

Abstract
Objective
Magnetic resonance imaging (MRI) often demonstrates brain lesions in neuropsychiatric systemic lupus erythematosus (NPSL). The present study compared post-mortem histopathology with pre-mortem MRI in NPSL.
Methods
200 subjects with NPSLE were studied prospectively with MRI over a 10-year period during which 22 subjects died. In 14 subjects, a brain autopsy with histopathology that permitted direct comparison with pre mortem MRI was successfully obtained. Surface anatomy was used to determine the approximate location of individual lesions.
Results
Pre mortem MRI findings in fatal NPSLE were small focal white matter lesions (100%), cortical atrophy (64%), ventricular dilation (57%), cerebral edema (50%), diffuse white matter abnormalities (43%), focal atrophy (36%), cerebral infarction (29%), acute leukoencephalopathy (25%), intracranial hemorrhage (21%), and calcifications (7%). Microscopic findings in fatal NPSLE included global ischemic changes (57%), parenchymal edema (50%), microhemorrhages (43%), glial hyperplasia (43%), diffuse neuronal/axonal loss (36%), resolved cerebral infarction (33%), microthomboemboli (29%), blood vessel remodeling (29%), acute cerebral infarction (14%), acute macrohemorrhages (14%), and resolved intracranial hemorrhages (7%). Cortical atrophy and ventricular dilation seen by MRI predicted brain mass at autopsy (r = -0.72, p = 0.01, and r = -0.77, p =0.01, respectively). Cerebral autopsy findings, including infarction, cerebral edema, intracranial hemorrhage, calcifications, cysts, and focal atrophy were also predicted accurately by pre mortem MRI.
Conclusion
Brain lesions in NPSLE detected by MRI accurately represent serious underlying cerebrovascular and parenchymal brain injury on pathology.
Keywords: SLE, Neuropsychiatric, Magnetic Resonance, NPSLE, MRI, Autopsy
Introduction
Neuropsychiatric systemic lupus erythematosus (NPSLE) is associated with both discrete and generalized brain lesions seen on neuroimaging, but the etiology and basis for these NPSLE-associated brain lesions remain uncertain (1-5). Lesions on magnetic resonance imaging (MRI) may be observed in 25-75% of NPSLE patients, and increase with disease severity, disease activity, patient age, and neurologic events (3-14). The significance of MRI-visible lesions in NPSLE generally remains speculative; however, recent evidence suggests that focal lesions in NPSLE represent neuronal injury from various etiologies (6-15). Except for a limited number of paired imaging-autopsy case reports, a major problem with past neuroimaging studies in NPSLE has been the general lack of histopathologic correlates to assist in interpretation. To address this deficiency, the present study compared prospective pre mortem MRI to post mortem histopathologic findings obtained at autopsy in each of 14 subjects.
Materials and Methods
Study Design
This study was approved by the institutional review board (IRB) and complied with the Declaration of Helinski. Each participant provided a priori written informed consent for both the clinical studies and the post mortem autopsy. The diagnosis of SLE was established in each subject using the American Rheumatism Association 1982 and American College of Rheumatology (ACR) 1997 revised criteria for systemic lupus erythematosus (SLE) (17,18). A rheumatologist confirmed the diagnosis of SLE after an in-depth face-to-face interview, medical history, physical examination, chart-review, and appropriate laboratory testing. Every 3 months and during NPSLE episodes, SLE disease activity was determined with the Systemic Lupus Erythematosus Disease Activity Index (SLEDAI) (19) and SLE disease severity (damage index) was measured with Systemic Lupus Erythematosus International Collaboarting Clinics/American College of Rheumatology Damage Index (SLICC/ACRDI) (20). Each of these was further subcategorized into Neuro-SLEDAI consisting of the neurologic components of SLEDAI (seizures, psychosis, organic brain syndrome, visual abnormality, headache, cerebral infarct) and Neuro-SLICC consisting of the neurologic components of SLICC/ACRDI (retinal pathology, optic atrophy, cognitive disorder, psychosis, seizures, stroke, neuropathy, transverse myelitis) as described previously (21). NPSLE was characterized by the ACR nomenclature and case definitions for NPSLE (22). Clinical characteristics are shown in Tables 1 and 2. 200 subjects with NPSLE were prospectively studied with MRI over a 10-year period during which 22 subjects died.
Table 1. Subject Characteristics.
| Subject | Sex | Age (years) | Ethnicity | NPSLE manifestations | Non-neurologic manifestations | SLEDAI | Neuro-SLEDAI | SLICC | Neuro-SLICC |
|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 44 | Spanish-American |
|
|
57 | 24 | 19 | 4 |
| 2 | M | 56 | White |
|
|
65 | 24 | 18 | 3 |
| 3 | F | 22 | Black |
|
|
24 | 8 | 11 | 3 |
| 4 | F | 43 | White |
|
|
12 | 8 | 8 | 4 |
| 5 | F | 19 | Spanish-American |
|
|
44 | 24 | 6 | 1 |
| 6 | F | 42 | Spanish-American |
|
|
12 | 8 | 7 | 4 |
| 7 | F | 45 | Spanish-American |
|
|
35 | 24 | 10 | 2 |
| 8 | F | 11 | Spanish-American |
|
|
33 | 24 | 10 | 3 |
| 9 | F | 27 | Spanish-American |
|
|
16 | 8 | 5 | 1 |
| 10 | F | 65 | Spanish-American |
|
|
38 | 16 | 18 | 1 |
| 11 | F | 48 | Black |
|
|
33 | 16 | 11 | 2 |
| 12 | F | 19 | Spanish-American |
|
|
41 | 24 | 5 | 2 |
| 13 | F | 36 | White |
|
|
32 | 16 | 12 | 2 |
| 14 | F | 13 | Spanish-American |
|
|
33 | 16 | 7 | 1 |
|
| |||||||||
| 93% women | 35±17 | 34±15 | 17±7 | 10±5 | 3±2 | ||||
Table 2. Autoantibody Profiles of NPSLE Subjects.
| Subject | ANA Titer | DNA Titer | Smith | RNP | SSA | SSB | Anti-ribosomal P | ACA IgG | ACA IgM | ACA IgA | LLI |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1:320 | 1:160 | Positive | Negative | Positive | Negative | Negative | 45 | 65 | 18 | Positive |
| 2 | 1:640 | 1:640 | Negative | Negative | Negative | Negative | Negative | 65 | 12 | 7 | Positive |
| 3 | 1:640 | 1:640 | Positive | Negative | Positive | Negative | Positive | 80 | 34 | 5 | Positive |
| 4 | 1:80 | Negative | Negative | Negative | Negative | Negative | Negative | 12 | 45 | 9 | Positive |
| 5 | 1:640 | 1:640 | Positive | Negative Depression | Positive | Negative | Negative | 32 | 28 | 12 | Negative |
| 6 | 1:320 | Negative | Positive | Positive | Positive | Negative | Negative | 14 | 54 | 18 | Positive |
| 7 | 1:320 | 1:40 | Positive | Negative | Negative | Negative | Positive | 8 | 2 | 9 | Negative |
| 8 | 1:320 | 1:320 | Negative | Negative | Negative | Negative | Negative | 36 | 23 | 4 | Positive |
| 9 | 1:640 | 1:320 | Negative | Negative | Positive | Negative | Negative | 13 | 8 | 7 | Negative |
| 10 | 1:160 | Negative | Negative | Negative | Positive | Negative | Negative | 0 | 4 | 5 | Negative |
| 11 | 1:640 | 1:640 | Positive | Negative | Positive | Positive | Negative | 75 | 24 | 32 | Positive |
| 12 | 1:320 | 1:160 | Negative | Negative | Negative | Negative | Negative | 32 | 12 | 4 | Negative |
| 13 | 1:640 | 1:160 | Positive | Negative | Positive | Negative | Positive | 32 | 12 | 4 | Negative |
| 14 | 1:640 | 1:640 | Positive | Negative | Positive | Negative | Positive | 45 | 54 | 14 | Positive |
|
| |||||||||||
| Summary | 100% | 79% | 57% | 7% | 64% | 7% | 29% | 64% | 57% | 36% | 64% |
ANA = antinuclear antibody; DNA = anti-double stranded DNA antibody, Smith = anti-Smith antibody, RNP = anti-ribonucleoprotein antibody, SSA = anti-soluble substance A antibody, SSB = anti-soluble substance B antibody, Anti-ribosomal P = Anti-ribosomal P antibody, ACA IgG - anticardiolipin IgG antibodies (GPL units), ACA IgM - anticardiolipin IgM antibodies (MPL units), ACA IgA - anticardiolipin IgA antibodies (APL units), LLI = lupus-like inhibitor (lupus anticoagulant).
Magnetic Resonance Imaging protocol
The study design was to obtain a baseline MRI at study entry, a repeat MRI in those subjects with active NPSLE episodes, and another MRI at resolution defined 3 months after the NPSLE episode. In 14 subjects a brain autopsy that permitted comparison of MRI obtained pre mortem with post mortem brain histopathology was obtained.
Pre mortem MRI was acquired at 1.5 Tesla with a General Electric Signa clinical scanner (GE Medical Systems, Waukesha, WI) using a transmit/receive head coil (33-37). Proton density (PD)/T2-weighted (T2) MR images (TR=3,000 ms; TE=30/100 ms; field of view=24 cm × 24 cm; slice thickness/gap = 5/1 mm), fluid attenuated inversion recovery (FLAIR) images (TR=10,002 ms, TE = 145 ms, TI = 2200 ms; slice thickness/gap = 5/0 mm), and T1-weighted (TE = 9 ms, TR = 550 ms) were obtained in the axial plane (14,23). To confirm or exclude acute cerebral infarct, diffusion weighted imaging (DWI) was obtained (24,25). In each case, neuroimaging was obtained either during the evaluation of the fatal episode for hospitalized subjects, or within a year of an unobserved death outside the hospital (Table 1).
MRI Data Analysis
Brain atrophy and lesions were quantified using previously described methods (7,21). Atrophy was characterized by grading each of cerebral atrophy and ventricular dilation using categorical scales where 0 = none, 1 = mild, 2 = moderate, and 3 = severe (7,21). Lesions were classified as follows: a) normal scan (defined as no focal or diffuse lesions on PD/T2 and FLAIR imaging), b) abnormal (any focal or diffuse lesion on PD/T2 and FLAIR imaging), c) small focal lesions (hyperintense focal lesions less than 3mm in diameter on PD/T2 and FLAIR imaging not associated with local encephalomalacia), d) resolved infarcts (hyperintense lesions greater than 3 mm in diameter on PD/T2 imaging associated with local encephalomalacia and typical changes on T1, e) acute infarcts (hyperintense lesions on PD/T2 imaging associated with restricted diffusion by DWI, but not associated with local encephalomalacia), and f) acute lupus leukoencephalopathy (hyperintense lesions on PD/T2 and FLAIR imaging in gray and white matter with poorly defined borders, often following the gyri, but frequently extensive and occasionally involving deep white matter, but that resolve with time) (21).
Pathologic Examination
Brain autopsy was obtained in each case and gross pathological changes were described. After fixation in 10% buffered formalin for 2 weeks, the brain was then weighed. The brain was examined for gross pathological changes, and then sectioned into standard coronal planes. Each coronal section was then examined for macroscopic changes, including obvious hemorrhage, focal atrophy, cyst formation, calcifications, and meningeal abnormalities. After inspection for obvious macroanatomic pathology, standard regions of brain were sampled in cerebral lobes and lesions. These tissue blocks were embedded in paraffin wax and the sections stained with hematoxylin and eosin (HE), Luxol fast blue-periodic acid Schiff (LFB/PAS), and other stains as indicated. The neuropathologist then prepared a formal report detailing histopathologic changes in each sampled area (26-42).
Statistical Analysis
All data were entered into Excel (Version 5, Microsoft, Seattle, WA), and were analyzed using StatView SE+Graphics, version 1.04 (Abacus Concepts, Inc, Berkeley, CA). Individual relationships between neuroimaging and histopathology were determined with Kendall rank correlations.
Results
Summary demographic and clinical data are included in Table 1 and SLE-related autoantibody data in Table 2. The MRI findings and histopathology are summarized in Tables 3 and 4. The most common pre mortem MRI findings in fatal NPSLE were small focal white matter lesions (seen in 100% of subjects), moderate to severe cortical atrophy (64%), moderate to severe ventricular dilation (57%), acute cerebral edema and/or acute leukoencephalopathy (50%), chronic diffuse white matter abnormalities (43%), post-infarction or hematoma focal atrophy with or without cyst formation (36%), cerebral infarction (29%), acute or resolved intracranial hemorrhage (21%), and obvious parenchymal calcifications (7%).
Table 3. Neuroimaging Findings and Histopathology.
| Subject | Terminal NPSLE Event | MRI to Death Interval | Cause of Death | Neuroimaging | Brain Histopathology |
|---|---|---|---|---|---|
| 1 | Acute ischemic stroke associated with accelerated SLE | 2 weeks |
|
|
Brain 1145 gms
|
| 2 | Acute confusional state, Fahr's disease, seizure disorder (associated with accelerated SLE) | 1 month |
|
|
Brain 1540 gms
|
| 3 | Acute confusional state, seizure disorder (associated with accelerated SLE) | 4 days |
|
|
Brain 1160 gms
|
| 4 | Chronic multifocal disease with dementia | 1 month |
|
|
Brain 1080 gms
|
| 5 | Acute confusional state and seizure disorder, (associated with accelerated SLE) | 2 weeks |
|
|
Brain 1280 gms
|
| 6 | Seizure disorder (epilepsy) | 1 year |
|
|
Brain 1080 gms
|
| 7 | Seizure disorder (associated with accelerated SLE) | 2 months |
|
|
Brain 1400 gm
|
| 8 | Seizure disorder (epilepsy), headache, and acute confusional state. | 6 months |
|
|
Brain 900 gms
|
| 9 | Acute confusional state | 2 weeks |
|
|
Brain 1250 gms
|
| 10 | Acute confusional state | 5 months |
|
|
Brain 1250 gms
|
| 11 | Acute confusional state, and lupus headache. | 3 weeks |
|
|
Brain 950 gms
|
| 12 | Headache. seizure disorder, and acute confusional state. | 1 week |
|
|
Brain 1230 gms
|
| 13 | Acute confusional state, seizure disorder, and headache. | 6 months |
|
|
Brain 1380 gms
|
| 14 | Status epilepticus Brain death | 1 week |
|
|
Brain 1100 gms
|
Table 4. Sensitivity of MRI and Histopathology.
| NPSLE Entity | Subjects affected | MRI Finding | Histopathologic Correlate | MRI Sensitivity (%/n) | Histo-pathology Sensitivity (%/n) |
|---|---|---|---|---|---|
| Cerebral atrophy | 1-6, 8, 11, & 14 (9 cases) |
|
|
100 (9/9) | 78 (7/9) |
| Large acute/subacute cerebral infarction | 1 (1 lesion) |
|
|
100 (1/1) | 100 (1/1) |
| Small to microscopic acute/subacute infarctions | 1, 3, 5, & 11 (25 lesions) |
|
|
52 (13/25) | 100 (25/25) |
| Resolved large cerebral infarction | 1, 2, 4, 6, & 8 (16 lesions) |
|
|
100 (16/16) | 100 (16/16) |
| Small punctate white matter lesions | 1-14 (14 cases) |
|
|
100 (14/14) | 64 (9/14) |
| Cyst | 1, 6, & 8 (6 lesions) |
|
|
100 (6/6) | 100 (6/6) |
| Acute or subacute large hemorrhage | 12 (1 lesion) |
|
|
100% (1/1) | 100 (1/1) |
| Small punctuate hemorrhage | 3, 5, 12, & 14 (19 lesions) |
|
|
47 (8/19) | 100 (19/19) |
| Acute lupus leukoencephalopathy | 3 & 14(9 lesions) |
|
|
100 (9/9) | 33 (3/9) |
| Cerebrocalcinosis (Fahrs Disease) | 2 (1 case) |
|
|
100 (1/1) | 100 (1/1) |
| Generalized bland acute ischemic injury | 3, 5, 6, & 12-14 (6 cases) |
|
|
66 (4/6) | 100 (6/6) |
In general, gross cerebral autopsy findings, including cerebral infarction, cerebral edema, intracranial hemorrhage, intracranial calcifications, cyst formation, and focal atrophy was predicted by pre mortem MRI (Table 4). Microscopic findings in fatal NPSLE were consistent with acute and chronic vascular and parenchymal injury and included global ischemic changes (57%), parenchymal edema (50%), acute microhemorrhages (43%), glial hyperplasia (43%), diffuse neuronal/axonal loss (36%), old cerebral infarction (33%), microthomboemboli (29%), blood vessel remodeling (29%), acute cerebral infarction (14%), acute macrohemorrhages (14%), extensive vascular and parenchymal calcification (7%), and resolved macrohemorrhages (7%). Examples of paired MRI and histopathologic finding are shown in Figures 1- 9 and are discussed in detail below. Brain mass was correlated with MRI assessments of cortical atrophy by (r = -0.72, p = 0.01) and ventricular dilation (r= -0.77, p =0.01).
Figure 1. NPSLE with Thrombotic Cerebrovascular Disease.
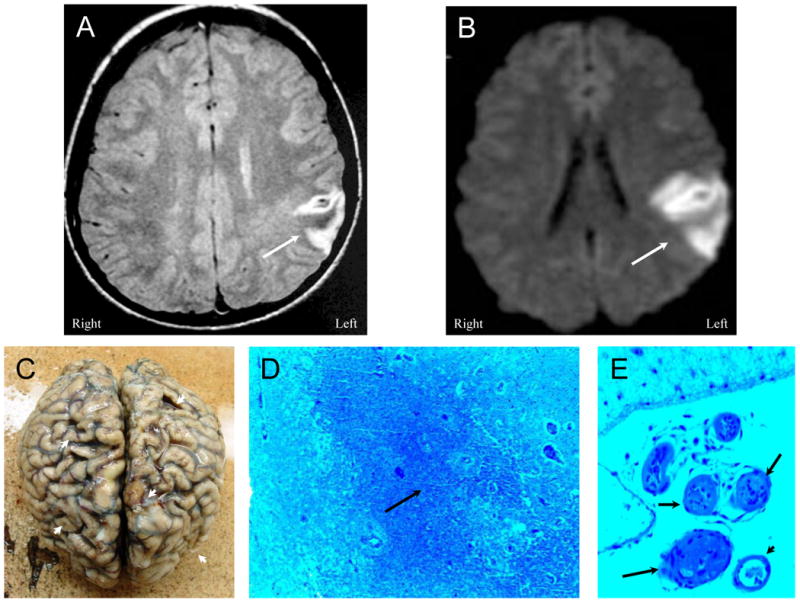
Figure 1A (Patient 1). A proton density MR image demonstrating an acute frontoparietal infarction (arrow) confirmed by a diffusion-weighted image (DWI) showing an area of restricted diffusion (Figure 1B, arrow). Figure 1C. After multiple other cerebral infarctions and death, autopsy demonstrates multiple old and new cortical infarcts with extensive cortical atrophy (arrowheads). Histology reveals extensive ischemic coagulation necrosis, microglial activation, and proliferation (Figure 1D, arrow) associated with frequent thromboembolic vasculopathy characterized by fibrin and platelet thromboembolism that obstructed blood vessels (Figure 1E, arrows) (LFB/PAS stain, Magnification X 50). A solitary non-thrombosed vessel remains in the field of view (arrowhead).
Figure 9. NPSLE with Accelerated SLE, Hypertension, and Intra-cerebral Hemorrhage.
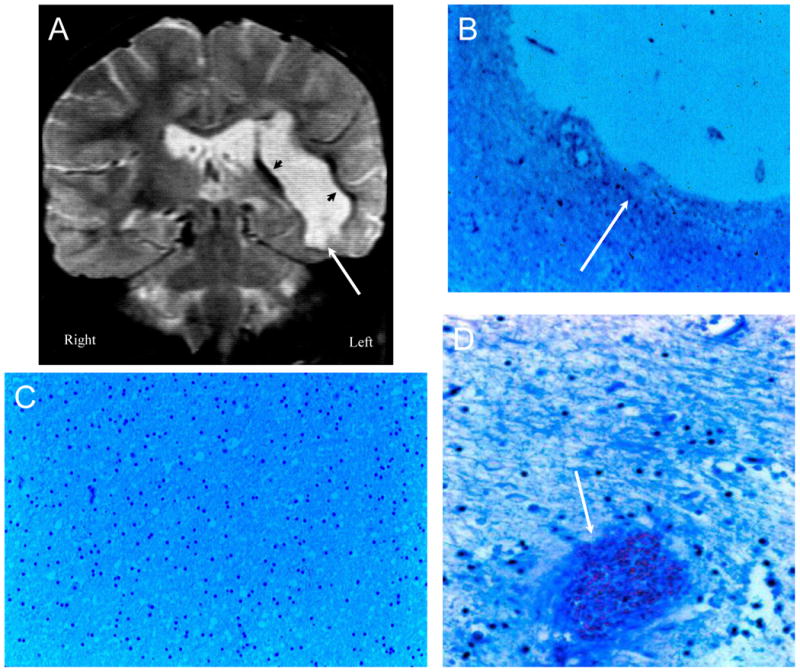
Figure 9A (Subject 8). The T2-weighted MRI demonstrates a large resolved intracerebral hemorrhage of the right hemisphere with formation of a cerebrospinal fluid filled cyst that appears as a hyperintense fluid mass next to the ventricles (arrow). A thin dark rim of hypointensities around the cyst represents dense connective tissue and hemosiderin-laden macrophages (arrowheads). Figure 9B (Subject 8). The cyst is lined with cortical tissue with marked gliosis and hemosiderin-laden macrophages (arrow; LFB/PAS stain, Magnification X 100. Figure 9C (Subject 8). The remainder of the cortex is relative unremarkable (LFB/PAS stain, Magnification X 100). Figure 9D (Subject 12). Sections of the corpus striatum demonstrate focal perivascular siderophages without vasculitis, and satellite hemorrhages (arrow) and edema in the parenchyma surround the hematoma (LFB/PAS stain, Magnification X 100).
Subject 1
This 44 year old (yo) woman with a 14 year history of SLE was increasingly disabled from multiple cerebral infarctions, resulting in an expressive aphasia. Past medical history included photosensitive dermatitis, mouth ulcers, malar rash, pericarditis, glomerulonephritis, renal insufficiency, hypertension, epilepsy, multiple strokes, arthritis, and thrombocytopenia. Physical examination demonstrated extensive livedo reticularis over the extremities, a vasculitic eruption, and an apparent expressive aphasia. The patient suffered a new right frontoparietal stroke resulting in tonsillar herniation and death several days later (Figures 1 and 2).
Figure 2. NPSLE with Thrombotic Cerebrovascular Disease.

Figure 2A (Patient 1). A T2-weighted MRI demonstrating multiple old cortical and white matter infarcts (arrows), small white matter lesions, and a large left sided post infarctive cyst. Figure 2B. Histopathology confirms a large post-infarctive cyst (arrow) consisting of cerebrospinal fluid, loss of neurons and axons, glial proliferation, and debris (LFB/PAS stain, Magnification X 10). Figure 2C demonstrates a resolving ischemic infarct with neuronal loss, gliosis, and resolving infarct with necrotic debris (arrow) (LFB/PAS stain, Magnification X 75). Figure 2D shows blood vessels with complete obliteration of certain small arterioles with fibrin and platelet debris, intimal hyperplasia, and vessel remodeling (arrows), while other adjacent blood vessels remain patent (arrowheads) (LFB/PAS stain, Magnification X 150). There is sparse inflammatory infiltrate around certain of the blood vessels without invasion of the vessel wall.
The final diagnosis was acute stroke superimposed on chronic multifocal disease secondary to active systemic lupus, tonsillar herniation, Libman-Sacks endocarditis, and antiphospholipid antibody syndrome.
Subject 2
This 56 yo man with a 10 year history of SLE experienced multiple hospital admissions complicated by recurrent bouts of systemic infection, leukopenia, thrombocytopenia, peripheral (digital) vasculitis, serositis, acute confusional state, and progressive neurologic deterioration, characterized by hyperreflexia, gait disturbance, dysarthria, emotional lability, depression, cognitive decline, and physical debilitation. The terminal episode was characterized by an acute confusional state, leukopenia, thrombocytopenia, depressed complements, elevated DNA, serositis, and coma followed by death (Figures 3 and 4).
Figure 3. NPSLE with Thrombotic Vasculopathy and Cerebral Calcinosis.
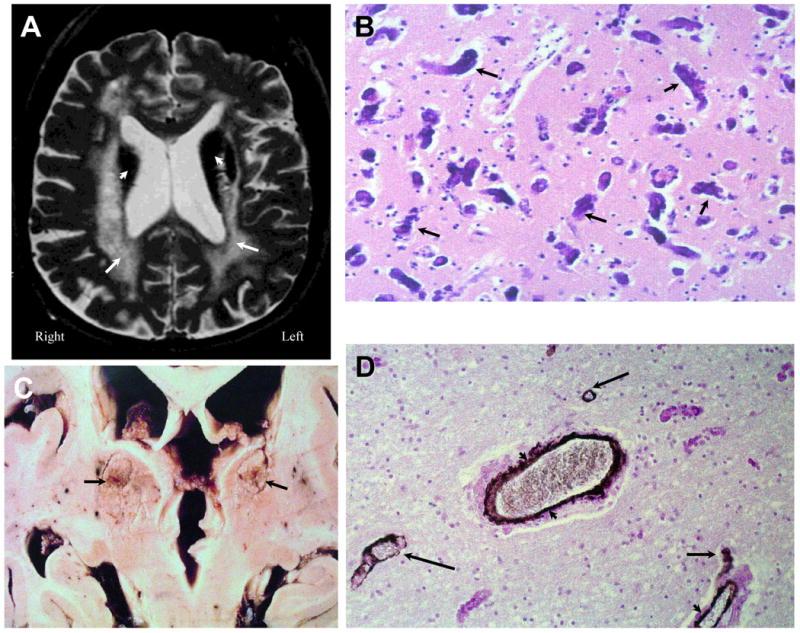
Figure 3A (Subject 2). A T2-weighted image demonstrating cerebral atrophy, ventricular dilation, diffuse periventricular white matter abnormalities (arrows), and severe hypointensities (arrowheads) in the thalamus, putamen, and caudate nucleus. Figure 3B. Histopathology demonstrated extensive heterotrophic calcifications in the form of spheroids (dark irregular concretions; arrows) in frontal gray matter with neuronal loss and minimal gliosis (H&E stain, Magnification X 150). Figure 3C demonstrates gross calcium deposits in the caudate nuclei, thalami, and putamen (arrows). Figure 3D demonstrates white matter with axonal loss, thinning and remodeling of the vascular wall, and calcifications through the adventia and medial of the blood vessels (arrowheads); sparse calcium spheroids are also observed (arrows; H&E stain with anti-actin antibody IHC, Magnification X 200). These findings are diagnostic of Fahr's Disease secondary to SLE.
Figure 4. NPSLE with Thrombotic and Cerebral Calcinosis.
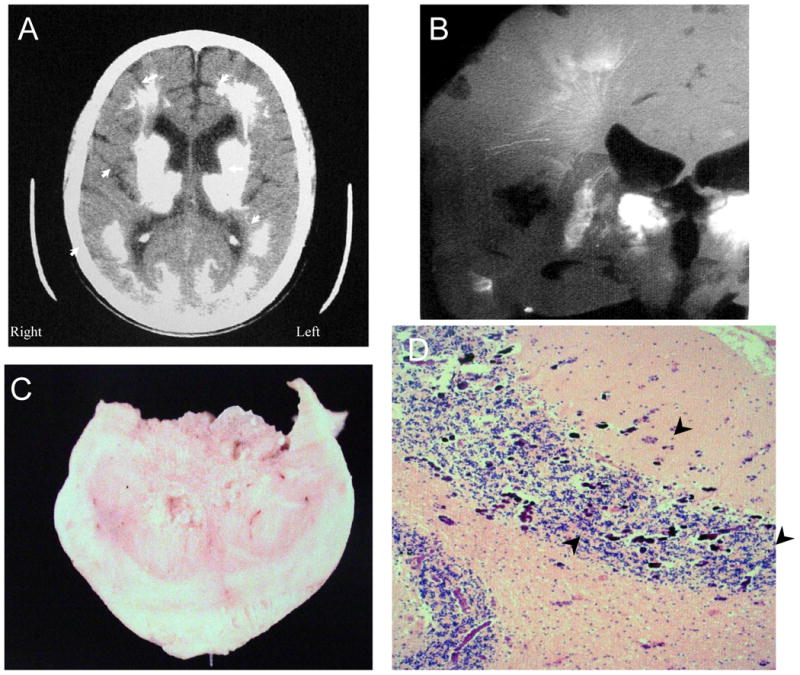
Figure 4 shows further features of the same individual as in Figure 3 (Subject 2). Figure 4A. The computed tomographic (CT) image demonstrates extensive calcification in the thalamus, putamen, caudate nucleus, white matter, and posterior gray matter (arrowheads). Figure 4B. A radiograph of the brain slice at autopsy shows lacey linear and flowering calcifications that follow the arteriolar and venular vasculature, as well as complete calcification of the caudate nucleus (arrowhead). Figure 4C shows one of the calcific spheroids after excision. Figure 4D demonstrates extensive microcalcifications at the gray matter-white matter junctions in the cerebellum (arrowheads; H&E stain, Magnification X 100).
The final diagnosis was cerebrocalcinosis (Fahr's disease) with severe neurologic impairment associated with active SLE and antiphospholipid antibody syndrome with terminal sepsis.
Subject 3
This 22 yo African-American woman with a 3 year history of SLE had been successfully treated with corticosteroids and pulsed cyclophosphamide. Past medical history included acute seizures, photosensitive dermatitis, mouth ulcers, malar rash, pericarditis, glomerulonephritis, hypertension, arthritis, and leukopenia. The patient's family brought her to the clinic because of increasing confusion. Physical examination demonstrated an apprehensive, confused woman with motor retardation and vasculitic lesions in the digits. After admission, the patient rapidly deteriorated and suffered a generalized tonic-clonic seizure followed by coma and necrosis of her fingers and toes. She progressed to respiratory failure and was intubated, experienced further seizures, and developed fatal brain edema with tonsillar herniation and death (Figure 5).
Figure 5. NPSLE with Accelerated SLE Activity and Confusional State.
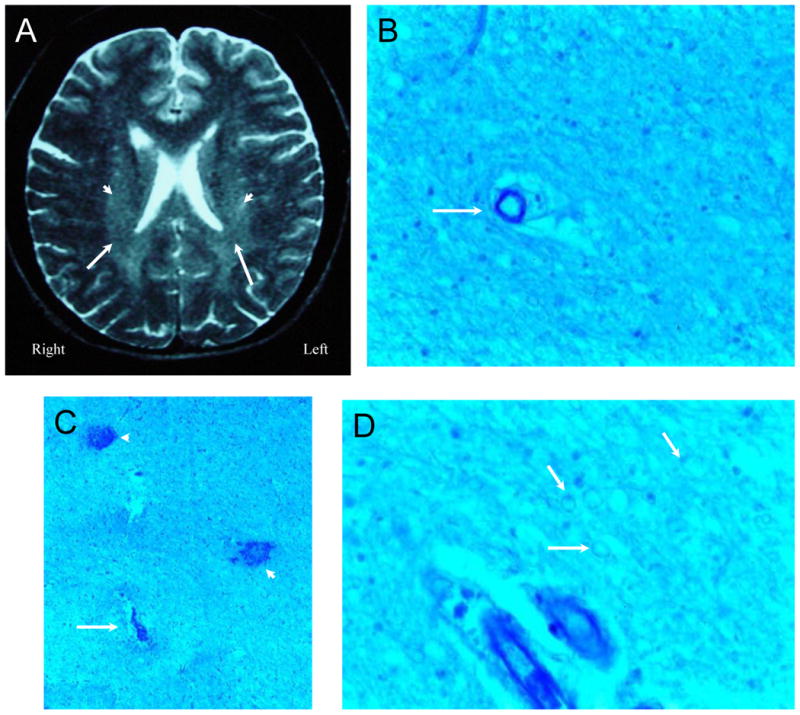
Figure 5A (Subject 3) is a T2-weighted MRI demonstrating vague periventricular white matter hyperintensities (arrowheads), foci of reduced signal in white matter, and scattered focal white matter lesions. Histological changes consisted of ill-defined areas of pallor commonly accentuated in the immediate vicinity of blood vessels and showing average reduction in numbers of oligodendrocytes with generally non-thrombosed blood vessels (Figure 5B, arrow; LFB/PAS stain, Magnification X 150). Figure 5C. In each putamen and left thalamus multiple small areas of necrosis (arrow), frequently hemorrhagic, contained ischemic neurons and showed mild gliosis and sparse macrophages at their periphery with frequent microhemorrhages (arrowheads; LFB/PAS stain, Magnification X 100). Figure 5D. The density of myelinated fibers was diminished and under high magnification, the fibers had a beaded appearance (arrows; LFB/PAS stain, Magnification X 200). The intrinsic and extrinsic cerebral blood vessels showed no abnormalities.
The final diagnosis was cerebral edema, diffuse ischemic encephalopathy, and peripheral necrotizing vasculitis associated with active SLE, Libman-Sacks endocarditis, and antiphospholipid antibody syndrome.
Subject 4
This 43 yo woman with a 14 year history of SLE was characterized by pericarditis, arthritis, multiple cerebral infarcts, depression, rash, headaches, and livedo reticularis. Neurologically, she demonstrated hyperreflexia, decreased cognition, and severe unremitting headaches. She suffered a spontaneous unobserved cardiopulmonary arrest at home and underwent an autopsy by order of the medical examiner (Figure 6).
Figure 6. NPSLE with Chronic Thrombotic Cerebrovascular Disease.
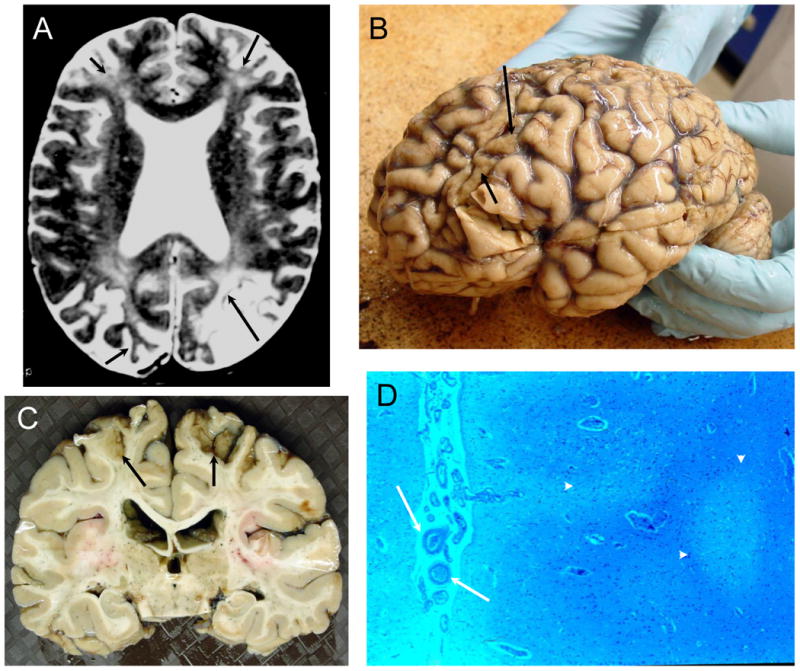
Figure 6A (Subject 4). The T2-weighted MRI demonstrates severe cortical atrophy, severe ventricular dilation, multiple small focal white matter abnormalities, diffuse hyperintensities in deep white matter, and old cortical infarcts with focal atrophy in both frontal lobes, left occipital lobe, and right occipital lobe (arrows). Figure 6B (Subject 4). The frontal lobes were flattened, and the cerebral gyri demonstrated sickle-shaped atrophic post-ischemic bands in the frontal and occipital areas (arrows) with evident generalized atrophy. Figure 6C (Subject 4). The brain slice demonstrates the focal frontoparietal post-ischemic atrophy superiorly (arrows). Figure 6D (Subject 4). The cortex is unevenly depleted of neurons with evident atrophy (arrowheads), sometimes laminar in distribution, with increases in astrocytes nuclei and adjacent reductions in white matter and axons with some glial hyperplasia and evidence of thromboembolic vasculopathy (arrows; LFB/PAS stain, Magnification X 50).
The final diagnosis was chronic multifocal disease, respiratory arrest, and diffuse ischemic encephalopathy due to excessive narcotic analgesic use in the setting of active SLE, Libman-Sacks endocarditis, and antiphospholipid antibody syndrome.
Subject 5
This 19 yo woman had SLE of 3 years duration complicated by glomerulonephritis, isolated seizures, arthritis, and recurrent headaches. She developed acute seizures and encephalopathy, and was found to have progressive Libman-Sacks valvular heart disease. Following mitral valve replacement, she developed further seizures and confusion, sepsis, and experienced respiratory arrest and coma (Figures 7 A and B). The terminal event was brain edema with tonsillar herniation.
Figure 7. NPSLE with Accelerated SLE Activity and Isolated Seizures.
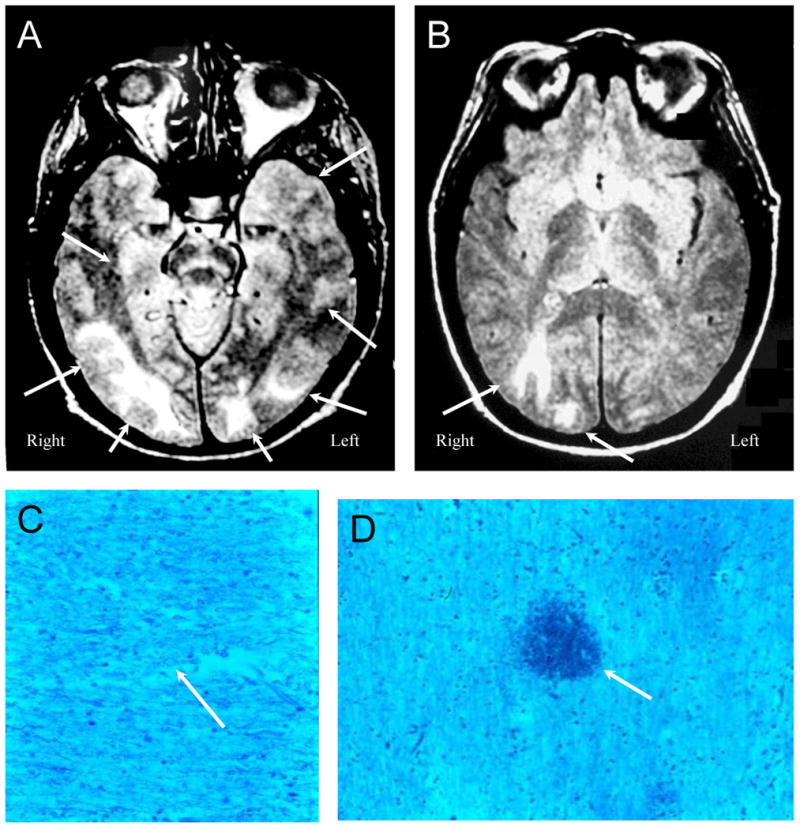
Figures 7A and B (Subject 5). Proton density images demonstrate multiple white matter lesions in the frontal, parietal, occipital, and temporal lobes consistent with acute lupus leukoencephalopathy (“posterior” leukoencephalopathy, arrows) characterized by seizures and accelerated SLE disease activity. Figures 7C and D (Subject 5) demonstrate bland global ischemic changes. The density of myelinated fibers is diminished, and under high magnification the fibers have a beaded appearance typical of various stages of ischemic degeneration (Figure 7C, arrow) with interspersed microhemorrhages (Figure 7D, arrow; LFB/PAS stain, Magnification X 100).
The final diagnosis was active SLE, cerebral edema with tonsillar herniation, diffuse ischemic encephalopathy, thromboembolic ischemic brain disease, and Libman-Sacks endocarditis complicated by sepsis.
Subject 6
This 42 yo woman had a 17 year history of SLE complicated by multiple cerebral infarctions, glomerulonephritis, and epilepsy. Pre mortem MRI demonstrated diffuse cortical atrophy, multiple punctate hyperintense lesions on T2/FLAIR MR images, and multiple resolved cortical infarcts consisting of focal cortical atrophy with underlying hyperintense white matter changes (Tables 3 and 4). She suffered an epileptic attack at home and developed irreversible fatal anoxic brain damage. Autopsy demonstrated atrophy of cerebral cortex with parenchymal loss, small cysts, adjacent areas of macrophages and reactive astrocytosis, as well as focal areas of hippocampal neuronal loss (Tables 3 and 4).
The final diagnosis was global ischemic encephalopathy due to epilepsy, chronic multifocal disease, SLE, and antiphospholipid antibody syndrome exacerbated by non-compliance with medications.
Subject 7
This 45 yo woman with a 20 year history of SLE complicated by epilepsy, autoimmune hepatitis, pancreatitis, and cognitive difficulties had been doing well. The patient experienced a generalized tonic-clonic seizure and suffered irreversible anoxic brain damage and death (Figure 8 A-C).
Figure 8. NPSLE with Epilepsy and Sudden Death.
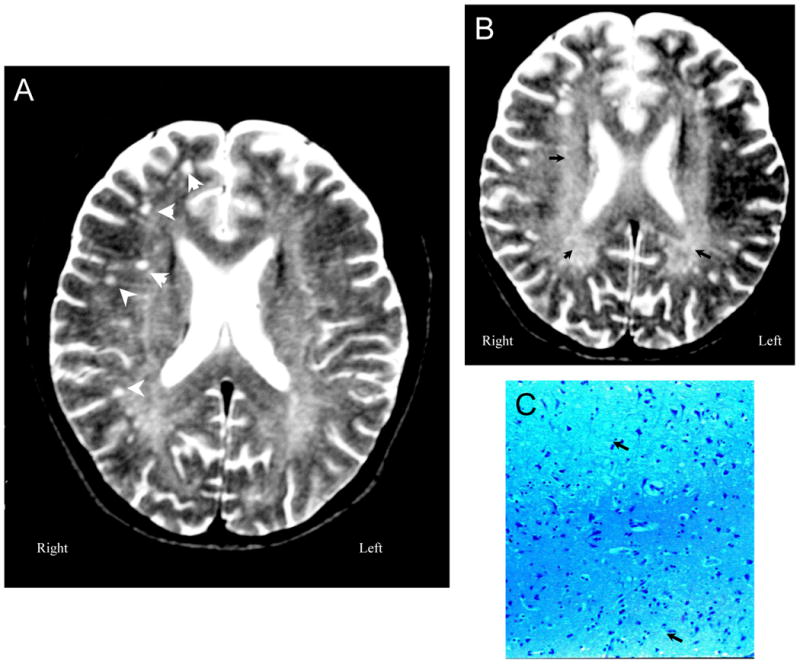
Figure 8A (Subject 7). T2-weighted MRI demonstrates moderate cerebral atrophy and ventricular dilation with multiple small focal white matter lesions in frontoparietal lobes (arrowheads). Figure 8B (Subject 7) shows diffuse generalized deep white matter abnormalities with increased signal (arrowheads). Figure 8C (Subject 7). Sections through white and gray matter demonstrate minimal changes with occasional vague reductions in neuron and axon numbers in a patchy distribution (arrowheads;LFB/PAS stain, Magnification X 100).
The final diagnosis was global ischemic encephalopathy due to seizure activity in the setting of SLE.
Subject 8
This 11 yo girl with a 4 year history of SLE complicated by glomerulonephritis, epilepsy, a large intracerebral hemorrhage (Figure 9 A-C), renal failure, and renal transplant rejection died abruptly from cardiac arrhythmias.
The final diagnosis was diffuse ischemic encephalopathy due to cardiac arrest, myocarditis with involvement of the conduction system, resolved massive intracerebral hemorrhage, and resolved cerebellar infarct.
Subject 9
This 27 yo woman with a 5 year history of SLE characterized by arthritis, pericarditis, hematological abnormalities, and lupoid hepatitis, was treated successfully with azathioprine and prednisone. She abruptly stopped her immunosuppression, and 2 weeks later developed chest pain, abdominal pain, fever, and hypotension. Pre mortem MRI demonstrated minimal cerebral atrophy, and a few hyperintense white matter lesions on T2/FLAIR imaging (Tables 3 and 4). She was admitted to the intensive care unit, but progressed to an acute confusional state with hypotension and died. The final diagnosis was diffuse global ischemic encephalopathy, active SLE, and pneumonia.
The diagnosis was active SLE, pneumonia, and sepsis.
Subject 10
This 65 yo woman had a 20 year history of SLE characterized by cognitive complaints, arthritis, headache, and recurrent serositis. Pre mortem MRI demonstrated minimal cerebral atrophy, septa in the lateral ventricles, and moderate hyperintense focal white matter lesions on T2/FLAIR imaging (Tables 3 and 4). She suddenly developed severe abdominal pain, and was found to have evidence of an acute myocardial infarction. Subsequently, she developed an acute confusional state followed by cardiopulmonary arrest and death. Brain autopsy found atherosclerosis of the basilar, carotid, and anterior cerebral arteries. There were a few areas of resolved infarction with reduced numbers of neurons and axons, and choroid plexus cysts in the lateral ventricles.
The final diagnosis was acute myocardial infarction with hypotension, atherosclerotic cerebrovascular disease, and resolved cerebral infarctions in the setting of SLE.
Subject 11
This 48 yo woman with an 18 year history of SLE characterized by psychosis, depression, headache, seizures, and glomerulonephritis, became depressed and confused, was admitted to the hospital and developed pericarditis, pneumonitis, and pancreatitis. Pre mortem MRI demonstrated severe cortical atrophy and ventricular dilation, multiple extensive focal white matter abnormalities, chronic diffuse white matter abnormalities, and late in the course, cerebral edema (Tables 3 and 4). She was treated with corticosteroids and cyclophosphamide, and initially improved, but then developed confusion, coma, and sepsis and died. Autopsy revealed small lesions (<0.5 cm) consistent with acute infarcts in frontal cortex, caudate nucleus, and right parietal white matter with peripheral zones of edema and necrotic core with macrophages, gliosis, and swollen axons. There were also areas of reduced neuronal density consistent with small prior infarcts. There was frontal cortical atrophy, moderate gliosis of the thalami, and diffuse loss of Purkinje cells consistent with chronically recurrent vascular brain injury.
The final diagnosis was active SLE with multiple cerebral infarcts complicated by sepsis.
Subject 12
This 19 year old woman with 3 year history of SLE was 34 weeks pregnant and developed peripheral edema, hypertension, and proteinuria. She abruptly experienced a seizure and became unresponsive, and was resuscitated and intubated. The infant was delivered by Caesarian section; however, the patient did not improve. On MRI she was found to have multiple intracerebral hemorrhages with surrounding parenchymal edema (Tables 3 and 4) that progressed to global cerebral edema, tonsillar herniation, and death. On autopsy, the brain was edematous with diffuse ischemic changes. There was subfalcian and cerebellar tonsillar herniation, two large intraparenchymal hemorrhages, mild perivascular inflammation with perivascular siderophages, and small satellite hemorrhages (Figure 9 D).
The final diagnosis was active SLE, lupus encephalopathy, hypertensive encephalopathy, cerebral edema, tonsillar herniation, and intracranial hemorrhage.
Subject 13
This 36 yo woman with an 8 year history of SLE complicated by serositis, hepatitis, glomerulonephritis, depression, and memory complaints experienced increasing confusion after seizure-like activity. Pre mortem MRI demonstrated minimal cortical atrophy and a few hyperintense focal white matter lesions (Tables 3 and 4). She subsequently experienced cardiopulmonary arrest followed by death. Brain histopathology demonstrated generalized diffuse ischemic changes, but otherwise was unremarkable.
The final diagnosis was active SLE, Libman-Sacks endocarditis, seizure disorder, pneumonia, and sepsis.
Subject 14
This 13 yo girl with a 5 year history of SLE characterized by arthritis, serositis, glomerulonephritis, and epilepsy, suffered active SLE with glomerulonephritis culminating in an intractable seizure that developed into unremitting status epilepticus. Pre mortem MRI demonstrated a few hyperintense focal white matter lesions, acute leukoencephalopathy in the putamen, pons, and right cerebellar areas with late generalized cerebral edema but minimal cortical atrophy (Tables 3 and 4). She eventually succumbed to cerebral edema with tonsillar herniation. Brain autopsy demonstrated bland global ischemic changes characterized by ill-defined areas of pallor commonly accentuated in the immediate vicinity of blood vessels. In Luxol fast blue stain, the density of myelinated fibers was diminished and under high magnification, the fibers had a beaded appearance with interspersed microhemorrhages.
The final diagnosis was status epilepticus, active SLE, glomerulonephritis, and acute lupus leukoencephalopathy.
Discussion
There are a number of prior paired MRI-autopsy case reports and a few classic SLE autopsy series (26-42). However, this is the first prospective study of NPSLE to systematically examine the histopathologic basis for the MRI findings in NPSLE by comparing pre mortem MRI with histopathology at autopsy (Tables 3 and 4).
The present study demonstrates that thromboembolism and hypercoagulability are dominant mechanisms for fatal NPSLE and are manifested histologically by the frequent presence of arterial macro- and microthrombi, focal lesions diagnostic of infarct, vascular remodeling, the presence of antiphospholipid antibodies, and the high incidence of Libman-Sacks endocarditis, a known source of thromboemboli in NPSLE (43-46). Diffuse endothelial injury is confirmed by the frequent involvement of small vessels, endothelial hyperplasia, the presence of microthrombi, and the frequent obvious focal or generalized brain edema, suggesting breakdown of the blood-brain barrier. These histopathologic findings of the present study indicate that the basic underlying pathologic process of NPSLE is cerebrovascular injury associated with disease activity and thromboembolism, resulting in focal and diffuse brain ischemia, small and large brain infarcts, focal and diffuse brain edema, brain hemorrhage, and focal and diffuse parenchymal injury (Tables 3 and 4).
Despite the obvious and pervasive histopathologic cerebrovascular changes in the present autopsy series, excitotoxicity may also be suggested by the high incidence of fatal or intractable seizures, diffuse cerebral atrophy without obvious infarct, and the frequent diffuse and patchy areas of neuronal loss without necrosis (47-49). Thus, although cerebrovascular injury appears to be the dominant underlying histopathologic process of NPSLE, the present study does not definitively exclude multiple coexisting pathogenic mechanisms underlying NPSLE - including thromboembolism, hypercoagulability, diffuse endothelial injury, and excitotoxicity (1-3,10,43-49). The present study design did not specifically address the role of antineuronal, excitotoxic, or anti-N-methyl-D-aspartic acid (NMDA) receptor antibodies (anti-NR2 antibodies); however, if present, it is likely that these antibodies would amplify the neuronal injury initiated by the primary vascular insult (31, 34-36, 47-49).
MRI is currently the anatomic imaging modality of choice in NPSLE (3,7,10-13). MRI is exceptionally sensitive for cerebral infarcts, central nervous system (CNS) hemorrhage, and transverse myelitis in NPSLE and can help exclude certain confounding disorders including infectious meningitis, brain abscess, and mycotic aneurysms (10). In the present study MRI was 100% sensitive for large anatomic lesions including cerebral infarct, focal edema, cyst, and macrohemorrhage when compared with brain histopathology at autopsy (Table 4). Specificity for individual lesions could not be accurately determined in the present study, as the number of gross lesions of each type was limited, and there were no comparison groups, including controls and subjects with confounding disease to determine true sensitivity.
Cerebral Atrophy
Atrophy on MRI, a common finding in NPSLE, was present in the majority of our subjects. The histopathologic findings associated with MRI-visible cerebral atrophy were highly variable, and included multiple infarcts and reduced neuronal density suggesting that atrophy in NPSLE may be associated with both generalized and focal brain injury (Tables 3 and 4). However, normal histological appearance was also noted in some atrophic brains. Although FLAIR imaging is often used to detect common NPSLE abnormalities, we found that conventional T2-weighted images were the most striking for visual detection of cortical atrophy and ventricular dilation, because cerebrospinal fluid is markedly hyperintense relative to skull and brain parenchyma on T2-weighted images (Figure 6A). Although brain mass (at autopsy) was significantly correlated with both cortical atrophy and ventricular dilation determined by MRI, MRI was more sensitive than brain histopathology for the presence of cerebral atrophy in NPSLE (Table 4). This is not surprising since there is variable brain shrinkage during formalin fixation making volume determination on autopsy challenging. Moreover, MRI in situ provides more global information than does the fixed brain since the volume of cerebrospinal fluid and brain tissue can be independently measured. Hence, with MRI it is possible to assess cerebral atrophy by comparison of brain volume to intracranial volume, including the amount of ventricular and pericortical cerebrospinal fluid and the relationship of the intracranial volume to brain volume permitting a more accurate estimate of prior brain volume and present brain volume, and thus atrophy.
Lesions
Acute lesions on T2-weighted, FLAIR, or diffusion-weighted images include new infarct, discrete gray matter lesions, diffuse grey matter hyperintensities, and cytotoxic edema (21,23-25). In the present study, MRI and DWI found 100% of large acute focal infarcts that were confirmed by histopathology (Table 4). Similarly, MRI was 100% sensitive to detecting resolved large infarcts, characterized by focal atrophy, cyst formation, and adjacent white matter changes, as confirmed by histopathology (Table 4). Thus, MRI is sensitive for large acute or resolved cerebral infarcts in NPSLE. In contrast, microinfarcts that were commonly noted on brain histopathology were often not obvious by MRI - probably because of inherent resolution limitations of the MRI technique. Thus, it should be recognized that more advanced high-resolution MRI approaches now available might be more sensitive to small lesions including microinfarcts.
Previous studies indicate that MRI may detect chronic focal lesions in 25-50% of patients with the number of these lesions increasing with SLE severity, patient age, and a history of NPSLE (3-7). In our study, 100% of the subjects demonstrated some form of chronic lesion. Small punctate focal lesions in white matter are most common (15-60%), followed in prevalence by cortical atrophy, ventricular dilation, periventricular white matter changes, diffuse white matter changes, and gross infarct (4-7,12). The present study of severely ill patients generally confirms these previous reports (Table 4). Small focal lesions are concentrated in subcortical white matter, especially in the frontoparietal regions, but may be seen elsewhere (11-16). The small punctate focal lesions visible on T2-weighted and FLAIR MRI in NPSLE appear similar to those reported in normal aging, although they occur much earlier in SLE subjects and in greater numbers (1-6,58-60).
Histopathology and neuroimaging from prior studies have suggested that the small focal lesions in NPSLE are a vascular phenomenon representing small infarcts, and the present study is broadly confirmatory (Table 4), although on autopsy individual small lesions on MRI may be difficult to identify exactly on histology due to imperfect registration (26-42). In this context, the present study suggests that MRI is more sensitive than brain histology for small focal white matter lesions, in part because it is easier to sample the entire brain with MRI as compared to histopathology that usually only samples selected areas (Table 4). The present study also suggests that small focal white matter lesions on T2-weighted or FLAIR imaging are usually small resolved infarcts or focal areas of reduced neuronal density, but in some cases they may be acute infarcts, focal edema, or even acute microhemorrhages (Table 4). Thus, small focal white matter lesions should not be viewed in the setting of NPSLE as a benign or incidental finding. On the other hand, these lesions should also not be viewed as a de facto sign of active brain disease since most are chronic and persist over many years; rather, small focal white matter lesions on T2-weighted or FLAIR imaging should be viewed as tendency towards acute and chronic cerebrovascular injury that require further evaluation as to etiology and prevention (3,4,12-16).
Seizures
Generalized isolated seizures in particular may be accompanied by reversible focal high intensity lesions on T2-weighted or FLAIR imaging in both white and gray matter (Figure 7). In the present study, such seizures often directly preceded fatal NPSLE, indicating that acute seizures in NPSLE should be viewed ominously (Tables 1 and 2). Nonetheless, if the patient survives the acute seizure episode, these lesions generally resolve within four weeks (4,8,10,13). Thus, MRI studies may show extensive bilateral, potentially reversible, white-matter abnormalities in the cerebral hemispheres, the brain stem, or the cerebellum usually associated with active NPSLE -the so-called “acute posterior leukoencephalopathy” as shown in Figure 7(10,13,14). In our study, histopathology suggested the reversible lesions of acute leukoencephalopathy of NPSLE were due to focal cerebral edema associated with blood vessel injury and microhemorrhages, although in many cases histopathology did not demonstrate specific lesions (Table 4). This is not surprising since histopathologic detection of blood-brain barrier breakdown in autopsy specimens requires specific stains for relevant serum proteins in brain tissues that were not used in the present study; thus, the presence of multifocal edema is unlikely to have been detected reliably.
Cerebral calcinosis
Fahr's disease (cerebral calcinosis) is known to complicate SLE resulting in a distinct form of NPSLE characterized by progressive movement disorder, Parkinsonian features, dysarthria, disability, and dementia associated with progressive calcinosis of the brain parenchyma, nuclei, and arterial media (61-64). Fahr's disease in the setting of NPSLE should be viewed as a progressive, disabling and eventually fatal condition. The etiology of the calcifications in Fahr's disease of NPSLE has been associated with antiphospholipid antibodies, antibodies to glial fibrillary acidic protein, and chronic vascular injury although there is likely a genetic aspect that predisposes to this unusual complication (65-67). Subject 2 (Figures 3 and 4) is a classic case of Fahr's disease associated with SLE. In this case, besides the heterotrophic calcifications, the most prominent finding was histological evidence of chronically recurrent vascular injury typical of NPSLE.
Hemorrhage
MRI was 100% sensitive for acute or resolved large hemorrhages by histopathology (Table 4). Resolved intraparenchymal hemorrhages created cysts (Figure 9A), similar to those resulting from large intraparenchymal infarcts (Figure 2A). The present study suggests post-hemorrhagic cysts can be differentiated from post-infarct cysts by the markedly hypointense hemosiderin-laden lining at the cyst/parenchyma interface on MRI (Figures 2A and 9A). Benign congenital cysts, which were also observed in this study, are usually differentiated by their anatomic position, and the lack of surrounding parenchymal injury. Microhemorrhages on histopathology corresponded to small foci of altered (reduced or increased) intensity on MRI or to normal-appearing brain (Table 4, Figure 7). However, in contrast to macrohemorrhages, microhemorrhages were often present by histopathology, but not recognized on MRI (Table 4). We suspect this may be another consequence of the limited resolution of MRI and to the variable relaxation of extravasated blood, which can cause increased signal with intact red blood cells, normal signal with partially-lysed red blood cells, and reduced signal with completely-lysed red blood cells and end-stage hemosiderin (68). As with micro-infarcts discussed above, the availability of higher-resolution imaging, perhaps including susceptibility weighted imaging, might yield improved sensitivity for some microhemorrhages (69).
Although magnetization transfer imaging (MTI), diffusion tensor imaging (DTI), or MR perfusion weighted imaging (MR PWI) were not used in the present study, the histopathologic results of the present study confirm the presence of extensive gross and subtle parenchymal and cerebrovascular injury that has been suggested by these advanced MR techniques (50-56). The present study suggests that brain lesions by MRI represent both current NPSLE activity and prior brain damage caused by previous episodes NPSLE (21,57). Recent studies have suggested several patterns of cerebrovascular disease NPSLE: 1) an antiphospholipid antibody cerebrovasculopathy characterized by bland thromboses, thrombotic microangiopathy and arterial intimal fibrous hyperplasia, 2) a diffuse cerebrovasculopathy characterized by endothelial injury associated with increased SLE disease activity, glomerulonephritis, hypertension, and perhaps neuroexcitotoxic antibodies, 3) thromboembolic NPSLE directly caused by cardiac valvular lesions, and 4) mixed cerebrovascular NPSLE with simultaneous aspects of antiphospholipid-associated thrombosis, increased disease activity and thromboembolic valvular lesions (1-3,10,43-46). Immune deposits and classic vasculitis (inflammatory or necrotic involvement of the vessel wall) are rare in the cerebral vessels in NPSLE (3-5%), and the present study confirms the rarity of true CNS system vasculitis in NPSLE (32).
In summary, fatal NPSLE is characterized by variable pre mortem MRI findings of small focal white matter lesions, cortical atrophy, ventricular dilation, cerebral edema, acute leukoencephalopathy, diffuse white matter abnormalities, focal atrophy, cyst formation, cerebral infarction, intracranial hemorrhage, and occasionally, extensive parenchymal calcifications consistent with Fahr's disease. The present paired neuroimaging-autopsy study in NPSLE demonstrates that brain abnormalities apparent by MRI represent serious underlying anatomic brain injury characterized by acute and chronic cerebrovascular and parenchymal brain injury.
Acknowledgments
Sources of Support: This work was supported by research grants from the US National Institutes of Health including R01 HL077422 to Dr. Roldan, R01 NS039123 to Dr. Brooks, and R01 NS035708 to Dr. Sibbitt.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Wilmer L. Sibbitt, Jr., Department of the University of New Mexico Heath Sciences Center, Albuquerque, NM, USA.
William M. Brooks, Department of Hoglund Brain Imaging Center, University of Kansas Medical Center, Kansas City, KS, USA.
Mario Kornfeld, Department of the University of New Mexico Heath Sciences Center, Albuquerque, NM, USA.
Blaine L. Hart, Department of the University of New Mexico, Albuquerque, NM, USA.
Arthur D. Bankhurst, Department of the University of New Mexico Heath Sciences Center, Albuquerque, NM, USA.
Carlos A. Roldan, Department of the University of New Mexico Heath Sciences Center, Albuquerque, NM, USA.
References
- 1.Hanly JG, Urowitz MB, Su L, Sanchez-Guerrero J, Bae SC, Gordon C, Wallace DJ, et al. Systemic Lupus International Collaborating Clinics. Short-term outcome of neuropsychiatric events in systemic lupus erythematosus upon enrollment into an international inception cohort study. Arthritis Rheum. 2008;59:721–9. doi: 10.1002/art.23566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanly JG. The neuropsychiatric SLE SLICC inception cohort study. Lupus. 2008;17:1059–63. doi: 10.1177/0961203308097568. [DOI] [PubMed] [Google Scholar]
- 3.McCune WJ, MacGuire A, Aisen A, Gebarski S. Identification of brain lesions in neuropsychiatric systemic lupus erythematosus by magnetic resonance scanning. Arthritis Rheum. 1988;31:159–166. doi: 10.1002/art.1780310202. [DOI] [PubMed] [Google Scholar]
- 4.Brooks WM, Sabet A, Sibbitt WL, Jr, Barker PB, van Zijl PCM, Duyn JH, et al. Neurochemistry of brain lesions determined by spectroscopic imaging in systemic lupus erythematosus. J Rheum. 1997;24:2323–2329. [PubMed] [Google Scholar]
- 5.Sundgren PC, Jennings J, Attwood JT, Nan B, Gebarski S, McCune WJ, et al. MRI and 2D-CSI MR spectroscopy of the brain in the evaluation of patients with acute onset of neuropsychiatric systemic lupus erythematosus. Neuroradiology. 2005;47:576–85. doi: 10.1007/s00234-005-1371-y. [DOI] [PubMed] [Google Scholar]
- 6.Friedman SD, Stidley C, Brooks WM, Hart BL, Sibbitt WL., Jr Brain injury and neurometabolic abnormalities in systemic lupus erythematosus. Radiology. 1998;209:79–8. doi: 10.1148/radiology.209.1.9769816. [DOI] [PubMed] [Google Scholar]
- 7.Sibbitt WL, Jr, Haseler L, Griffey RH, Hart B, Sibbitt RR, Matwiyoff N. Analysis of cerebral structural changes in systemic lupus erythematosus by MR proton spectroscopy. Am J Neurorad. 1994;15:923–928. [PMC free article] [PubMed] [Google Scholar]
- 8.Sibbitt WL, Jr, Brooks WM, Haseler LJ, Griffey RH, Frank LM, Hart BL, et al. Spin-spin relaxation of brain tissues in systemic lupus erythematosus. Arthritis Rheum. 1995;38:810–818. doi: 10.1002/art.1780380615. [DOI] [PubMed] [Google Scholar]
- 9.Sibbitt WL, Jr, Haseler LJ, Griffey RR, Friedman SD, Brooks WM. Neurometabolism of active neuropsychiatric lupus determined by proton MR spectroscopy. AJNR. 1997;18:1271–1277. [PMC free article] [PubMed] [Google Scholar]
- 10.Sibbitt WL, Jr, Sibbitt RR, Brooks WM. Neuroimaging in neuropsychiatric SLE. Arthritis Rheum. 1999;42:2026–2038. doi: 10.1002/1529-0131(199910)42:10<2026::AID-ANR2>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 11.Sabet A, Sibbitt WL, Jr, Stidley CA, Danska J, Brooks WM. Neurometabolite markers of cerebral injury in the antiphospholipid antibody syndrome of systemic lupus erythematosus. Stroke. 1998;29:2254–2260. doi: 10.1161/01.str.29.11.2254. [DOI] [PubMed] [Google Scholar]
- 12.Ishikawa O, Ohnishi K, Miyachi Y, Ishizaka H. Cerebral lesions in systemic lupus erythematosus detected by magnetic resonance imaging. Relationship to anticardiolipin antibody. J Rheumatol. 1994;21:87–90. [PubMed] [Google Scholar]
- 13.Jarek M, West SG, Baker MR, Rak KM. Magnetic resonance imaging in systemic lupus erythematotus patients without a history of neuropsychiatric lupus erythematosus. Arthritis Rheum. 1994;37:1609–1613. doi: 10.1002/art.1780371108. [DOI] [PubMed] [Google Scholar]
- 14.Sibbitt WL, Jr, Sibbitt RR, Griffey RH, Eckel CG, Bankhurst AD. Magnetic resonance and CT imaging in the evaluation of acute neuropsychiatric disease in systemic lupus erythematosus. Ann Rheum Dis. 1989;48:1014–1022. doi: 10.1136/ard.48.12.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kozora E, West SG, Kotzin BL, Julian I, Porter S, Bigler E. Magnetic resonance imaging abnormalities and cognitive defects in systemic lupus erythematosus patients without overt central nervous system disease. Arthritis Rheum. 1998;41:41–47. doi: 10.1002/1529-0131(199801)41:1<41::AID-ART6>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 17.Tan EM, Cohen AS, Fries JF, Masi AT, McShane DJ, Rothfield NF, et al. 1982 Revised criteria for the classification of systemic lupus erythematosus. Arth Rheum. 1982;25:1271–1277. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 18.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725–26. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 19.Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH, editors. The Committee on Prognosis Studies in SLE: Derivation of the SLEDAI. A disease activity index for lupus patients. Arthritis Rheum. 1992;35:630–640. doi: 10.1002/art.1780350606. [DOI] [PubMed] [Google Scholar]
- 20.Gladman D, Ginzler E, Goldsmith C, Fortin P, Liang M, Urowitz M, et al. The development and initial validation of the Systemic Lupus International Collaborating Clinics/American College of Rheumatology damage index for systemic lupus erythematosus. Arthritis Rheum. 1996;39:363–369. doi: 10.1002/art.1780390303. [DOI] [PubMed] [Google Scholar]
- 21.Sibbitt WL, Jr, Schmidt PJ, Hart BL, Brooks WM. Fluid Attenuated Inversion Recovery (FLAIR) imaging in neuropsychiatric systemic lupus erythematosus. J Rheumatol. 2003;30:1983–9. [PubMed] [Google Scholar]
- 22.The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999;42:599–608. doi: 10.1002/1529-0131(199904)42:4<599::AID-ANR2>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 23.Walecki J, Sierakowski S, Lewszuk A, Sulik A, Tarasow E, Lebkowska U. MR in neurological syndromes of connective tissue diseases. Med Sci Monit. 2002;8:MT105–11. [PubMed] [Google Scholar]
- 24.Moritani T, Shrier DA, Numaguchi Y, Takahashi C, Yano T, Nakai K, Zhong J, Wang HZ, Shibata DK, Naselli SM. Diffusion-weighted echo-planar MR imaging of CNS involvement in systemic lupus erythematosus. Acad Radiol. 2001;8:741–53. doi: 10.1016/S1076-6332(03)80581-0. [DOI] [PubMed] [Google Scholar]
- 25.Bosma GP, Huizinga TW, Mooijaart SP, Van Buchem MA. Abnormal brain diffusivity in patients with neuropsychiatric systemic lupus erythematosus. AJNR Am J Neuroradiol. 2003;24:850–4. [PMC free article] [PubMed] [Google Scholar]
- 26.Funata N. Cerebral vascular changes in systemic lupus erythematosus. Bull Tokyo Med Dent Univ. 1979;26:91–112. [PubMed] [Google Scholar]
- 27.Ellison D, Gatter K, Heryet A, Esiri M. Intramural platelet deposition in cerebral vasculopathy of systemic lupus erythematosus. J Clin Pathol. 1993;46:37–40. doi: 10.1136/jcp.46.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shiozawa S, Kuroki Y, Kim M, Hirohata S, Ogino T. Interferon-alpha in lupus psychosis. Arthritis Rheum. 1992;35:417–422. doi: 10.1002/art.1780350410. [DOI] [PubMed] [Google Scholar]
- 29.Johnson R, Richardson EP. The neurological manifestations of systemic lupus erythematosus. A clinical-pathological study of 24 cases and review of the literature. Medicine. 1968;47:337–369. doi: 10.1097/00005792-196807000-00002. [DOI] [PubMed] [Google Scholar]
- 30.Ellis SG, Verity MA. Central nervous systemic involvement in systemic lupus erythematosus: a review of neuropathological findings in 57 cases, 1955-1977. Semin Arthritis Rheum. 1979;8:212–221. doi: 10.1016/s0049-0172(79)80009-8. [DOI] [PubMed] [Google Scholar]
- 31.Hanly JG, Walsh N, Sangalang V. Brain pathology in systemic lupus erythematosus. J Rheumatol. 1992;19:732–741. [PubMed] [Google Scholar]
- 32.Devinsky O, Petito CK, Alonso DR. Clinical and neuropathological findings in systemic lupus erythematosus: The role of vasculitis, heart emboli, and thrombotic thrombocytopenic purpura. Ann Neurol. 1988;23:380–384. doi: 10.1002/ana.410230411. [DOI] [PubMed] [Google Scholar]
- 33.Sakaki T, Morimoto T, Utsumi S. Cerebral transmural angiitis and ruptured cerebral aneurysms in patients with systemic lupus erythematosus. Neurochirurgia (Stuttg) 1990;33:132–135. doi: 10.1055/s-2008-1053572. [DOI] [PubMed] [Google Scholar]
- 34.Hammad A, Tsukada Y, Torre N. Cerebral occlusive vasculopathy in systemic lupus erythematosus and speculation on the part played by complement. Ann Rheumatic Dis. 1992;51:550–552. doi: 10.1136/ard.51.4.550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hopkins P, Belmont HM, Buyon J, Philips M, Weissman G, Abramson SB. Increased levels of plasma anaphylatoxins in systemic lupus erythematosus predict flares of the disease and may elicit vascular injury in lupus cerebritis. Arthritis Rheum. 1988;31:632–641. doi: 10.1002/art.1780310508. [DOI] [PubMed] [Google Scholar]
- 36.Belmont HM, Abramson SB, Lie JT. Pathology and pathogenesis of vascular injury in systemic lupus erythematosus: Interactions of inflammatory cells and activated endothelium. Arthritis Rheum. 1996;39:9–22. doi: 10.1002/art.1780390103. [DOI] [PubMed] [Google Scholar]
- 37.Falk RJ, Dalmasso AP, Kim Y, Lam S, Michael A. Radioimmunoassay of the attack complex of complement in serum from patients with systemic lupus erythematosus. N Engl J Med. 1985;312:1594–1599. doi: 10.1056/NEJM198506203122502. [DOI] [PubMed] [Google Scholar]
- 38.Mitsias P, Levine SR. Large cerebral vessel occlusive disease in systemic lupus erythematosus. Neurology. 1994;44:385–393. doi: 10.1212/wnl.44.3_part_1.385. [DOI] [PubMed] [Google Scholar]
- 39.Hughson MD, McCarty GA, Sholer CM, Brumback RA. Thrombotic cerebral arteriopathy in patients with the antiphospholipid syndrome. Mod Pathol. 1993;6:644–653. [PubMed] [Google Scholar]
- 40.Levine SR, Deegan MJ, Futrell N, Welch KM. Cerebrovascular and neurologic disease associated with antiphospholipid antibodies: 48 cases. Neurology. 1990;40:1181–1189. doi: 10.1212/wnl.40.8.1181. [DOI] [PubMed] [Google Scholar]
- 41.Bruyn GA. Controversies in lupus: nervous system involvement. Ann Rheum Dis. 1995;54:159–16. doi: 10.1136/ard.54.3.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shintaku M, Matsumoto R. Disseminated perivenous necrotizing encephalomyelitis in systemic lupus erythematosus: report of an autopsy case. Acta Neuropathol (Berl) 1998;95:313–317. doi: 10.1007/s004010050804. [DOI] [PubMed] [Google Scholar]
- 43.Roldan CA, Gelgand EA, Qualls CR, Sibbitt WL., Jr Valvular heart disease as a cause of cerebrovascular disease in patients with systemic lupus erythematosus. Am J Cardiol. 2005;95:1441–7. doi: 10.1016/j.amjcard.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 44.Roldan CA, Gelgand EA, Qualls CR, Sibbitt WL. Valvular heart disease is associated with non-focal neuropsychiatric systemic lupus erythematosus. J Clin Rheumatol. 2006;12:3–10. doi: 10.1097/01.rhu.0000200378.42836.7f. [DOI] [PubMed] [Google Scholar]
- 45.Roldan CA, Gelgand EA, Qualls CR, Sibbitt WL., Jr Valvular heart disease by transthoracic echocardiography is associated with focal brain injury and central neuropsychiatric systemic lupus erythematosus. Cardiology. 2007;108:331–7. doi: 10.1159/000099104. [DOI] [PubMed] [Google Scholar]
- 46.Roldan CA, Qualls CR, Sopko KS, Sibbitt WL., Jr Transthoracic versus transesophageal echocardiography for detection of Libman-Sacks endocarditis: a randomized controlled study. J Rheumatol. 2008;35:224–9. [PubMed] [Google Scholar]
- 47.DeGiorgio LA, Konstantinov KN, Lee SC, Hardin JA, Volpe BT, Diamond B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat Med. 2001;7:1189–93. doi: 10.1038/nm1101-1189. [DOI] [PubMed] [Google Scholar]
- 48.Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–22. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- 49.Kowal C, DeGiorgio LA, Nakaoka T, Hetherington H, Huerta PT, Diamond B, et al. Cognition and immunity; antibody impairs memory. Immunity. 2004;21:179–88. doi: 10.1016/j.immuni.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 50.Emmer BJ, Steens SC, Steup-Beekman GM, van der Grond J, Admiraal-Behloul F, Olofsen H, et al. Detection of change in CNS involvement in neuropsychiatric SLE: a magnetization transfer study. J Magn Reson Imaging. 2006;24:812–6. doi: 10.1002/jmri.20706. [DOI] [PubMed] [Google Scholar]
- 51.Steens SC, Steup-Beekman GM, Bosma GP, Admiraal-Behloul F, Olofsen H, Doornbos J, et al. The effect of corticosteroid medication on quantitative MR parameters of the brain. AJNR Am J Neuroradiol. 2005;26:2475–80. [PMC free article] [PubMed] [Google Scholar]
- 52.Bosma GP, Steens SC, Petropoulos H, Admiraal-Behloul F, van den Haak A, Doornbos J, et al. Multisequence magnetic resonance imaging study of neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. 2004;50:3195–202. doi: 10.1002/art.20512. [DOI] [PubMed] [Google Scholar]
- 53.Steens SC, Admiraal-Behloul F, Bosma GP, Steup-Beekman GM, Olofsen H, Le Cessie S, et al. Selective gray matter damage in neuropsychiatric lupus. Arthritis Rheum. 2004;50:2877–81. doi: 10.1002/art.20654. [DOI] [PubMed] [Google Scholar]
- 54.Steens SC, Bosma GP, Steup-Beekman GM, le Cessie S, Huizinga TW, van Buchem MA. Association between microscopic brain damage as indicated by magnetization transfer imaging and anticardiolipin antibodies in neuropsychiatric lupus. Arthritis Res Ther. 2006;8:R38. doi: 10.1186/ar1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dehmeshki J, Van Buchem MA, Bosma GP, Huizinga TW, Tofts PS. Systemic lupus erythematosus: diagnostic application of magnetization transfer ratio histograms in patients with neuropsychiatric symptoms--initial results. Radiology. 2002;222:722–8. doi: 10.1148/radiol.2223010413. [DOI] [PubMed] [Google Scholar]
- 56.Steens SC, Bosma GP, ten Cate R, Doornbos J, Kros JM, Laan LA, et al. A neuroimaging follow up study of a patient with juvenile central nervous system systemic lupus erythematosus. Ann Rheum Dis. 2003;62:583–6. doi: 10.1136/ard.62.6.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sanna G, Piga M, Terryberry JW, Peltz MT, Giagheddu S, Satta L, et al. Central nervous system involvement in systemic lupus erythematosus: cerebral imaging and serological profile in patients with and without overt neuropsychiatric manifestations. Lupus. 2000;9:573–83. doi: 10.1191/096120300678828695. [DOI] [PubMed] [Google Scholar]
- 58.Braffman BH, Zimmerman RA, Trojanowski JQ, Gonatas NK, Hickey WF, Schlaepfer WW. Brain MR: pathologic correlation with gross and histopathology. 1. Lacunar infarction and Virchow-Robin spaces AJR. Am J Roentgenol. 1988;151:551–8. doi: 10.2214/ajr.151.3.551. [DOI] [PubMed] [Google Scholar]
- 59.Braffman BH, Zimmerman RA, Trojanowski JQ, Gonatas NK, Hickey WF, Schlaepfer WW. Brain MR: pathologic correlation with gross and histopathology. 2. Hyperintense white-matter foci in the elderly. AJR Am J Roentgenol. 1988;151:559–66. doi: 10.2214/ajr.151.3.559. [DOI] [PubMed] [Google Scholar]
- 60.Grossman RI, Braffman BH, Brorson JR, Goldberg HI, Silberberg DH, Gonzalez-Scarano F. Multiple sclerosis: serial study of gadolinium-enhanced MR imaging. Radiology. 1988;169:117–22. doi: 10.1148/radiology.169.1.3420246. [DOI] [PubMed] [Google Scholar]
- 61.Man'kovskii NB. Diencephalic pathology in the clinical picture of certain collagenoses in young and middle aged patients. Gerontol Clin. 1969;11:231–8. [PubMed] [Google Scholar]
- 62.Anderson JR. Intracerebral calcification in a case of systemic lupus erythematosus with neurological manifestations. Neuropathol Appl Neurobiol. 1981;7:161–6. doi: 10.1111/j.1365-2990.1981.tb00085.x. [DOI] [PubMed] [Google Scholar]
- 63.Nagaoka S, Matsunaga K, Chiba J, Ishigatsubo Y, Tani K. Five cases of systemic lupus erythematosus with intracranial calcification. Rinsho Shinkeigaku. 1982;22:635–43. [PubMed] [Google Scholar]
- 64.Yokota S, Mori T, Kosuge K, Takahashi K, Nishiyama Y, Uechi M, et al. Basal ganglia calcification in two children with systemic lupus erythematosus and neuropsychiatric manifestations. Ryumachi. 1985;25:115–22. [PubMed] [Google Scholar]
- 65.Stuart BM, Gregson NA. Cerebral calcification in a patient with systemic lupus erythematosus and a monoclonal IgG reactive with glial fibrillary acidic protein. Br J Rheumatol. 1998;37:1355–7. doi: 10.1093/rheumatology/37.12.1355. [DOI] [PubMed] [Google Scholar]
- 66.Shimojima Y, Gono T, Hoshi K, Yamamoto K, Yoshida K, Matsuda M, Ikeda S. Neuropsychiatric systemic lupus erythematosus associated with anti-phospholipid syndrome, showing massive intracranial calcifications. No To Shinkei. 2003;55:885–8. [PubMed] [Google Scholar]
- 67.Cañas CA, Tobón GJ. Multiple brain calcifications in a patient with systemic lupus erythematosus. Clin Rheumatol. 2008;27(2):S63–5. doi: 10.1007/s10067-008-0933-x. [DOI] [PubMed] [Google Scholar]
- 68.Viswanathan A, Chabriat H. Cerebral microhemorrhage. Stroke. 2006;37:550–5. doi: 10.1161/01.STR.0000199847.96188.12. [DOI] [PubMed] [Google Scholar]
- 69.Haacke EM, Xu Y, Cheng YC, Reichenbach JR. Susceptibility weighted imaging (SWI) Magn Reson Med. 2004;52:612–8. doi: 10.1002/mrm.20198. [DOI] [PubMed] [Google Scholar]