

Abstract
Alzheimer's disease (AD) and cerebral amyloid angiopathy (CAA) are two common pathologies associated with β‐amyloid (Aβ) accumulation and inflammation in the brain; neither is well understood. The objective of this study was to evaluate human post‐mortem brains from AD subjects with purely parenchymal pathology, and those with concomitant CAA (and age‐matched controls) for differential expression of microglia‐associated Aβ ligands thought to mediate Aβ clearance and the association of these receptors with complement activation. Homogenates of brain parenchyma and enriched microvessel fractions from occipital cortex were probed for levels of C3b, membrane attack complex (MAC), CD11b and α‐2‐macroglobulin and immunoprecipitation was used to immunoprecipitate (IP) CD11b complexed with C3b and Aβ. Both C3b and MAC were significantly increased in CAA compared to AD‐only and controls and IP showed significantly increased CD11b/C3b complexes with Aβ in AD/CAA subjects. Confocal microscopy was used to visualize these interactions. MAC was remarkably associated with CAA‐affected blood vessels compared to AD‐only and control vessels. These findings are consistent with an Aβ clearance mechanism via microglial CD11b that delivers Aβ and C3b to blood vessels in AD/CAA, which leads to Aβ deposition and propagation of complement to the cytolytic MAC, possibly leading to vascular fragility.
Keywords: ApoE E4, CR1, CR3, Mac‐1, morphology, αMβ2
Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disorder among people over the age of 65 and is accompanied by some degree of the microvasculopathy cerebral amyloid angiopathy (CAA) in 75–90% of cases 15. CAA is characterized by the deposition of amyloid beta (Aβ) in the tunica adventitia and media of leptomeningeal and penetrating cortical arteries and degeneration of medial vascular smooth muscle, resulting in vascular fragility and intracerebral hemorrhage 42, 45. Evidence is mounting that CAA plays an important role in the pathogenesis of cognitive deficits associated with AD, sometimes through the mechanism of microbleeds or microinfarcts 19, 29, 39. However, the mechanisms responsible for this vascular Aβ accumulation, smooth muscle loss and fragility have remained speculative and defied therapeutic intervention.
Microglia are the resident immune and phagocytic cells of the nervous system and are known effectors of brain Aβ clearance 17, 47. There is an extensive literature describing the chemotactic and phagocytic response of microglia to Aβ from studies on human autopsy material, as well as animal and in vitro studies. Microglia are capable of binding and phagocytosing Aβ and removing its breakdown products from the brain parenchyma. In the normal brain, microglia exist in a highly ramified phenotype, constantly surveying their environment, probing for tissue architecture abnormalities and foreign infiltrates 13. After a pathological insult, microglia shift their phenotype to an “activated” amoeboid‐like morphology, expressing a variety of inflammatory and receptor proteins 11, 17. Recent studies of human autopsy material suggest that microglia become dystrophic and functionally impaired with aging, resulting in immune dysfunction and improper expression of cytokine and receptor profiles 23, 40. This theme of age‐related alterations in microglial function led us to examine the possible role of microglia in the pathogenesis of CAA.
In addition to the role of microglia, there is evidence for late‐complement activation on the microvasculature in CAA 25, 34, 36. Complement activation involves a series of coordinated protein cleavages and interactions ultimately activating and depositing the terminal lytic membrane attack complex (MAC). The MAC is a macromolecular protein complex consisting of single components of C5b, C6, C7, C8 and multiple C9 components forming a pore in target cell membranes, inducing cell lysis. MAC formation is initiated by the formation of a C5‐convertase (any of a variety of complexes containing C3b). Complement C3b, a cleavage product of the central C3 component, is a high‐affinity ligand for β‐amyloid and can act as an opsonin of Aβ plaques 7, 35. This complex (C3b/Aβ) binds to complement receptor 1 (CR1) on erythrocytes, which appears to be involved in clearance of Aβ in the peripheral circulation 35. CD11b (CR3) a homolog of CR1, which is selectively expressed on microglia in the brain, binds to C3b and is implicated in the adhesive interactions of monocytes, macrophages and granulocytes 17, 30 as well as complement coated particles, and binds Aβ in a yeast model 8. We hypothesized that CD11b may serve as a cell‐surface receptor for the complex of Aβ and C3b and may be a mechanism of Aβ clearance from the parenchyma and complement activation on vascular elements. Finally, apolipoprotein E ε4 (ApoE ε4), a genetic risk factor for AD, is known to activate the complement cascade 26, so we additionally asked whether or not the apoE E4/CD11b/C3b/Aβ complex occurred on human microglia in human post‐mortem tissue. This study of human, post‐mortem brains with CAA has led to the identification of a microglial cell‐surface complex that appears to be involved in Aβ clearance in CAA and may explain the late‐complement activation on the vessel wall at the site of Aβ deposition.
Methods
Tissue selection
Post‐mortem tissue was obtained from the Alzheimer's Disease Research Center Brain Bank at the University of California, Los Angeles. All patients or their surrogates consented to participate in research protocols prior to tissue donation, and the study was approved by the Institutional Review Board of Loma Linda University Medical Center (approval #54174). Patient demographics are described in Table 1. Both frozen tissue specimens and fixed tissues were available for study. Neuropathologic examination at the time of autopsy included Braak and Braak staging of AD and Vonsattel grading of CAA pathology 6, 12. CAA staging of CAA severity is as follows: stage 1—β‐amyloid deposition limited to the basement membrane of arterioles and primarily involving leptomeningeal vessels; stage 2—β‐amyloid deposited between vascular smooth muscle cells and pathology extends to penetrating arterioles; and stage 3—β‐amyloid largely replaces vascular smooth muscle in arterioles. Specimens were graded based on the majority of vessels displaying the respective Vonsattel stage. Microaneurysms and microhemorrhages often occur with Vonsattel stage 3 CAA; however, microhemorrhages were rare in this cohort. Additionally, no patients were included in this study with fatal lobar or intracerebral hemorrhage.
Table 1.
Patient demographics. Abbreviations: NP = no pathological abnormality; PMI = post‐mortem interval; y = years; h = hours; M = male; F = female
| Case# | Sex | Braak and Braak stage | APOE status | CR1 status§ | Age (y) | PMI (h) |
|---|---|---|---|---|---|---|
| Controls | ||||||
| 1 | F | NP | ε3/ε4 | A/G | 47 | 7 |
| 2 | F | NP | ε3/ε3 | A/G | 42 | 22 |
| 3 | F | NP | ε3/ε3 | A/G | 81 | 23 |
| 4 | M | NP | ε3/ε3 | A/G | 70 | 23 |
| AD only | ||||||
| 5 | M | VI | ε3/ε4 | A/G | 80 | 22 |
| 6 | F | VI | ε3/ε3 | A/G | 81 | 6.5 |
| 7 | M | VI | ε3/ε3 | A/G | 89 | 15 |
| 8 | M | VI | ε3/ε4 | A/A | 81 | 9.5 |
| AD mild CAA | ||||||
| 9 | F | V | ε3/ε3 | A/G | 79 | 6 |
| 10 | F | V/VI | ε3/ε3 | A/A | 91 | 3.5 |
| 11 | F | VI | ε3/ε3 | A/G | 89 | 24 |
| 12 | M | V/VI | ε3/ε3 | A/A | 82 | 16 |
| 13 | F | VI | ε3/ε4 | A/A | 97 | 12 |
| 14 | F | VI | ε3/ε3 | A/A | 73 | 13 |
| 15 | F | VI | ε3/ε4 | A/G | 81 | 24 |
| 16 | M | VI | ε3/ε3 | A/A | 95 | 22 |
| AD moderate CAA | ||||||
| 17 | M | V/VI | ε3/ε3 | A/A | 82 | 5 |
| 18 | F | VI | ε3/ε3 | A/A | 96 | 7 |
| 19 | F | VI | ε3/ε3 | A/A | 81 | 11 |
| 20 | M | VI | — | — | 83 | 12 |
| AD severe CAA | ||||||
| 21 | F | VI | ε3/ε4 | A/A | 88 | 38 |
| 22 | F | VI | ε4/ε4 | A/A | 71 | 20 |
| 23 | F | VI | ε3/ε3 | A/A | 83 | 6 |
| 24 | F | VI | ε3/ε3 | A/A | 69 | 18 |
§The CR1 A/A allele was significantly associated with CAA cases vs. non‐CAA cases, P = 0.006.
Frozen tissue was isolated from the occipital lobe for five groups of patients; four samples were obtained from an aged, neurological control group (n = 4), along with four AD samples without evidence of significant CAA (Vonsattel grades 0–1) and 16 AD samples with varying degrees of CAA, grouped into mild (n = 8), moderate (n = 4) and severe (n = 4) CAA (grade 3) (ADmiCAA, ADmodCAA and ADsevCAA, respectively).
Microvessel enrichment
Brain microvessels were isolated from the occipital lobe of frozen post‐mortem brain tissue from four cases in each group. A homogenization buffer was prepared over ice consisting of 50 mM Tris‐HCl (pH 7.4), 150 mM NaCl, 300 mM sucrose, 2 mM ethylenediaminetetraacetic acid and protease inhibitor cocktail. Samples were homogenized with three to four vertical strokes of a Teflon pestle in a glass tube, the homogenate then poured over a 72 μm mesh sieve (Gelman Sciences, Ann Arbor, MI, USA) with microvessels collected by flushing the sieve with ice‐cold homogenization buffer. Homogenates containing microvessels were then poured into 20 mL Falcon tubes, centrifuged for 15 minutes at 2000 rpm, and vessels concentrated into homogenization buffer and verified by examining a sample fixed with 100% ethanol on charged glass slides (Fisher Scientific, Waltham, MA, USA). Samples were sonicated three times to further homogenize blood vessels and protein quantification cached by the Bradford assay (Pierce Laboratories, Rockford, IL, USA).
Western blot analysis
Protein levels of complement components and regulators and α2M on occipital lobe vessels and occipital lobe whole‐brain tissue were determined by denaturing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) and Western blot. Briefly, tissue samples were isolated containing roughly equal proportions of white and gray matter from the occipital lobe, and homogenized in an ice‐cold sucrose buffer containing a protease inhibitor cocktail. Tissue was further homogenized in a fitted Teflon pestle in a glass homogenization tube. Nuclear debris was cleared from the suspension by centrifugation at 1000 × g for 15 minutes and total protein concentration was determined in the supernatant by the Bradford assay (Pierce Laboratories). Samples were prepared for electrophoresis by adding 25% v/v loading buffer (Life Technologies, Grand Island, NY, USA) and 10% v/v beta‐mercaptoethanol and heating at 95°C for 10 minutes. Ten micrograms of total protein were loaded into each lane of a 10% polyacrylamide gel and electrophoresed at 100 V in a MiniProtean Tetra System (Bio‐Rad Laboratories, Irvine, CA, USA). The proteins were then transferred to a nitrocellulose membrane at 30 V for 4 h. The resulting blot was blocked in an albumin‐based blocking buffer (Invitrogen) and probed with either beta‐actin (mouse monoclonal; 1:1000), α2M (rabbit polyclonal; 1:500), CD11b (rabbit polyclonal; 1:500), C3b (mouse monoclonal; 1:500), C6 (mouse monoclonal; 1:500), C5b‐9 (rabbit polyclonal; 1:500), CD59 (mouse monoclonal; 1:500) and apoE E4 (mouse monoclonal; 1:500) antibodies (all from Abcam, Cambridge, MA, USA) and β‐amyloid 1–16 (clone AB 10; mouse monoclonal; 1:1000; Millipore, Billerica, MA, USA). After an overnight incubation at 4°C, blots were thoroughly washed with Tris‐buffered saline (TBS) buffer containing 0.05% Tween‐20 and a fluorescently labeled goat anti‐mouse or rabbit secondary (IRDye, Licor, Lincoln, NE, USA) was applied. After 2‐h incubation, blots were washed and visualized on an Odyssey Infrared Imaging System (Licor Biosciences, NE, USA). Optical density was determined using Odyssey 2.0 software. Data was collected as the relative intensity of the band of interest compared to the corresponding loading control band of β‐actin.
Immunohistochemistry
Paraffin‐embedded, formalin‐fixed sections of occipital lobe tissue were deparaffinized in two exchanges of xylene and rehydrated in serial exchanges of ethanol. Antigen retrieval was performed by heating sections in a microwave on high power (3× at 90 s each) in a 10 mM citrate buffer. The sections were then treated with 1% hydrogen peroxide in phosphate‐buffered saline (PBS) to block endogenous peroxidase activity, incubated with a blocking solution of 4% normal serum in PBS and incubated with the primary antibody of choice overnight at 4°C. After washing with PBS, sections were incubated with a horseradish peroxidase‐conjugated secondary antibody for 2 h at 4°C, washed in additional exchanges of PBS and treated with diaminobenzidine/hydrogen peroxide (DAB) for 10 minutes (Vector Laboratories, Burlingame, CA, USA), followed by hematoxylin as a counterstain. They were then rinsed for 3 minutes in PBS, dehydrated through serial alcohols, cleared in xylene and coverslipped with mounting resin (Permount, Fisher). The antibody used was against C6 (mouse monoclonal 1:500; Abcam).
Co‐immunofluorescent studies were prepared on sections as previously mentioned. After blocking with 4% normal serum in PBS, slides were incubated with primary antibodies against alpha smooth muscle actin (αSMA; mouse polyclonal; 1:200), C5b‐9 (rabbit polyclonal; 1:500) (Abcam), CD11b (rabbit polyclonal; 1:500; Abcam) and an antibody against Aβ40 (mouse monoclonal; 1:1000; Abbiotec, San Diego, CA, USA). Sections were similarly prepared with 0.01% Thioflavin S, but washed in two exchanges of 70% ethanol before application of 4',6‐diamidino‐2‐phenylindole (DAPI). After overnight incubation at 4°C, slides were washed and incubated with secondary antibody conjugated to Dylight 550, Texas Red or fluoroscein isothiocyanate (FITC) (all Abcam) for 2 h at 4°C and then washed. Sections were stained with DAPI (Vector Laboratories) and visualized by a LSM 710 confocal microscope (Zeiss, Thornwood, NY, USA). Negative controls were prepared in the same fashion, but lacked the incubation step with primary antibody.
Immunoprecipitation
Co‐immunoprecipitation was done on occipital lobe whole‐brain homogenates; groups consisted of controls, AD only and AD with severe CAA (n = 4 in all groups). Briefly, magnetic Dynabeads (Invitrogen) were resuspended and 50 μL was transferred to 12 tubes. Beads were separated from supernatant on a magnet and the supernatant was removed. 5 μL of anti‐CD11b antibody (1 μg/μL; Abcam) was diluted in 200 μL 1× PBS with 0.02% Tween‐20 (pH 7.4) and each tube was incubated for 10 minutes with rotation at room temperature. Tubes were then placed on the magnet and the supernatant was removed. The bead‐antibody complex was resuspended in 200 μL PBS with Tween‐20 and washed with gentle agitation. Tubes were placed on the magnet and the supernatant was removed. Samples from control, AD only and AD with CAA (n = 4 in each group) were added to the bead‐antibody complex at 150 μL and incubated with gentle agitation at room temperature for 30 minutes. Tubes were then placed on the magnet and the supernatant was transferred to a clean tube for further analysis. The beads‐antibody‐antigen complex was washed three times using 200 μL of 1× PBS with gentle agitation. The complex was resuspended in 100 μL of 1× PBS and transferred to clean tubes to avoid co‐elution of proteins bound to the tube wall. Tubes were placed on the magnet and the supernatant removed, followed by addition of 20 μL non‐denaturing elution buffer (50 mM glycine pH 2.8) and incubated for 2 minutes. Tubes were placed on the magnet and the supernatant was transferred to a clean tube and prepared for Western blot as described in the Western blot section.
Statistical analysis
Data are reported as mean ± standard error. Because of the small sample size, the data did not fit a normal distribution, so differences between groups were assessed by Kruskal–Wallis nonparametric ANOVA. Significant differences between groups were then reported by post hoc analysis. Genotyping analysis was assessed with Fischer's exact test for categorical data. α < 0.05 was taken as a significant result and statistical analysis was done on SPSS 20 software (SPSS Inc., Chicago, IL, USA).
Results
APOE4 and CR1 genotyping analysis
Genotyping of our patient cohort did not show the usual trend for risk of AD or CAA with APOE4, in which even only one ε4 allele can significantly increase the risk of both AD and CAA. The majority of patients in all groups were homozygous for the ε3 allele, with at least one case harboring one ε4 allele in each group. CR1 genotyping was done in light of recent findings by Biffi et al 5, in which individuals homozygous for the A allele showed a higher risk for intracerebral hemorrhage (ICH) caused by CAA compared to heterozygous (A/G) or homozygous neutral (G/G). The product of the CR1 gene, CR1, binds to and cleaves C3b into inactive components iC3b and C3c 18. In our cohort, 12/16 (75%) of CAA cases were homozygous A/A (Table 1), the major risk genotype for CAA‐associated ICH 5. On the other hand, only one case was homozygous A/A in the AD‐only group and none of the controls were homozygous A/A (Table 1). When all CAA cases were compared to all non‐CAA cases, the association of CAA pathology to the A/A genotype was highly significant (P = 0.006). Genotype data could not be obtained for patient 20.
α2M and C3b levels in occipital lobe
Two different Aβ receptor/ligand complexes were studied in our post‐mortem brains; the well‐recognized LRP1/α2M/Aβ 14, 27, 28, 37 and a new cell‐surface complex, CD11b/C3b/Aβ. A role for these adhesion molecules (α2M and C3) in amyloid clearance is recognized, but yet to be fully elucidated. We report for the first time that levels of both α2M and C3b, two previously indicated Aβ chaperones 28, 44, differ in the AD‐only and AD/CAA brain, suggesting different phenotypes between the two diseases. α2M showed a significant difference between groups (H = 13.5, 4 d.f., P = 0.009; Figure 1A) and was significantly decreased in ADmiCAA, ADmodCAA and ADsevCAA as compared to controls (P = 0.04, P = 0.02 and P = 0.02, respectively) and to AD only (P = 0.01, P = 0.02 and P = 0.02, respectively; Figure 1A). C3b also showed a significant difference between groups (H = 12.5, 4 d.f., P = 0.01) and was significantly increased in the same cases compared to controls (P = 0.006, P = 0.02 and P = 0.02; Figure 1B) and only marginally increased in ADmodCAA and ADsevCAA compared to AD only (Figure 1B). Changes in both α2M and C3b additionally showed cross‐sectional trends as CAA pathology progressed: trending decrease for α2M (Figure 1A) and trending increase for C3b, although this tapered off in ADsevCAA cases (Figure 1B).
Figure 1.
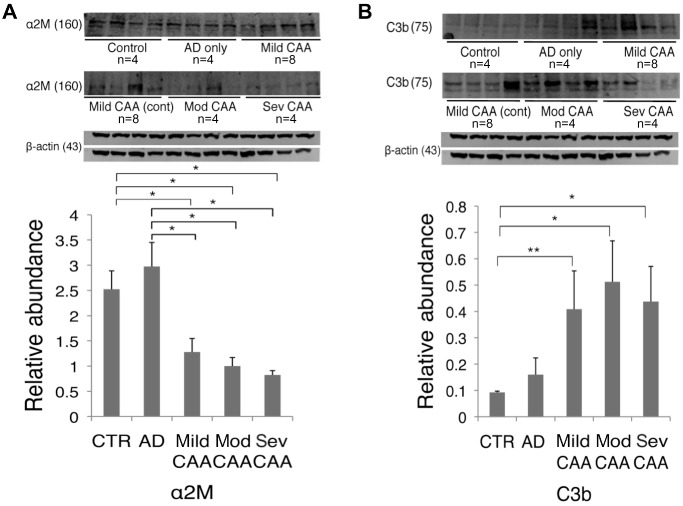
Western blot analysis of human occipital lobe parenchyma (whole‐brain homogenate) in cases classified as control (n = 4), AD‐only (n = 4) and AD/CAA. AD/CAA cases were further categorized into ADmiCAA (n = 8), ADmodCAA (n = 4) and ADsevCAA (n = 4). A. Analysis of α2M, a major chaperone of the Aβ peptide, showed a significant decrease in mild, moderate and severe CAA compared to control and pure AD. B. Analysis of C3b, the ligand for CD11b and opsonin of the Aβ peptide, was significantly increased across the spectrum of CAA cases from mild, moderate and severe compared to controls, while moderate and severe CAA were both significantly increased from pure AD. *P < 0.05; **P < 0.01. Data represented as mean ± SEM. Relative abundance is the ratio of α2M and C3b: beta‐actin as optical density.
Microglial Aβ binding via CD11b
Confocal fluorescent evaluations of fixed occipital lobe gray matter from each of the 12 brains (four each of AD with severe CAA, AD only and control) showed a consistent pattern of CAA‐specific microglial Aβ protein : protein interactions. Interactions were identified by FITC‐conjugated secondary antibodies to localize CD11b (green; Figure 2A) and a Texas Red‐conjugated secondary antibody to localize Aβ40 (red). Nuclei are identified by DAPI (blue). All AD/CAA CD11b+ microglia colocalized with Aβ40 on ramified processes (Figure 2A insets) in contrast to the primarily intracellular colocalization of CD11b and Aβ in the AD‐only cases. Only minimal colocalization on process‐like projections in control brains was observed. Identical patterns of colocalization were found in all brains (n = 4 for each group). This consistent observation has led to the hypothesis that a microglial Aβ‐binding mechanism causes downstream focal Aβ vascular complement activation 3 and MAC generation in AD/CAA. Representative 3D z‐stack micrographs show the consistently different Aβ distribution found on microglial processes colocalized with CD11b in AD/CAA (arrowheads) compared to AD only and controls (Figure 2B). Negative controls done with the same protocol, but without primary antibody validated the staining (Supporting Information Figure S1).
Figure 2.
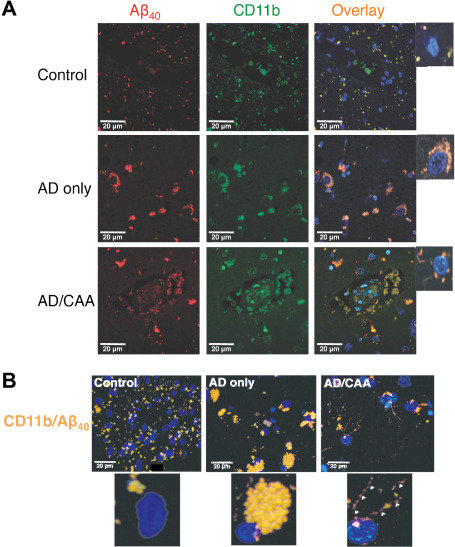
Representative confocal micrographs of fixed human occipital lobe tissue. Images were taken at 630× magnification and immunostained for (A) CD11b (n = 4 for each group) and colocalized with an anti‐Aβ40 antibody. A FITC‐conjugated secondary antibody was used for CD11b (green) and a Texas Red‐conjugated secondary antibody was used for Aβ40 (red). Nuclei were stained with DAPI (blue). (B). Representative Zstack images of microglia colocalized for CD11b and Aβ40. Panels show colocalized images in which microglial nuclei are stained with DAPI and colocalization of CD11b and Aβ40 is stained yellow/orange. Note the colocalization of CD11b with Aβ40 in punctate densities at the plasma membrane in AD/CAA (arrowheads), rather than the perinuclear, phagocytic vesicle distribution as seen in AD‐only cases. Controls show only faint colocalization for CD11b and Aβ40 that appear to localize to microglial processes.
Co‐immunoprecipitation of CD11b
Co‐immunoprecipitation of CD11b from occipital lobe whole‐brain homogenate yielded relatively equal abundance of CD11b protein on SDS‐PAGE Western blot analysis in all three groups (Figure 3A). C3b was significantly increased when CD11b‐containing supernatant was analyzed with Western blot analysis in cases of AD/CAA compared to both controls and AD only (H = 9.8, 2 d.f., P = 0.007; P = 0.02 and P = 0.02, respectively; Figure 3A,B).
Figure 3.
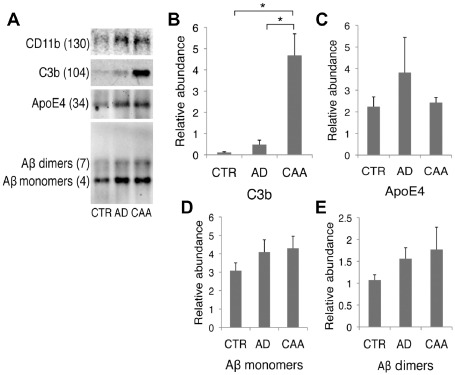
Co‐immunoprecipitation of CD11b. After CD11b immobilization and incubation with occipital lobe whole‐brain homogenate, Western blots were performed on CD11b‐containing supernatant and probed for (A) C3b, ApoE4, Aβ to measure the relative abundance of the CD11b/C3b/Aβ complex in each case. (B) AD/CAA had significantly higher C3b bound to CD11b compared to both controls and AD‐only. (C) ApoE4 showed no difference between groups. (D, E) Western analysis shows a nonsignificant increasing trend in the binding of Aβ monomers or dimers by CD11b in AD/CAA compared to controls and AD only. *P < 0.05. Data represented as mean ± SEM Relative abundance is the ratio of the target protein: CD11b‐containing supernatant as optical density.
As the APOE ε4 allele is the major substantiated genetic risk factor for nonhereditary AD and CAA and the E4 isotype has been shown to potentiate complement activation in vitro 26, we chose to determine if it was part of the CD11b/C3b complex. We found that apoE4 was bound to CD11b upon immunoprecipitation; however, it showed no statistical significance between groups (Figure 3B). This will need further testing in culture in order to confirm that when apoE E4 is complexed with CD11b and C3b, potentiation of complement activation occurs and that this is what causes increased vascular fragility secondary to MAC deposition.
Further analysis of CD11b‐containing supernatant showed increased (albeit not significant) levels of monomeric and dimeric Aβ bound to microglial CD11b in AD/CAA compared to AD only and controls (Figure 3A,D,E). These data are in agreement with our confocal data, where we observed that in every case, CD11b colocalizes with Aβ40. However, it appears that only in AD/CAA does CD11b significantly bind to Aβ through C3b, thus delivering active complement to the cerebral vasculature, setting up downstream MAC activation.
In order to confirm binding, we performed the reverse immunoprecipitation on the same samples for C3b, apoE4 and Aβ and performed Western blots for CD11b (Supporting Information Figure S2).
MAC on occipital lobe cerebral vasculature
The evidence for binding of Aβ via the CD11b/C3b complex leading to MAC activation on vascular smooth muscle cells at the vessel wall during microglial clearance in AD/CAA was further evaluated on CAA‐affected microvasculature by both Western blot and immunofluorescence for C5b‐9 deposition. C5b‐9 was significantly different between groups (H = 6, 2 d.f., P = 0.05). C5b‐9 was significantly increased when compared to both control and AD‐only vessels in a microvessel enrichment preparation from the occipital lobe (P = 0.02 and P = 0.03; Figure 4A). Additionally, we probed the enriched microvessel fraction for CD59, an endogenous regulator of late‐complement activation by inhibiting the binding of C8 and C9 of the MAC 20, which has shown to be decreased in the brain parenchyma in AD 49. We found a decreasing trend in CD59 on blood vessels of AD/CAA cases compared to AD‐only cases, which parallels the increased MAC deposition on the same AD/CAA vessels (Figure 4B).
Figure 4.
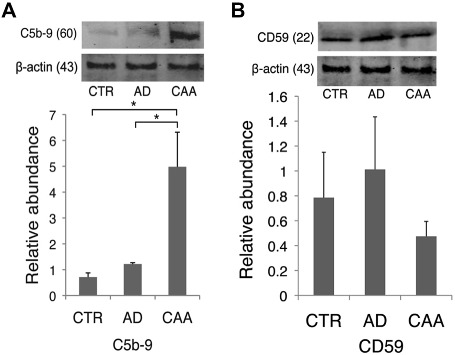
Western blot for C5b‐9 and CD59 on isolated cerebral blood vessels. A. C5b‐9 was significantly increased on isolated AD/CAA vessels compared to both controls and AD‐only vessels. B. CD59 showed a trending decrease on occipital blood vessels of AD/CAA cases compared to the control and AD‐only groups. *P < 0.05; **P < 0.01. Data represented as mean ± SEM. Relative abundance is the ratio of C5b‐9 and CD59:beta‐actin as optical density.
Immunofluorescent studies showed C5b‐9 on AD/CAA vessels colocalized with thioflavin staining, where as control and AD‐only vessels lacked such staining (Figure 5). Negative controls without primary antibody were used to verify the specificity of fluorescent staining (Supporting Information Figure S3). Additionally, MAC localized to the vicinity of smooth muscle cells in AD/CAA with co‐immunofluorescence staining of C5b‐9 and αSMA (Supporting Information Figure S4). Western blot analysis for C6, an integral component of the MAC, showed significantly increased levels in the enriched microvessel fraction in AD/CAA cases compared to controls (Figure 6A). Immunohistochemical analysis showed robust staining on AD/CAA vessels (Figure 6B), which selectively localized to the muscular layer of leptomeningeal arteries (Figure 6C). This staining is almost exclusively limited to leptomeningeal arteries and cortical arterioles in the gray matter, while the white matter is almost completely spared (Figure 6D,E)—a finding that matches the pattern of CAA involving leptomeningeal and penetrating cortical arterioles, sparing deeper white matter arterioles 43.
Figure 5.
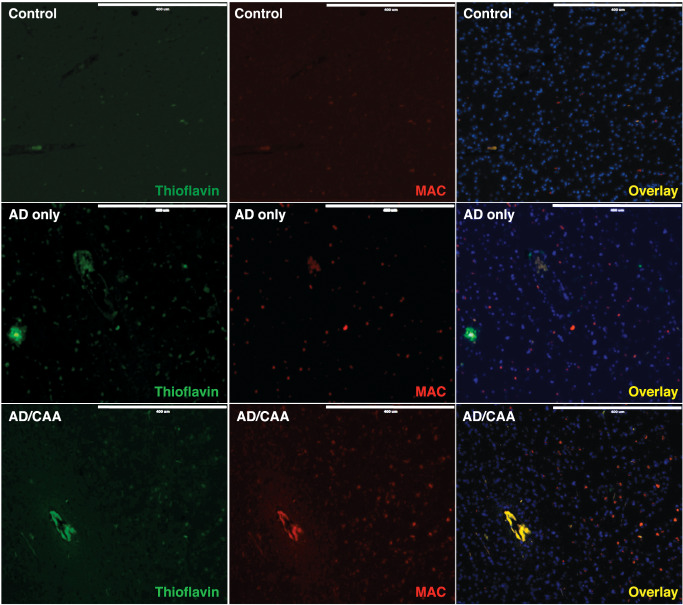
Representative confocal micrographs of fixed human occipital lobe tissue for C5b‐9 and fibrillar amyloid on cerebral blood vessels. Images were acquired at 100× and immunostained for C5b‐9 (MAC). A DyLight 550 secondary was used for visualization of MAC (red) and 0.01% Thioflavin S was used to visualize amyloid (green). Nuclei were stained with DAPI (blue). MAC and amyloid staining colocalize on cortical blood vessels in AD/CAA, which is absent in controls and AD‐only sections. Scale bar = 400 μm.
Figure 6.
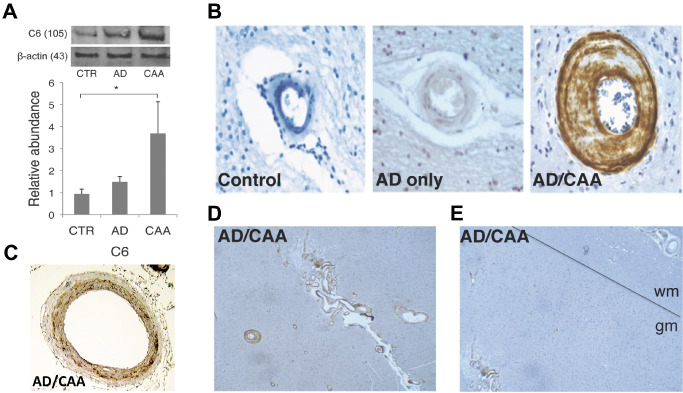
Western blot and immunohistochemistry for C6 on cerebral blood vessels. A. C6 is significantly increased on AD/CAA occipital blood vessels compared to controls (n = 4 for each group). B. Representative image of fixed human brain shows robust C6 staining of cortical arterioles in gray matter (400× magnification). C. C6 selectively localizes to the muscular layer of leptomeningeal arteries (dark brown). D. Representative image of C6 staining on gray matter arterioles in a case of AD/CAA (100 × magnification). E. Representative image of difference in C6 staining on gray matter arterioles (GM) vs. lack of staining on white matter arterioles delineated by the black line (40× magnification). Molecular weights of proteins are in parentheses. *P < 0.05. Data represented as mean ± SEM. Relative abundance is the ratio of the C6:beta‐actin as optical density.
Discussion
In this study, we present the hypothesis that Aβ clearance in AD subjects with CAA is distinct from that in AD subjects lacking CAA, and is mediated by microglial CD11b binding to extracellular C3b/Aβ complexes and delivering both active C3b and Aβ to the microvasculature, resulting in late‐complement activation. We evaluated whether observations from human post‐mortem occipital lobe tissue from subjects with varying degrees of CAA were consistent with this hypothesis. The complex CD11b/C3b/Aβ is found only in cases of AD/CAA and not in control and AD cases lacking CAA. This process occurs with a concomitant reduction in α2M, a known Aβ chaperone and likely a major mediator of microglial Aβ clearance in normal brain and in AD only 14. This CAA‐specific microglia shift in Aβ binding/clearance seems to be associated with cytolytic MAC deposition on leptomeningeal and penetrating cortical arterioles in the pattern of vascular Aβ deposition.
Several studies have implicated the LRP1/α2M complex in normal Aβ binding and clearance from the brain 21, 27, 28. Additionally, it has been shown that activated α2M is capable of blocking the formation of Aβ fibrils in vitro 51. In comparison to the observation of an age‐dependent decrease in LRP1 (with no change in α2M levels) on vessels in an AD mouse model 37 and in human AD postmortem tissue 4, 9, we found a significant decrease in α2M as the severity of CAA worsened compared to AD only and controls. The results from these studies and ours suggest a faulty Aβ clearance mechanism that involves both vascular (LRP1) and parenchymal (α2M) entities in the pathogenesis of cerebral vascular accumulation of Aβ.
The complement cascade and in particular, the C3b cleavage product of C3, has also been speculated to be involved in Aβ clearance and degradation 2. Blocking the complement cascade at C3 is thought to result in increased AD pathology, neurodegeneration and an altered microglial phenotype 24, 48. Interestingly, with a decrease in α2M in our cohort of AD with CAA cases, we also observed a concomitant increase in C3b in the same cases compared to both controls and pure AD cases. This is mirrored by an increase in the C3b receptor, CD11b, on microglia in both human AD tissue 3 and mouse models of AD 10. This seems to be a compensatory mechanism, as if the decrease in LRP1/α2M is driving a shift toward increased CD11b/C3b to mediate Aβ clearance.
The unique mechanism implied from this human post‐mortem study could explain vascular Aβ40 deposition based on the cell‐surface localization of CD11b/Aβ40 on microglia in CAA cases rather than the intracellular localization of this complex in AD‐only cases, as also demonstrated by Adolfsson et al 1. This suggests the complex may be subject to lysosomal degradation before it reaches the vascular tree. Aβ and C3b retained on the cell surface are presumably transported to the vasculature intact in CAA, facilitating both amyloid deposition and late‐complement activation. Control brain microglia showed an even more intriguing Aβ distribution. CD11b/Aβ colocalization seemed to appear at the ends of microglial processes, which is suggestive of unchallenged microglial phagocytosis in a healthy brain 38, 41. Although it has not been confirmed experimentally, this “ball and chain” mechanism of endocytosis by ramified microglia may represent efficient Aβ removal in the healthy brain 41. This observation is consistent with work by Liu et al 22 who also demonstrated Aβ42 localizing to vesicular structures within microglial processes.
The implications of our findings of CD11b binding Aβ in the presence of C3b in cases with CAA may explain the visualization of MAC on penetrating cortical arterioles and correlation of CAA‐affected vessels and microbleeds 36. If microglia in AD cases with CAA are binding amyloid through the CD11b receptor via C3b on their cell surface, this would allow the delivery of activated complement to the vessel wall allowing for the propagation toward MAC deposition on vascular smooth muscle cells. The immune system has several mechanisms by which to check the activation of complement. CD59 is a glycosylphosphatidylinositol (GPI) anchored glycoprotein that binds the C8 component and inhibits addition of C9 and thus MAC deposition. Several studies have shown that CD59 is decreased in the AD brain 49, 50. We found a nonsignificant decrease in CD59 on isolated vessels from AD/CAA cases, which could explain the inability of CAA‐affected vessels to protect against MAC deposition and why vessels in AD cases lacking CAA are not affected despite increased parenchymal MAC loads.
Additionally, validation of our biochemical findings can be found in a recent genetic study. A variation at rs6656401 on the A allele of the CR1 gene seems to confer risk for CAA‐associated ICH 5. 75% of the CAA cases in our cohort were homozygous for the risk allele, to which our protein data matches well with respect to parenchymal C3b levels and MAC levels on cerebral blood vessels. Patients with the CR1 variant might be at an increased risk for CAA‐associated ICH because of the inability of CR1 to effectively inactivate C3b, thus allowing its uninhibited propagation to the terminal lytic MAC. This has been shown experimentally in a mouse model of multiple sclerosis. Ramaglia et al 33 demonstrated that microglia became primed and pro‐inflammatory as a result of CD11b/C3b binding after knockout of the mouse homolog of CR1. Increased levels of activated C3b could then provide a ligand for the binding of Aβ to CD11b on microglia. Upon clearance of this complex, microglia deposit both Aβ and C3b near the vascular smooth muscle cells resulting in CAA and propagation of the complement cascade to MAC and thus the generation of focal microbleeds (Figure 7).
Figure 7.
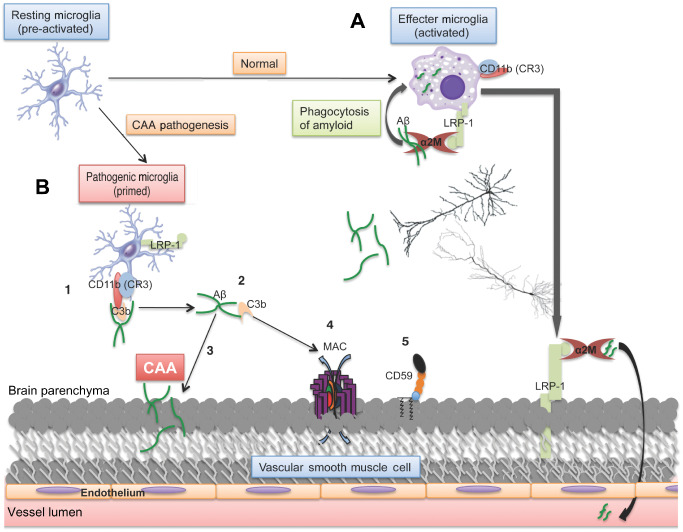
Proposed mechanism of MAC and Aβ deposition in CAA pathogenesis. A. In the healthy brain, activated microglia become amoeboid and clear Aβ through phagocytosis via α2M and LRP‐1. B. In AD with CAA, LRP‐1 and α2M are both decreased in human tissue, resulting in the compensatory mechanism of clearance involving C3b‐mediated opsonization of Aβ and binding by CD11b on microglia (1). This could be exacerbated if the variant in the CR1 gene negatively effects C3b homeostasis, which could result in the inability to inactivate C3b, leading to its accumulation. Upon delivery of the complex to cerebral blood vessels for clearance (2), Aβ may become deposited in the vessel wall caused by lack of proper phagocytosis (3) and C3b is allowed to propagate activation to the MAC (4), which would be even more pronounced with decreased defense mechanisms such as CD59 (5). The ultimate result is deposition of amyloid on the blood vessel wall and complement‐mediated cell lysis, both leading to hallmarks of this vasculopathy, CAA and microbleeds, respectively.
A final point of interest is the relationship of these findings to the topic of Aβ immunotherapy. Immunotherapy has been tested in numerous preclinical studies and a clinical trial. While it appears to be successful in clearing parenchymal Aβ deposits, it exacerbates vascular Aβ pathology and has an equivocal effect on cognition 31, 32, 46. The introduction of an anti‐Aβ antibody to the microglial Aβ clearance pathways discussed here would be expected to favor clearance through classical complement activation and the C3b/CD11b pathway. This may be well tolerated in subjects who primarily clear Aβ through noncomplement‐activating mechanisms, but in subjects who phenotypically accumulate Aβ in the vasculature, this would be expected to worsen the pathology. If this hypothesis is accurate, it is potentially relevant therapeutically because possible CAA can be diagnosed based on the extent of Pittsburgh compound B retention in the occipital lobe on positron emission tomography imaging 16 and subjects with evidence of significant CAA could be removed from future trials of anti‐Aβ immunotherapy.
Although this study is observational and therefore cannot conclusively define disease mechanisms, these data point to possible explanations for (i) complement activation on CAA‐affected vasculature, (ii) vascular fragility in CAA, (iii) immunotherapy exacerbation of the CAA phenotype and (iv) the importance of understanding and reporting pathologic phenotypes in studies of human AD. These data also point to the late‐complement cascade as a potential therapeutic target for the subtype of AD with CAA. More work is planned to delineate these mechanisms in vitro and determine how apoE4 may play a role in directing enhanced complement activation to cerebral blood vessels. Additionally, follow‐up studies are warranted in order to better understand the potential mechanisms described here in other types of CAA, such as the less‐frequent inflammatory CAA and cases of pure CAA without AD changes, and if the large lobar hemorrhages associated with CAA pathology are the result of complement‐mediated vascular fragility.
Conflict of Interest
The authors are aware of no real or potential conflict of interest.
Supporting information
Figure S1. Negative controls for CD11b and Aβ confocal images. Images were taken at 400× magnification and followed a similar protocol, and were incubated in FITC and Texas Red conjugated secondary antibodies without primary antibody.
Figure S2. Immunoprecipitation was performed on all target proteins from Figure 3. Western blots for CD11b were done on precipitates of a C3b, b ApoE4, c Aβ40/42 to confirm findings in Figure 3.
Figure S3. Negative controls for C5b‐9 (MAC) staining on fixed occipital lobe sections. Images were taken at 200× magnification and were prepared using a similar protocol and were incubated in Dylight 550 conjugated secondary antibody without primary antibody. A negative control image for Thioflavin S was taken without staining on the green channel. Scale bar = 200 μm.
Figure S4. Representative images of C5b‐9 (MAC) staining (red) colocalized with αsma (green) on a cortical arteriole in a case of AD/CAA. Scale bar = 30 μm.
Acknowledgments
The authors would like to acknowledge April Dickson for lab assistance and Jackie Knecht for her support and the assistance of Dr. Sean Wilson on the Zeiss LSM 710 confocal microscope. This material is funded by NIH grant AG20948 and NSF grant no. MRI‐DBI 0923559 (S.M. Wilson) and by Loma Linda University School of Medicine. Harry V. Vinters and Spencer Tung are supported in part by P50AG16570 and PO1AG12435.
References
- 1. Adolfsson O, Pihlgren M, Toni N, Varisco Y, Buccarello AL, Antoniello K et al (2012) An effector‐reduced anti‐beta‐amyloid (Abeta) antibody with unique abeta binding properties promotes neuroprotection and glial engulfment of Abeta. J Neurosci 32:9677–9689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM et al (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21:383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Akiyama H, McGeer PL (1990) Brain microglia constitutively express beta‐2 integrins. J Neuroimmunol 30:81–93. [DOI] [PubMed] [Google Scholar]
- 4. Bell RD, Deane R, Chow N, Long X, Sagare A, Singh I et al (2009) SRF and myocardin regulate LRP‐mediated amyloid‐beta clearance in brain vascular cells. Nat Cell Biol 11:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Biffi A, Shulman JM, Jagiella JM, Cortellini L, Ayres AM, Schwab K et al (2012) Genetic variation at CR1 increases risk of cerebral amyloid angiopathy. Neurology 78:334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 7. Bradt BM, Kolb WP, Cooper NR (1998) Complement‐dependent proinflammatory properties of the Alzheimer's disease beta‐peptide. J Exp Med 188:431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Choucair‐Jaafar N, Laporte V, Levy R, Poindron P, Lombard Y, Gies JP (2010) Complement receptor 3 (CD11b/CD18) is implicated in the elimination of beta‐amyloid peptides. Fundam Clin Pharmacol 25:115–122. [DOI] [PubMed] [Google Scholar]
- 9. Donahue JE, Flaherty SL, Johanson CE, Duncan JA, 3rd , Silverberg GD, Miller MC et al (2006) RAGE, LRP‐1, and amyloid‐beta protein in Alzheimer's disease. Acta Neuropathol 112:405–415. [DOI] [PubMed] [Google Scholar]
- 10. Gallagher JJ, Finnegan ME, Grehan B, Dobson J, Collingwood JF, Lynch MA (2011) Modest amyloid deposition is associated with iron dysregulation, microglial activation, and oxidative stress. J Alzheimers Dis 28:147–161. [DOI] [PubMed] [Google Scholar]
- 11. Graeber MB, Streit WJ (2010) Microglia: biology and pathology. Acta Neuropathol 119:89–105. [DOI] [PubMed] [Google Scholar]
- 12. Greenberg SM, Vonsattel JP (1997) Diagnosis of cerebral amyloid angiopathy. Sensitivity and specificity of cortical biopsy. Stroke 28:1418–1422. [DOI] [PubMed] [Google Scholar]
- 13. Hanisch UK, Kettenmann H (2007) Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 10:1387–1394. [DOI] [PubMed] [Google Scholar]
- 14. Hughes SR, Khorkova O, Goyal S, Knaeblein J, Heroux J, Riedel NG, Sahasrabudhe S (1998) Alpha2‐macroglobulin associates with beta‐amyloid peptide and prevents fibril formation. Proc Natl Acad Sci U S A 95:3275–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jellinger KA, Attems J (2005) Prevalence and pathogenic role of cerebrovascular lesions in Alzheimer disease. J Neurol Sci 229–230:37–41. [DOI] [PubMed] [Google Scholar]
- 16. Johnson KA, Gregas M, Becker JA, Kinnecom C, Salat DH, Moran EK et al (2007) Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol 62:229–234. [DOI] [PubMed] [Google Scholar]
- 17. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011) Physiology of microglia. Physiol Rev 91:461–553. [DOI] [PubMed] [Google Scholar]
- 18. Khera R, Das N (2009) Complement receptor 1: disease associations and therapeutic implications. Mol Immunol 46:761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kirsch W, McAuley G, Holshouser B, Petersen F, Ayaz M, Vinters HV et al (2009) Serial susceptibility weighted MRI measures brain iron and microbleeds in dementia. J Alzheimers Dis 17:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kolev MV, Tediose T, Sivasankar B, Harris CL, Thome J, Morgan BP, Donev RM (2010) Upregulating CD59: a new strategy for protection of neurons from complement‐mediated degeneration. Pharmacogenomics J 10:12–19. [DOI] [PubMed] [Google Scholar]
- 21. Lauer D, Reichenbach A, Birkenmeier G (2001) Alpha 2‐macroglobulin‐mediated degradation of amyloid beta 1–42: a mechanism to enhance amyloid beta catabolism. Exp Neurol 167:385–392. [DOI] [PubMed] [Google Scholar]
- 22. Liu Z, Condello C, Schain A, Harb R, Grutzendler J (2010) CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid‐beta phagocytosis. J Neurosci 30:17091–17101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lopes KO, Sparks DL, Streit WJ (2008) Microglial dystrophy in the aged and Alzheimer's disease brain is associated with ferritin immunoreactivity. Glia 56:1048–1060. [DOI] [PubMed] [Google Scholar]
- 24. Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA (2008) Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci 28:6333–6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McGeer PL, McGeer EG (2002) The possible role of complement activation in Alzheimer disease. Trends Mol Med 8:519–523. [DOI] [PubMed] [Google Scholar]
- 26. McGeer PL, Walker DG, Pitas RE, Mahley RW, McGeer EG (1997) Apolipoprotein E4 (ApoE4) but not ApoE3 or ApoE2 potentiates beta‐amyloid protein activation of complement in vitro. Brain Res 749:135–138. [DOI] [PubMed] [Google Scholar]
- 27. Mikhailenko I, Battey FD, Migliorini M, Ruiz JF, Argraves K, Moayeri M, Strickland DK (2001) Recognition of alpha 2‐macroglobulin by the low density lipoprotein receptor‐related protein requires the cooperation of two ligand binding cluster regions. J Biol Chem 276:39484–39491. [DOI] [PubMed] [Google Scholar]
- 28. Narita M, Holtzman DM, Schwartz AL, Bu G (1997) Alpha2‐macroglobulin complexes with and mediates the endocytosis of beta‐amyloid peptide via cell surface low‐density lipoprotein receptor‐related protein. J Neurochem 69:1904–1911. [DOI] [PubMed] [Google Scholar]
- 29. Nicoll JA, Yamada M, Frackowiak J, Mazur‐Kolecka B, Weller RO (2004) Cerebral amyloid angiopathy plays a direct role in the pathogenesis of Alzheimer's disease. Pro‐CAA position statement. Neurobiol Aging 25:589–597. discussion 603–4. [DOI] [PubMed] [Google Scholar]
- 30. Perry VH, O'Connor V (2008) C1q: the perfect complement for a synaptic feast? Nat Rev Neurosci 9:807–811. [DOI] [PubMed] [Google Scholar]
- 31. Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M et al (2002) Cerebral hemorrhage after passive anti‐Abeta immunotherapy. Science 298:1379. [DOI] [PubMed] [Google Scholar]
- 32. Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK et al (2005) Exacerbation of cerebral amyloid angiopathy‐associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J Neurosci 25:629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ramaglia V, Hughes TR, Donev RM, Ruseva MM, Wu X, Huitinga I et al (2012) C3‐dependent mechanism of microglial priming relevant to multiple sclerosis. Proc Natl Acad Sci U S A 109:965–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD et al (1992) Complement activation by beta‐amyloid in Alzheimer disease. Proc Natl Acad Sci U S A 89:10016–10020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rogers J, Li R, Mastroeni D, Grover A, Leonard B, Ahern G et al (2006) Peripheral clearance of amyloid beta peptide by complement C3‐dependent adherence to erythrocytes. Neurobiol Aging 27:1733–1739. [DOI] [PubMed] [Google Scholar]
- 36. Schrag M, McAuley G, Pomakian J, Jiffry A, Tung S, Mueller C et al (2009) Correlation of hypointensities in susceptibility‐weighted images to tissue histology in dementia patients with cerebral amyloid angiopathy: a postmortem MRI study. Acta Neuropathol 119:291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B et al (2000) Clearance of Alzheimer's amyloid‐ss(1‐40) peptide from brain by LDL receptor‐related protein‐1 at the blood‐brain barrier. J Clin Invest 106:1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet‐Wadiche LS et al (2010) Microglia shape adult hippocampal neurogenesis through apoptosis‐coupled phagocytosis. Cell Stem Cell 7:483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Soontornniyomkij V, Lynch MD, Mermash S, Pomakian J, Badkoobehi H, Clare R, Vinters HV (2010) Cerebral microinfarcts associated with severe cerebral beta‐amyloid angiopathy. Brain Pathol 20:459–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Streit WJ, Braak H, Xue QS, Bechmann I (2009) Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol 118:475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A (2011) The role of microglia in the healthy brain. J Neurosci 31:16064–16069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vinters HV (1987) Cerebral amyloid angiopathy. A critical review. Stroke 18:311–324. [DOI] [PubMed] [Google Scholar]
- 43. Vinters HV, Gilbert JJ (1983) Cerebral amyloid angiopathy: incidence and complications in the aging brain. II. The distribution of amyloid vascular changes. Stroke 14:924–928. [DOI] [PubMed] [Google Scholar]
- 44. Webster S, Bradt B, Rogers J, Cooper N (1997) Aggregation state‐dependent activation of the classical complement pathway by the amyloid beta peptide. J Neurochem 69:388–398. [DOI] [PubMed] [Google Scholar]
- 45. Weller RO, Boche D, Nicoll JA (2009) Microvasculature changes and cerebral amyloid angiopathy in Alzheimer's disease and their potential impact on therapy. Acta Neuropathol 118:87–102. [DOI] [PubMed] [Google Scholar]
- 46. Wilcock DM, Colton CA (2009) Immunotherapy, vascular pathology, and microhemorrhages in transgenic mice. CNS Neurol Disord Drug Targets 8:50–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wirenfeldt M, Babcock AA, Vinters HV (2011) Microglia—insights into immune system structure, function, and reactivity in the central nervous system. Histol Histopathol 26:519–530. [DOI] [PubMed] [Google Scholar]
- 48. Wyss‐Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E (2002) Prominent neurodegeneration and increased plaque formation in complement‐inhibited Alzheimer's mice. Proc Natl Acad Sci U S A 99:10837–10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yang LB, Li R, Meri S, Rogers J, Shen Y (2000) Deficiency of complement defense protein CD59 may contribute to neurodegeneration in Alzheimer's disease. J Neurosci 20:7505–7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yasojima K, McGeer EG, McGeer PL (1999) Complement regulators C1 inhibitor and CD59 do not significantly inhibit complement activation in Alzheimer disease. Brain Res 833:297–301. [DOI] [PubMed] [Google Scholar]
- 51. Yerbury JJ, Kumita JR, Meehan S, Dobson CM, Wilson MR (2009) alpha2‐Macroglobulin and haptoglobin suppress amyloid formation by interacting with prefibrillar protein species. J Biol Chem 284:4246–4254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Negative controls for CD11b and Aβ confocal images. Images were taken at 400× magnification and followed a similar protocol, and were incubated in FITC and Texas Red conjugated secondary antibodies without primary antibody.
Figure S2. Immunoprecipitation was performed on all target proteins from Figure 3. Western blots for CD11b were done on precipitates of a C3b, b ApoE4, c Aβ40/42 to confirm findings in Figure 3.
Figure S3. Negative controls for C5b‐9 (MAC) staining on fixed occipital lobe sections. Images were taken at 200× magnification and were prepared using a similar protocol and were incubated in Dylight 550 conjugated secondary antibody without primary antibody. A negative control image for Thioflavin S was taken without staining on the green channel. Scale bar = 200 μm.
Figure S4. Representative images of C5b‐9 (MAC) staining (red) colocalized with αsma (green) on a cortical arteriole in a case of AD/CAA. Scale bar = 30 μm.