

Abstract
The oxidative coupling of enolates, enol silanes, and enamines provides a direct method for the construction of useful 1,4-dicarbonyl synthons. Despite being first reported in 1935, with subsequent important advances beginning in the 1970’s, the development of this powerful reaction into a reliable methodology was somewhat limited.
In recent years, there have been a number of reports from several research groups demonstrating advances in several neglected areas of oxidative coupling. This microreview summarizes these new advances in methodology and provides an overview of recent natural product syntheses that showcase the power of these transformations.
Keywords: Oxidative Coupling, Enolates, Enol Silanes, Enamines, Natural Product Synthesis
Introduction
The oxidative coupling of two enolates provides a direct and convergent method for the construction of 1,4-dicarbonyl motifs (Figure 1). At the strategic level, application of such transforms during retrosynthetic planning can allow for the concise synthesis of complex molecules by the efficient, and often stereocontrolled, formation of a σ-bond between two functional groups arranged in an otherwise dissonant relationship. One advantage of such a strategy is that it can provide a more streamlined synthesis by avoiding functional group Umpolung or the use of operational equivalents that in many cases may require additional synthetic manipulations. On a tactical level, oxidative coupling represents a powerful methodology for the construction of 1,4-diketones that are highly useful precursors to a wide range of useful structures, such as pyrroles and furans, for example. The earliest report of such an oxidative enolate coupling dates back to 1935,[i] when Ivanoff and Spasoff demonstrated that the enolate of phenylacetic acid undergoes an oxidative dimerization when exposed to dioxygen or molecular iodine. The reported reactions were plagued by low yields and the formation of multiple unwanted side products, and it was not until the 1970s that synthetically useful methods for preparative oxidative enolate coupling were pioneered. In 1971, Rathke and coworkers reported the oxidative dimerization of ester enolates using Cu(II) bromide or Cu(II) valerate to afford substituted succinate esters.[ii] In 1975, Saegusa and coworkers reported the use of Cu(II) chloride as an effective stoichiometric oxidant for the dimerization of ketone enolates, and further demonstrated that intramolecular variants were quite efficient.[iii] They also reported the cross-coupling of two different enolates to generate unsymmetrical 1,4-diketones, but this required the use of one ketone enolate in a three-fold excess over the other. Following these reports, various other oxidants were established for the oxidative coupling of enolates, such as Cu(II) triflate,[iv] Fe(III) chloride, [v] iodine, [vi] Ti(IV) chloride, [vii] and Ag(I) chloride. [ viii] Contemporaneously, the use of non-enolate carbonyl derivatives for oxidative coupling was also pioneered. This research led to effective methods for the oxidative coupling of enol silanes, [ix ] enamines, [x ] and enol acetates. [xi] In several instances, cross-coupling could also be achieved, but as with lithium enolates this typically came at the expense of one reaction partner.
Figure 1.
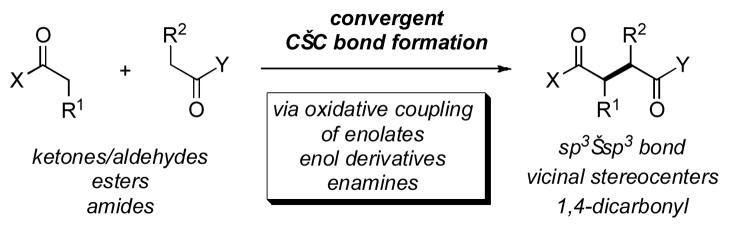
Oxidative Enolate Coupling: An Overview
The synthetic utility of these powerful bond constructions has been demonstrated through application in the total synthesis of several structurally diverse natural products, including lamellarin G trimethyl ether (1), [xii] rac-chimonanthine (2), [xiii] rac-folicanthine (3), [xiv] cis-jasmone (4) [3] and rac-hirsutene (5). [xv] Notably, the groups of both Kise and Helmchen developed useful strategies for the stereocontrolled enantioselective synthesis of several lignans, such as ent-hinokinin (6), through the oxidative dimerization of Evans oxazolidinones. [xvi] In addition to natural product synthesis, Paquette and coworkers made use of an intramolecular oxidative enolate coupling in their elegant synthesis of the structurally unusual hemispheric hydrocarbon, C16 hexaquinacene (7). [xvii]
Despite the body of literature pertaining to oxidative enolate coupling that appeared last century, it remained a relatively under investigated research area. In terms of methodology development, cross-coupling with equal stoichiometry of reacting partners remained a significant challenge, and systems that enabled uniformly high levels of diastereocontrol were underexplored. Moreover, no straightforward catalytic and enantioselective methods for oxidative enolate coupling were available. Beginning with the turn of the new millennium, however, there have been significant advances made in all of these areas that have led to several powerful new methods for oxidative enolate coupling. The purpose of this microreview is to summarize these contributions, and to provide recent examples of strategic applications of oxidative enolate coupling in the realm of natural product total synthesis. This microreview will not cover related oxidative bond forming reactions that involve enolates (or derivatives thereof) with aromatic systems, such as electron rich aryl groups, pyrroles, indoles and furans.[xviii]
Methodology Development
Recent methodological developments in oxidative enolate coupling (the term is used here to broadly describe alkali metal enolate, enol silane and enamine-based reactions) have focused on solving the cross-coupling problem, while simultaneously addressing the question of stereocontrol, both relative and absolute. In recent years, several complementary methods for oxidative enolate coupling have been reported and this work is summarized in the following subsections.
Lithium Metal Enolates
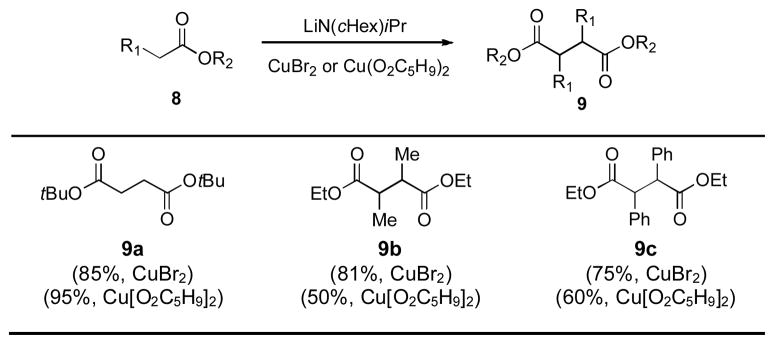
In 1971, Rathke and Lindert disclosed a significant advance in the area of oxidative enolate coupling. They showed that when treated with either Cu(II) bromide or Cu(II) valerate, the lithium enolates of several esters underwent an efficient oxidative coupling to generate succinate derivatives (Table 1).[2]
Table 1.
Rathke and Lindert’s Oxidative Dimerization[a]
|
Yields refer to isolated yields following vacuum distillation. cHex = cyclohexyl.
While only six substrates were reported in total, and diastereoselectivities were poor (for 9b and 9c, for example), this work provided much precedent for subsequent work in the field.
In 1975, Saegusa and coworkers reported the use of Cu(II) chloride to bring about the oxidative coupling of ketone-derived lithium enolates. [3] In this early report, they demonstrated that cross-coupled products could be obtained in synthetically useful yields when one enolate is used in excess (Table 2).
Table 2.
Saegusa and Coworkers Intermolecular Cross-Coupling[a]

|
Yields refer to isolated yields following chromatography.
While the power of this approach is clear for the synthesis of simple unsymmetrical 1,4-diketones, the necessary use of a large excess of one ketone limited its use in more complex settings. The enolate used in excess is lost due to competitive dimerization; this situation is of little consequence for the case of acetone, but would become prohibitive for advanced ketone intermediates en route to complex natural products. It would be 30 years until synthetic chemists returned to this problem.
In 2006, Baran and DeMartino reported the development of a system that enabled selective intermolecular oxidative coupling of lithium enolates using equal stoichiometry of the reacting partners for the first time (Table 3).[xix]
Table 3.
Baran and DeMartino’s Intermolecular Cross-Coupling[a]

|
Yields refer to isolated yields following chromatography.
A subsequent paper detailing the full scope of the reaction was later published in 2008.[xx] Using LDA to generate the appropriate enolates, they found that subsequent addition of Fe(III) or Cu(II) oxidants enabled the formation of cross-coupled 1,4-dicarbonyls derived from oxazolidinones or oxindoles with ketones or esters (Table 3). Good yields were obtained across a range of diverse substrates, and in some cases preparatively useful levels of diastereocontrol were observed. The cross-coupling reactions of Evans oxazolidinones offer an especially concise route to enantioenriched products that are useful precursors to natural products (see later for example), while the use of α-substituted oxindoles afforded products containing a newly forged quaternary stereocenter. Along with further establishing the range of useful compounds that could be prepared by this powerful reaction, the 2008 publication also provided some insight into the reaction. The stereochemical outcome for the Fe(II)-mediated oxidative coupling of phenylacetyl oxazolidinones (i.e., Table 3, 15a–d) was opposite to that obtained for related dihydrocinnamyl derivatives under Cu(II)-promoted conditions (i.e., Table 3, 15g–i); a somewhat surprising outcome. For enolate alkylations of Evans oxazolidinones, the expected relationship between the auxiliary and the α-stereocenter is syn, as is observed for the Cu(II)-promoted reactions. In an effort to reconcile the anti stereochemistry observed for the Fe(III)-mediated reactions with the expected outcome for Evans auxiliaries, Baran and coworkers showed that exposure of the “syn” isomer 16 to LDA followed by acidic enolate quenching led to complete inversion of the Ph-bearing stereocenter to produce anti isomer 16 (Scheme 1).
Scheme 1.
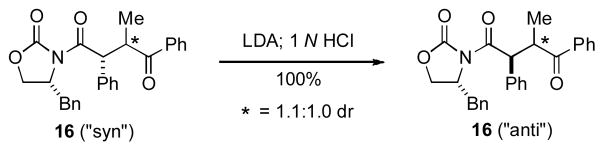
Anomalous Stereochemical Outcome Rationalized
Thus, it appeared for phenylacetyl adducts that the α-stereocenter is sufficiently acidic that the presumed initial syn adduct of oxidative coupling undergoes base-promoted epimerization to the observed anti configuration.
In addition to these interesting substrate based stereochemical quandaries, different reaction outcomes were found depending upon the oxidant employed, which was interpreted as being due to differing mechanisms for Cu(II) vs Fe(III) oxidations. For the Fe-based oxidations, it appeared that the ketone enolate was more readily oxidized leading to preferential coupling with the oxazolidinone enolate via an open transition state akin to intermediate 20 (Figure 3). Carbon bond formation, followed by oxidation of the intermediate radical 21, provides the product 22 in good yield (although typically this would undergo epimerization to the “anti” isomer). In contrast, for the Cu-based oxidations, mechanistic studies revealed little preference for oxidation of the ketone enolate over the oxazolidinone enolate. Therefore, they proposed selective coupling as a consequence of favorable formation of a Cu-bound bis-enolate intermediate such as 23 (Figure 3). Once again, the cross-coupled product was isolated in good yield, with the major diastereomer consistent with the intermediacy of a species such as bisenolate 23. Together, these models also serve to rationalize the higher than statistical yields for the cross-coupled products over dimers.
Figure 3.

Baran’s Mechanistic and Stereochemical Model
While the studies of Baran and coworkers provided important insights into the cross-coupling of lithium enolates, subsequent work reported by Flowers and Casey in 2011 presented spectroscopic and mechanistic results showing that the selective generation of cross-coupled products for the ketones they investigated was the result of heteroaggregation of the lithium enolates (Table 4).[xxi]
Table 4.
Casey and Flowers’ Studies on Li-Enolate Aggregation Effects of Oxidative Cross-Coupling[a]

| |||||
|---|---|---|---|---|---|
| Entry | Ketone A | Product 26 | A2B2:A4 + B4 | Product ratio | Yield (%) |
| 1 |
|
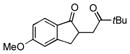
|
15.7:1 | 13.8:1 | 62 |
| 2 |

|
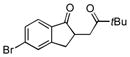
|
14.7:1 | 12.8:1 | 58 |
| 3 |
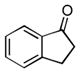
|
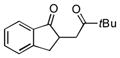
|
14:3.1 | 12.4:1 | 62 |
| 4 |

|

|
8.5:1 | 7.0:1 | 46 |
| 5 |

|
|
4.1:1 | 3.0:1 | 47 |
Ratios and yields determined by NMR spectroscopy. CTAN = ceric tetra-n butylammonium nitrate.
Using 7Li NMR spectroscopy, they showed that 1:1 mixtures of different Li-enolates (i.e., A and B) tended to preferentially form heteroaggrerates (A2B2) and that the ratio of the heteroaggregate to the monomeric aggregates (A4 + B4) correlated well with the ratio of cross-coupled to homo-coupled products obtained upon oxidative coupling (Table 4). These fascinating and important studies revealed a level of complexity to the situation that had not previously been recognized, and provided key insights for future development of cross-coupling reactions (and the interpretation of results).
In 2011, we reported the use of stereoselective oxidative enolate dimerization of enantioenriched monoketal dihydroquinones for the assembly of non-racemic biphenols (Scheme 2). [xxii] Treatment of the readily prepared enone precursors (27a or 27b) [xxiii] with LDA followed by Cu(II) chloride forged the hindered central σ-bond linking the two rings within the dione products (28a or 28b). We had reasoned that conformational rigidity about this bond would enable transfer of the point chirality present in the dimers to the axial chirality present in the aromatized biphenol products (i.e., 29). Thus, we found that Lewis acid induced loss of methanol with simultaneous keto to enol tautomerization afforded the desired biphenols in good yield and with complete transfer of stereochemical information from starting material to product. The dimerization process itself is interesting in that an amplification of enantiopurity from the monomer is observed due to loss of the minor enantiomer through formation of a meso-isomer.[xxiv]
Scheme 2. Traceless Stereochemical Transfer from 1,4-Diketones.
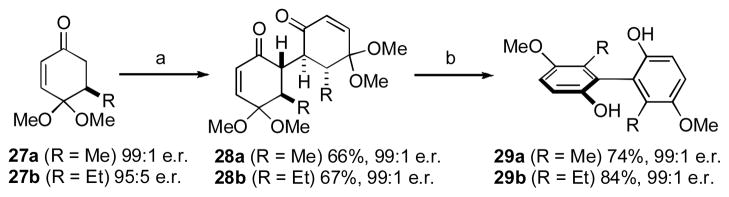
[a] LDA (1.1 equiv), CuCl2 (1.15 equiv), THF/DMF, − 78°C to r.t., 1.5 h. [b] BF3•OEt2 (15.0 equiv), toluene, 110 °C, 12 h. Reprinted from L. C. Konkol, F. Guo, R. J. Thomson J. Am. Chem. Soc. 2011, 133, 18–20. Copyright 2011 American Chemical Society.
In an effort to expand the scope of this novel method for the synthesis of non-racemic biphenols, we devised an enantioselective synthesis of aryl-substituted enones (i.e., 30) by modifying chemistry reported by Hayashi and coworkers[ xxv ] for the asymmetric conjugate addition of aryl zinc reagents. Thus, we prepared a selection of aryl-substituted monomers and demonstrated that the oxidative dimerization/stereochemical transfer process proceeded as well as for the alkyl-derivatives (Table 5).[22]
Table 5.
Elaboration of Traceless Stereochemical Transfer[a]

| |||
|---|---|---|---|
| Entry | Ar | Yield 31 (e.r.) | Yield 32 (e.r.) |
| 1 | Ph (30a) | 56 (99:1) | 77 (99:1) |
| 2 | 2-Naphthyl (30b) | 51 (99:1) | 82 (99:1) |
| 3 | 4-OMe-Ph (30c) | 50 (99:1) | 86 (99:1) |
| 4 | 4-Me-Ph (30d) | 63 (99:1) | 89 (99:1) |
| 5 | 4-F-Ph (30e) | 66 (99:1) | 88 (99:1) |
| 6 | 3,5-di-Me-Ph (30f) | 73 (99:1) | 85 (99:1) |
| 7 | 3,5-di-OMe-Ph (30g) | 48 (99:1) | 75 (99:1) |
| 8 | 3,4-methylenedioxy-Ph (30h) | 52 (99:1) | 78 (99:1) |
Yields refer to isolated yields following chromatography. Enantiomeric ratios determined by HPLC.
This new method for the concise preparation of enantioenriched axially chiral biphenols has potential applications for the synthesis of new ligands for asymmetric catalysis, and has already found use in natural product synthesis (see later).
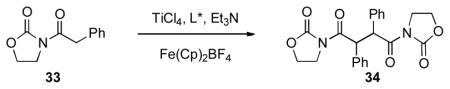
We conclude this section by describing attempts by Nguyen and Schäfer to develop an enantioselective oxidative coupling. [xxvi] In their report from 2001, Nguyen and Schäfer showed that generation of the titanium enolate of acyl oxazolidinone 33 with Fe(Cp)2BF4 as the stoichiometric oxidant allows for effective oxidative dimerization to produce 34 (Table 6). The use of chiral additives in stoichiometric quantities provided the product in good yields, but with varying levels of diastereo- and enantioselectivity. Unfortunately, while the best additive for enantioselectivity (i.e., G, Table 5, entry 7) provided the product in 76% ee, the ratio of dl:meso isomers was only 1:3. Additional substrates were not reported, and while this reaction is somewhat limited it does provide a possible template for future efforts in the design of methods for the enantioselective oxidative coupling of enolates.
Table 6.
Nguyen and Schäfer’s Enantioselective Dimerization[a]
| ||||
|---|---|---|---|---|
| Entry | Additive L* | Yield 34 | dl:meso | %ee |
| 1 | A | 67 | 54:46 | 39 |
| 2 | B | 63 | 31:69 | 5 |
| 3 | C | 52 | 24:76 | 16 |
| 4 | D | 59 | 35:65 | 6 |
| 5 | E | 81 | 45:55 | 24 |
| 6 | F | 58 | 48:52 | 46 |
| 7 | G | 91 | 25:75 | 76 |
Ratios determined by NMR spectroscopy.

Silyl Bis-Enol Ethers
In 1998, Schmittel and coworkers reported that silyl bis-enol ether 35 undergoes intramolecular oxidative coupling when treated with Ce(IV) ammonium nitrate (CAN) to generate diketone 36 in good yield with high levels of diastereocontrol (Scheme 3). [xxvii]
Scheme 3.
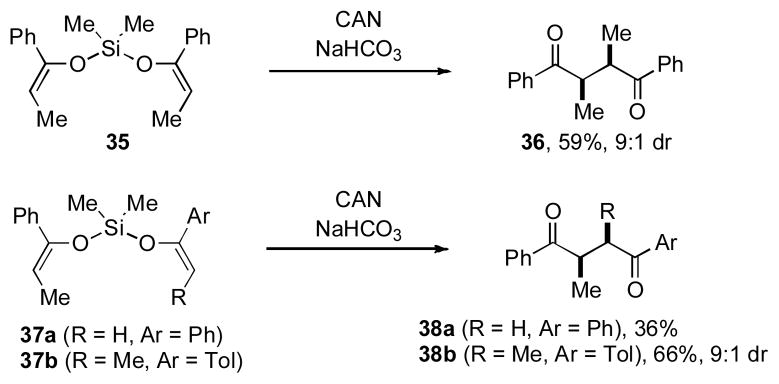
Schmittel’s Report of Silyl Bis-Enol Ether Coupling
In addition, they showed that cross-coupling could be achieved when unsymmetrical silyl bis-enol ethers were utilized; thus bis-enol ethers 37a and 37b gave rise to diketones 38a and 38b, respectively. While only four substrates were examined in total, the formation of diketone 38b from bis-enol ether 37b represented the first time that highly stereoselective cross-coupling had been achieved. As such, the concept of silicon-tethering represents a powerful strategy for developing a general method for controlled oxidative coupling that should have broad utility in natural product synthesis. Our own research group sought to expand upon these preliminary findings of Schmittel with this long-term goal in mind.
In 2007, we reported the development of a general method for the oxidative cross-coupling of 2-methyltetralone (39) to afford unsymmetrical 1,4-diketones (i.e., 42) with simultaneous generation of a quaternary stereocenter (Table 7). [xxviii] While modification of the tetralone component was somewhat limited, the reaction displayed good substrate scope for the methyl ketone component (representative examples shown), yielding 1,4-diketones in good yield. As had been a criterion from the outset, the synthesis of the unsymmetrical silyl bis-enol ethers could be conducted using only 1.1 equivalents of 40, negating the need for a large excess of one reacting partner.
Table 7.
Silyl Bis-Enol Ether Cross Coupling of 2-Methyltetralone[a]

|
Yields refer to isolated yields following chromatography. LDA = lithium diisopropylamide; CAN = ceric ammonium nitrate.
Following these initial studies, we investigated the diastereoselective synthesis of linked bicyclic diketones using silyl bis-enol ethers. Initial studies focused on the dimerization of cyclohexanone via bis-enol ether 43 (Table 8).[xxix]
Table 8.
The Influence of Silicon Substituents on Diastereselectivity[a]

| |||
|---|---|---|---|
| Entry | R2Si | dr (44:45) | Yield of 44 |
| 1 | Me (43a) | 2.4:1 | 29 |
| 2 | Et (43b) | 5:1 | 47 |
| 3 | Ph (43c) | 5:1 | 29 |
|
| |||
| 4 | iPr (43d) | 10:1 | 57 |
|
| |||
| 5 | –C4H8– (43e) | 3:1 | 31 |
| 8 | –C5H10– (43f) | 6:1 | 56 |
Yields refer to isolated yields following chromatography. Diastereomeric ratios determined by 1H NMR spectroscopy. CAN = ceric ammonium nitrate.
As an important benchmark, it was known from the literature that dimerization of the lithium enolate of cyclohexanone gave low yields and poor diastereocontrol slightly favoring the meso-isomer 45. [xxx ] We prepared several silyl bis-enol ethers with varying silicon substituents, and noted enhanced bias for the chiral diastereomer 44 with larger substituents. Ultimately, we found diisopropyl silyl bis-enol ether 43d gave the best combination of high diastereoselectivity and chemical yield (entry 4, Table 7).
We rationalized the inherent preference for the chiral isomer over the meso isomer by invoking two possible reactive conformations during carbon–carbon bond formation of the intermediate radical-cation species (Figure 4). Conformation I minimizes eclipsing interactions between the two rings when compared to conformer II, thus providing a selection bias for generation of the chiral diastereomer. This destablizing effect within II is further enhanced with larger substituents of silicon; a Thorpe-Ingold effect would reduce the size of angle γ when R = i-Pr versus R = Me, thereby creating a larger energy difference between I and II that is reflected in the product distribution.
Figure 4. Model for Stereoselectivity.

Reprinted with permission from C. T. Avetta, Jr., L. C. Konkol, C. N. Taylor, K. C. Dugan, C. L. Stern, R. J. Thomson Org. Lett. 2008, 10, 5621–5624. Copyright 2008 American Chemical Society.
The developed reaction conditions proved remarkably general for the stereoselective dimerization of cyclic ketones (47a–c, Table 9).
Table 9.
Diastereoselective Dimerization and Cross-Coupling[a]

|
Yields refer to isolated yields following chromatography. Diastereomeric ratios determined by 1H NMR spectroscopy. CAN = ceric ammonium nitrate.
Highly diastereoselective cross-coupling could also be achieved (47d–i, Table 9), and for the cases where one reaction partner possessed a β-substituent (such as carvone) an added element of facial control was observed. As expected, the newly forged σ-bond formed opposite to the β-disposed substituent (47g–i, Table 9). The uniformly high diastereoselectivity observed for both dimerization and cross-coupling are the highest yet reported and should pave the way for future applications in complex molecule synthesis.
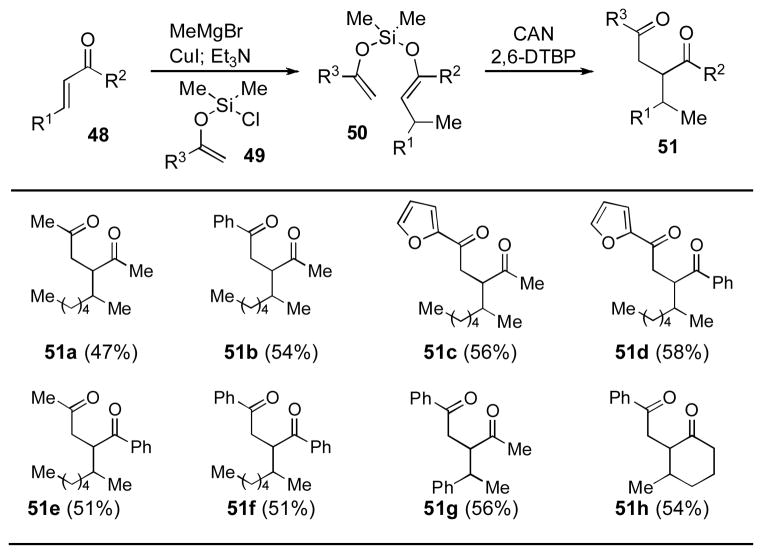
For all the cases discussed thus far, the silyl bis-enol ether was formed from the corresponding enolate generated by deprotonation of the immediate ketone precursor. We speculated that it should be possible to form silyl bis-enol ethers (i.e., 50) by the trapping of an enolate generated through a conjugate addition to an appropriate enone starting material (i.e., 48). In this way, 1,4-addition would allow for a merged three-component sequence when coupled to a subsequent oxidative carbon–carbon bond-forming event (Table 10).[xxxi]
Table 10.
Merged Conjugate Addition/ Oxidative Cross-Coupling[a]
|
Yields refer to isolated yields following chromatography. CAN = ceric ammonium nitrate; DTBP = di-tert-butylpyridine.
The reaction displayed generality for a variety of different substrates, allowing for a concise entry into structurally diverse 1,4-diketones in reasonably good yields over the two-step merged sequence (i.e., 48 → 51). A subsequent Paal–Knorr pyrrole synthesis demonstrated the usefulness of this methodology for the synthesis of substituted pyrroles and led to application of this approach to the total synthesis of prodigiosin alkaloids (see later).
Enamine Catalysis
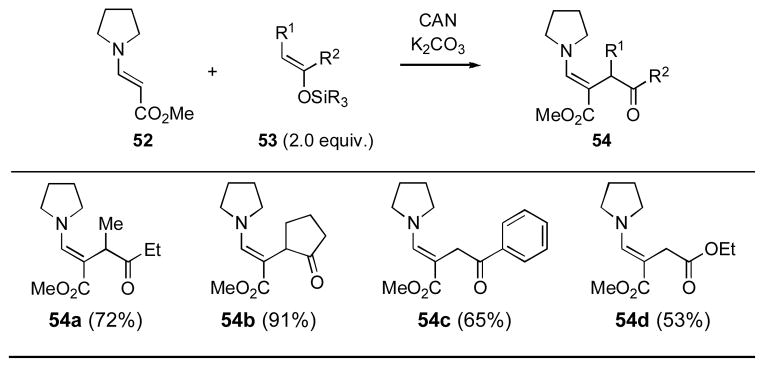
In 1992, Narasaka and coworkers reported the efficient oxidative cross-coupling of preformed enamines with enol silanes upon treatment with Ce(IV) ammonium nitrate (Table 11). [9] While the majority of the examples in this publication reported cross-coupling with enamine 52 to generate stable enamine products (i.e., 54), there was one isolated example of a cross-coupling with a morpholine-derived enamine that afforded the corresponding 1,4-diketone.
Table 11.
Narasaka and Coworkers Oxidative Cross Coupling[a]
|
Yields refer to isolated yields following chromatography. CAN = ceric ammonium nitrate.
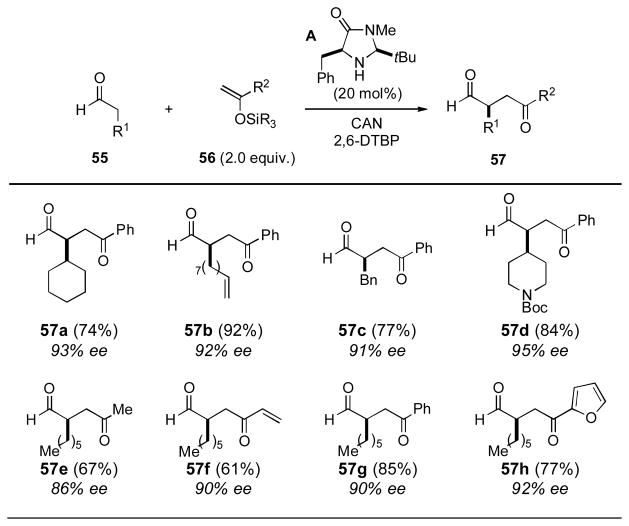
The scope of this chemistry went largely unexplored until the MacMillan group creatively leveraged their expertise in enamine organocatalysis to develop the first, and to date the only, method for catalytic enantioselective oxidative coupling (Table 12). [xxxii] Under the reported reaction conditions, two equivalents of an enol silane (56) will undergo Ce(IV) ammonium nitrate promoted oxidative bond formation with an aldehyde (55) in the presence of 20 mol% of an organocatalyst (A). For the wide variety of substrates reported, the yields of the 1,4-dicarbonyl adduct 57 were high and the enantioselectivities obtained were uniformly excellent.
Table 12.
MacMillan and Coworkers Enantioselective Coupling[a]
|
Yields refer to isolated yields following chromatography. Enantiomeric excess determined by GC or SFC analysis. CAN = ceric ammonium nitrate; DTBP = di-tert-butylpyridine.
This report is especially promising for new directions in oxidative enolate coupling as it demonstrates that catalyst controlled enantioselective variants are possible and paves the way for further exciting developments in the future.
In 2010, the Jia group developed a new method for the direct construction of symmetrical pyrroles through the oxidative dimerization of aldehydes in the presence of amines (Table 13). [xxxiii] In these reactions, the amine (59) serves not only to facilitate pyrrole formation via Paal–Knorr pyrrole synthesis, but also promotes oxidative coupling through initial enamine formation. Optimum yields were obtained using silver acetate as the stoichiometric oxidant. Although currently limited to the construction of symmetrical pyrroles, the Jia group has successfully employed this methodology in the rapid total synthesis of numerous pyrrole-containing natural products (see later section).
Table 13.
Jia and Coworkers Oxidative Dimerization[a]

|
Yields refer to isolated yields following chromatography. OAc = acetate, PMP = 4-methoxyphenyl.
Natural Product Synthesis
Paralleling the development of new methods for oxidative coupling there have been a number of significant applications of oxidative coupling to the successful synthesis of a variety of different natural products. The range of structures targeted and methods employed, serve to demonstrate the great power of oxidative coupling reactions when they are deployed strategically in total synthesis. The following section summarizes this recent work and is divided into sections dealing with dimerizations, cross-coupling reactions and intramolecular versions.
Oxidative Dimerization
The Lomaiviticin Aglycone
Lomaivaticin A (61) and B (62) are two incredibly complex glycosylated natural products isolated from a fermentation broth of a species of actinomycete (Figure 5). [xxxiv] Their potent DNA-strand cleaving ability arises due to the presence of the rather unusual diazo functionality; a feature they share with the biogenetically related kinamycin molecules. [xxxv] Structurally, the aglycone core of the lomaiviticins is dimeric, and it is therefore most likely that the biosynthesis of these compounds occurs through the oxidative dimerization of a suitable monomeric precursor, such as 63.
Figure 5.

Lomaiviticin A (61) and B (62)
From a strategic standpoint, when contemplating a total synthesis of the lomaiviticins, this symmetry leads to the logical conclusion that such a dimerization strategy would enable the most efficient synthesis. The question is then one of tactics to deal with the complexity of this approach. Major issues facing this oxidative dimerization strategy via an enolate are the possibility of elimination of the β-hydroxyl group and the question of diastereofacial control. In their work published towards the lomaiviticins, Shair and coworkers utilized tricyclic ketone 64 to deal with each of these issues (Scheme 4). [xxxvi] The oxo-bridged [2.2.1] bicyclic ketone 64 ensures excellent diastereocontrol for the exo-face, while geometric constraints limit the facility of E1CB decomposition of the intermediate enolate under the reaction conditions. Thus, the complex polycyclic 1,4-diketone 65 was formed in 45–51% yield as a single stereoisomer. Further elaboration of dione 65 gave rise to the hydrate 66, which underwent a controlled fragmentation of the oxo-bridge under basic conditions to install the requisite tertiary alcohol with simultaneous displacement of the sulfone with methoxide. The central core of the lomaiviticin aglycon (i.e., 67) was thus constructed with complete stereocontrol in only four steps from the monomeric ketone 64.
Scheme 4. The Shair Group’s Approach to the Lomaiviticin Core.

[a] LiHMDS (1.7 equiv), HMPA (5 equiv), THF, −78 °C, 1.5 h; then [Cp2Fe]PF6 (3 equiv), −20 °C, 20 h, 45–51%; [b] 48% aq. HF (20 equiv), MeCN, 23 °C, 3 d; [c] DMP (3 equiv), CH2Cl2, 23 °C, 3 h, 26% over 2 steps; [d] K2CO3 (6 equiv), MeOH, 0 °C to 23 °C, 3 h, 14%. [Cp2Fe]PF6 = ferrocenium hexafluorophosphate, HMDS = hexamethyldisilazane, HMPA = hexamethylphosphoramide, TBS = tert-butyldimethylsilyl, DMP = Dess-Martin periodinane.
In a subsequent publication in 2010, Shair and coworkers reported the successful oxidative coupling of a more fully elaborated precursor that contained all the required rings associated with the lomiaviticin aglycon (Scheme 5). [xxxvii] Synthesis of the precursor for oxidative coupling (i.e., 68) proceeded using an approach related to that of the less complex monomer 64 shown in Scheme 4. Under similar conditions to the successful dimerization of 64, ketone 68 underwent a highly stereoselective and high yielding oxidative dimerization to generate the complex dimer 69 in 72% yield after allyl group removal. The stereochemistry of adduct 69 was confirmed by X-ray crystal analysis. The combination of these two reports by the Shair lab suggest that a robust route to the natural product is in place, and highlights the power of oxidative coupling to forge sterically congested bonds.
Scheme 5. The Shair Group’s Second Approach to the Lomaiviticin Core.
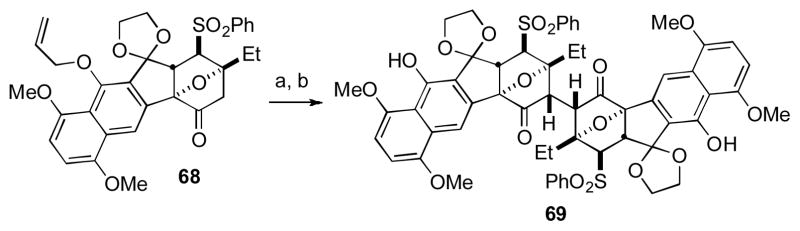
[a] LiHMDS (1.7 equiv), HMPA (5 equiv), THF, −78 °C, 2 h; then [Cp2Fe]PF6 (3 equiv), −60 °C, 72 h, 80%; [b] [PdCl2(PPh3) 2], Bu3SnH, AcOH, CH2Cl2, 0 °C, 90%. [Cp2Fe]PF6 = ferrocenium hexafluorophosphate, HMDS = hexamethyldisilazane, HMPA = hexamethylphosphoramide.
The Herzon group opted to take a more direct approach and tackle the problem of dimerization of a more fully elaborated monomeric precursor already containing the diazo functionality (akin to 63 in Figure 5). This maximally convergent approach led to the first synthesis of the lomaiviticin aglycone in 2011 (Scheme 6).[xxxviii]
Scheme 6. The Herzon Group’s Synthesis of the Lomaiviticin Aglycon.

[a] TMSOTf, E t3N, CH2Cl2, 0 ° C; [b] Manganese tris(hexafluoroacetylacetonate), PhH, 21 °C, 26%; [c] TFA, TBHP, CH2Cl2, −35 °C, 12 h, 39%. TMS = trimethylsilyl, Tf = trifluoromethylsulfonyl, TFA = trifluoroacetic acid, TBHP = tert-butylhydroperoxide.
Preparation of the requisite monomeric ketone 70 was carried out using methods related to the Herzon labs preparation of the kinamycins. [ xxxix ] After extensive studies into conditions for oxidative dimerization (>1500 experiments), they found that treatment of the TMS enol ether 71 with Mn(III) hexafluoroacteoacetate produced 26% yield of the desired 1,4-diketone 72, along with 12% of the undesired isomer possessing the bis-epimeric stereochemistry across the linking σ-bond. Use of the exo-mesityl acetal proved crucial in obtaining a facial preference for 72. Deprotection of the diol function produced the aglycon of lomaitviticin B (i.e., 74) in 39% yield. Isolation of the keto form corresponding to lomaiviticin A (i.e., 73) was also possible, although this was shown to convert rapidly into 74 on standing in chloroform. While the yields for the transformations reported remain modest, the rapid escalation of complexity that is obtained over this two-step sequence of oxidative coupling and deprotection is truly impressive, and serves once more to demonstrate the power of oxidative coupling.
Scytonemin
Scytonemin (77) is a unique cyanobacteria-produced alkaloid comprising a dimeric 1,1′ linked cyclopent[b]indole-2(1H)-one nucleus. [ xl ] Inspired by the biosynthetic studies by Balskus and Walsh, [xli] Mårtensson and coworkers prepared ketone 75 in seven steps from 3-indole acetic acid and showed that dimer 76 is readily generated when the lithium enolate of 75 is treated with FeCl3 (Scheme 7). [xlii] The diastereoselectivity of this coupling was inconsequential; after cleavage of the methyl ethers with BBr3, DDQ-mediated oxidation afforded the natural product 77 (28% over two steps from 76).
Scheme 7. The Mårtensson Group’s Synthesis of the Scytonemin.

[a] LDA, FeCl3, THF, −78 °C to r.t., 70%; [b] BBr3, CH2Cl2,, −78 °C to 0 °C; [c] DDQ, THF, 0 °C, 28% over two steps. DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone.
Purpurone and Related Alkaloids
Alkaloids of the lamellarin family have been heavily pursued by synthetic chemists owing to their diverse array of biological activities including antitumor activity, HIV-1 integrase inhibition activity, and reversal of multidrug resistance.[xliii] Of particular interest are those members that are potent inhibitors of ATP-citrate lyase (ACL) as these compounds have incredible therapeutic potential in cancer treatment. Included among these alkaloids is the ACL inhibitior purpurone (81). [xliv] Recognising the importance of this family of alkaloids, the Jia group developed a general method for the direct construction of dimeric pyrroles from simple amine and aldehyde precursors (see previous section) and initially targeted purpurone for total synthesis using this method (Scheme 8).
Scheme 8. The Jia Group’s Synthesis of Purpurone.
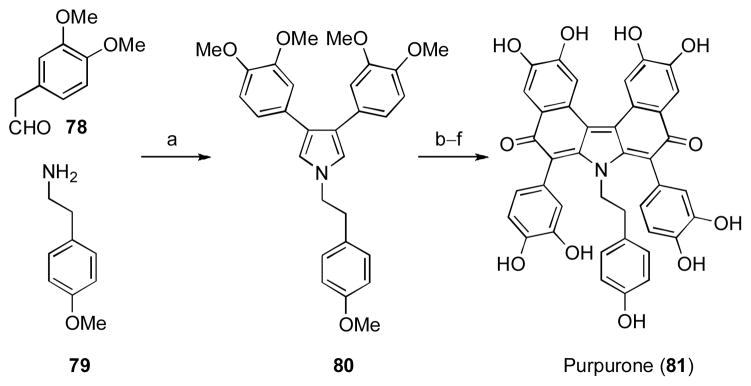
[a] AgOAc (2.0 equiv.), NaOAc (2.0 equiv.), THF, r.t. to 60 °C, 59%; [b] 2,2,2-trichloroethyl 2-bromo-2-(3,4-dimethoxyphenyl)acetate, Al2O3, DCM, reflux, 63%; [c] Zinc (50 equiv.), NH4OAc (4.0 equiv.), THF, r.t.; [d] Ac2O (solvent), KOAc (2.6 equiv.), reflux, 58% (two steps); [e] NaOH (excess), MeOH, air, 55 °C, 66%; [f] BBr3 (20 equiv.), cyclohexene (40 equiv.), DCM, −78 to −10 °C, 90%. OAc = acetate, THF = tetrahydrofuran, DCM = dichloromethane.
The Jia synthesis began with this key transformation, wherein silver(I)-mediated oxidative dimerization of the enamine derived from condensation of amine 79 with aldehyde 78 provided direct access to the key pyrrole intermediate (80).[33] The remainder of the synthesis followed methods developed by Steglich, [xlv] allowing the authors to complete a concise six step synthesis of purpurone that proceeded in 13% overall yield, making this the most rapid and efficient synthesis of this alkaloid reported to date.
Following the completion of this work, Jia and coworkers were able to demonstrate the versatility of their method by employing it in the total syntheses of four related natural products (Figure 6).[xlvi] In each case, the central pyrrole could be accessed in rapid fashion using this powerful method, allowing each natural product to be constructed in seven or fewer synthetic manipulations and with high overall efficiency.
Figure 6.

Related Natural Products Prepared by the Jia Group
Bismurrayaquinone A
The curry leaf plant has been the source of numerous biologically active carbazole natural products. Amongst these compounds are a number of dimeric carbazoles, such as the unusual biquinone, bismurrayaquinone A (87). [xlvii] Biosynthetically, such compounds are hypothesized to derive from oxidative coupling of monomeric phenols, and this approach was successfully employed by Bringmann and coworkers in their inaugural synthesis of racemic 87. [xlviii] While this strategy is optimally convergent, control of absolute stereochemistry is challenging. Our previous methodology study on the dimerization of ketones as precursors to enantioenriched biphenols (see previous section) served as the primary driving force behind our bidirectional approach to bismurrayaquinone A (87, Scheme 9).[xlix]
Scheme 9. The Thomson Group’s Synthesis of Bismurrayaquinone A.

[a] LDA (1.1 equiv); CuCl2 (1.1 equiv), 63%; [b] BF3•OEt2 (10 equiv), toluene, reflux, 87%; [c] Br2 (2.0 equiv), 90%; [d] MeI (10 equiv), KOH (3.0 equiv), 84%; [e] 2-chloroaniline (3.0 equiv), Pd(OAc)2 (8 mol%), [HPtBu3][BF4] (11 mol%), NaOtBu (11 equiv), toluene, reflux, 90%; [f] Pd(OAc)2 (20 mol%), [HPtBu3][BF4] (28 mol%), NaOtBu (7.0 equiv), 160 °C, microwave, 73%; [g] CAN (10 equiv), MeCN/H2O, 95%. LDA = lithium diisopropylamide, CAN = ceric ammonium nitrate.
Oxidative dimerization of enantioenriched monomer 27a proceeded to afford the 1,4-diketone 28a, which was converted to the biphenol 29a with no loss of optical purity upon treatment with BF3•OEt2. Conversion of 29a to the bis-aniline derivative 86 by a Buchwald–Hartwig amination set the stage for the crucial Pd-catalyzed carbazole forming reaction, which after oxidative demethylation led to bismurrayaquinone A (87).
An earlier approach had formed the carbazolequinone ring system from a biquinone, but afforded racemic 87. These observations led to a comprehensive study into the configurational study of axially chiral biquinones, and established the barrier to racemization of 87 to be 54 kcal•mol–1 (the natural product is configurationally stable at room temperature). Unfortunately, at this time no data exists into whether the naturally derived material exists in enantiomerically enriched form or whether it is a racemate. Regardless, the synthesis served to provide a demonstration of the usefulness of oxidative coupling for the synthesis of axially chiral natural products.
Oxidative Cross-Coupling
Bursehernin
The dimeric lignan natural products have been attractive targets for total synthesis by way of oxidative dimerization. For example, the concise enantioselective synthesis of hinokinin (see Figure 2) was achieved through the oxidative dimerization of the appropriate Evans oxazolidinone. [16] Application of such approaches to the synthesis of unsymmetrical lignans remained elusive, however, until the contributions of Baran and coworkers. Using their methodology that enables oxidative cross-coupling, they were able to conduct the efficient synthesis of enantioenriched bursehernin (91, Scheme 10).[19, 20] The key cross-coupling event took place using an excess of the less “expensive” ester 89 to afford the unsymmetrical 1,4-dicarboxylate 90 as a 1.6:1 mixture of stereoisomers. This modest diastereocontrol adjacent to the tert-butyl ester was inconsequential; reduction of the unpurified mixture and treatment of the resulting alcohol with DBU induced lactonization and equilibration of the diastereomeric mixture to afford the target 91 in 41% yield as a single diastereomer. This synthesis served to demonstrate the strategic utility of this powerful method for intermolecular cross-coupling.
Figure 2.

Natural Products Prepared by Oxidative Enolate Coupling
Scheme 10. The Baran Group’s Synthesis of Bursehernin.

[a] 88, LDA (THF, 1.15 equiv), LiCl (5.0 equiv), PhMe, −78 to 0 to −78 °C (10 min each), 89 (1.75 equiv), PhMe, LDA (THF, 1.85 equiv), −78 °C, 30 min, then Cu(II) 2-ethylhexanoate, −78 to 23 °C, 20 min; [b] LiBH4 (10 equiv), MeOH (5.0 equiv), THF, −78 to −10 °C, 1.5 h; [c] DBU (10 equiv), PhMe, 110 °C, 24 h, 41% (3 steps). DBU=1,8-diazabicyclo[5.4.0]undec-7-ene.
Metacycloprodigiosin and Prodigiosin R1
The prodigiosins are a family of polypyrrole natural products that have garnered much interest amongst the chemical community due to their interesting structures, pronounced biological activity and fascinating biosynthesis. [l] As part of our development of the merged 1,4-addition/oxidative coupling sequence of silyl bis-enol ethers (see Table 10), we targeted the 12-membered cyclophane prodigiosin, metacycloprodigiosin (98). [29] Our synthesis commenced with an enantioselective conjugate addition of EtMgBr to enone 92, followed by trapping with chloroenol silane 93 (Scheme 11).
Scheme 11. The Thomson Group’s Synthesis of Metacycloprodigiosin.

[a] EtMgBr, CuBr•DMS (5 mol%), (R,S)-JosiPhos (6 mol%), then 93; [b] CAN, 2,6-di-tert-Bu-Py, MeCN/EtCN, 35% from 92; [c] Grubbs II (10 mol%), dichloromethane, 69% (82% brsm); [d] Pd(OAc)2, H2, EtOH, then NH4OAc, 87%; [e] DDQ, H2O, MeCN, 69%; [f] (i) 4-Methoxyl-1,5-dihydro-2H-pyrrol-2-one, TMSOTf, iPr2NEt, dichloromethane; (ii) HCl, THF, 98% (2 steps); [g] Tf2O, dichloromethane, −50 °C to 0 °C, 1h; [h] (i) Pd(PPh3)4 ( 3 mol%), 1-(tert-butoxycarbonyl)pyrrole-2-boronic acid, Na2CO3, DME, 80 °C; (ii) NaOMe, MeOH, r.t., 76% (3 steps). CAN = ceric ammonium nitrate. Tf = triflouromethanesulfonyl.
Exposure of unpurified bis-enol ether 94 to CAN induced the desired oxidative coupling to generate 1,4-diketone 95 in 35% yield over the two-step sequence. The yield for this sequence was a little disappointing since related substrates lacking the terminal alkenes gave yields greater that 50%; we were happy to press forward due to the efficient nature of the sequence that readily allowed access to the otherwise challenging unsymmetrical diketone. Ring-closing metathesis to form the 12-membered ring followed by condensation with ammonium acetate gave rise to the pyrrole nucleus 96 in good yield. From here, a selective oxidation of the methyl group within 96 set the stage for a vinylogous aldol condensation to afford 97, which underwent Suzuki cross-coupling reaction with 1-(t-butoxycarbonyl)pyrrole-2-boronic acid to give the natural product (98). This short sequence thus enabled the first enantioselective synthesis of metacycloprodigiosin (98) and established its absolute configuration for the first time. [ li] The usefulness of this silicon-tethered method for oxidative cross-coupling was further demonstrated by the synthesis of prodigiosin R1 (99) through the same strategy.
Maoecrystal V
The complex polycyclic norditerpene maoecrystal V (106) [lii] poses several challenges to any synthetic attempt and has led to a number of creative approaches,[liii] and one completed total synthesis to date.[liv] Perhaps foremost amongst the challenges is how to devise a convergent fragment coupling about the centrally located carbon–carbon bond possessing vicinal quaternary substitution. In their model study directed towards maoecrystal V (106), Baran and coworkers utilized an intermolecular oxidative cross-coupling of enol 100 and cross-conjugated siloxydiene 101 to produce 1,4-diketone 102 in 46% yield (Scheme 12).[53b] Redox state adjustments led to the synthesis of linked bicyclic dione 104 in five steps from 102 and set the stage for an intramolecular Diels–Alder reaction to deliver the core features of the nature product (i.e., 105). The cycloadduct 105 was shown to possess the incorrect relative stereochemistry required for the natural product, which led to the development of a second model system that corrected this issue (but did not use oxidative enolate coupling). While compound 105 was not taken forward to a completed total synthesis, this study demonstrated the feasibility of the cycloaddition approach to maoecrystal V and provides an excellent example of forming a quaternary stereocenter through oxidative enolate coupling.
Scheme 12. The Baran Model Study Towards Maoecrystal V.

[a] CAN, NaHCO3, MeOH, 0 °C, 46%; [b] Li(t-\BuO)3AlH, THF, −78 °C, 53%; [c] acryloyl chloride, Et3N, DMAP, DCM, 0 °C, 36%; [d] TBSOTf, Et3N, DCM, 63%; [e] Pd/C, O2, PhMe, 110 °C, 99%; [f] Pb(OAc)4, AcOH, 59%; [g] 165 °C, o-DCB, 60%; CAN = ceric ammonium nitrate, DMAP = 4-dimethylaminopyridine, o-DCB = ortho-dichlorobenzene.
Intramolecular Oxidative Coupling
Stephacidins A and B, and Avrainvillamide
Stephacidin B (107), a unique fungi-derived metabolite, is one of the most structurally complex natural products. [lv] Formally, 107 is a dimer of the simpler compound, avrainvillamide (109), which arises from oxidation of stephacidin A (108). Common to these alkaloids is the presence of the bicyclo[2.2.2]diazaoctane core, which is proposed to be derived biosynthetically from an intramolecular Diels–Alder reaction. While such approaches have enabled the total synthesis of related compounds, [lvi] Baran and coworkers sought to apply a different strategy.
Rather than form the common bicyclo[2.2.2]diazaoctane core through the more precedented Diels–Alder reaction, Baran and coworkers viewed these compounds as ideal targets to explore the strategic application of oxidative enolate coupling (Scheme 13). [lvii] Diketopiperazine 110 could be readily prepared in a minimal number of operations, setting the stage for the key oxidative coupling event. At that time, there were no examples of ester enolates effectively coupling with enolates derived from amides. After screening a number of oxidizing reagents, Fe(III) acetoacetate was found to be the most effective; thus, exposure of diketopiperazine 110 to LDA and Fe(acac)3 under the optimal conditions afforded the desired product 111 as a single diastereomer in 61% yield.
Scheme 13. The Baran Group’s Synthesis of Stephacidins A and B, and Avrainvillamide.

[a] LDA (2.2 equiv), THF, −78 °C, 5 min; then Fe(acac)3 (2.2 equiv), −78 °C, 5 min; then −78 to 20 °C, 20 min, 61%; [b] B-bromocatecholborane (1.5 equiv), CH2Cl2, 0 °C, 10 min, 63%; [c] MeMgBr (6.0 equiv), toluene, 20 °C, 10 min; [d] Burgess reagent (2.0 equiv), benzene, 50 °C, 30 min, 88% yield over two steps; (e) 200 °C, sulfolane, 1 h, 28–45%; (f) NaBH3CN (40 equiv), AcOH, 20 °C, 24 h, >95%; (g) SeO2 (0.25 equiv), 35% aqueous H2O2 (50 equiv), 1,4-dioxane, 20 °C, 40 h, 27%.
The power of the reaction is evident in that it could be conducted on a gram-scale without diminished yields. MOM-deprotection and subsequent conversion of the methyl ester to the corresponding disubstituted alkene 112 was carried out as a prelude to the final ring-closing step. Thermal Boc-deprotection and cyclization at 240 °C furnished ent-stephacidin A (ent-108) in 45% yield. Partial reduction of the indole ring within ent-108 followed by oxidation with selenium dioxide then provided ent-avranvillamide (ent-109), which was shown to undergo a spontaneous and reversible dimerization to afford ent-stephacidin B (ent-107). [lviii]
Access to these compounds in optically enriched form enabled the Baran lab to establish the absolute configuration of the natural material to be opposite to their synthetic compounds (i.e., as shown in Figure 7 above). From a purely synthetic viewpoint, the successful implementation of an intramolecular oxidative enolate coupling between two different functionalities embedded within a complex precursor amply serves as an example of the utility of such reactions in total synthesis.
Figure 7.
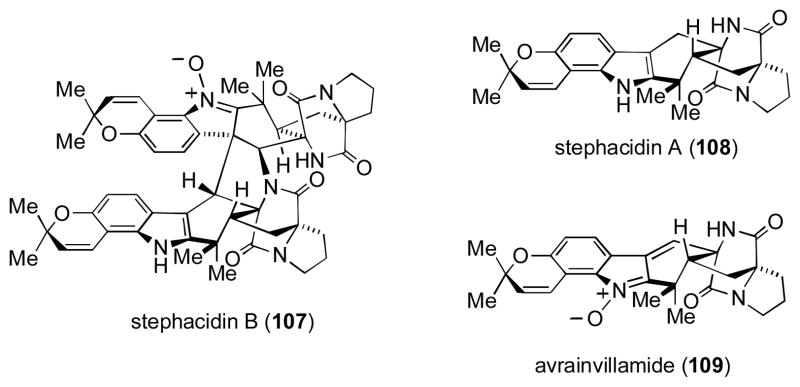
Stephacidins A and B, and Avrainvillamide
Actinophyllic Acid
In 2005, Quinn and co-workers reported the structure of actinophyllic acid (119). [ lix ] This indole alkaloid, isolated from the leaves of Alstonia actinophylla, was shown to be an inhibitor of carboxypeptidase U and was thus of interest for its potential in treating cardiovascular disorders. From a structural standpoint, the unprecedented combination of both a 1-azabicyclo[4.4.2]dodecane and 1-azabicyclo[4.2.1]nonane ring system is unique amongst the indole alkaloids. This mixture of promising biological profile and impressive molecular complexity spurred Overman and coworkers to develop a concise route to 119 amenable to multi-gram scale-up for future biological testing.
The crux of the synthesis began with the key intramolecular oxidative enolate coupling between the malonate and ketone functionalities within readily prepared indole 113 (Scheme 14).[lx]
Scheme 14. Overman and Coworkers Synthesis of Actinophyllic Acid.

[a] LDA (3.2 equiv), [Fe(DMF)3Cl2][FeCl4] (3.5 equiv), THF, − 78 °C to r.t., 60–63%; [b] CeCl3, CH2=CHMgBr, THF, − 78 °C, 99%; [c] (i) TFA, dichloromethane, 0 °C; (ii) (CH2O)n, CSA, PhH, 70 °C; [d] TFA, r.t., 76% (3 steps); [g] LDA, CH2O, THF, − 78 °C, TFA; [h] 4 M HCl, 70 °C, 50% (2 steps).
Exposure of 113 to LDA, followed by the addition of [Fe(DMF)3Cl2][FeCl4] forged the key keto-bridged hexahydroazocino[4,3-b]indole ring system in 60–63% yield. Noteworthy aspects of this particular bond construction are that it did not require protection of the indole subunit, and could be performed effectively on a 10 gram scale. 1,2-Addition of a vinyl Grignard reagent into the bridging carbonyl group within 114 takes place with complete diastereocontrol due to the shielding afforded by the exo tert-butyl ester substituent to deliver alcohol 115. Cleavage of the N-Boc group, followed by exposure to paraformaldehyde in the presence of camphorsulfonic acid initiated the planned aza-Cope/Mannich cascade, to cleanly provide 117 via intermediate 116. Exposure of 117 to neat TFA, followed by treatment of the resulting acid with methanolic HCl enabled the generation of methyl ester 118, which was isolated as its TFA salt. Ultimately, this sequence was reduced to a single one-pot transformation of allylic alcohol 115 to ester 118 in 62% yield. Completion of the synthesis entailed a stereoselective aldol reaction of the lithium enolate of 118 with monomeric formaldehyde to install the furan ring, followed by acidic hydrolysis of the ester. Thus, actinophylic acid was prepared in just 5 steps from the readily accessed indole 113 (or 8 steps if the aza-Cope Mannich sequence of 115 → 118 was carried out in a stepwise fashion). Subsequent work detailed the enantioselective synthesis of actinophylic acid (119) from enantioenriched indole 113.[60b] The tactical combination of intramolecular oxidative enolate coupling and the aza-Cope/Mannich reaction seen in this concise synthesis is a particularly effective example of seamless synthetic planning involving very few function group manipulations between key bond forming events.
Axinellamine A
The intricate structures of the marine organism-derived pyrrole alkaloids, such as axinellamine A (124), have caught the imagination of synthetic chemists across the globe. [lxi] In 2012, Harran and coworkers reported an approach to the core structure of axinellamine A that takes advantage of hidden symmetry within the compound to significantly simply the approach (Scheme 15).[lxii]
Scheme 15. Harran and Coworkers Approach to Axinellamine A.

[a] (i-, PrCp)2TiCl2 (1.05 equiv), THF, −78 °C, 10 min, then KHMDS (1.2 equiv), − 78 °C, 30 min, Cu(OTf)2 (1.5 equiv, slurry in THF), − 78 °C, 3 h, 55%, d.r. = 1.2:1; [b] [ClRh(PPh3)3] (0.6 equiv), H2 (600 psi), THF, 40 °C, 3d, 67%; [c] NBS (4.0 equiv), THF, 0 °C, 45 min, 57%; [d] [18]crown-6 (3.5 equiv), KHMDS (4.0 equiv), THF, −78 °C, dilute with EtOAc, quench with phosphate buffer (pH = 7.8), 54% (mixture of diastereomers); [e] TBD (1.2 equiv), THF, r.t., 1h; [f] aq. NH4OH, 1,2-dimethoxyethane/H2O (4:1), 120 °C, 90 min, then TFA, CH2Cl2, r.t., 1 h, followed by Et3N, MeOH; 43% (2 steps); [g] Li(NH2BH3) (excess), THF, 60 °C, 10 hr, TFA/H2O (1:9), 60 °C, 4 h, 39% (mixture of epimers); [h] 3-(3-nitrophenyl)-2-(phenylsulfonyl) oxaziridine (1.4 equiv), TFA/H2O (1:9), 55 °C, 5 H, 26% (dr = 4:1); [i] SmI2 (excess), THF/H2O, −40 °C to r.t., 45%.
Readily prepared tetracyclic compound 120 was treated with KHMDS and diisopropyltitanocene dichloride to generate a putative extended titanocene dienolate, which underwent γ-selective oxidative coupling when treated with Cu(II) triflate. Thus, the dimer 121 was produced in 55% yield as a 1.2:1 mixture of C2-symmetric and meso-diastaereomers that were readily separable. Alkene reduction and halogenation of the acyl pyrrole groups set the stage for an intriguing base-promoted ring contraction to afford the spirocyclic species 122 as a mixture of isomers. This mixture could be funnelled successfully into bisguandine 123 by a five-step sequence involving oxidation state adjustment and intramolecular cyclization. This concise route to the core structure of these fascinating natural products demonstrates the power of symmetry within synthetic planning, which in this case is made possible through an efficient oxidative dimerization of an extended dienolate.
Conclusion
Recent developments in oxidative enolate coupling have seen the advent of new highly effective methods that allow for the controlled cross-coupling of enolates, often with high levels of stereocontrol. Highly diastereoselective processes are now possible through the use of silicon tethers, and for the case of aldehydes as substrates high enantioselectivity can now be achieved through the use of organocatalysis. Mechanistic insights into the nature of lithium enolate aggregation in solution have begun to answer questions as to why certain conditions allow for selective cross-coupling beyond what would be statistically expected. The combination of these methodological and mechanistic advances will pave the way for future advances into this important reaction class.
The state of the field as it currently stands has already seen impressive applications of oxidative enolate coupling in the realm of natural product synthesis. The diversity of complex substances outlined in this review highlights the versatility of oxidative cross-coupling and showcases the power of the reaction when utilized strategically in synthetic planning. Future applications within the context of increasingly more complex molecules will further drive the advent of more general and useful methodology.
Acknowledgments
The authors gratefully acknowledge support from the National Institutes of Health (NIH/NIGMS R01GM085322) and Northwestern University.
Biographies

Dr. Fenghai Guo studied chemistry at Shanghai Jiaotong University in Shanghai, China. He then moved to Clemson University, South Carolina, where he completed his Ph.D. with Prof. Karl Dieter in 2009 studying organometallic reagent-mediated synthesis of heterocycles. Since 2010, he has been a postdoctoral research associate with Prof. Regan J. Thomson at Northwestern University, working on the enantioselective synthesis of biaryls through traceless stereochemical exchange and the total synthesis of axially chiral alkaloids such as bismurrayaquinone A.

Dr. Michael D. Clift was born in Bellevue, Michigan. He received his B.S. in Chemistry from Western Michigan University in 2005 working with Dr. James J. Kiddle. He graduated with a Ph. D. in Chemistry from Northwestern University in 2010, where he worked in the laboratory of Prof. Regan J. Thomson. His graduate studies focussed on the development of silyl bisenol ethers as useful species for the controlled oxidative cross-coupling of ketones, and on the total synthesis of alkaloids. He is currently a post-doctoral research fellow in the labs of Prof. David W. C. MacMillan at Princeton University.

Prof. Regan J. Thomson was born in Balclutha, New Zealand. After completing three years of his undergraduate studies at The University of Auckland, New Zealand, he moved to Australia where he completed his B.Sc. (First Class Honours) at The Australian National University (ANU), Canberra, in 1998. He received his Ph.D. in 2003 at The Research School of Chemistry at ANU working with Professor Lewis N. Mander. Following postdoctoral studies with Professor David A. Evans at Harvard University, he began his independent career at Northwestern University in 2006. His research interests are broadly focussed on reaction methodology and natural product total synthesis.
References
- 1.Ivanoff D, Spasoff A. Bull Soc Chim Fr. 1935;2:76–78. [Google Scholar]
- 2.Rathke MW, Lindert A. J Am Chem Soc. 1971;93:4605–4606. [Google Scholar]
- 3.a) Ito Y, Konoike T, Saegusa T. J Am Chem Soc. 1975;97:2912–2914. [Google Scholar]; b) Ito Y, Konoike T, Harada T, Saegusa T. J Am Chem Soc. 1977;99:1487–1493. [Google Scholar]
- 4.a) Kobayashi Y, Taguchi T, Tokuno E. Tetrahedron Lett. 1977;42:3741–3742. [Google Scholar]; b) Kobayashi Y, Taguchi T, Morikawa T, Tokuno E, Sekiguchi S. Chem Pharm Bull. 1980;28:262–267. [Google Scholar]
- 5.Frazier RH, Jr, Harlow RL. J Org Chem. 1980;45:5408–5411. [Google Scholar]
- 6.Brocksom TJ, Petragnani N, Rodrigues R, LaScala Teixeira H. Synthesis. 1975:396–397. [Google Scholar]
- 7.Ojima I, Brandstadter SM, Donavan RJ. Chem Lett. 1992:1591–1594. [Google Scholar]
- 8.Babler JH, Haack RA. Synth Commun. 1983;13:905–911. [Google Scholar]
- 9.a) Ito Y, Konoike T, Saegusa T. J Am Chem Soc. 1975;97:649–651. [Google Scholar]; b) Baciocchi E, Casu A, Ruzziconi R. Tetrahedron Lett. 1989;30:3707–3710. [Google Scholar]; c) Fujii T, Hirao T, Ohshiro Y. Tetrahedron Lett. 1992;33:5823–5826. [Google Scholar]; d) Paolobelli AB, Latini D, Ruzziconi R. Tetrahedron Lett. 1993;34:721–724. [Google Scholar]; e) Ryter K, Livinghouse T. J Am Chem Soc. 1998;120:2658–2659. [Google Scholar]
- 10.Narasaka K, Okauchi T, Tanaka K, Murikami M. Chem Lett. 1992:2099–2103. [Google Scholar]
- 11.Heiba EI, Dessau RM. J Org Chem. 1974;39:3457–3459. [Google Scholar]
- 12.Heim A, Terpin A, Steglich W. Angew Chem Int Ed. 1997;36:155–156. [Google Scholar]
- 13.Scott AI, McCapra F, Hall ES. J Am Chem Soc. 1964;86:302–303. [Google Scholar]
- 14.Fang CL, Horne S, Taylor N, Rodrigo R. J Am Chem Soc. 1994;116:9480–9486. [Google Scholar]
- 15.Ramig K, Kuzemko MA, McNamara K, Cohen T. J Org Chem. 1992;57:1968–1969. [Google Scholar]
- 16.a) Kise N, Tokioka K, Aoyama Y. J Org Chem. 1995;60:1100–1101. [Google Scholar]; b) Langer T, Illich M, Helmchen G. Synlett. 1996:1137–1139. [Google Scholar]
- 17.Paquette LA, Degenhardt CR, Berk HC. J Am Chem Soc. 1978;100:1600–1602. [Google Scholar]
- 18.For an earlier review of stereoselective enolate coupling, see: Csákÿ AG, Plumet J. Chem Soc Rev. 2001;30:313–320.For a recent review of oxidative dehydrogenative coupling that includes sections on oxidative arylation and related transformations, see: Yeung CS, Dong VM. Chem Rev. 2011;111:1215–1292. doi: 10.1021/cr100280d.
- 19.Baran PS, DeMartino MP. Angew Chem Int Ed. 2006;45:7083–7086. doi: 10.1002/anie.200603024. [DOI] [PubMed] [Google Scholar]
- 20.DeMartino MP, Chen K, Baran PS. J Am Chem Soc. 2008;130:11546–11560. doi: 10.1021/ja804159y. [DOI] [PubMed] [Google Scholar]
- 21.Casey BM, Flowers RA., II J Am Chem Soc. 2011;133:11492–11495. doi: 10.1021/ja205017e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo F, Konkol LC, Thomson RJ. J Am Chem Soc. 2011;133:18–20. doi: 10.1021/ja108717r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Imbos R, Brilman MHG, Pineschi M, Feringa BL. Org Lett. 1999;1:623–625. [Google Scholar]
- 24.Rautenstrauch V. Bull Soc Chim Fr. 1994;131:515–524. [Google Scholar]
- 25.Shintani R, Tokunaga N, Doi H, Hayashi T. J Am Chem Soc. 2004;126:6240–6241. doi: 10.1021/ja048825m. [DOI] [PubMed] [Google Scholar]
- 26.Nguyen PQ, Schäfer HJ. Org Lett. 2001;3:2993–2995. doi: 10.1021/ol016326+. [DOI] [PubMed] [Google Scholar]
- 27.a) Schmittel M, Burghart A, Malisch W, Reising J, Sollner R. J Org Chem. 1998;63:396–400. [Google Scholar]; b) Schmittel M, Haeuseler A. J Orgmet Chem. 2002;661:169–179. [Google Scholar]
- 28.Clift MD, Taylor CT, Thomson RJ. Org Lett. 2007;9:4667–4669. doi: 10.1021/ol702306z. [DOI] [PubMed] [Google Scholar]
- 29.Avetta CA, Jr, Konkol LC, Taylor CN, Dugan KC, Stern CL, Thomson RJ. Org Lett. 2008;10:5621–5624. doi: 10.1021/ol802516z. [DOI] [PubMed] [Google Scholar]
- 30.Hoffmann HMR, Münnich I, Nowitzki O, Stucke H, Williams DJ. Tetrahedron. 1996;52:11783–11798. [Google Scholar]
- 31.Clift MD, Thomson RJ. J Am Chem Soc. 2009;131:14579–14583. doi: 10.1021/ja906122g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jang HY, Hong JB, MacMillan DWC. J Am Chem Soc. 2007;129:7004–7005. doi: 10.1021/ja0719428. [DOI] [PubMed] [Google Scholar]
- 33.Li Q, Fan A, Lu Z, Cui Y, Lin W, Jia Y. Org Lett. 2010;12:4066–4069. doi: 10.1021/ol101644g. [DOI] [PubMed] [Google Scholar]
- 34.He H, Ding WD, Bernan VS, Richardson AD, Ireland CM, Greenstein M, Ellestad GA, Carter GT. J Am Chem Soc. 2001;123:5362–5363. doi: 10.1021/ja010129o. [DOI] [PubMed] [Google Scholar]
- 35.Ito S, Matsuya T, Omura S, Otani M, Nakagawa A. J Antibiot. 1970;23:315–317. doi: 10.7164/antibiotics.23.315.Hata T, Omura S, Iwai Y, Nakagawa A, Otani M. J Antibiot. 1971;24:353–359. doi: 10.7164/antibiotics.24.353.Omura S, Nakagawa A, Yamada H, Hata T, Furusaki A, Watanabe T. Chem Pharm Bull. 1971;19:2428–2430.Omura S, Nakagawa A, Yamada H, Hata T, Furusaki A. Chem Pharm Bull. 1973;21:931–940. doi: 10.1248/cpb.21.931.Seaton PJ, Gould SJ. J Antibiot. 1989;42:189–197. doi: 10.7164/antibiotics.42.189.For a review of kinamycin biosynthesis, see: Gould SJ. Chem Rev. 1997;97:2499–2509. doi: 10.1021/cr9600215.
- 36.Krygowski ES, Murphy-Benenato K, Shair MD. Angew Chem Int Ed. 2008;47:1680–1684. doi: 10.1002/anie.200704830. [DOI] [PubMed] [Google Scholar]
- 37.Lee HG, Ahn JY, Lee AS, Shair MD. Chem Eur J. 2010;16:13058–13062. doi: 10.1002/chem.201002157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herzon SB, Lu L, Woo CM, Gholap SL. J Am Chem Soc. 2011;133:7260–7263. doi: 10.1021/ja200034b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woo CM, Lu L, Gholap SL, Smith DR, Herzon SB. J Am Chem Soc. 2010;132:2540–2541. doi: 10.1021/ja910769j. [DOI] [PubMed] [Google Scholar]
- 40.Proteau PJ, Gerwick WH, Garcia-Pichel F, Castenholz R. Experientia. 1993;49:825–829. doi: 10.1007/BF01923559. [DOI] [PubMed] [Google Scholar]
- 41.a) Balskus EP, Walsh CT. J Am Chem Soc. 2008;130:15260–15261. doi: 10.1021/ja807192u. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Balskus EP, Walsh CT. J Am Chem Soc. 2009;131:14648–14649. doi: 10.1021/ja906752u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ekebergh A, Karlsson I, Mete R, Pan Y, Börje A, Mårtensson J. Org Lett. 2011;13:4458–4461. doi: 10.1021/ol201812n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.For a recent review, see: Fan J, Peng J, Hamann MT, Hu J. Chem Rev. 2008;108:264–287. doi: 10.1021/cr078199m.
- 44.Chan GW, Francis T, Thureen DR, Offen PH, Pierce NJ, Westley JW, Johnson RK, Faulkner DJ. J Org Chem. 1993;58:2544–2546. [Google Scholar]
- 45.Peschko C, Steglich W. Tetrahedron Lett. 2000;41:9477–9481. [Google Scholar]
- 46.Li Q, Jiang J, Fan A, Cui Y, Jia Y. Org Lett. 2011;13:312–315. doi: 10.1021/ol1027877. [DOI] [PubMed] [Google Scholar]
- 47.Ito C, Thoyama Y, Omura M, Kajiura I, Furukawa H. Chem Pharm Bull. 1993;41:2096–2100. [Google Scholar]
- 48.a) Bringmann G, Ledermann A, Stahl M, Gulden KP. Tetrahedron. 1995;51:9353–9360. [Google Scholar]; b) Bringmann G, Tasler S. Tetrahedron. 2001;57:331–343. [Google Scholar]
- 49.Konkol LC, Guo F, Sarjeant AA, Thomson RJ. Angew Chem Int Ed. 2011;50:9931–9934. doi: 10.1002/anie.201104726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wasserman HH, Rodgers GC, Keith DD. J Am Chem Soc. 1969;91:1263–1264. doi: 10.1021/ja01033a065.Wasserman HH, Keith DD, Nadelson J. J Am Chem Soc. 1969;91:1264–1265. doi: 10.1021/ja01033a066.Wasserman HH, Keith DD, Rodgers GC. Tetrahedron. 1976;32:1855–1861.Wasserman HH, Keith DD, Nadelson J. Tetrahedron. 1976;32:1867–1871.Kawasaki T, Sakurai F, Hayakawa Y. J Nat Prod. 2008;71:1265–1267. doi: 10.1021/np7007494.For reviews of the chemistry and biology of the prodigiosins, see: Fürstner A. Angew Chem, Int Ed. 2003;42:3582–3603. doi: 10.1002/anie.200300582.Williamson NR, Fineran PC, Leeper FJ, Salmond GPC. Nature Rev Microbiol. 2006;4:887–899. doi: 10.1038/nrmicro1531.Williamson NR, Fineran PC, Gristwood T, Chawrai SR, Leeper FJ, Salmond GPC. Future Microbiol. 2007;2:605–618. doi: 10.2217/17460913.2.6.605.
- 51.Hu DX, Clift MD, Lazarski KE, Thomson RJ. J Am Chem Soc. 2011;133:1799–1804. doi: 10.1021/ja109165f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li SH, Wang J, Niu XM, Shen YH, Zhang HJ, Sun HD, Li ML, Tian QE, Lu Y, Cao P, Zheng QT. Org Lett. 2004;6:4327–4330. doi: 10.1021/ol0481535. [DOI] [PubMed] [Google Scholar]
- 53.a) Gong J, Lin G, Li CC, Yang Z. Org Lett. 2009;11:4770–4773. doi: 10.1021/ol9014392. [DOI] [PubMed] [Google Scholar]; b) Krawczuk PJ, Schöne N, Baran PS. Org Lett. 2009;11:4774–4776. doi: 10.1021/ol901963v. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Peng F, Yu ML, Danishefsky SJ. Tetrahedron Lett. 2009;50:6586–6589. doi: 10.1016/j.tetlet.2009.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Nicolaou KC, Dong L, Deng LJ, Talbot AC, Chen DY-K. Chem Commun. 2010;46:70–72. doi: 10.1039/b917045f. [DOI] [PubMed] [Google Scholar]; e) Singh V, Bhalerao P, Mobin SM. Tetrahedron Lett. 2010;51:3337–3339. [Google Scholar]; f) Lazarski KE, Hu DX, Stern CL, Thomson RJ. Org Lett. 2010;12:3010–3013. doi: 10.1021/ol101025r. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Baitinger I, Mayer P, Trauner D. Org Lett. 2010;12:5656–5659. doi: 10.1021/ol102446u. [DOI] [PubMed] [Google Scholar]; h) Gu Z, Zakarian A. Org Lett. 2011;13:1080–1082. doi: 10.1021/ol1031238. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Peng F, Danishefsky SJ. Tetrahedron Lett. 2011;52:2104–2106. doi: 10.1016/j.tetlet.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Dong L, Deng L, Lim YH, Leung GYC, Chen DYK. Chem Eur J. 2011;17:5778–5781. doi: 10.1002/chem.201100232. [DOI] [PubMed] [Google Scholar]
- 54.Gong J, Lin G, Sun W, Li CC, Yang Z. J Am Chem Soc. 2010;132:16745–16746. doi: 10.1021/ja108907x. [DOI] [PubMed] [Google Scholar]
- 55.Qian-Cutrone J, Huang S, Shu YZ, Vyas D, Fairchild C, Menendez A, Krampitz K, Dalterio R, Klohr SE, Gao Q. J Am Chem Soc. 2002;124:14556–14557. doi: 10.1021/ja028538n. [DOI] [PubMed] [Google Scholar]
- 56.a) Artman GD, Grubbs AW, Williams RM. J Am Chem Soc. 2007;129:6336–6342. doi: 10.1021/ja070259i. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Greshock TJ, Grubbs AW, Jiao P, Wicklow DT, Gloer JB, Williams RM. Angew Chem Int Ed. 2008;47:3573–3577. doi: 10.1002/anie.200800106. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) McAfoos T, Williams RM. Heterocycles. 2010;82:461–472. doi: 10.3987/COM-10-S(E)19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.a) Baran PS, Guerrero CA, Ambhaikar NB, Hafensteiner BD. Angew Chem Int Ed. 2005;44:606–609. doi: 10.1002/anie.200461864. [DOI] [PubMed] [Google Scholar]; b) Baran PS, Guerrero CA, Hafensteiner BD, Ambhaikar NB. Angew Chem Int Ed. 2005;44:3892–3895. doi: 10.1002/anie.200500655. [DOI] [PubMed] [Google Scholar]; c) Baran PS, Hafensteiner BD, Ambhaikar NB, Guerrero CA, Gallagher JD. J Am Chem Soc. 2006;128:8678–8693. doi: 10.1021/ja061660s. [DOI] [PubMed] [Google Scholar]
- 58.Herzon SB, Myers AG. J Am Chem Soc. 2005;127:5342–5344. doi: 10.1021/ja0510616. [DOI] [PubMed] [Google Scholar]
- 59.Carroll AR, Hyde E, Smith J, Quinn RJ, Guymer G, Foster PI. J Org Chem. 2005;70:1096–1099. doi: 10.1021/jo048439n. [DOI] [PubMed] [Google Scholar]
- 60.a) Martin CL, Overman LE, Rohde JM. J Am Chem Soc. 2008;130:7568–7569. doi: 10.1021/ja803158y. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Martin CL, Overman LE, Rohde JM. J Am Chem Soc. 2010;132:4894–4906. doi: 10.1021/ja100178u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.a) Forte B, Malgesini B, Piutti C, Quartieri F, Scolaro A, Papeo G. Mar Drugs. 2009;7:705–753. doi: 10.3390/md7040705. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Al-Mourabit A, Zancanella MA, Tilvi S, Romo D. Nat Prod Rep. 2011;28:1229–1260. doi: 10.1039/c0np00013b. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang J, Zhan Y, Jiang B. Prog Chem. 2011;23:2065–2078. [Google Scholar]; d) Hoffmann H, Lindel T. Synthesis. 2003:1753–1783. [Google Scholar]
- 62.Ding H, Roberts AG, Harran PG. Angew Chem Int Ed. 2012;51:4340–4343. doi: 10.1002/anie.201200205. [DOI] [PMC free article] [PubMed] [Google Scholar]