead). D, E, E13.5, 72 h after BrdU injection, Hu/BrdU cells (D) and GnRH-1/BrdU-positive cells (E) were detected in the nasal area (NA). F, Second experimental paradigm: pregnant mice were injected with a single BrdU injection (50 μg/g) at the indicated embryonic stage and the BrdU incorporation in migratory neurons and in non-migratory VNO neurons was analyzed at E14.5. Schematic shows BrdU incorporation in migratory neurons declined as VNO neurogenesis increased. Sections of VNO immunolabeled for BrdU after single BrdU pulse at E10.5 (G, H), E11.5 (I), and E12.5 (J). G, Double labeling for GnRH-1 (brown) and BrdU (black) at E14.5 after BrdU pulse at E10.5 showed migratory cells and double-labeled BrdU/GnRH-1 (arrowheads) in the nasal area (NA), whereas only a few heavy BrdU-labeled cells (arrow) were detected in the VNO. Inset, Double-labeled BrdU/GnRH-1 neuron. H, I, Few heavy labeled BrdU-positive cells (black) were detected (arrow) in the VNO after BrdU injections at E10.5 and E11.5. J, Heavy labeled cells were detected in the VNO after BrdU injection at E12.5, and the pattern of these BrdU labeled cells was similar to postmitotic neurons stained for Hu (brown, K) at E14.5 within the VNO (arrow), although Hu-positive cells were still detected migrating out of (arrowhead) the VNO. Red dashed line, VNO. Scale bars: A, 50 μm; C, inset in G, 25 μm; G, 150 μm.
A single BrdU injection at E10.5, E11.5, or E12.5, followed by analysis at E14.5, indicated that neurogenesis of the migratory neurons originating from the proliferative compartment of the VNO preceded the massive neurogenesis of the non-migratory vomeronasal neurons that reside in the VNO and progressively declined as the latter started (Fig. 8F–J).
The increase in GnRH-1 cells in FE65 mutants is associated with an increase in total migratory neuron cell number
As described above, the pan-neuronal marker Hu labeled maturating GnRH-1 precursors together with other migratory neuronal cells types negative for GnRH-1 expression (Fig. 5). At E14.5, the plateau in GnRH-1 number was reached in both controls and FE65 mutants (Fig. 2G). At this stage, GnRH-1 neurons and Hu cells, negative for GnRH-1, were distributed in the nasal area. To determine whether the larger number of GnRH-1 neurons in FE65 mutants could be attributable to an increase in GnRH-1 cell specification associated with a reduction in number of Hu-positive/GnRH-1-negative cells, these two cell types were quantified (Fig. 9). Quantification in FE65 mutants and controls at E14.5 indicated significantly higher numbers for both Hu-positive/GnRH-1-negative and GnRH-1-positive cells emerging from the VNO. The higher total number of migratory neurons (GnRH-1-positive and Hu-positive/GnRH-1-neagtive neurons) found in FE65 mutants indicates that there is a general increase in migratory neurons originating in the VNO.
Figure 9.
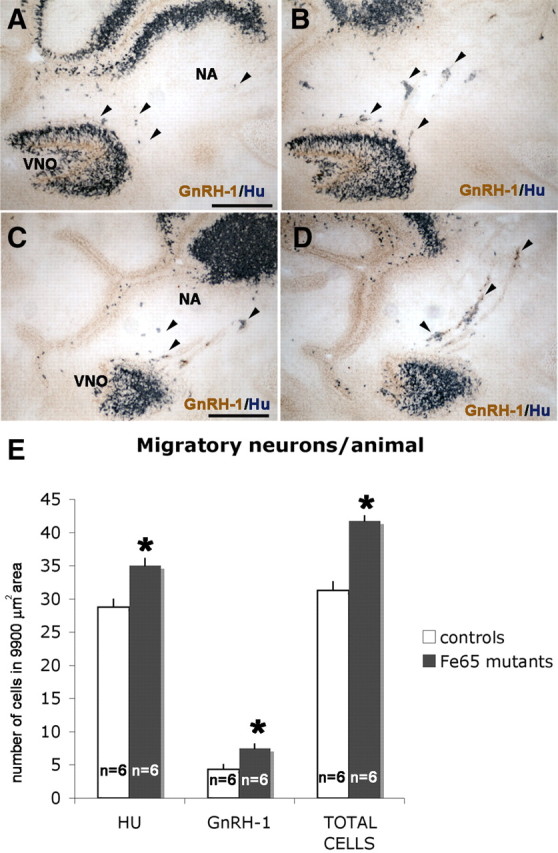
Increase in total migratory neuron cell number in FE65 mutants. Immunolabeling for pan-neuronal marker Hu (blue) and GnRH-1 (brown) on WT (A, C) and FE65 KO (B, D) 14.5 sagittal sections. E, Quantification of migratory neurons (Hu-positive/GnRH-1-negative and GnRH-1-positive) indicated more migratory neurons were present in the nasal area (NA) of FE65 mutants. n = animals, mean values ± SE. *p < 0.05, t test.
Increased neurogenesis in FE65 mutants is not associated with changes in proliferation or cell death
From immunolabeling at E11.5 (Fig. 7), a pattern emerged between the FE65-negative proliferative portion of the developing VNO and the presence of FE65-positive newly formed neurons. This pattern is consistent with previous results that indicated that FE65 is not expressed or expressed at low levels by proliferating neuronal progenitors (Simeone et al., 1994; Bruni et al., 2002). To evaluate whether the increase in GnRH-1 neurogenesis observed in FE65 mutants was attributable to differences in the number of proliferative progenitors or to differences in cell cycle progression, cells in active S and M phase were immunolabeled and quantified. At E11.5, cells in S phase were quantified after a short BrdU pulse (Fig. 10A–C), whereas cells in M phase were quantified by labeling for phospho-histone-3 (Fig. 10D–F). No differences between genotypes in either the number of proliferating cells or in cell cycle distribution was detected (Fig. 10C,F). In contrast, counts of premigratory Hu-positive cells in the VNO anlage on E11.5 serial sections revealed a significantly higher number of Hu-positive cells/VNO in p97FE65 mutants, which accounted for 100 cells per animal (Fig. 10G–I).
Figure 10.

Increased neurogenesis without changes in proliferation or cell death. A–C, Representative VNO section of control (A) and FE65 mutant (B) littermates stained for BrdU after short BrdU pulse. C, Means of BrdU-positive cells per section at E11.5. The number of proliferating cells in S phase was similar in FE65 mutants and controls. D–F, Sections of control (D) and FE65 mutant (E) after phospho-histone-3 immunostaining. E, Means of phospho-histone-3-positive cells per section at E11.5; no significant differences were found in number of cells in M phase. G–I, An increased number of Hu-positive precursors was detected in FE65 mutants. Representative VNO sections of control (G) and FE65 mutant (H) littermates stained for Hu (brown). I, Means of total Hu-positive cells in VNO per animal at E11.5. J–L, The number of apoptotic cells was similar in controls and FE65 mutants. Representative VNO sections of control (J) and FE65 mutant (K) littermates stained for active caspase-3 (black). Values ± SE, n = number of animals analyzed. *p < 0.05, t test. Scale bar, 50 μm.
During this early stage of development, no glial markers were found expressed in cell soma in the developing VNO but were in cells/processes distributed along nasal sensory fibers and around the developing olfactory bulb (data not shown). As a result of the lack of specific markers for VNO/olfactory epithelium-derived glial cells (Murdoch et al., 2010), reliable quantifications could not be performed to determine whether a corresponding decrease in VNO glial cells occurred as GnRH-1 cell number increased. However, because significantly more premigratory neurons were observed in FE65 mutants without changes in proliferation, differences in apoptosis (Cao et al., 2000) were examined. Quantification of cells immunolabeled for the activated form of caspase-3 at E11.5 did not reveal significant differences in cell death in the VNO anlage (Fig. 10J–L). Together, these data are consistent with a role for FE65 in the control and timing of GnRH-1 neuronal cell fate and suggest that progenitor cells of migratory neurons and GnRH-1 progressively shift toward the generation of other cell types, as normal GnRH-1 neurogenesis declines.
Discussion
The genesis of GnRH-1 neurons is one of the earliest events during olfactory placode invagination and development of the VNO. The definition of the neurogenic compartment and lineage of the GnRH-1 progenitors, as well as the identification of the pre-GnRH-1 neurons has remained unsolved. In this paper, FE65, a multimodular adaptor protein known to influence APP processing and gene transcription, is shown to have a role in suppressing GnRH-1 neurogenesis within the developing nasal area.
Modulation of neurogenesis, which implies proliferation, cell fate commitment, and differentiation, are tightly regulated by both extrinsic and intrinsic signals (Miller and Gauthier, 2007). By genetic screening on single-cell cDNAs the expression of FE65 was identified in newly formed GnRH-1 neurons. Analyzing p97FE65 KO, a mouse line missing the 97 kDa form of FE65, a transient increase in GnRH-1 neurogenesis during embryonic development (∼25%) was found, whereas no changes in GnRH-1 neuronal migration were detected. RT-PCR on laser captured tissue indicated that, as described previously for the developing brain (Wang et al., 2004), p97FE65 KO express the shorter p60 isoform of FE65 in the VNO. To confirm that the increase in GnRH-1 cell number in p97FE65 KO was strictly dependent on disruption of the N-terminal portion of FE65 or whether a more severe phenotype could be found in the absence of both p97 and p60FE65 isoforms (Wang et al., 2004; Guénette et al., 2006) GnRH-1 development was analyzed in a second mice model lacking both the 97 and 60 kDa isoforms of FE65. A similar increase in GnRH-1 cell number was found in the second line, without additional phenotypes. The increase in GnRH-1 neurogenesis found in our study (25%) provides in vivo data that are consistent with findings by Ma et al. (2008) that showed increased neurogenesis in cultured cortical neural progenitor cells of p97FE65-deficient mice, with the number of TUJ1-positive cells increasing by 38%. However, no differences in brain or VNO size were detected in the mice examined in the present study (data not shown). This suggests that FE65 may alter neurogenesis in multiple regions but that each region needs to be examined during a specific temporal window.
FE65 has been described to regulate cell motility, actin dynamics, and neuronal migration (Sabo et al., 2001; Wolfe and Guénette, 2007). In FE65 mutants, a progressive increase in GnRH-1 number was observed; when in control littermates, a plateau in GnRH-1 neurons was reached. However, no defects in GnRH-1 neuronal migration into the brain were observed in FE65 mutants. Comparable numbers of GnRH-1 cells accessed the brain per day in both genotypes (200 ± 50) and similar numbers of GnRH-1 neurons were counted in the brains of FE65 KOs and controls throughout development and in the postnatal stages. This indicates that the extra GnRH-1 cells (up to ∼200) were found in the nasal area, and, independent of age, more cells did not access the brain. Even at E17.5, after the “normal” migratory windows ends, a significantly larger number of GnRH-1 neurons were still detectable in the nasal area of FE65 KO compared with controls. This observation indicates that the number of GnRH-1 cells able to migrate into the brain per day is independent of the total number of GnRH-1 cells “available” in the nasal area and confirms that the time of access to the brain is finely regulated over time by the cellular environment (Tobet and Schwarting, 2006). Thus, no differences in the number of GnRH-1 neurons were observed between FE65 mutants and controls in the brain.
Birth-date tracing experiments in the early neurogenic peak (E9.5–E10.5) and in the declining period of GnRH-1 neurogenesis (E11–E12) indicated that, in the FE65 KOs, a higher percentage of GnRH-1 cell form during the neurogenic declining phases. These data suggest a prolonged timing in GnRH-1 neurogenesis in the absence of FE65. Prolonged GnRH-1 neurogenesis is consistent with the progressive increase in GnRH-1 cell number observed in FE65 mutants after the normal GnRH-1 neuronal plateau was reached in control animals. Examining FE65 expression during the GnRH-1 neurogenic window indicated that FE65 is expressed at low or undetectable levels by proliferating neural progenitor cells (Sox2, Nestin, Ki67, APP, Notch-3 positive) facing the lumen of the developing VNO and that FE65 expression rapidly increased in nonproliferative daughter cells from the first postmitotic phase (Sox2 positive, Ki67 negative) (Simeone et al., 1994; Stevanovic, 2003). This is in agreement with work in Caenorhabditis elegans, in which the FE65 homolog feh-1 is detectable unambiguously only after proliferation has ceased (Zambrano et al., 2002). Whether the identified progenitors facing the lumen of the invaginating VNO anlage were the origin of GnRH-1 neurons was further investigated by BrdU incorporation tracing experiments. These experiments showed that these neural progenitors gave birth to migratory Hu-positive precursors, some of which started to express GnRH-1, between 48 and 72 h postmitosis, while migrating or still within the VNO.
Vomeronasal neurons express Tag1 (Wolfer et al., 1994; Yoshida et al., 1999). Tag1 has been identified as a trigger for the APP/FE65-mediated suppression of neurogenesis by increasing APP processing and APP/FE65 signaling (Ma et al., 2008a). Using BrdU labeling, we determined a decline in GnRH1 neurogenesis while vomeronasal/olfactory neurogenesis increased. An increase of Tag1-expressing neurons in the GnRH-1 neurogenic niche could constitute an environmental activator for the suppression of GnRH-1 neurogenesis (Miller and Gauthier, 2007). The TAG1/APP/FE65 system has been suggested to have a role in controlling progenitor self-renewal (Mattson and van Praag, 2008). At E11.5, when GnRH-1 neurogenesis is still active, no differences in proliferation or cell death were found in the anlage of the VNO, but a significantly greater number of Hu-positive neuronal precursors were detected (12%). Together, these data indicate similar cell cycle progression with no changes in the number of progenitor self-renewal in the VNO of FE65 mutants. These data are consistent with the increase in GnRH-1 neurons being the result of differences in neurogenesis over time (Kim et al., 2005).
Both APP and Notch-3 were expressed in cells in the GnRH-1 neurogenic niche. APP and Notch are type-1 transmembrane proteins (Sotthibundhu et al., 2009), and their intracellular signaling depends on cleavage by γ-secretase, which liberates their intracellular portions NICD and AICD, respectively (De Strooper et al., 1999; De Strooper, 2000). FE65 can stimulate γ-secretase cleavage of APP (Wiley et al., 2007) and participate in APP signaling by binding AICD. FE65 was also recently described to bind NICD, suggesting a multifunctional role for FE65 in modulating both APP and Notch activity in cell fate determination (Fischer et al., 2005; Konietzko et al., 2010). A series of studies have shown roles for APP and FE65 in modulating both proliferation and cell fate determination of neuronal precursor cells (Bruni et al., 2002; Caillé et al., 2004; López-Toledano and Shelanski, 2007; Ma et al., 2008b; Sugaya, 2008; Chen and Dong, 2009; Sotthibundhu et al., 2009). FE65 is able to modulate the secretion of the cleaved forms of APP: Aβ40 and Aβ42 (Wiley et al., 2007). Biochemical assays on primary neuronal cultures obtained from p97FE65 KO crossed with APP overexpressors (APPswe transgenics) revealed a 58% reduction of Aβ40 and a 68% reduction of Aβ42 secretion (Wang et al., 2004), whereas quantification on brain lysates from adult p97/60FE65 KO suggested a role for FE65 mainly in Aβ42 formation (Guénette et al., 2006). Recent work indicated Aβ40 as a key factor leading to neuronal cell fate determination and Aβ42 as a facilitator toward astro-gliogenesis (Chen and Dong, 2009). Counts of GnRH-1 and of other migratory GnRH-1-negative neurons originating from the VNO revealed significant increase for both cell types, indicative of a general increase neurogenesis for migratory neurons originating from VNO. Glial labeling in the nasal region was performed during early development and highlighted a variety of cells distributed along sensory fibers in the nasal area and within the developing olfactory bulb. Because of the lack of specific markers (Murdoch and Roskams, 2008; Murdoch et al., 2010) for VNO/olfactory epithelium-derived glial cells, reliable quantifications could not be performed. Additional lineage tracing studies are needed to determine whether a specific glial cell population originates from the anlage of the VNO and whether a corresponding decrease in this population occurs in FE65 KOs, as GnRH-1 cells increases (Murdoch et al., 2010).
In summary, the data in this report indicate a role for FE65 in suppressing the GnRH-1 neurogenic program and highlight an important role for the N-terminal portion of the protein that includes the WW domain of p97FE65. Analysis of FE65 expression indicated that FE65 is expressed at low levels by neurogenic proliferative cells and that the lack of p97FE65 expression did not affect normal proliferation rate or cell death. The GnRH-1 neurons, unlike other cell types originating in the olfactory area, have a short and limited neurogenic window (Wray et al., 1989a; Murdoch et al., 2010). The GnRH-1 neurogenic increase observed in FE65 mutants is consistent with GnRH-1 neurons originating from multipotent progenitor cells able to generate other cell types in response to environmental changes as the GnRH-1 neurogenesis declines.
Footnotes
This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Neurological Disorders and Stroke. We thank Drs. H. Arnheiter for critical review of this manuscript and G. M. Martins for providing p97FE65 animals.
References
- Bruni P, Minopoli G, Brancaccio T, Napolitano M, Faraonio R, Zambrano N, Hansen U, Russo T. Fe65, a ligand of the Alzheimer's beta-amyloid precursor protein, blocks cell cycle progression by down-regulating thymidylate synthase expression. J Biol Chem. 2002;277:35481–35488. doi: 10.1074/jbc.M205227200. [DOI] [PubMed] [Google Scholar]
- Caillé I, Allinquant B, Dupont E, Bouillot C, Langer A, Müller U, Prochiantz A. Soluble form of amyloid precursor protein regulates proliferation of progenitors in the adult subventricular zone. Development. 2004;131:2173–2181. doi: 10.1242/dev.01103. [DOI] [PubMed] [Google Scholar]
- Cao H, Pratt N, Mattison J, Zhao Y, Chang NS. Characterization of an apoptosis inhibitory domain at the C-termini of FE65-like protein. Biochem Biophys Res Commun. 2000;276:843–850. doi: 10.1006/bbrc.2000.3527. [DOI] [PubMed] [Google Scholar]
- Cao X, Südhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- Chen Y, Dong C. Abeta40 promotes neuronal cell fate in neural progenitor cells. Cell Death Differ. 2009;16:386–394. doi: 10.1038/cdd.2008.94. [DOI] [PubMed] [Google Scholar]
- Deschênes-Furry J, Perrone-Bizzozero N, Jasmin BJ. The RNA-binding protein HuD: a regulator of neuronal differentiation, maintenance and plasticity. BioEssays. 2006;28:822–833. doi: 10.1002/bies.20449. [DOI] [PubMed] [Google Scholar]
- De Strooper B. Alzheimer's disease. Closing in on gamma-secretase. Nature. 2000;405:627–629. doi: 10.1038/35015193. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- Fischer DF, van Dijk R, Sluijs JA, Nair SM, Racchi M, Levelt CN, van Leeuwen FW, Hol EM. Activation of the Notch pathway in Down syndrome: cross-talk of Notch and APP. FASEB J. 2005;19:1451–1458. doi: 10.1096/fj.04-3395.com. [DOI] [PubMed] [Google Scholar]
- Fornaro M, Geuna S, Fasolo A, Giacobini-Robecchi MG. HuC/D confocal imaging points to olfactory migratory cells as the first cell population that expresses a post-mitotic neuronal phenotype in the chick embryo. Neuroscience. 2003;122:123–128. doi: 10.1016/j.neuroscience.2003.07.004. [DOI] [PubMed] [Google Scholar]
- Forni PE, Scuoppo C, Imayoshi I, Taulli R, Dastrù W, Sala V, Betz UA, Muzzi P, Martinuzzi D, Vercelli AE, Kageyama R, Ponzetto C. High levels of Cre expression in neuronal progenitors cause defects in brain development leading to microencephaly and hydrocephaly. J Neurosci. 2006;26:9593–9602. doi: 10.1523/JNEUROSCI.2815-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fueshko S, Wray S. LHRH cells migrate on peripherin fibers in embryonic olfactory explant cultures: an in vitro model for neurophilic neuronal migration. Dev Biol. 1994;166:331–348. doi: 10.1006/dbio.1994.1319. [DOI] [PubMed] [Google Scholar]
- Giacobini P, Kopin AS, Beart PM, Mercer LD, Fasolo A, Wray S. Cholecystokinin modulates migration of gonadotropin-releasing hormone-1 neurons. J Neurosci. 2004;24:4737–4748. doi: 10.1523/JNEUROSCI.0649-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham V, Khudyakov J, Ellis P, Pevny L. SOX2 functions to maintain neural progenitor identity. Neuron. 2003;39:749–765. doi: 10.1016/s0896-6273(03)00497-5. [DOI] [PubMed] [Google Scholar]
- Guénette S, Chang Y, Hiesberger T, Richardson JA, Eckman CB, Eckman EA, Hammer RE, Herz J. Essential roles for the FE65 amyloid precursor protein-interacting proteins in brain development. EMBO J. 2006;25:420–431. doi: 10.1038/sj.emboj.7600926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q, Hearn MG, Jin LW, Bressler SL, Martin GM. Alternatively spliced isoforms of FE65 serve as neuron-specific and non-neuronal markers. J Neurosci Res. 1999;58:632–640. doi: 10.1002/(sici)1097-4547(19991201)58:5<632::aid-jnr4>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Jasoni CL, Porteous RW, Herbison AE. Anatomical location of mature GnRH neurons corresponds with their birthdate in the developing mouse. Dev Dyn. 2009;238:524–531. doi: 10.1002/dvdy.21869. [DOI] [PubMed] [Google Scholar]
- Kallmann FJ, Schönfeld WA, Barrera SE. The genetic aspects of primary eunuchoidism. Am J Ment Defic. 1944;48:203–236. [Google Scholar]
- Kesavapany S, Banner SJ, Lau KF, Shaw CE, Miller CC, Cooper JD, McLoughlin DM. Expression of the Fe65 adapter protein in adult and developing mouse brain. Neuroscience. 2002;115:951–960. doi: 10.1016/s0306-4522(02)00422-0. [DOI] [PubMed] [Google Scholar]
- Kim J, Wu HH, Lander AD, Lyons KM, Matzuk MM, Calof AL. GDF11 controls the timing of progenitor cell competence in developing retina. Science. 2005;308:1927–1930. doi: 10.1126/science.1110175. [DOI] [PubMed] [Google Scholar]
- King GD, Scott Turner R. Adaptor protein interactions: modulators of amyloid precursor protein metabolism and Alzheimer's disease risk? Exp Neurol. 2004;185:208–219. doi: 10.1016/j.expneurol.2003.10.011. [DOI] [PubMed] [Google Scholar]
- Konietzko U, Goodger ZV, Meyer M, Kohli BM, Bosset J, Lahiri DK, Nitsch RM. Co-localization of the amyloid precursor protein and Notch intracellular domains in nuclear transcription factories. Neurobiol Aging. 2010;31:58–73. doi: 10.1016/j.neurobiolaging.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer PR, Wray S. Novel gene expressed in nasal region influences outgrowth of olfactory axons and migration of luteinizing hormone-releasing hormone (LHRH) neurons. Genes Dev. 2000;14:1824–1834. [PMC free article] [PubMed] [Google Scholar]
- Kramer PR, Wray S. Nasal embryonic LHRH factor (NELF) expression within the CNS and PNS of the rodent. Brain Res Gene Expr Patterns. 2001;1:23–26. doi: 10.1016/s1567-133x(01)00004-7. [DOI] [PubMed] [Google Scholar]
- Krüger M, Ruschke K, Braun T. NSCL-1 and NSCL-2 synergistically determine the fate of GnRH-1 neurons and control necdin gene expression. EMBO J. 2004;23:4353–4364. doi: 10.1038/sj.emboj.7600431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusano K, Fueshko S, Gainer H, Wray S. Electrical and synaptic properties of embryonic luteinizing hormone-releasing hormone neurons in explant cultures. Proc Natl Acad Sci U S A. 1995;92:3918–3922. doi: 10.1073/pnas.92.9.3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- López-Toledano MA, Shelanski ML. Increased neurogenesis in young transgenic mice overexpressing human APP(Sw, Ind) J Alzheimers Dis. 2007;12:229–240. doi: 10.3233/jad-2007-12304. [DOI] [PubMed] [Google Scholar]
- Ma QH, Bagnard D, Xiao ZC, Dawe GS. A TAG on to the neurogenic functions of APP. Cell Adh Migr. 2008a;2:2–8. doi: 10.4161/cam.2.1.5790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma QH, Futagawa T, Yang WL, Jiang XD, Zeng L, Takeda Y, Xu RX, Bagnard D, Schachner M, Furley AJ, Karagogeos D, Watanabe K, Dawe GS, Xiao ZC. A TAG1-APP signalling pathway through Fe65 negatively modulates neurogenesis. Nat Cell Biol. 2008b;10:283–294. doi: 10.1038/ncb1690. [DOI] [PubMed] [Google Scholar]
- Mattson MP, van Praag H. TAGing APP constrains neurogenesis. Nat Cell Biol. 2008;10:249–250. doi: 10.1038/ncb0308-249. [DOI] [PubMed] [Google Scholar]
- Miller FD, Gauthier AS. Timing is everything: making neurons versus glia in the developing cortex. Neuron. 2007;54:357–369. doi: 10.1016/j.neuron.2007.04.019. [DOI] [PubMed] [Google Scholar]
- Miller NL, Wevrick R, Mellon PL. Necdin, a Prader-Willi syndrome candidate gene, regulates gonadotropin-releasing hormone neurons during development. Hum Mol Genet. 2009;18:248–260. doi: 10.1093/hmg/ddn344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch B, Roskams AJ. A novel embryonic nestin-expressing radial glia-like progenitor gives rise to zonally restricted olfactory and vomeronasal neurons. J Neurosci. 2008;28:4271–4282. doi: 10.1523/JNEUROSCI.5566-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch B, DelConte C, García-Castro MI. Embryonic Pax7-expressing progenitors contribute multiple cell types to the postnatal olfactory epithelium. J Neurosci. 2010;30:9523–9532. doi: 10.1523/JNEUROSCI.0867-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano HJ, Darnell RB. A hierarchy of Hu RNA binding proteins in developing and adult neurons. J Neurosci. 1997;17:3024–3037. doi: 10.1523/JNEUROSCI.17-09-03024.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J Cell Biol. 2001;153:1403–1414. doi: 10.1083/jcb.153.7.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The amyloid precursor protein and its regulatory protein, FE65, in growth cones and synapses in vitro and in vivo. J Neurosci. 2003;23:5407–5415. doi: 10.1523/JNEUROSCI.23-13-05407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifi N, Reuss AE, Wray S. Prenatal LHRH neurons in nasal explant cultures express estrogen receptor beta transcript. Endocrinology. 2002;143:2503–2507. doi: 10.1210/endo.143.7.8897. [DOI] [PubMed] [Google Scholar]
- Simeone A, Duilio A, Fiore F, Acampora D, De Felice C, Faraonio R, Paolocci F, Cimino F, Russo T. Expression of the neuron-specific FE65 gene marks the development of embryo ganglionic derivatives. Dev Neurosci. 1994;16:53–60. doi: 10.1159/000112088. [DOI] [PubMed] [Google Scholar]
- Sotthibundhu A, Li QX, Thangnipon W, Coulson EJ. Abeta(1-42) stimulates adult SVZ neurogenesis through the p75 neurotrophin receptor. Neurobiol Aging. 2009;30:1975–1985. doi: 10.1016/j.neurobiolaging.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Standen CL, Perkinton MS, Byers HL, Kesavapany S, Lau KF, Ward M, McLoughlin D, Miller CC. The neuronal adaptor protein Fe65 is phosphorylated by mitogen-activated protein kinase (ERK1/2) Mol Cell Neurosci. 2003;24:851–857. doi: 10.1016/j.mcn.2003.07.002. [DOI] [PubMed] [Google Scholar]
- Stevanovic M. Modulation of SOX2 and SOX3 gene expression during differentiation of human neuronal precursor cell line NTERA2. Mol Biol Rep. 2003;30:127–132. doi: 10.1023/a:1023961009869. [DOI] [PubMed] [Google Scholar]
- Sugaya K. Mechanism of glial differentiation of neural progenitor cells by amyloid precursor protein. Neurodegener Dis. 2008;5:170–172. doi: 10.1159/000113693. [DOI] [PubMed] [Google Scholar]
- Telese F, Bruni P, Donizetti A, Gianni D, D'Ambrosio C, Scaloni A, Zambrano N, Rosenfeld MG, Russo T. Transcription regulation by the adaptor protein Fe65 and the nucleosome assembly factor SET. EMBO Rep. 2005;6:77–82. doi: 10.1038/sj.embor.7400309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobet SA, Schwarting GA. Minireview: recent progress in gonadotropin-releasing hormone neuronal migration. Endocrinology. 2006;147:1159–1165. doi: 10.1210/en.2005-1275. [DOI] [PubMed] [Google Scholar]
- Wang B, Hu Q, Hearn MG, Shimizu K, Ware CB, Liggitt DH, Jin LW, Cool BH, Storm DR, Martin GM. Isoform-specific knockout of FE65 leads to impaired learning and memory. J Neurosci Res. 2004;75:12–24. doi: 10.1002/jnr.10834. [DOI] [PubMed] [Google Scholar]
- Wiley JC, Smith EA, Hudson MP, Ladiges WC, Bothwell M. Fe65 stimulates proteolytic liberation of the beta-amyloid precursor protein intracellular domain. J Biol Chem. 2007;282:33313–33325. doi: 10.1074/jbc.M706024200. [DOI] [PubMed] [Google Scholar]
- Wolfe MS, Guénette SY. APP at a glance. J Cell Sci. 2007;120:3157–3161. doi: 10.1242/jcs.03481. [DOI] [PubMed] [Google Scholar]
- Wolfer DP, Henehan-Beatty A, Stoeckli ET, Sonderegger P, Lipp HP. Distribution of TAG-1/axonin-1 in fibre tracts and migratory streams of the developing mouse nervous system. J Comp Neurol. 1994;345:1–32. doi: 10.1002/cne.903450102. [DOI] [PubMed] [Google Scholar]
- Wray S. Development of gonadotropin-releasing hormone-1 neurons. Front Neuroendocrinol. 2002;23:292–316. doi: 10.1016/s0091-3022(02)00001-8. [DOI] [PubMed] [Google Scholar]
- Wray S, Gähwiler BH, Gainer H. Slice cultures of LHRH neurons in the presence and absence of brainstem and pituitary. Peptides. 1988;9:1151–1175. doi: 10.1016/0196-9781(88)90103-9. [DOI] [PubMed] [Google Scholar]
- Wray S, Grant P, Gainer H. Evidence that cells expressing luteinizing hormone-releasing hormone mRNA in the mouse are derived from progenitor cells in the olfactory placode. Proc Natl Acad Sci U S A. 1989a;86:8132–8136. doi: 10.1073/pnas.86.20.8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray S, Nieburgs A, Elkabes S. Spatiotemporal cell expression of luteinizing hormone-releasing hormone in the prenatal mouse: evidence for an embryonic origin in the olfactory placode. Brain Res Dev Brain Res. 1989b;46:309–318. doi: 10.1016/0165-3806(89)90295-2. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Rutishauser U, Crandall JE, Schwarting GA. Polysialic acid facilitates migration of luteinizing hormone-releasing hormone neurons on vomeronasal axons. J Neurosci. 1999;19:794–801. doi: 10.1523/JNEUROSCI.19-02-00794.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano N, Bimonte M, Arbucci S, Gianni D, Russo T, Bazzicalupo P. feh-1 and apl-1, the Caenorhabditis elegans orthologues of mammalian Fe65 and beta-amyloid precursor protein genes, are involved in the same pathway that controls nematode pharyngeal pumping. J Cell Sci. 2002;115:1411–1422. doi: 10.1242/jcs.115.7.1411. [DOI] [PubMed] [Google Scholar]
- Zimmerman L, Parr B, Lendahl U, Cunningham M, McKay R, Gavin B, Mann J, Vassileva G, McMahon A. Independent regulatory elements in the nestin gene direct transgene expression to neural stem cells or muscle precursors. Neuron. 1994;12:11–24. doi: 10.1016/0896-6273(94)90148-1. [DOI] [PubMed] [Google Scholar]