

Abstract
Chitin is a polymer of N-acetylglucosamine with the ability to regulate innate and adaptive immune responses. However, the detailed mechanisms of chitin-mediated regulation of intestinal inflammation are only partially known. In this study, Chitin-microparticles (CMPs) or PBS were orally administered to acute and chronic colitis models every three days for six consecutive weeks beginning at weaning age. The effects of this treatment were evaluated by histology, cytokine production, co-culture study and enteric bacterial analysis in DSS-induced colitis or TCRα knockout chronic colitis models. Histologically, chitin-treated mice showed significantly suppressed colitis as compared to PBS-treated mice in both animal models. The production of IFNγ was upregulated in the mucosa of chitin-treated mice compared to control mice. The major source of IFNγ-producing cells was CD4+ T cells. In mouse dendritic cells (DCs), we found that CMPs were efficiently internalized and processed within 48 hours. Mesenteric lymph nodes (MLNs) CD4+ T cells isolated from chitin-treated mice produced 7-fold higher amount of IFNγ in the culture supernatant after being co-cultured with DCs and chitin as compared to the control. Proliferation of CFSElow CD4+ T cells in MLNs and enteric bacterial translocation rates were significantly reduced in chitin-treated mice when compared to the control. In addition, CMPs improved the imbalance of enteric bacterial compositions and significantly increased IL-10-producing cells in non-inflamed colon, indicating the immunoregulatory effects of CMPs in intestinal mucosa. In conclusion, CMPs significantly suppress the development of inflammation by modulating cytokine balance and microbial environment in colon.
Keywords: chitin, chitinase, colitis, cytokine, animal model
Introduction
Chitin is a polymer of β-1, 4 N-acetylglucosamine (GlcNAc), which represents the second most abundant polysaccharide in nature next to cellulose and is produced by fungi, insects, and crustaceans [1, 2]. In contrast, mammals and bacteria do not possess chitin as a structural component [2]. The ability of chitin to modulate immune responses is critically determined by the size of chitin and/or by the route of administration [3–7]. Recent studies have shown that orally administered chitin-microparticles (CMPs) with a restricted phagocytosable size, ranging from 1 to 10 μm, efficiently inhibited allergen-induced IgE production, lung inflammation and macrophage activation in experimental model of asthma [3]. Reese et al demonstrated that intraperitoneally injected chitin (presumably medium-size [>40 μm] chitin particles) induces the accumulation of IL-4-expressing eosinophils and basophils to the lungs as early as 6 hours [4]. Another group also showed that chitin has size-dependent effects on macrophage stimulation; medium-size chitin activated macrophage IL-17 production and induced acute tissue inflammation via TLR2, MyD88, and IL-17A-dependent mechanisms [5]. Of note, intranasal-administered small (<40 μm, largely 2–10 μm), but not intermediate (40–70 μm) or large-size (70–100 μm) chitin particles, are a strong stimulator of IL-10 production in lung macrophage [6]. Some immune regulatory function of chitin may be mediated by surface receptors including mannose receptors and TLR2 [6, 8]. However, mammalian chitinases and chitinase-like proteins with a chitin-binding motif may mediate the detection of chitin (presumably <40 μm in size) in inflammatory conditions [9, 10].
In this study, we determined whether chitin itself can mediate immune regulation during the initiation of colitis and mucosal recovery. We propose that oral administration of CMPs can contribute to the suppression of both innate-immune mediated acute colitis and Th2-mediated chronic colitis. CMPs enhance acute mucosal defense responses by initially upregulating IFN-γ expression but subsequently promoting mucosal recovery through the recruitment of IL-10-producing cells into the colon. These findings suggest that the processing and recognition of CMPs can modulate mucosal inflammation by regulating mucosal defense and recovery mechanisms.
Materials & Methods
Cell culture
JAWSII, mouse bone marrow-derived dendritic cells (DCs) established from C57Bl/6 (H-2b) mice were purchased from the American Type Culture Collection (Rockville, MD) and were maintained in alpha minimum essential medium with L-glutamine, ribonucleoside and deoxyribonucleosides supplemented with 20% (vol/vol) fetal bovine serum (Atlanta biological, Lawrenceville, GA), 2.5% 1M HEPES buffer1% antibiotics mixture (penicillin G, streptomycin and amphotericin B), 5 ng/ml murine GM-CSF (Bio Vision, Mountain View, CA), 1 mM sodium pyruvate (Sigma). Cells were incubated at 37°C in 5% CO2. All tissue culture items and reagents were purchased from Fisher Scientific (Fair Lan, NJ) unless specified.
Mice
C57Bl/6 WT and C57Bl/6 TCRα KO mice were purchased from the Jackson Laboratory (Bar Harbor, ME). C57Bl/6 IL-10 transcriptional reporter mice [11] were a gift from Dr. Christopher L. Karp (Cincinnati, OH). These mice were maintained under specific Helicobacter free SPF conditions at Massachusetts General Hospital. The animal care and procedure of the experiments in this study were approved by the Subcommittee on Research Animal Care, Massachusetts General Hospital.
Reagents
CMPs (1–10 μm in size) were provided by Dr. Y. Shibata (Florida Atlantic University, Boca Raton, FL) [12]. Anti-BrdU antibody and FITC-chitin-binding probe (1:500 dilutions) were purchased from SeroTec (Oxford, England) and New England Biolabs (Ipswich, MA), respectively.
Oral administration of CMPs
Both TCRα KO and C57Bl/6 WT mice were orally gavaged with a 20–22 gauge curved needle (Harvard Apparatus, Holliston, MA) with CMPs (1.5 mg/day) once per every 3 days, beginning at weaning, for 6 consecutive weeks. The control group was fed with PBS.
Animal models of colitis
For DSS-induced colitis model, acute colitis was induced in chitin- or PBS-treated C57Bl/6 mice after their body weight reached 19–21 gram (around 8 weeks old) by administration of 4% DSS (MW 35–45 KDa, MP Biomedicals, Irvine, CA) into the drinking water for 5 days and then changed to regular drinking water, and mice were sacrificed on 12 days after DSS-treatment. For spontaneously developed colitis model, chitin- or PBS-treated TCRα KO mice were sacrificed at 20 weeks old.
Clinical and histological evaluation of colitis
H&E sections in both models were determined by two investigators in a blinded fashion as previously described [13–15].
Cell isolation method
Spleen and mesenteric lymph nodes (MLNs) cells were extracted as described previously [16]. Isolation method for colonic lamina propria (LP) cells has been described elsewhere [17, 18].
Flow cytometry analysis
Isolated spleen, MLNs and colonic cells were resuspended in FACS buffer (PBS with 1 % BSA and 0.1% sodium azide) and stained for 30 min at 4°C with fluorescence conjugated mAb against -CD4 (RM 4–5) and IFNγ (XMG1.2). Both antibodies were purchased from eBioscience (San Diego, CA). Isotype-matched IgG was used for the internal control of anti-IFNγ (eBioscience). For intracellular cytokine staining, BD cytofix/cytoperm plus fixation/permeabilization kit (BD Biosciences, San Diego, CA) were used according to the manufacturer’s instructions. Samples were acquired in a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) and analyzed by Flow Jo software (Tree Star, Inc, Ashland, OR).
Chitin internalization assay
JAWSII cells (0.2 million cells/well) were cultured in a 4 chamber tissue culture slide (BD Falcon, Bedford, MA) for 4–5 days, and incubated with 100 μg CMPs at 37°C for 6-different intervals. Cells were stained by a previously described method [12]. Briefly, JAWSII cells that phagocytosed CMPs were fixed with 100% acetone for 20 minutes at −20C, followed by permeabilization with 1% Triton-X100 in PBS for 15 minutes. Cells are then incubated in blocking buffer consisting of 10% goat serum for 30 minutes at room temperature, followed by incubation with FITC-chitin-binding probe (1:500 dilution, New England Bio Labs), anti-F4/80 Ab (1:100 dilution, AbD Serotec) or anti-CD11c Ab (1:100 dilution, BD Pharmingen) for 1 hour, and then with FITC-rabbit anti-rat IgG (Vector) or Texas red-goat anti-hamster IgG at room temperature. Nuclear counter staining was performed by 1.5 μg/ml DAPI (Vector) staining for 30 minutes at room temperature. After each incubation, cells were washed with PBS three times. Floating cells were harvested to prepare cytosmears to perform the same staining as described above. Images were analyzed by fluorescent microscopy (Olympus AX70, Shinjuku, Tokyo, Japan).
Cytokine enzyme-linked immunosorbent assay (ELISA)
Tissues homogenate were centrifuged, and cytokine levels in the supernatants of homogenate were measured by ELISA following the manufacturer’s instructions (R&D Systems, Minneapolis, MN).
5-bromo-2′-deoxyuridine (BrdU) incorporation assay
BrdU incorporation assay was performed as previously described [13].
Bacterial translocation assay
Tissues were homogenized in Hank’s balanced salt solution as previously described [19]. Serial dilutions of the tissue-homogenates were plated on Luria-Bertani (LB) agar plate and incubated at 37°C over night. Colony-forming units (CFU) were counted as previously described [20].
RT-PCR
16s rDNA-specific primers [Table 1] were used to analyze stool bacterial composition as previously described [21]. Quantitative PCR analysis was performed by the MX3000p quantitative PCR machine (Agilent Technologies, Santa Clara, CA), and the cDNA concentration was normalized further as the cycle threshold value of β-actin.
Table 1.
Sequences of 16S rDNA-specific primers in this study
| Group | Primer Sequence | PCR product |
|---|---|---|
| All Bacteria | F: 5′-ACTCCTACGGGAGGCAGCAGT R: 5′-ATTACCGCGGCTGCTGGC |
175 bp |
| Bacteroidetes | F: 5′-GGTTCTGAGAGGAAGGTCCC R: 5′-GCTGCCTCCCGTAGGAGT |
54 bp |
| Enterobacteriaceae | F: 5′-GTGCCAGCAGCCGCGGTAA R: 5′-GCCTCAAGGGCACAACCTCCAAG |
335 bp |
| Lactobacillaceae | F: 5′-CACCGCTACACATGGAG R: 5′-AGCAGTAGGGAATCTTCCA |
316 bp |
| Clostridiales | F: 5′-ACTCCTACGGGAGGCAGC R: 5′-GCTTCTTAGTCAGGTACCGTCAT |
154 bp |
Ex vivo cell culture and proliferation assays
For purifying CD4+ cells, isolated cells were incubated with mouse CD4 (L3T4) microbeads (Miltenyi Biotec, Auburn, CA) at 4°C for 30 minutes. After washing, the labeled cells were isolated using MACS system (Miltenyi Biotec). T cells were resuspended at 107 cells/ml in DMEM with 1% FBS and 20 mM Hepes, and the CD4+ cells (3x105 cells/well) were cultured in flat bottom 24-well plate coated with anti-CD3ε (5 μg/ml) and anti-CD28 (2 μg/ml) antibodies (BD Biosciences) for 3 days with or without mouse DCs in a 1:1 ratio with or without CMPs (100 μg/ml). Purified CD4+ T cells were incubated with 2.5 mM CFSE (Invitrogen, Carlsbad, CA) for 20 min at 37°C and then washed using FBS gradient. JAWSII cells were used as mouse-derived DCs in this study. CFSE-labeled CD4+ T cells (3–4 x105 cells/well) were co-cultured for 3 days with or without mouse DCs (CD4+ T cells: DCs = 2:1) and CMPs as described above. Samples were acquired in a FACSCalibur flow cytometer and supernatant was accessed by cytokine ELISA.
Confocal microscopic analysis
GFP-IL-10 reporter mice were anesthetized with intraperitoneal injection of 2.5% Avertin. After 5 minutes, wheat germ agglutinin (WGA)-Texas Red (1 mg/ml) was intravenously injected to each mouse 7 minutes later the colon was removed, cut transversely, spread on a slide glass facing up, and covered by a cover glass. Images were analyzed with a confocal microscope (model Radiance 2000; Bio-Rad Laboratories, Hercules, CA) using multi-tracking (line switching) for two-color imaging. Image acquisition was performed with LaserSharp Scanning software (Bio-Rad Laboratories)
Statistics
The statistical significance was evaluated by Student t test for parametric data, which are shown as the mean SD. Nonparametric data were analyzed by using the Mann-Whitney test (e.g. clinical and histological data). The median SEM was determined for these data. Differences were considered statistically significant at a P value of less than .05.
Results
CMPs can be internalized and processed in a time dependent manner by dendritic cells (DCs) in vitro
DCs are professional antigen-presenting cells (APC), which play central and critical roles in the regulation of immune responses in mucosal tissues. To confirm the biological significance of CMPs in antigen presenting cells, we initially performed an in vitro experiment utilizing a JAWSII mouse bone-marrow derived DC line. The size (less than 10 μm) of CMPs is phagocytosable and much smaller than that of DCs. In the culture of JAWSII cells, CMPs were added on different time intervals from 0 to 48 hours. The majority of adherent DCs were positively stained with both anti-CD11c and anti-F4/80 antibodies, while floating cells were of the F4/80+ but not the CD11c+ phenotype [Supplemental Figure 1]. Internalization of CMPs by DCs can be observed about 15 minutes after adding the particles in the culture supernatant [Figure 1A], and the increased number of internalized CMPs, as detected by FITC-chitin-binding protein, was positively associated with the incubation period for up to 24 hours [Figures 1A and B]. After 48 hours, a reduction of internalized CMPs was observed and internalized CMPs were presumably processed by DCs [Figures 1A and B]. This result suggests that mouse DCs efficiently internalize and process CMPs in the cytoplasmic compartment.
Figure 1.

CMPs are efficiently internalized and presented by dendritic cells. (A) JAWSII cells cultured with CMPs (100 μg/ml) for different time intervals. Immunofluorescent staining was performed to detect CMPs (FITC), CD11c+ cells (Texas red) and nucleic compartment (DAPI) of JAWSII cells. (B) CMPs-internalized cell number was count at 6 different time points in 10 different fields within the 1 mm square under x40, objective. #P <.01, ##P <.001, ###P <.0001 (versus 1 hour) and *P<10−6, **P<10−7, ***P<10−8 (versus 0 hour). (C) CD4+ T cells were isolated from MLNs of TCRα KO mice then co-cultured with or without CMPs (100 μg/ml) and mouse dendritic cells on anti-CD3ε/anti-CD28 coated 24-well plate for 72 hours (triplicate two times, n=4 in each group). PBS was used as a control for CMPs. Cytokine productions were examined by ELISA. *P <.05, **P <.01, ***P<.005.
INFγ production and proliferation of MLN CD4+ T Cells are regulated by oral chitin administration in TCRα KO Mice
We further examined CMP-mediated cellular biological effects in a colitis model of TCRα KO mice. Our laboratory has previously reported that chronic colitis in TCRα KO mice is mediated by TCRα−β+ CD4+ T cells in MLNs and colon, which actively produce the Th2-type cytokine IL-4 [17, 22]. Oral administration of CMPs (1.5 mg in 200 ul PBS) was started from the weaning age and repeated every three days for 6 weeks, and mice were sacrificed at 20 weeks old. A control group of mice received same volume of PBS. MLN CD4+ T cells from CMP-treated TCRα KO mice produced 7-fold higher amount of IFNγ in the culture supernatant after being co-cultured with mouse DCs and CMPs as compared to the PBS-treated control mice, while there was no significant difference in the production of IL-10 or IL-4 between the two groups [Figure 1C]. In contrast, the production of IL-10 was unchanged without CMPs or DCs in the culture [Figure 1C]. The production of IL-4 from MLNs CD4+ T cells with or without co-cultured APC was significantly (p<0.05) reduced in the CMP-treated group compared to control group [Figure 1C]. Since the CD4+ T cells from the MLN were the major source of IFNγ in both TCRα KO or DSS-treated mice with colitis, we employed an in vitro culture system to evaluate the proliferative response of CD4+ T cells in the presence or absence of CMPs and DCs. CFSE-labeled MLN CD4+ T cells from CMP-treated mice showed less proliferative response to chitin as compared to PBS-fed mice, while there was no difference in the proliferation of CFSE-labeled CD4+ T cells from the spleen [Figure 2]. These results suggest that MLNs CD4+ T cells in CMP-treated mice produced IFNγ with chitin-dependent manner.
Figure 2.
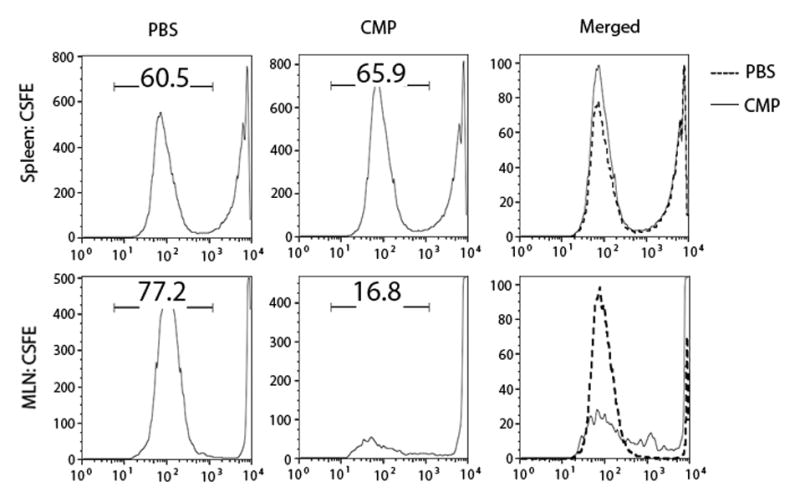
Proliferation of MLN CD4+ T cells were suppressed by CMP- but not PBS-treatment. CD4+ T cells isolated from MLNs and spleen from CMP- or PBS-treated mice were labeled with CFSE and co-cultured with CMPs and dendritic cells for 72 hours on antiCD3ε/antiCD28 antibodies coated 24-well plate. Data are representative of 2 independent experiments. n=4 mice per group.
Oral administration of CMPs ameliorates DSS-induced acute colitis by upregulating IFNγ production
To examine the in vivo effect of chitin fragments in intestinal inflammation, we next tested whether CMPs administration ameliorates dextran sulfate sodium (DSS)-induced colitis. Oral administration of CMPs (1.5 mg in 200 ul PBS) or PBS (control) was started from the weaning age and repeated every three days for 6 weeks, and 5 days DSS-treatment was started around 8–11 weeks old (19–21 gram in body weight). CMP-administered mice showed significantly enhanced recovery from the acute colitis as indicated by the improvement of body weight loss and clinical score as compared to the PBS-treated mice [Figure 3A]. Histological analysis confirmed that CMP-treated mice exhibit much less epithelial damage with significantly (p<.01) lower histological score and less inflammatory cell infiltration compared to the PBS-treated mice [Figures 3B and C].
Figure 3.

Oral CMP administration significantly enhances the recovery from acute DSS-induced colitis. (A) Body weight (left) and clinical scores (right) were shown during the course of 4% DSS-induced colitis in C57BL/6 mice that were orally administered CMPs or PBS. (B, C) Representative H&E staining (B: objective, 20x) or histology score (C) of the middle part of the colon of DSS-treated mice (day 12) that received PBS (left) or CMPs (right) treatment were shown. (D) Cytokine levels of spleen (Sp), mesenteric lymph nodes (MLN) or colon (Co) homogenates were analyzed by ELISA on DSS day 12. Each value is the average of 6 mice in each group. *P <.05; and **P <.01 (CMP-treated versus PBS-treated mice in each tissue).
The reduced inflammatory response in CMP treated mice was associated with a reduced production of TNFα and IL-4 in MLNs [Figure 3D]. Surprisingly, however we found the IFNγ production in MLNs, but not spleen or colon, significantly increased in chitin-treated mice at day 12 after DSS treatment as compared to PBS-treated mice [Figure 3D].
As IFNγ was the only increased cytokine by oral CMPs administration, we further analyzed the source of this cytokine by flow cytometric analysis. Relative percentage of IFNγ+ cells within CD4+ T cells was significantly increased in the MLNs but not in spleen or colonic lamina propria [Figures 4A and B]. However, total numbers of CD4+ IFNγ+ cells were only marginally (p=.07) increased in MLNs [Figure 4C], suggesting the presence of another source of IFNγ. We further analyzed IFNγ producing CD11c+, NK1.1+, CD8+, F4/80+ or CD11b+ in spleen, MLN or colon in each group of DSS-induced colitis model, but we could not identify any alternative source by flow cytometric analysis [data not shown].
Figure 4.

CMP-treatment significantly enhances IFNγ production in a DSS-induced colitis model. (A–C) Cells in spleen, mesenteric lymph nodes (MLNs) or colonic lamina propria (LP) of 4% DSS-treated WT mice on Day 12 were stained with FITC-anti CD4 and APC-IFNγ, antibodies, and were assessed by flowcytometry as shown in A. The Numbers indicate the percent of CD4 and IFNγ double-positive cells within the indicated gates (A). The numbers of percent IFNγ positive cells within the total CD4+ T cells (B) or absolute number of CD4 and IFNγ double-positive cells (C) in each group of mice are shown. *P <.05 (CMP-treated versus PBS-treated mice in each tissue).
CMPs enhance the IL-10 production in the inflamed colon
Although our ELISA result suggests that IL-10 production was not significantly but marginally upregulated after CMPs administration in DSS-induced colitis, we tested the ability of CMPs to enhance IL-10 production during this colitis and mucosal recovery since IL-10 has been reported as a critical regulator in chitin-mediated suppression in lung inflammation [6]. We utilized an IL-10 reporter (GFP-IL-10 reporter) mouse system, which was genetically engineered to carry a neomycin-IRES-eGFP cassette between the endogenous stop site and the poly(A) site of Il10 locus [11]. In normal steady state, no IL-10 GFP positive cells were detectable in the colon of GFP-IL-10 reporter mice. After administration of DSS, GFP-IL-10-reporter mice showed similar course of colitis as seen in WT mice [Supplementary Figure 2]. Again, CMPs treatment enhanced recovery from DSS colitis in GFP-IL-10-reporter mice [Supplementary Figure 2]. In both groups of mice, GFP-expressing cells could be observed in spleen, MLNs and colon by flow cytometry [Figures 5A and B]. During the recovery from colitis, CMP treatment enhanced the number of IL-10-GFP+ CD4+ cells recruitment in the colon in comparison to the control mice [Figure 5A]. This response was specific to the large intestine, since no significant difference in the percentage of IL-10 expressing cells were found in spleens and MLNs [Figure 5B]. The increase in IL-10 expressing cells was (p<.05) most evident in non-inflamed area as compared to inflamed regions of analyzed colons [Figures 5C and D]. In addition, total IL-10 production was increased in colon, but not in spleen or MLNs, in CMP-treated mice as compared to PBS-treated mice [Figure 5E]. These results suggest that CMPs treatment enhances the recruitment of IL-10-producing cells in intestine, and IL-10 may be partially involved in the CMP-mediated immunoregulatory function in acute intestinal inflammation.
Figure 5.

IL-10 producing cells migrate into the colonic LP in association with inflammation in both CMP- and PBS-treated mice on DSS day 10. (A, B) Cells in spleen, mesenteric lymph nodes (MLNs) or colon of 4% DSS-treated IL-10-GFP reporter mice (n=6 in each group) on DSS day 10 were stained with PE-anti-CD4 antibody, and were assessed by flowcytometry (A). The number of percent IL-10-GFP cells within the total CD4+ cells (upper panel) and absolute number of lymphoid-type IL-10-GFP positive cells, which were specifically selected by size, in each organ (lower panel) were shown (B). (C) Representative confocal fluorescence microscopic images of IL-10-GFP+ cells in the inflamed or non-inflamed colon of IL-10-GFP reporter mice were shown. Multiple images of colon were collected from PBS- (n=5) or chitin- (n=4) treated mice. Inserted white scale bars indicate 50 μm. (D) Graphic representation of green fluorescence positive cells enumerated in multiple fields (0.1 mm2) of colonic sections of PBS- (n=4) or CMP- (n=4) treated IL-10-GFP reporter mice were analyzed. (E) Tissue homogenate IL-10 levels in spleen, mesenteric lymph nodes (MLN) and colon were examined by ELISA (n=4 in each group). *P <.05.
The development of chronic colitis in TCRα KO mice was suppressed with CMPs by upregulating IFNγ production
We next examined the therapeutic potential of CMPs in a Th2-mediated chronic colitis developing in TCRα KO mice [17, 22]. Of note, treatment with CMPs significantly improved the disease severity as judged by histological evaluation. The disease score in CMPs-treated TCRα KO mice was 2.35 ± 2.56 (p<.05) and that of PBS-treated control mice was 4.69 ± 2.71 [Figure 6A]. Enhanced CEC proliferation as judged by BrdU-incorporation has been shown to correlate with the severity of colitis in TCRα KO mice [23]. Indeed, significantly less (p<.01) proliferative response of CEC was observed in CMPs-treated mice as compared to control mice [Figures 6A and B]. In addition, oral CMPs-treatment significantly (p<.05) suppressed the productions of IL-4 and TNFα in the MLNs, whereas this treatment enhanced the production of IFN-γ [Figure 6C]. Interestingly, the effect of CMPs on cytokine responses was somehow tissue specific as indicated by no significant difference in the production of IL-4, IFNγ, TNFα in spleen, cecum and colon between CMPs-treated and control groups [Figure 6C].
Figure 6.

CMP-treatment significantly suppresses chronic colitis development in TCRαKO mice by upregulating IFNγ production. (A) Histology score of PBS-fed (n=13) and chitin-fed (n=14) TCRα KO colitis mice was evaluated according to previous criteria. (B) BrdU was injected intraperitoneally 1 hour prior to the sacrifice of mice (n=6) and staining were done on the colonic tissue sections followed by positive cell numbers count per crypt. (C)Tissue homogenate cytokine levels in cecum, colon, spleen and mesenteric lymph nodes (MLN) were examined by ELISA (n=6). *P <.05; **P <.01.
Oral chitin administration mediated immune modulation affect the microbial composition in the two models of colitis
Chitin has been proposed to possess several biological functions for wound healing, and anti-microbial and anti-inflammatory activities [24, 25]. Indeed, chitin has already been applied to treatment of humans with wounds infected by antibiotic resistant microorganisms [24–26]. Therefore, this raised a possibility that CMPs-mediated suppression of chronic colitis in TCR α KO mice is due to an immunoregulation of host/microbial interaction by suppressing the systemic invasion of local bacteria. Indeed, the total number of bacteria in contents obtained from proximal part of colon was significantly reduced in CMPs-treated group as compared to control group [Figure 7A].
Figure 7.

CMP-treatment modulates the bacterial loading, commensal bacterial components and bacterial translocation. (A) Bacterial number in cecum and colon in TCRα KO mice was examined. (B, C): Stool sample in cecum from TCRα KO mice were collected and DNA were extracted for RT-PCR analysis using selected 16s rDNA-specific primers as shown in Table 1 (B) or analyze the Clostridiales group of bacteria by quantitative-PCR (C). (D) Number of translocated bacteria in the liver of TCR α KO mice was assessed by LB-agar culture. Each value is the average of 5 mice in each group. *P <.05, **P<.01 (CMP-treated versus PBS-treated mice). (E) Number of translocated bacteria in spleen, liver or MLN was shown on DSS day 12 in C57Bl/6 WT mice. Each value is the average of 6 mice in each group. *P <.05; and **P <.01 (CMP-treated versus PBS-treated mice in each tissue).
To further confirm the CMPs-mediated regulation in host/microbial interaction, we next examined the impact of CMP-treatment on the bacterial commensals in TCR α KO mice. RT-PCR was performed on bacterial 16S rDNA using primers targeting the universal bacteria (whole bacterial kingdom) or major groups of intestinal bacterial commensals as listed on Table 1. In CMPs-treated mice, the composition of bacteria was evenly distributed without obvious expansion of bacterial sub-groups [Figure 7B]. In contrast, one of the sub-groups of commensals such as Clostridiales (including Clostridium, Eubacterium, Epulopicium, Dorea, and Ruminococcus) were reduced by 2–5 fold in PBS-treated group as compared to CMPs-treated group [Figure 7B and data not shown]. Lactobacillaceae (including Lactobacillus, Pediococcus, and Bacteriodes), Bacteroides, or Enterobacteriaceae (including Escherichia, Salmonella and Yersinia) did not show the differential expression between the two groups [Figure 7B]. We confirmed these finding by quantitative PCR that Clostridiales group was significantly (p<0.001) reduced by 4.59 ± 1.18 fold in PBS-treated mice as compared to CMPs-treated mice (n=8 in each group) [Figure 7C]. Furthermore, the rate of bacterial translocation in peripheral blood and lymphoid organs, including spleen, MLNs, liver, in CMPs- or PBS-treated TCRα KO mice were examined. As shown in Figure 7D, oral CMPs treatment significantly (p=0.027) suppressed the bacterial translocation in liver as compared to PBS treated mice. None of the bacteria were detected from peripheral blood, spleen, or MLNs of either group of mice [data not shown]. These data indirectly suggest that oral CMPs-treatment may have an influence for the bacterial composition in intestinal commensals.
We next tested the possibility if CMPs can control intestinal microflora in DSS-induced colitis. CMPs treatment significantly enhanced intestinal barrier function and reduced the tissue-translocation of enteric bacteria into the spleen (P<.001) and liver (P<.0001) as compared to mock (PBS) treatment [Figure 7E]. In contrast, the bacterial transport into the MLNs (P=.23) was not changed by CMPs treatment [Figure 7E] although IFNγ production was increased only in MLNs [Figures 3D and 4A and B]. The less number of bacteria translocation in spleen and liver may be associated with the CMPs-mediated bacterial flora modulation and/or enhanced barrier function of CECs.
Discussions
Large chitin particles (diameter larger than 100 μm) and medium chitin particles (40 to 70 μm) are believed to be non-functional and proinflammatory, respectively, while small 1 to 10 μm CMPs play a regulatory role in respiratory inflammation [7, 27]. In addition to particle size-dependent inflammatory effects, recent report suggests that the effectiveness of non-phagocytosable sizes of chitin particles (>40 μm) in vivo is independent of its carbohydrate composition/structure [12]; It has been shown that non-phagocytosable Sephadex G 100 beads (>40 μm) were capable of efficiently inducing innate eosinophilia in mice [12]. In contrast, it is of particular importance that immunoregulatory effects induced by phagocytosable sizes of CMP are dependent on chitin chemical composition/structure [28]. In this study, we performed experiments utilizing two models of acute and chronic colitis to address the immunological and biological effects of CMPs in the course of intestinal inflammation. We demonstrate here the unique and novel aspects of CMP-mediated immunoregulation during the development of intestinal inflammation. Oral administration of CMPs significantly enhanced the recovery of DSS-induced acute colitis as well as spontaneously developed Th2-type chronic colitis in TCRα KO mice as compared to the age-matched PBS-administered control mice. Interestingly and unexpectedly, significantly enhanced IFNγ production was observed in MLNs but not in the colon or spleen of CMPs-treated mice compared to the control in both colitis models. It was believed that CD4+ Th1 cells, CD8+ cytotoxic T cells, and NK cells exclusively produced IFNγ [29, 30], but recently it has been found that other types of cells including B cells, NKT cells, and professional APCs also secrete this cytokine [31–34]. In this study, we identified that the DCs efficiently take up and process CMPs in a time dependent manner. Furthermore, in TCRα KO mice, the production of IFNγ by MLN CD4+ T cells were CMPs-dependent, suggesting that DCs may play a critical role in CMP-mediated immunoregulation during the development of spontaneous colitis. The major sources of IFNγ after CMPs-administration was CD4+ T cells in DSS-induced acute colitis model. In addition, our data suggests that IFN γ produced by MLN CD4+ T cells seems to be important for adaptive immune responses in negatively regulating colonic TNFα production.
It is well known that inbred mouse strains vary in their ability to secrete IFN γ: for example, T cells of C57BL/6 and C3H mice secrete significantly higher amount of IFNγ as compared to those of BALB/c and B10.D2 mice [35]. Increased IFNγ production in these strains is positively associated with increased resistance to certain type of bacteria, such as Yersinia enterocolitica [36, 37]. IFNγ is also known to enhance the microbicidal effecter functions of macrophages [38] and neutrophils [39]. It has been also reported that IFN γ can promote microbial destruction by upregulating surface expression of high-affinity Fc γRI and complement receptor on mononuclear phagocytes, which further promote antibody-dependent, cell mediated cytotoxicity, and complement-mediated phagocytosis, respectively [40, 41]. Furthermore, Cole et al reported that IFNγ-inducible tripeptide motif Glu-Leu-Arg (ELR)− CXC chemokines were defensin-like antimicrobial against Escherichia coli and Listeria monocytogenes [42]. Taken together, IFNγ seems to augment microbicidal and anti-microbial activities in many cell types during the protective immune responses. In our present study, we found increased IFN γ production in MLNs and less bacterial translocation in liver in oral CMPs-treated acute and chronic colitis models as compared to PBS-treated mice. However, so far it is still unclear these two findings are mutually exclusive observations or related phenomena each other.
The commensal microbiota is important for toning and shaping both local and systemic immune responses [43, 44]. Recent report also suggests that oral inoculation of 2-week-old neonatal SPF mice with feces from Clostridium-treated mice resulted in resistance to DSS-induced colitis [45]. In this study, we revealed that oral CMPs administration efficiently modulated the balance in intestinal microflora, in particular Clostridiales group (e.g. Clostridium, Eubacterium, Epulopiscium, Dorea, Ruminococcus). CMP-mediated normalization of Clostridium strains in TCRα KO mice may be one of factors in normalizing intestinal inflammation. However, further extensive studies will be required to completely prove the possibility by analyzing the number of mucosal regulatory T cells and change of microbiota during the course of intestinal inflammation with or without CMPs treatment.
The recent reports from Elias’s group demonstrated that small-size chitin displayed immunoregulatory effect by enhancing the IL-10 and TNFα productions of macrophages in a murine asthma model [5, 6]. They identified that CMPs stimulate TNFα production via TLR2, dectin-1 and NF-κB-mediated signaling pathways, while CMPs stimulate IL-10 production via dectin-1-dependent and TLR-2-dependent and-independent pathways that involved mannose receptor and spleen tyrosine kinase (Syk) [6]. Some chitin-derivatives are known to be non-allergic or non-toxic, however, it has been reported that chitin shows size-dependent effects in vivo and in vitro [7]. For example, medium sized (40–70 μm) chitin particles efficiently trigger inflammatory reactions by triggering the productions of TNFα, IL-17, and IL-23 via the pattern recognition receptor TLR2 and MyD88 (myeloid differentiation primary response gene 88) [5]. In contrast, smaller sized (<40 μm) chitin particles showed anti-inflammatory effect by stimulating IL-10 production of murine lung macrophages via the stimulations of TLR-2-, dectin-1-and/or mannose receptor in asthma model [6, 7]. In our current study revealed that oral administration of CMPs (<10 μm) enhanced the IL-10 production in the colons of DSS-induced colitis model. Of note, CMPs-treated mice significantly or marginally enhanced the recruitment of IL-10 producing cells in the non-inflamed- or inflamed-area of DSS-induced colitis (DSS day 10), respectively, as compared to PBS-treated control in GFP-IL-10-reporter mice. In this model, the total tissue levels of IL-10 in colon were significantly increased in CMP-treated mice as compared to PBS-treated mice. As previously reported by other groups, the efficient recruitment of IL-10 producing CD4+ T cells in the colon of DSS-induced colitis may be orchestrated by IFNγ in MLNs, which enables to control the trafficking and migration of specific immune cells to sites of inflammation presumably through up-regulating expression of chemokines and adhesion molecules [36, 40]. In addition to this IFNγ/IL-10-mediated immunoregulatory effects, CMP-treatment significantly suppress the proliferation of CD4+ T cells (which includes the major pathogenic population of TCRα−β+ T cells) in MLNs but not spleen in TCRα KO mice.
In summary, our results provide evidence that CMPs have anti-inflammatory and anti-microbial effects in the acute and chronic types of intestinal inflammatory conditions. These results suggest that CMPs as a strong potential therapeutic agent in the context of intestinal inflammation in clinical settings.
Supplementary Material
Acknowledgments
The authors are grateful to Drs. Daniel K. Podolsky, Ramnik J. Xavier, Elke Cario, Deanna Nguyen and Scott B. Snapper for their helpful discussions and advices. We would like to thank Mr. Terry Danford Lott for his excellent secretarial assistance in preparing this manuscript, Dr. Christopher L. Karp (Cincinnati Children’s Hospital medical Center, Cincinnati, OH) for the kind gift of IL-10-GFP reporter mice, and Drs. Sayeda N. Alam, Matsuka Murakami, Mayumi Kawada, Chun-Chuan Chen, and Joo-Hye Song for their professional assistances in performing some experiments in this study.
Grant Supports: This work has been supported by National Institute of Health (DK 80070, DK74454, DK64289 and DK43351), and grants from the Eli and Edythe L. Broad Medical Foundation and American Gastroenterological Association Foundation to EM.
Abbreviations used in this paper
- Ab
antibody
- AIEC
adherent-invasive Escherichia coli
- APC
antigen-presenting cells
- BrdU
5-bromo-2′-deoxyuridine
- CEC
colonic epithelial cells
- CFSE
carboxyfluorescein succinimidyl ester
- CFU
colony-forming unit
- CHI3L1
chitinase 3-like-1
- CMPs
chitin microparticles
- DSS
dextran sulfate sodium
- IBD
inflammatory bowel disease
- IFN
interferon
- IL
interleukin
- MLN
mesenteric lymph nodes
- PBS
phosphate buffered saline
- TCRα KO
T-cell receptor alpha-chain knock out
- TLR
toll-like receptor
- TNF
tumor necrosis factor
Footnotes
Financial Disclosure: All authors have nothing to disclose any potential conflicts that are relevant to this manuscript.
References
- 1.Debono M, Gordee RS. Antibiotics that inhibit fungal cell wall development. Annu Rev Microbiol. 1994;48:471–497. doi: 10.1146/annurev.mi.48.100194.002351. [DOI] [PubMed] [Google Scholar]
- 2.Hakala BE, White C, Recklies AD. Human cartilage gp-39, a major secretory product of articular chondrocytes and synovial cells, is a mammalian member of a chitinase protein family. J Biol Chem. 1993;268:25803–25810. [PubMed] [Google Scholar]
- 3.Shibata Y, Foster LA, Bradfield JF, et al. Oral administration of chitin down-regulates serum IgE levels and lung eosinophilia in the allergic mouse. J Immunol. 2000;164:1314–1321. doi: 10.4049/jimmunol.164.3.1314. [DOI] [PubMed] [Google Scholar]
- 4.Reese TA, Liang HE, Tager AM, et al. Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature. 2007;447:92–96. doi: 10.1038/nature05746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Da Silva CA, Hartl D, Liu W, et al. TLR2 and IL-17A in chitin-induced macrophage activation and acute inflammation. J Immunol. 2008;181:4279–4286. doi: 10.4049/jimmunol.181.6.4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Da Silva CA, Chalouni C, Williams A, et al. Chitin is a size-dependent regulator of macrophage TNF and IL-10 production. J Immunol. 2009;182:3573–3582. doi: 10.4049/jimmunol.0802113. [DOI] [PubMed] [Google Scholar]
- 7.Lee CG, Da Silva CA, Lee JY, et al. Chitin regulation of immune responses: an old molecule with new roles. Curr Opin Immunol. 2008;20:1–6. doi: 10.1016/j.coi.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shibata Y, Metzger WJ, Myrvik QN. Chitin particle-induced cell-mediated immunity is inhibited by soluble mannan: mannose receptor-mediated phagocytosis initiates IL-12 production. J Immunol. 1997;159:2462–2467. [PubMed] [Google Scholar]
- 9.Elias JA, Homer RJ, Hamid Q, et al. Chitinases and chitinase-like proteins in T(H)2 inflammation and asthma. J Allergy Clin Immunol. 2005;116:497–500. doi: 10.1016/j.jaci.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 10.Anderson OA, Dixon MJ, Eggleston IM, et al. Natural product family 18 chitinase inhibitors. Nat Prod Rep. 2005;22:563–579. doi: 10.1039/b416660b. [DOI] [PubMed] [Google Scholar]
- 11.Madan R, Demircik F, Surianarayanan S, et al. Nonredundant roles for B cell-derived IL-10 in immune counter-regulation. J Immunol. 2009;183:2312–2320. doi: 10.4049/jimmunol.0900185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kogiso M, Nishiyama A, Shinohara T, et al. Chitin particles induce size-dependent but carbohydrate-independent innate eosinophilia. J Leukoc Biol. 2011;90:167–176. doi: 10.1189/jlb.1110624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mizoguchi E. Chitinase 3-like-1 exacerbates intestinal inflammation by enhancing bacterial adhesion and invasion in colonic epithelial cells. Gastroenterol. 2006;130:398–411. doi: 10.1053/j.gastro.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 14.Mizoguchi A, Mizoguchi E, Takedatsu H, et al. Chronic intestinal inflammatory condition generates IL-10-producing regulatory B cell subset characterized by CD1d upregulation. Immunity. 2002;16:219–30. doi: 10.1016/s1074-7613(02)00274-1. [DOI] [PubMed] [Google Scholar]
- 15.Matharu KS, Mizoguchi E, Cotoner CA, et al. Toll-like receptor 4-mediated regulation of spontaneous Helicobacter-dependent colitis in IL-10-deficient mice. Gastroenterol. 2009;137:1380–90. doi: 10.1053/j.gastro.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mizoguchi A, Mizoguchi E, Chiba C, et al. Role of appendix in the development of inflammatory bowel disease in TCR-alpha mutant mice. J Exp Med. 1996;184:707–715. doi: 10.1084/jem.184.2.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mizoguchi A, Mizoguchi E, Chiba C, et al. Cytokine imbalance and autoantibody production in TCRα−/− mice with inflammatory bowel disease. J Exp Med. 1996;183:847–856. doi: 10.1084/jem.183.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimomura Y, Ogawa A, Kawada M, et al. A unique B2 B cell subset in the intestine. J Exp Med. 2008;205:1343–1355. doi: 10.1084/jem.20071572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizoguchi E, Hachiya Y, Kawada M, et al. TNF receptor I-dependent activation of innate responses to reduce intestinal damage-associated mortality. Gastroenterol. 2008;134:470–480. doi: 10.1053/j.gastro.2007.11.055. [DOI] [PubMed] [Google Scholar]
- 20.Kawada M, Chen CC, Arihiro A, et al. Chitinase 3-like-1 enhances bacterial adhesion to colonic epithelial cells through the interaction with bacterial chitin-binding protein. Lab Invest. 2008;88:883–95. doi: 10.1038/labinvest.2008.47. [DOI] [PubMed] [Google Scholar]
- 21.Bouskra D, Brezillon C, Berard M, et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. 2008;456:507–10. doi: 10.1038/nature07450. [DOI] [PubMed] [Google Scholar]
- 22.Mizoguch A, Mizoguchi E, Bhan AK. The critical role of interleukin-4 but not interferone-γ in the pathogenesis of colitis in T cell receptor α mutant mice. Gastroenterol. 1999;116:320–326. doi: 10.1016/s0016-5085(99)70128-9. [DOI] [PubMed] [Google Scholar]
- 23.Mizoguchi E, Mizoguchi A, Bhan AK. Role of cytokines in the early stages of chronic colitis in TCRα-mutant mice. Lab Invest. 1997;76:385–397. [PubMed] [Google Scholar]
- 24.Lee SB, Kim YH, Chong MS, et al. Preparation and characteristics of hybrid scaffolds of β-chitin and collagen. Biomaterials. 2004;25:2309–2317. doi: 10.1016/j.biomaterials.2003.09.016. [DOI] [PubMed] [Google Scholar]
- 25.Jayakumar R, Nwe N, Tokura S, Tamura H. Sulfated chitin and chitosan as novel biomaterials. Int J Biol Macromol. 2007;40:175–181. doi: 10.1016/j.ijbiomac.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 26.Lee YM, Kim SS, Park MH, Song KW, Sung YK, Kang IK. β-chitin-based wound dressing containing silver sulfurdiazine. J Mater Sci: Mater Med. 2000;11:817–823.33. doi: 10.1023/a:1008961730929. [DOI] [PubMed] [Google Scholar]
- 27.Lee CG, Silva CD, Dela Cruz CS, et al. Role of chitin and chitinase/Chitinase-like proteins in inflammation, tissue remodeling, and injury. Ann Rev Physiol. 2011;73:479–501. doi: 10.1146/annurev-physiol-012110-142250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shibata Y, Foster LA, Metzger WJ, et al. Alveolar macrophage priming by intravenous administration of chitin particles, polymers of N-acetyl-D-Glucosamine, in mice. Infect Immun. 1997;65:1734–1741. doi: 10.1128/iai.65.5.1734-1741.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bach E, Aguet A, Schreiber RD. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol. 1997;15:563–591. doi: 10.1146/annurev.immunol.15.1.563. [DOI] [PubMed] [Google Scholar]
- 30.Young HA. Regulation of interferon-gamma gene expression. J Interferon Cytokine Res. 1996;16:563–568. doi: 10.1089/jir.1996.16.563. [DOI] [PubMed] [Google Scholar]
- 31.Carnaud C, Lee D, Donnars O, et al. Cutting edge: cross-talk between cells of the innate immune system: NKT cells rapidly activate NK cells. J Immunol. 163:4647–4650. [PubMed] [Google Scholar]
- 32.Frucht DM, Fukao T, Bogdan C, et al. IFN-gamma production by antigen-presenting cells: mechanisms emerge. Trends Immunol. 2001;22:556–560. doi: 10.1016/s1471-4906(01)02005-1. [DOI] [PubMed] [Google Scholar]
- 33.Yoshimoto T, Takeda K, Takeda T, et al. IL-12 up-regulates IL-18 receptor expression on T cells. Th1 cells, and B cells: synergism with IL-18 for IFN-gamma production. J Immunol. 1998;161:3400–3407. [PubMed] [Google Scholar]
- 34.Harris DP, Haynes I, Sayles PC, et al. Reciprocal regulation of polarized cytokine production by effector B and T cells. Nat Immunol. 2000;1:475–482. doi: 10.1038/82717. [DOI] [PubMed] [Google Scholar]
- 35.Schroder K, Hertzog PJ, Ravasi T, et al. Interferone-γ: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 36.Autenrieth IB, Reissbrodt R, Saken E, et al. Desferrioxaminepromotes virulence of Yersinia enterocolitica in mice depends on both desferrioxamine type and mouse strain. J Infect Dis. 1994;169:562–567. doi: 10.1093/infdis/169.3.562. [DOI] [PubMed] [Google Scholar]
- 37.Autenrieth IB, Beer M, Bohn E, et al. Immune responses to Yersinia enterocolitica in susceptible BALB/c and resistant C57Bl/6 mice: an essential role for gamma interferon. Infect Immun. 1994;62:2590–2599. doi: 10.1128/iai.62.6.2590-2599.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gordon MA, Gordon SB, Musaya L, et al. Primary macrophages from HIV-infected adults show dysregulated cytokine responses to salmonella, but normal internalization and killing. AIDS. 21:2399–2408. doi: 10.1097/QAD.0b013e3282f25107. [DOI] [PubMed] [Google Scholar]
- 39.Boehm U, Klamp T, Groot M, et al. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- 40.Erbe DV, Collins JE, Shen L, et al. The effect of cytokine on the expression and function of Fc receptors for IgG on human myeloid cells. Mol Immunol. 1990;27:57–67. doi: 10.1016/0161-5890(90)90060-d. [DOI] [PubMed] [Google Scholar]
- 41.Strunk RC, Cole FS, Perlmutter DH, et al. Gamma-interferone increases expression of class III complement genes C2 and factor B in human monocytes and in murine fibroblasts transfected with human C2 and factor B genes. J Biol Chem. 1985;260:15280–15285. [PubMed] [Google Scholar]
- 42.Cole AM, Ganz T, Liese AM, et al. IFN-inducible ELR−CXC chemokine display defensin-like antimicrobial activity. J Immunol. 2001;167:623–627. doi: 10.4049/jimmunol.167.2.623. [DOI] [PubMed] [Google Scholar]
- 43.Garrett WS, Gordon JI, Glimcher LH. Homeostasis and inflammation in the intestine. Cell. 2010;140:859–870. doi: 10.1016/j.cell.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sartor RB. Key questions to guide a better understanding of host-commensal microbiota interactions in intestinal inflammation. Mucosal Immunol. 2011;4:127–132. doi: 10.1038/mi.2010.87. [DOI] [PubMed] [Google Scholar]
- 45.Atarashi K, Tanoue T, Shima T, et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331:337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.