

Abstract
Acid treatment of densely substituted 2-silyl-1,2-dihydropyridines provides a new and convenient entry to reactive azomethine ylides that can (1) be protonated and reduced with high stereoselectivity to give piperidines, (2) participate in [3+2] dipolar cycloaddition to give tropanes, and (3) undergo a Nazarov-like 6-π electrocyclization that upon reduction give 2-azabicyclo[3.1.0] systems.
Nonaromatic nitrogen heterocycles such as piperidines, tropanes and pyrrolidines are valuable, ubiquitous structural motifs in biologically active alkaloids as well as drugs that have had a major impact on disease.1 While C-H bond functionalization strategies have been extensively applied to the synthesis and elaboration of aromatic nitrogen heterocycles,2 few applications to nonaromatic frameworks have been reported.3 Recently, we disclosed a highly diastereoselective process that enables the formation of densely substituted tetrahydropyridines4,5 from α,β-unsaturated imines and alkynes using Rh(I) catalyzed C–H activation6 (Scheme 1). A current limitation of this process is the requirement that internal alkynes be used, which necessarily introduces substitution at the 6-position. Terminal alkynes would be attractive coupling partners but are not viable due to competitive homocoupling leading to complex mixtures of products.7 Herein, we report a strategy for using TMS acetylenes as terminal alkyne surrogates for the convergent assembly of diverse tetrahydropyridines with high stereo- and regiocontrol. This class of alkynes provides access to piperidines with a substitution pattern commonly found in drugs and natural products.8 Moreover, mechanistic inquiry reveals that the intermediate silyldihydropyridine can function as a new and convenient azomethine ylide precursor enabling rapid entry into highly substituted tropanes, and through an unprecedented rearrangement, 2-azabicyclo[3.1.0] systems.
Scheme 1.

Rapid entry into substituted piperidine, tropane and 2-azabicyclo[3.1.0] frameworks
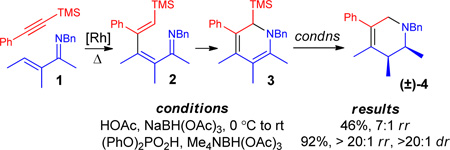
Treatment of imine 1 (eq 1) with 1.5 equiv of 1-phenyl-2-trimethylsilylacetylene, 2.5 mol % of [RhCl(coe)2]2 and 5 mol % of 4-Me2N-C6H4-PEt2 in toluene (1 M) at 100 °C for 1 h resulted in alkenylation to give azatriene 2 that undergoes a 6-π electrocyclization in situ leading to the clean formation of silyldihydropyridine 3 as a single detectable regioisomer, as assessed by 1H NMR. When the reduction sequence was carried out according to previously optimized conditions (HOAc and NaBH(OAc)3 in MeOH/PhCH3 from 0 °C to rt),4 desired product (±)-4 was formed in modest yield and with a considerable amount of olefin isomerization (7:1 mixture of inseparable isomers).
Investigation of different acids and hydride sources established that direct addition of Me4NBH(OAc)3 in CH2Cl2 followed by (PhO)2PO2H to the Rh(I)-catalyzed reaction mixture provided clean formation of (±)-4 in excellent overall yield as a single observable diastereomer and regioisomer (eq 1). The relative configuration of (±)-4 was determined by single crystal X-ray analysis of the hydrochloride salt.4 This stereoselective protonation/reduction sequence proceeds with concomitant cleavage of the trimethysilyl moiety to formally introduce a terminal acetylene unit with complete regiocontrol in one pot from the TMS acetylene and imine starting materials. This strategy consequently addresses a major synthetic limitation to our previously published reports.
 |
(1) |
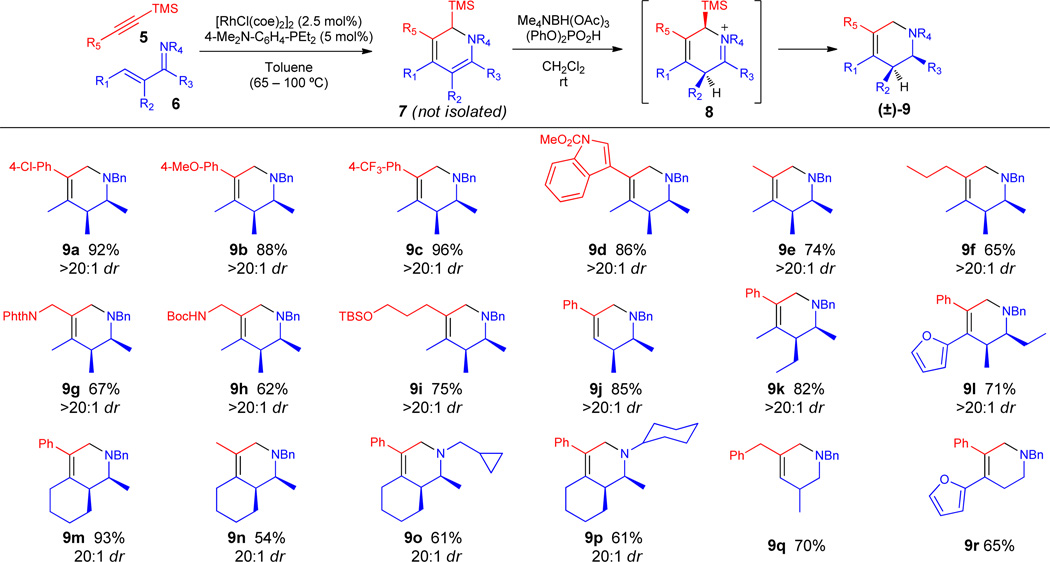
An important feature of this approach is the vast number of readily available TMS acetylene and α,β-unsaturated ketone inputs that enable the rapid assembly of a range of piperidine analogs. To this end, a number of TMS acetylenes and α,β-unsaturated imines have been surveyed to establish broad reaction scope (Table 1). Alkyne components containing electron-rich and -deficient aromatic as well as heteroaromatic groups are tolerated and result in the desired tetrahydropyridines in excellent yield and diastereoselectivity (9a–d). Alkynes bearing aliphatic groups also react efficiently under the reaction conditions (9e,f). Furthermore, functional groups such as Boc-protected primary amines (9h), phthalimides (9g), and silyl ethers (9i) that are sensitive to either strongly acidic or reducing conditions are viable coupling partners for this process and provide a platform for further diversification.
Table 1.
Convergent assembly of piperidines with terminal acetylene equivalent.a
 |
Yields correspond to isolated product after silica gel chromatography and represent overall yields from the imine precursor 6. Diastereoselectivity was determined by 1H NMR of the crude reaction mixture.
Conducted using HF-pyridine in THF at 0 °C.
The substitution pattern of the α,β-unsaturated imine was also explored. Imines bearing no substitution in the β-position (9j) and differential substitution patterns (9k) react efficiently enabling the site-specific functionalization of the piperidine core. Furthermore, a piperidine bearing different substitution at each position of the ring (9l) could be constructed by appropriate choice of imine and acetylene inputs. Bicyclic tetrahydropyridines can be accessed from a cyclic α,β-unsaturated imine precursor (9m–p). Moreover, consistent with our previous work,4 this process is not restricted to benzyl imines as N-cyclopropylmethyl (9o) and N-cyclohexyl (9p) imines are also suitable directing groups.
While this method works exceedingly well for ketimine precursors, aldimine substrates underwent incomplete desilylation under the standard reaction conditions. We reasoned that the aldiminium intermediate is sufficiently reactive that hydride delivery to 8 (R3 = H) competes with desilylation (vide infra). For these substrates, we found it beneficial to conduct the reaction in THF using pyridinium fluoride as the acid source to give the desired tetrahydropyridines 9q,r with complete desilylation.
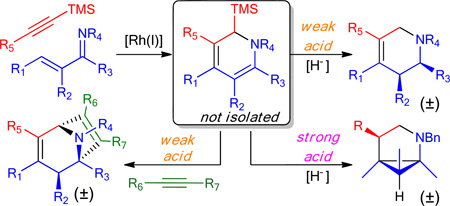
We envisioned that upon protonation of the dihydropyridine 7, desilylation could occur through two possible reaction pathways (Scheme 2). In scenario A, iminium 8 would be reduced to 10 followed by protonation/desilylation to give the observed product 9. Alternatively, 8 could undergo desilylation resulting in the intermediacy of unstabilized azomethine ylide 11. Subsequent protonation followed by reduction of 11 would also account for the formation of the desired product. In support of pathway B, Padwa has leveraged species 12 to access unstabilized azomethine ylides under acidic conditions for [3+2]-dipolar cycloadditions.10 This class of dipoles has been widely utilized for the synthesis of substituted pyrrolidines including those found in natural products and drugs.11,12 However, approaches for generating more highly substituted azomethine ylides typically require the presence of a stabilizing electron withdrawing group.
Scheme 2.

Proposed mechanistic pathways
To distinguish between the two proposed mechanisms, we first resubmitted a silyl-substituted tetrahydropyridine, isolated as a byproduct during early optimization experiments, to the (PhO)2PO2H and TMABH(OAc)3 reaction conditions (eq 2). Cleavage of the silyl group was not observed ruling out pathway A.
 |
(2) |
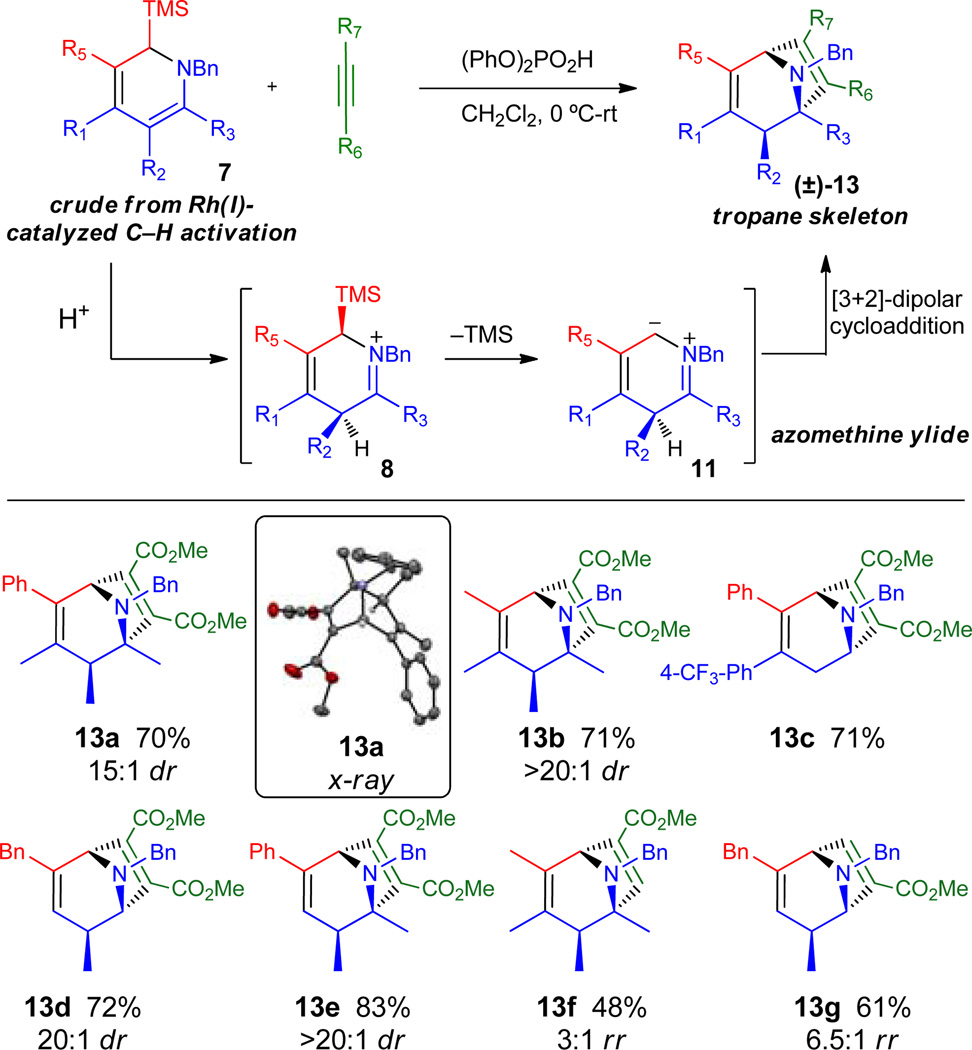
We therefore set out to intercept the transient azomethine ylide in pathway B with a dipolarophile. In support of its intermediacy, direct addition of the [Rh]-reaction mixture containing crude DHP 3 to a solution of 1.2 equiv of (PhO)2PO2H and 2 equiv of dimethylacetylene dicarboxylate (DMAD) at 0 °C resulted in the clean formation of tropane 13a (Table 2), which was characterized by 2D NMR spectroscopy and single crystal X-ray diffraction. While [3+2]-dipolar cycloadditions of azomethine ylides have been widely employed in pyrrolidine synthesis, relatively few examples that provide access to tropanes have been disclosed.13 Furthermore, dipole generation, which proceeds through protonation of the enamine followed by desilylation, represents a new entry into unstabilized azomethine ylides.
Table 2.
Tropane synthesis via azomethine
 |
Yields correspond to isolated product after silica gel chromatography and represent overall yields from the imine precursor 6. Diastereoselectivity was determined by 1H NMR of the crude reaction mixture.
Conducted at −78 °C in toluene.
A number of substituted tropanes were then constructed using this new transformation, showing broad scope in both the imine and trimethylsilyl alkyne inputs (Table 2, 13a–e). Utilization of the unsymmetrical alkyne methyl propiolate also resulted in moderate to good regioselectivity for the dipolar cycloaddition (13f and g), with the regioisomers readily separated by silica gel chromatography. This process represents a new strategy for rapid entry into structurally dense tropanes that would otherwise be challenging to prepare.
A subsequent investigation of the effect of acid strength on reaction yield uncovered an unanticipated reaction product (Scheme 3). Treatment of DHP-3 with PhSO3H in dichloromethane at room temperature resulted in an intractable mixture of products as determined by 1H NMR. However, by adding PhSO3H and Me4NBH(OAc)3 at −78 °C with slow warming to room temperature, a new product was formed in good yield. On the basis of extensive NMR spectroscopy, we assigned the structure as the 2-azabicyclo[3.1.0] system (±)-15a, which was unambiguously confirmed through single crystal X-ray diffraction of the hydrochloride salt. Use of an alkyl rather than an aryl substituted TMS acetylene input also provided the related bridged pyrrolidine 15b, in a one pot process. This core structural motif has significant relevance to drug discovery. For example, it was specifically introduced into the approved drug Saxagliptin to enhance metabolic stability.14
Scheme 3.

Nazarov-like electrocyclization
Variable temperature NMR was performed to gain insight into the formation of 15a from 3. The presence of 1.05 equiv of PhSO3H resulted in the rapid and persistent generation of the N-protonated dihydropyridine 16 between −78 °C and −20 °C. This was converted to a complex mixture of products upon gradual warming between −20 °C and +23 °C. In the presence of Me4NBH(OAc)3, we observed the formation of iminium 17 starting at −20 °C. Upon warming to +5 °C, we observed the gradual formation of 15a.
On the basis of these studies, we conclude that this reaction proceeds through N-protonation of the dihydropyridine followed by proton transfer to generate C-protonated ketiminium 17. Assisted desilylation, either by the sulfate counteranion or hydride, induces a torquoselective disrotatory Nazarov-like15 6-π electrocyclization to form enamine 18,16 which could undergo subsequent stereoselective protonation and reduction resulting in the desired product 15a. This serendipitous finding provides a new avenue for the preparation of highly substituted derivatives of this azabicyclic framework.
In summary, densely substituted 2-silyl 1,2-dihydropyridines are readily prepared from α,β-unsaturated imines and TMS acetylenes by a C-H bond functionalization and electrocyclization sequence. Without any purification or isolation, treatment with acid generates unstabilized azomethine ylides as versatile intermediates that lead to valuable nitrogen heterocycles. In situ protonation and reduction provides tetrahydropyridines with a substitution pattern matching that expected for terminal alkyne incorporation. Alternatively, azomethine generation in the presence of a dipolarophile provides highly substituted tropanes. Finally, by selectively tuning the acid in the protonation step, an unprecedented rearrangement was observed that can be used to produce 2-azabicyclo[3.1.0] systems.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by NIH Grant GM069559 (to J.A.E.). R.G.B. acknowledges funding from The Director, Office of Energy Research, Office of Basic Energy Sciences, Chemical Sciences Division, U.S. Department of Energy, under Contract DE-AC02-05CH11231. M.A.I. also acknowledges support from an NRSA postdoctoral fellowship (F32GM090661).
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information. Full experimental details and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Jordan AM, Roughly SD. J. Med. Chem. 2011;54:3451. doi: 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]
- 2.For recent general reviews on C-H functionalization with applications to heterocycle synthesis, see: Yamaguchi J, Yamaguchi AD, Itami K. Angew. Chem. Int. Ed. 2012;51:8960. doi: 10.1002/anie.201201666. Song G, Wang F, Li X. Chem. Soc. Rev. 2012;41:3651. doi: 10.1039/c2cs15281a. Wencel-Delord J, Droge T, Liu F, Glorius F. Chem. Soc. Rev. 2011;40:4740. doi: 10.1039/c1cs15083a. Colby DA, Bergman RG, Ellman JA. Chem. Rev. 2010;110:624. doi: 10.1021/cr900005n. Satoh T, Miura M. Chem.-Eur. J. 2010;16:11212. doi: 10.1002/chem.201001363. Lyons TW, Sanford MS. Chem. Rev. 2010;110:1147. doi: 10.1021/cr900184e. Chen X, Engle KM, Wang D-H, Yu J-Q. Angew. Chem. Int. Ed. 2009;48:5094. doi: 10.1002/anie.200806273. Seregin IV, Gevorgyan V. Chem. Soc. Rev. 2007;36:1173. doi: 10.1039/b606984n.
- 3.For leading references, see: Saget T, Lemouzy SJ, Cramer N. Angew. Chem. Int. Ed. 2012;51:2238. doi: 10.1002/anie.201108511. Hyster TK, Knörr L, Ward TR, Rovis T. Science. 2012;338:500. doi: 10.1126/science.1226132. Ye B, Cramer N. Science. 2012;338:504. doi: 10.1126/science.1226938. Guimond N, Gorelsky SI, Fagnou K. J. Am. Chem. Soc. 2011:6449. doi: 10.1021/ja201143v. Rakshit S, Grohmann C, Besset T, Glorius F. J. Am. Chem. Soc. 2011:2350. doi: 10.1021/ja109676d. Nakanish M, Katayev D, Besnard C, Kündig EP. Angew. Chem. Int. Ed. 2011;50:7438. doi: 10.1002/anie.201102639. Lewis JC, Bergman RG, Ellman JA. Acc. Chem. Res. 2008;41:1013. doi: 10.1021/ar800042p.
- 4.(a) Duttwyler S, Lu C, Rheingold AL, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2012;134:4064. doi: 10.1021/ja2119833. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Duttwyler S, Chen S, Takase MK, Wiberg KB, Bergman RG, Ellman JA. Science. doi: 10.1126/science.1230704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For recent elegant examples of diastereoselective, convergent assembly of complex tetrahydropiperidines, see: Yang D, Micalizio GC. J. Am. Chem. Soc. 2012;134:15237. doi: 10.1021/ja306362m. Wong H, Garnier-Amblard EC, Liebeskind LS. J. Am. Chem. Soc. 2011;133:7517. doi: 10.1021/ja201012p. Harrison DP, Iovan DA, Myers WH, Sabat M, Wang S, Zottig VE, Harman WD. J. Am. Chem. Soc. 2011;133:18378. doi: 10.1021/ja2075086.
- 6.Colby DA, Bergman RG, Ellman JA. J. Am. Chem. Soc. 2008;130:3645. doi: 10.1021/ja7104784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin RM, Bergman RG, Ellman JA. J. Org. Chem. 2012;77:2501. doi: 10.1021/jo202280e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For information regarding piperidine-containing drug and drug candidates, see PubChem (http://pubchem.ncbi.nlm.nih.gov/) for the structure, bioactivity, full list of published studies, and information regarding clinical trials, applications, and usage. Examples of top selling pharmaceuticals: plavix (CID 60606), oxycontin (CID 5284603), spiriva (5487426), suboxone (CID 56841117), combivent (CID 24847804), concerta (CID 9280). Select piperidine-containing drugs with multiple stereocenters: paroxetine (CID 43815), hydergine (CID 168870), moxifloxacin (CID 101526), nelfinavir (CID 64142), cabergoline (CID 83879), alvimopan (CID 5488547), morphine (CID 5464110).
- 9.(a) For a review on the synthesis and elaborations of dihydropyridine derivatives, see: Bull JA, Mousseau JJ, Pelletier G, Charette AB. Chem. Rev. 2012;112:2642. doi: 10.1021/cr200251d. For leading references on alternative methods for accessing substituted 1,2-dihydropyridines, see: Harrison DP, Sabat M, Myers WH, Harman WD. J. Am. Chem. Soc. 2010;132:17282. doi: 10.1021/ja107536w. Harschneck T, Kirsch SF. J. Org. Chem. 2011;76:2145. doi: 10.1021/jo102545m. Tejedor D, Mendez-Abt G, García-Tellado F. Chem.—Eur. J. 2010;16:428. doi: 10.1002/chem.200902140. Comins DL, Weglarz MA, O’Connor S. Tetrahedron Lett. 1988;29:1751.
- 10.Padwa A, Dent W. J. Org. Chem. 1987;52:235. [Google Scholar]
- 11.For reviews on [3+2]-azomethine ylide dipolar cycloadditions, see: Pandey G, Banerjee P, Gadre SR. Chem. Rev. 2006;106:4484. doi: 10.1021/cr050011g. Coldham I, Hufton R. Chem. Rev. 2005;105:2765. doi: 10.1021/cr040004c. Gothelf KV, Jørgensen KA. Chem. Rev. 1998;98:863. doi: 10.1021/cr970324e.
- 12.For recent elegant applications of azomethine ylides in complex synthesis, see: Belanger G, Boudreault J, Levesque F. Org. Lett. 2011;13:6204. doi: 10.1021/ol202629d. Coldham I, Burrell AJM, Guerrand HDS, Oram N. Org. Lett. 2011;13:1267. doi: 10.1021/ol102961x. Davoren J, Gray D, Harris A, Nason D, Xu W. Synlett. 2010;2010:2490. Peese KM, Gin DY. Chem. Eur. J. 2008;14:1654. doi: 10.1002/chem.200701290. Eckelbarger JD, Wilmot JT, Epperson MT, Thakur CS, Shum D, Antczak C, Tarassishin L, Djaballah H, Gin DY. Chem. Eur. J. 2008;14:4293. doi: 10.1002/chem.200701998. Coldham I, Burrell AJM, White LE, Adams H, Oram N. Angew. Chem. Int. Ed. 2007;46:6159. doi: 10.1002/anie.200701943. Su S, Porco JA., Jr J. Am. Chem. Soc. 2007;129:7744. doi: 10.1021/ja072737v. Eckelbarger JD, Wilmot JT, Gin DY. J. Am. Chem. Soc. 2006;128:10370. doi: 10.1021/ja063304f.
- 13.(a) Pandey G, Laha JK, Lakshmaiah G. Tetrahedron. 2002;58:3525. [Google Scholar]; (b) Pandey G, Sahoo AK, Bagul TD. Org. Lett. 2000;2:2299. doi: 10.1021/ol006070s. [DOI] [PubMed] [Google Scholar]; (c) Pandey G, Laha JK, Mohanakrishnan AK. Tetrahedron Lett. 1999;40:6065. [Google Scholar]; (d) Pandey G, Lakshmaiah G, Ghatak A. Tetrahedron Lett. 1993;34:7301. [Google Scholar]
- 14.(a) Jones GS, Savage SA, Ivy S, Benitez PL, Ramirez A. J. Org. Chem. 2011;76:10332. doi: 10.1021/jo202052a. [DOI] [PubMed] [Google Scholar]; (b) Savage SA, Jones GS, Kolotuchin S, Ramrattan SA, Vu T, Waltermire RE. Org. Proc. Res. Dev. 2009;13:1169. [Google Scholar]; (c) Augeri DJ, Robl JA, Betebenner DA, Magnin DR, Khanna A, Robertson JG, Wang A, Simpkins LM, Taunk P, Huang Q, Han S-P, Abboa-Offei B, Cap M, Xin L, Tao L, Tozzo E, Welzel GE, Egan DM, Marcinkeviciene J, Chang SY, Biller SA, Kirby MS, Parker RA, Hamann LG. J. Med. Chem. 2005;48:5025. doi: 10.1021/jm050261p. [DOI] [PubMed] [Google Scholar]
- 15.For recent reviews on the Nazarov reaction, see: Vaidya T, Eisenberg R, Frontier AJ. Chem Cat Chem. 2011;3:1531. Frontier AJ, Collison C. Tetrahedron. 2005;61:7577. Pellissier H. Tetrahedron. 2005;61:6479. For examples of 6-π anionic electrocyclizations, see: Baktharaman S, Afagh N, Vandersteen A, Yudin AK. Org. Lett. 2010;12:240. doi: 10.1021/ol902550q. Woo CM, Lu L, Gholap SL, Smith DR, Herzon SB. J. Am. Chem. Soc. 2010;132:2540. doi: 10.1021/ja910769j. Williams DR, Reeves JT. J. Am. Chem. Soc. 2004;126:3434. doi: 10.1021/ja049353e.
-
16.To the best of our knowledge, no other literature examples have been reported for intramolecular allylsilane additions to imines or carbonyls to give cyclopropyl amines or alcohols, respectively.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.