

Abstract
Recently, a hexanucleotide repeat expansion in the C9ORF72 gene has been identified to account for a significant portion of Caucasian families affected by frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS). Given the clinical overlap of FTD with Alzheimer disease (AD), we hypothesized that C9ORF72 expansions may contribute to AD. In Caucasians, we found C9ORF72 expansions in the pathogenic range of FTD/ALS (>30 repeats) at a rate of 0.76% in AD cases versus zero in controls (p=3.3E-03, 1,182 cases, 1,039 controls). In contrast, no large expansions were detected in individuals of African American ethnicity (291 cases, 620 controls). However, in the range of normal variation of C9ORF72 expansions (0–23 repeat copies), we detected significant differences in distribution and mean repeat counts between Caucasians and African Americans. Clinical and pathological reevaluation of identified C9ORF72 expansion carriers revealed nine clinical and/or autopsy confirmed AD and two FTD finial diagnoses. Thus, our results support the notion that large C9ORF72 expansions lead to a phenotypic spectrum of neurodegenerative disease including AD.
Keywords: C9ORF72, repeat expansion, Alzheimer Disease, genetic association, repeat-primed PCR, spectrum of neurodegenerative phenotypes
1. Introduction
Despite clinical and genetic heterogeneity, a number of molecular commonalities have long been recognized for disorders as diverse as Alzheimer disease (AD), Parkinson disease (PD), frontotemporal dementia (FTD), and amyotrophic lateral sclerosis (ALS). These diseases show extracellular and/or intracellular deposition of abnormal proteins, which has been attributed to a number of pathways, foremost the misfolded protein hypothesis (Taylor, et al., 2002). Shared molecular mechanisms are usually the result of overlapping genetic factors. This has indeed been documented by the AD&FTLD&PD Mutation Databases which curate mutations in the 15 established monogenetic disease genes for AD, FTD and PD: With the exception of the PD genes PARK2 and PINK1, all other genes, for instance the AD gene PSEN2 or the FTD gene MAPT, are associated with more than one clinical diagnosis or characteristics thereof (Cruts, et al., 2012). Further characterization of the phenotypic spectrum of genes involved in neurodegeneration does improve clinical and molecular disease models.
Genetic linkage with FTD and/or ALS was established to chromosome 9p21 (Luty, et al., 2008, Morita, et al., 2006, Vance, et al., 2006) and subsequently an intronic hexanucleotide (GGGGCC)n repeat expansion in the C9ORF72 gene [NM_145005] was identified as the underlying mutation (DeJesus-Hernandez, et al., 2011, Renton, et al., 2011). Expansions measured by repeat-primed PCR showing more than 30 repeats are considered pathogenic for FTD/ALS. Large samples of population controls have been reported with repeat counts ranging from 2 to 23 units (DeJesus-Hernandez, et al., 2011, Renton, et al., 2011). Studies of large Caucasian patient samples have shown that the C9ORF72 repeat expansion accounts for up to 50% of ALS/FTD families, 7% – 12% of FTD families, and 2% – 5% of sporadic FTD (Boeve, et al., 2012, Mahoney, et al., 2012, Simon-Sanchez, et al., 2012, Snowden, et al., 2012). Similar frequencies have been observed in ALS families and sporadic cases (Chio, et al., 2012, Cooper-Knock, et al., 2012).
Given the clinical, pathological, and genetic characteristics of FTD, we hypothesized that C9ORF72 expansions may also confer risk to the most common neurodegenerative disease, Alzheimer disease (AD). Clinically, both dementias show progressive impairments in cognition, memory, language, behavior and motor functions (Goedert, et al., 2012). Pathologically, atrophy in the frontal and temporal lobe is overlapping and a tau positive pathology is typical for AD and forms of FTD (Heutink, 2000). Genetically, mutations in the tau protein (MAPT) cause autosomal dominant inherited FTD as well as heterogeneous clinical phenotypes resembling AD (Goedert, et al., 2012, Rademakers, et al., 2003). In fact, Majounie et al. (2012a) recently reported C9ORF72 repeat expansions in 3 out of 342 screened families from an AD collection. Based on post-mortem brain studies that revealed FTD rather than AD in one family, the authors concluded that the most likely explanation is misclassification, rather than C9ORF72 being a direct cause for AD (Majounie, et al., 2012a). Another screen for C9ORF72 expansions in 568 AD cases did not detect any expansion carriers (Rollinson, et al., 2012).
Here we present a study of C9ORF72 repeat expansions in AD, comprising 1,184 unrelated cases and 1,039 controls of European decent. We also studied 291 African American AD cases and 620 controls to investigate on repeat expansion distributions between ethnicities. Each individual classified as an AD case met the NINCDS-ADRDA criteria for probable or definite AD (McKhann, et al., 1984). The results of this study likely expand the genetic basis of AD and contribute to the understanding of the phenotypic spectrum of C9ORF72 repeat expansions.
2 Methods
2.1 Study samples
Written informed consents were obtained from all participants, in accordance with Institutional Review Board protocols at each study center. The study included 1,184 unrelated European and 291 unrelated African American individuals with a clinical diagnosis of AD and 1,039 European and 620 African American cognitive controls. Supplementary Table 1 provides descriptive characteristics about these study samples. All cases and controls were clinically ascertained through the University of Miami at the John P. Hussman Institute for Human Genomics (HIHG), the Vanderbilt University Center for Human Genetics Research (CHGR), the Department of Biology at North Carolina Agricultural and Technical (NCA&T) State University, and were sampled from pathologically confirmed donors to the University of Miami Brain Endowment Bank (BEB). Each individual classified as an AD case met the NINCDS-ADRDA criteria for probable or definite AD (McKhann, et al., 1984). Where possible clinically ascertained and deceased participants had AD confirmed via autopsy. Individual’s pathological diagnoses most critical to the interpretation of this study outcome were reevaluated by two independent neuropathologists with long-standing experienced in AD, Drs. Carol Petito (University of Miami) and Christine Hulette (Duke University). Age-at-onset (AAO) was estimated as date of first onset of symptoms as reported by the patient, their informant, or abstracted from the patient’s medical records. Controls had a Mini-Mental State Exam (MMSE) scores ≥ 27 at an age at exam (AAE) ≥ 60 years. Genome-wide SNP data was available for the studied individuals, allowing principal components analysis on a sample of 20,000 SNPs to remove population outliers by using the top ten principal components over 5 iterations with a threshold of six standard deviations (Naj, et al., 2010). The top three principal component loadings were used to validate the ethnical background of self-reported ethnicity. The pedigrees presented in Fig. 1 have been partially altered and masked to hide identifying information. DNA samples of three ALS patients (M. Benatar), served as positive controls for C9ORF72 repeats expansions (> 50 repeats) (Renton, et al., 2011). Apolipoprotein E (APOE) genotype data were available on all participants and were determined as previously described (Naj, et al., 2011).
Fig. 1. Overview of families and sporadic individuals affected by C9ORF72 expansions.

(A) FTD reclassified index case/family, (B) AD/ALS families, (C) AD family and sporadic/isolated AD cases. Sporadic is defined as a single case with recorded family history, whereas an isolated case did not have family history available. Gender is not provided for privacy reasons.
2.2 Repeat-primed PCR assay
DNA samples of cases and controls were subjected to repeat-primed PCR to determine the number of hexanucleotide (GGGGCC)n repeat expansions in the C9ORF72 gene on chromosome 9p21. We used a previously described repeat-primed PCR assay (Renton, et al., 2011) by applying the same reaction mix protocol and primers, but optimized PCR cycling program to achieve robust results. Fragment length analysis was performed on an ABI 3730xl genetic analyzer (ABI), and data were analyzed using GeneMapper software (version 4, ABI). Our assay limits for the maximal number of detectable repeats was between 60 and 70 repeats which is in accordance with previous studies (Kobayashi, et al., 2011). To determine the assay’s precision in counting out repeat copies in the range of normal variation (≤4–23 repeats observed in our controls), we used synthetic ultramer DNA oligos (Integrated DNA Technologies, Inc., USA) with incorporated eight and sixteen repeat units in addition to the three units present in the human reference sequence. Moreover, in this range, we determined re-sampling reproducibility of repeat counts of the assay by running repeated measurements (N=260) of 100 randomly selected samples.
Each of the expansion carrying samples was genotyped by Sanger sequencing for the single nucleotide polymorphism (SNP) rs3849942, which serves as a surrogate marker for expanded C9ORF72 haplotypes in Europeans (DeJesus-Hernandez, et al., 2011).
2.3 Statistical Methods
We tested for an overrepresentation of large repeat expansions (>30 repeats) in the pathogenic range for ALS/FTD (Renton, et al., 2011) in AD cases versus cognitively normal elderly controls (AAE ≥60 years) by conducting one-sided Fisher’s exact tests in Caucasians and African Americans (Table 2). In the normal range of repeat copy variation (≤4–23 in this study’s controls), we tested for case-control and ethnic differences in repeat count distributions and means by applying the Kolmogorov-Smirnov test and a standard t-test, respectively (Table 3). All analyses were conducted using R software version 2.13.0 (http://www.r-project.org/).
Table 2.
Final diagnoses and further characterization of large C90RF72 expansion carriers and unaffected family members
| Phenotype | Family ID | Individual ID | Repeats | Final diagnosis | Relatedness | AAO | AAE | APOE |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| FTD, sporadic | 1 | 001 | 57 | Clinical FTD | index patient | 63 | 65 | 3/4 |
|
| ||||||||
| FTD/AD | 2 | 001 | 60 | Autopsy confirmed FTLD-U | index patient | 56 | 59 | 3/3 |
| 101 | 60 | unaffected | sibling | - | 54 | 3/3 | ||
| 1001 | ≤4 | unaffected | sibling | - | 81 | 3/3 | ||
| 1005 | - | Clinical AD | paternal uncle | - | - | - | ||
|
| ||||||||
| AD/ALS | 3 | 001 | 64 | Clinical AD | index patient | 66 | 74 | 3/3 |
| 101 | 8 | unaffected | sibling | - | 56 | 3/3 | ||
| 102 | 8 | unaffected | sibling | - | 54 | 3/4 | ||
| 104 | - | ALS | sibling | - | - | - | ||
|
| ||||||||
| AD/ALS | 4 | 001 | 63 | Clinical AD | index patient | 73 | 78 | 3/4 |
| 102 | - | ALS | sibling | |||||
|
| ||||||||
| AD | 5 | 101 | 62 | Clinical AD | index patient | 61 | 66 | 4/4 |
| 105 | ≤4 | unaffected | sibling | - | 83 | 3/4 | ||
| 108 | 56 | possible AD | sibling | - | 74 | 3/3 | ||
| 9017 | 8 | unaffected | nephew | - | 54 | 3/4 | ||
|
| ||||||||
| AD, sporadic | 6 | 001 | 43 | Autopsy confirmed AD | index patient | 91 | 93 | 3/3 |
|
| ||||||||
| AD, sporadic | 7 | 001 | 52 | Clinical AD | index patient | 71 | 74 | 2/3 |
|
| ||||||||
| AD, sporadic | 8 | 2023 | 68 | Clinical AD | index patient | 70 | 72 | 3/4 |
|
| ||||||||
| AD, sporadic | 9 | 4191 | 61 | Clinical AD | index patient | 72 | 72 | 3/3 |
|
| ||||||||
| AD, isolated | 10 | 98 | 42 | Autopsy confirmed AD | index patient | 77 | 82 | 3/4 |
|
| ||||||||
| AD, isolated | 11 | 21 | 42 | Autopsy confirmed AD | index patient | 74 | 80 | 4/4 |
(AAO: age at onset of disease, AAE: age at exam, APOE: Apolipoprotein E AD risk genotype).
Table 3.
C90RF72 repeat mean copy count and distribution comparisons in the range of normal repeat copy variation (≤4–23 repeats). (A) Case-control comparisons, (B) Ethnic comparisons
| Comparisons | N | Average repeats | KS-test | T-test | ||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| (A) Case-Control status * | cases | controls | cases | controls | Dn | p | t | P |
|
| ||||||||
| African American (AA) | 292 | 620 | 6.64 | 6.76 | 0.07 | 0.220 | −0.68 | 0.495 |
| European descent (ED) | 1257 | 1039 | 6.81 | 7.08 | 0.05 | 0.141 | −2.00 | 0.046 |
| All (AA+ED) | 1549 | 1659 | 6.78 | 6.96 | 0.05 | 0.073 | −1.71 | 0.087 |
|
| ||||||||
| (B) AA vs. EDethnicity * | AA | ED | AA | ED | Dn | p | t | P |
|
| ||||||||
| Controls | 620 | 1039 | 6.76 | 7.08 | 0.11 | 1.9E-04 | −2.17 | 0.0305 |
| Cases | 292 | 1257 | 6.64 | 6.81 | 0.10 | 0.015 | −1.02 | 0.3080 |
| All, (Cases+Controls) | 912 | 2296 | 6.72 | 6.93 | 0.11 | 1.5E-07 | −1.96 | 0.0498 |
samples with >30 repeats were excluded.
3. Results
3.1 A subset of Caucasian AD patients presents with C9ORF72 repeat expansions
We identified C9ORF72 expansions in the reported pathogenic range of FTD/ALS (>30 repeats) in cases that met the NINCDS-ADRDA criteria for probable or definite AD from European descent at a rate of 0.9% (11 out of 1,184 unrelated cases). In contrast, no expansions were identified in 1,039 dementia free elderly controls from the same ethnicity (one-sided Fisher exact test p=9.6E-04, Table 1). This case-control associations remained significant after exclusion of two C9ORF72 expansion carriers that were reclassified with autopsy-confirmed FTD and possible clinical FTD (p=3.3E-03, Table 1).
Table 1.
Case-Control (A) and ethnic comparisons (B) of AD case-control collections with and without C90RF72 expansions in the pathogenic range of ALS/FTD (>30 repeat copies)
| Comparisons | Cases | Controls | Fisher exact | ||
|---|---|---|---|---|---|
| (A) Case-Control | <30 repeats | ≥30 repeats | <30 repeats | ≥30 repeats | P, one-sided |
| African American (AA) | 291 | 0 | 620 | 0 | 1.0 |
| European descent (ED) | 1173 | 11 (9) | 1039 | 0 | 9.6E-04 (3.3E-03) |
| (B) AA vs. ED ethnicity | AA | ED | Fisher exact | ||
| <30 repeats | ≥30 repeats | <30 repeats | ≥30 repeats | P, one-sided | |
| Cases | 291 | 0 | 1173 | 11 (9) | 0.044 (0.069) |
| Cases + Controls | 911 | 0 | 2212 | 11 (9) | 0.023 (0.045) |
NOTE: Counts and corresponding p-values in brackets represent an analysis that excluded retrospectively reclassified FTDcases.
In African American individuals C9ORF72 expansions were not present (291 cases, 620 controls). Given the frequency of expansion carriers observed in samples of European descent we would have expected to detect 2.7 expansion carriers in the set of 291 African American cases. Formally, C9ORF72 expansions were significantly overrepresented in Europeans compared to African Americans (one-side Fisher exact p=0.023, Table 1).
Four index patients with expansions had family members with diagnosis of dementia or ALS, five were sporadic dementia cases with known family history, and two were isolated cases (no family history data available). Three of these families included 2–7 dementia cases with affected individuals in multiple generations (families 2, 3, 5, Fig. 1, Table 2). The C9ORF72 expansions co-segregated in these large families with the phenotype (Fig. 1).
The SNP marker rs389942 accurately tags the Northern Europe derived haplotype on which large C9ORF72 repeat expansion have been reported (DeJesus-Hernandez, et al., 2011). We confirmed the presence of the risk-haplotype associated A-allele in all of our expanded samples.
3.2 Clinical presentations of patients with expanded repeats
Clinical data was available for each of the 11 index patients with C9ORF72 expansions (>30 repeats, see supplementary information for detailed clinical reports). Four index patients obtained an autopsy confirmed diagnoses 2-001, 6-001, 10–98 and 11–21; Fig. 1 and 2, Table 2). For two of these cases histological slides were available for reevaluation of autopsy diagnoses (2-001, 6-001, see supplementary text for autopsy reports). We identified a clinical spectrum of phenotypes associated with C9ORF72 expansions in these families and patients (Fig 1). In brief, two families were characterized by index patients, for which reevaluation of clinical and neuropathological data resulted in their reclassification with FTD (1-001 and 2-001). Two additional families with C9ORF72 expansions presented with index patients with clinical AD who also had first-degree relatives with ALS (3 and 4). Finally, the large family number 5 consisted of an index patient and family members (N=5) that were exclusively diagnosed with clinical AD. Furthermore, we identified four sporadic and two isolated AD index patients with clinical diagnoses of AD (families 6–11) of which three had an autopsy confirmed diagnoses of AD (6-001, 10-098 and 11-021).
Fig. 2. Histopathological findings in C9ORF72 expansion carriers.
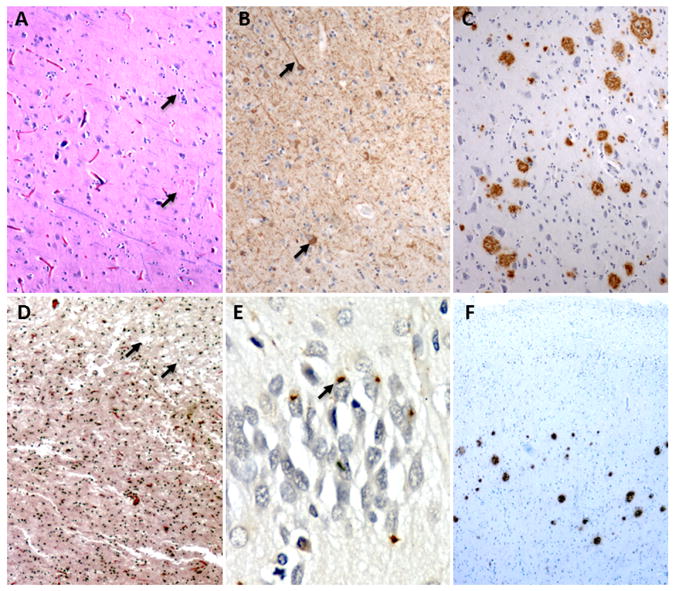
(A – C) The index case in family 6 showed classic AD changes including (A) neuronal loss and plaques (B) tau positive threads and tangles, and (C) amyloid plaques throughout the neocortex and hippocampus. (D – F) The index patient from family 2 showed (D) microvacuolation in the frontal cortex, (E) ubiquitin-immunoreactive intracellular inclusions in the hippocampus, and (F) cortical extracellular amyloid plaques. For more details see supplementary data.
3.3 C9ORF72 repeat expansions in the normal range of variation do not contribute to AD risk
The range of C9ORF72 repeat copies varied between ≤4 and 23 repeat units in our combined samples of 1,464 cases and 1,659 controls after exclusion of cases with repeat expansions in the pathogenic range (>30 copies, Fig. 3). In the normal range of C9ORF72 repeat expansions, the repeat-primed PCR assay applied to determine C9ORF72 repeat copy numbers was highly accurate and reproducible: The average re-sampling error rate was 0.16 repeat copies per measurement with a maximal observed deviation of two repeat units. We addressed the hypothesis of whether C9ORF72 repeats especially in the upper normal range of variation might already interfere with C9ORF72 gene function leading to AD risk. We tested for differences in mean repeat counts and repeat count distributions between AD cases and controls. Statistical test results are given in Table 3A and histograms of repeat distributions are shown in Fig. 3A/B. We did not observe a statistically significant difference of repeat count distribution between cases and controls in African Americans, Caucasians or the combined datasets. Case-control comparison of mean repeat copy numbers in Caucasians only reached nominal significance, but with a slightly higher mean in repeat copies in controls compared to cases.
Fig. 3.
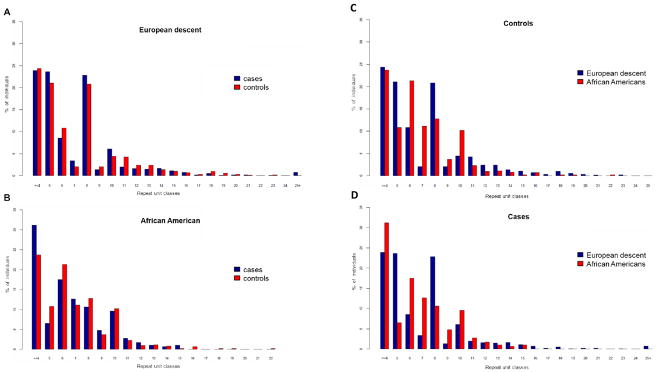
(A and B) Histograms of C9ORF72 repeat copies in AD case-control collections of (A) European and (B) African American descent. (C and D) Histograms of C9ORF72 repeat copies compared between European and African American ethnicities in (C) cases and (D) controls.
3.4 C9ORF72 repeat distributions vary between European and African American ethnicities
As we reported above, carriers of large C9ORF72 repeat expansions (> 30 repeats) were exclusively found in AD cases of European descent, but not in African Americans. Interestingly, we did observe significant differences in mean repeat copies and repeat distributions between samples of African American and European descent in the normal range of C9ORF72 repeat copy variation (≤4 and 23 repeats, Table 3B). The mean repeat length was slightly greater in European versus African American controls and in the combined case and control samples (6.97 versus 6.72 repeat copies, T-test p=0.023). The Kolmogorov-Smirnov test showed highly significant differences in the distribution of repeat counts in African Americans versus European samples. This was observed in controls, cases and the combined samples (p=5.9E-07, Table 3B). The histograms in Fig. 3C/D show that specific repeat copy number classes are more common in either African American or European samples.
4 Discussion
4.1 C9ORF72 repeat expansions underlie a spectrum of neurodegenerative phenotypes, including AD
Our study represents a sizable screen for C9ORF72 expansions in AD comprising more than 3,100 unrelated individuals from European and African American decent. We detected C9ORF72 expansion carriers at a rate of 0.9% exclusively among Caucasians with an initial clinical diagnosis of AD meeting NINCDS-ADRDA criteria for probable or definite AD (McKhann, et al., 1984). This case-control associations remained significant after removal of two cases that were reclassified as FTD (p=3.3E-03, 1-001 and 2-001, see supplementary text). These findings are in concordance with a recently reported C9ORF72 repeat expansion screen in another AD cohort, reporting one misclassified FTD family based on autopsy (Majounie, et al., 2012a). However, the remaining nine expansion carriers of this study presented with a final diagnosis of AD after careful clinical and pathological reevaluation of their diagnoses. Three of these index cases additionally obtained an autopsy confirmed diagnoses of AD (6-001, 10-098 and 11-021). None of the nine confirmed AD cases showed any signs of clinical or pathological FTD. The autopsy-confirmed AD diagnosis of index case 6-001, for which histological slides were available, was independently ascertained by two experienced neuropathologists (see methods). Furthermore, two clinically confirmed AD index cases belonged to AD families with first and/or second-degree relatives affected by AD. Importantly, repeat expansions in these families co-segregated with AD (family 3 and 5, fig. 1). Interestingly, two AD index cases had family members with a diagnosis of ALS (family 3 and 4). Although we have no knowledge on the C9ORF72 repeat status on these family members it is an intriguing observation that suggests additional environmental or genetic factors modulating the phenotypic outcome of expanded repeats.
AD is a genetically heterogeneous disease with the APOE ε4 allele constituting a considerable risk factor for late-onset AD (Corder, et al., 1993). However, the APOE ε4 allele was not overrepresented in C9ORF72 expansion carriers of this study. Five expansion carriers were also carrier for the APOE ε4 allele and four were not (Table 1, Fig. 1). Among the three C9ORF72 expansion carriers with a pathologically confirmed diagnosis of AD, two carried APOE ε4 risk alleles (ε3/ε4 and ε4/ε4 genotypes), but a third one did not (ε3/ε3). Further, mean disease onsets were not significantly different between APOE ε4 carriers and non-carriers.
In C9ORF72 FTD/ALS, average age of disease onset is in the fifties, but a very wide age range has been reported from 32 to 78 (Hodges, 2012). The condition is highly penetrant with 50% of carriers affected at 58 years of age and near 100% by age 80 (Majounie, et al., 2012b). The nine C9ORF72 carriers affected by AD in this study presented with a seemingly higher average disease onset of 77.8 years, although their number is not large enough for an adequately powered comparison. All of them had an onset of AD above 60 years of age, thus representing cases of late-onset AD (Corder, et al., 1993). Intriguingly, this higher disease onset observed in our index cases is consistent with findings on c9FTD/ALS cases of a previous pathological study where the subgroup of FTLD-TDP patients characterized by Mackenzie Type 1 TDP-43 pathology with neuronal intranuclear inclusions and hippocampal sclerosis presented with a similarly higher disease onset, and with some of them being thought to have an Alzheimer-type dementia (Murray, et al., 2011).
Although we definitely can exclude phenocopies for the three index AD cases that obtained an autopsy-confirmed diagnoses of AD, defined by the presence of extracellular neuritic plaques and intracellular neurofibrillary tangles (Heutink, 2000) which are absent in classic FTD, we cannot completely rule out concomitant AD causally unrelated to C9ORF72 expansions. However, we feel that this scenario is unlikely, given the nearly complete penetrance of C9ORF72 FTD/ALS at the age of our index patients together with the absence of clinical or neuropathological signs of FTD in nine AD index cases, co-segregation of expansions in two AD families and the significant case-control association between large C9ORF72 expansions and reevaluated AD cases compared to elderly neurologically healthy controls.
In conclusion, our data support the notion that large C9ORF72 expansions underlie a spectrum of neurodegenerative phenotypes, likely including AD, besides well-established FTD and ALS. As shown in the results part, we detected large C9ORF72 expansion carriers with a final diagnosis of AD at a rate of 0.76%. Although this portion seems small, it actually is of importance, since with the exception of rare mutations in the established early-onset AD genes (APP, PSEN1 and PSEN2) (Cruchaga, et al., 2012) monogenic disease genes have not yet been described for late-onset AD. Moreover, C9ORF72 expansions in AD at the observed rate would account for total AD cases at a similar rate as the three yet described AD genes combined lead to monogenic forms of AD (Brouwers, et al., 2008). However, further studies of even larger AD cohorts, preferably with neuropathologically confirmed diagnoses and segregation patterns in families are warranted to firmly establish the contribution of large C9ORF72 expansions to AD pathology. The broad phenotypic spectrum associated with C9ORF72 expansions offers an opportunity to further decipher the underlying molecular pathology of neurodegenerative disease.
4.2 C9ORF72 repeat count distributions vary between ethnicities of African and European origin
The prevalence of the C9ORF72 repeat expansion has been established in large studies of ALS/FTD, but is mainly based on samples of European descent. However, large expansion carriers have been reported in African Americans, Hispanic Americans and Japanese ALS patients (Ishiura, et al., 2012, Majounie, et al., 2012b). To our knowledge, our study represents the largest screen for C9ORF72 expansions in AD case-control cohorts in African American samples. We did not identify any carriers of large C9ORF72 expansions in African Americans. Although our study reached a significant underrepresentation of C9ORF72 expansion carriers in African American compared to Caucasian samples, this analysis was not statistically highly powered due to the smaller number of African American samples compared to those from European descent (N=292 vs. N=1,184) and low C9ORF72 expansion frequency in AD. However, our results suggest a lower frequency of large C9ORF72 expansion carriers in African Americans which is consistent with the proposed Northern European origin of the expansion haplotype. The large majority of expansion carriers, including non-Europeans, share the same ancestral founder haplotype (Ishiura, et al., 2012, Majounie, et al., 2012b, Mok, et al., 2012). In Caucasians, this haplotype is highly tagged by the A-allele of the surrogate marker, SNP rs3849942 (DeJesus-Hernandez, et al., 2011). However, in contrast to some African populations, the A-allele frequency of rs3849942 is only slightly lower in African Americans (18–20%) compared to 23.5% in Caucasians. Thus, C9ORF72 expansion carrier rates could potentially be similar, though not cohesively, due to varying linkage disequilibrium structures between both ethnicities (Gabriel, et al., 2002).
Interestingly, in the normal range of repeat copy variation (≥4–23 copies), we observed significant differences in mean C9ORF72 repeat copies and repeat copy distributions between samples of African American and European descent. Mean repeat copy numbers were significantly higher in individuals of European descent. The ethnic differences in repeat copy number distributions were most pronounced and reached significance already in case and control separate analyses with the lowest p-value in the combined analysis (p=1.5E-07, Table 3). The frequency distribution of large C9ORF72 expansions throughout European populations is in concordance with a single one-off expansion that occurred in Northern Europe about 1,500 years ago (Majounie, et al., 2012b, Mok, et al., 2012). Alternatively, it has been speculated that the expansion haplotype could be more prone to repeat expansions. In fact, the later has been proposed based on a study reporting a highly significant mean C9ORF72 repeat copy number difference in Caucasian controls being homozygous for the A-allele of rs3849942 (mean: ~8 repeat copies) in relation to controls being homozygous for the alternative G-allele (mean: ~3 repeat copies at population average) (DeJesus-Hernandez, et al., 2011). The on average already higher C9ORF72 repeat copy numbers on the ‘expansion’ haplotype might be at higher risk for further expansion during meiosis, a phenomenon referred to as genetic anticipation that has been reported for C9ORF72 expansions at least in the pathogenic range (Hodges, 2012). In this respect, the here reported ethnical differences in C9ORF72 repeat copy number distributions in the normal range of variation becomes important, especially since these differences are strongest in repeat copy number classes ranging from 3 to 10 repeat copies (fig. 3). Thus, the rather lower abundance of large C9ORF72 expansions in African Americans compared to Caucasians could be a consequence of different haplotype distributions between these ethnicities, including haplotypes at different risk for spontaneous expansion, consistent with their genetic history and a founder event in Northern Europe. Differences in the prevalence of C9ORF72 expansion carriers between ethnicities have the potential to increase our knowledge on the underlying mechanisms and prevent ethnically driven health disparities.
Supplementary Material
Acknowledgments
We thank Drs. Carol Petito (University of Miami) and Christine Hulette (Duke University) for their neuropathological expertise. We are grateful to the families and staffs who participated in this study. This work was supported by National Institute of Health grants R01 AG027944 (MPV), R01 AG019085, R01 AG028786-02 (MPV), RC2AG036528 (MPV; JLH), 5R01 AG028786-04 (MPV, JLH, GByrd), the Alzheimer’s Association grant IIRG09133827 (MPV), and the American Health Assistance Foundation grant A2011048 (MPV, SZ). Michael Benatar’s funding support relevant to this work are the MDA (Muscular Dystrophy Association), the ALS Association and the ALS Recovery Fund. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
5.1 Disclosure Statement
The authors of this manuscript have no potential conflicts of interests related to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Boeve BF, Boylan KB, Graff-Radford NR, DeJesus-Hernandez M, Knopman DS, Pedraza O, Vemuri P, Jones D, Lowe V, Murray ME, Dickson DW, Josephs KA, Rush BK, Machulda MM, Fields JA, Ferman TJ, Baker M, Rutherford NJ, Adamson J, Wszolek ZK, Adeli A, Savica R, Boot B, Kuntz KM, Gavrilova R, Reeves A, Whitwell J, Kantarci K, Jack CR, Jr, Parisi JE, Lucas JA, Petersen RC, Rademakers R. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain: a journal of neurology. 2012;135(Pt 3):765–83. doi: 10.1093/brain/aws004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwers N, Sleegers K, Van Broeckhoven C. Molecular genetics of Alzheimer’s disease: an update. Annals of Medicine. 2008;40(8):562–83. doi: 10.1080/07853890802186905. [DOI] [PubMed] [Google Scholar]
- Chio A, Borghero G, Restagno G, Mora G, Drepper C, Traynor BJ, Sendtner M, Brunetti M, Ossola I, Calvo A, Pugliatti M, Sotgiu MA, Murru MR, Marrosu MG, Marrosu F, Marinou K, Mandrioli J, Sola P, Caponnetto C, Mancardi G, Mandich P, La Bella V, Spataro R, Conte A, Monsurro MR, Tedeschi G, Pisano F, Bartolomei I, Salvi F, Lauria Pinter G, Simone I, Logroscino G, Gambardella A, Quattrone A, Lunetta C, Volanti P, Zollino M, Penco S, Battistini S, Renton AE, Majounie E, Abramzon Y, Conforti FL, Giannini F, Corbo M, Sabatelli M consortium I. Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain: a journal of neurology. 2012;135(Pt 3):784–93. doi: 10.1093/brain/awr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Knock J, Hewitt C, Highley JR, Brockington A, Milano A, Man S, Martindale J, Hartley J, Walsh T, Gelsthorpe C, Baxter L, Forster G, Fox M, Bury J, Mok K, McDermott CJ, Traynor BJ, Kirby J, Wharton SB, Ince PG, Hardy J, Shaw PJ. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain: a journal of neurology. 2012;135(Pt 3):751–64. doi: 10.1093/brain/awr365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science (New York, NY) 1993;261(5123):921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Cruchaga C, Haller G, Chakraverty S, Mayo K, Vallania FL, Mitra RD, Faber K, Williamson J, Bird T, Diaz-Arrastia R, Foroud TM, Boeve BF, Graff-Radford NR, St Jean P, Lawson M, Ehm MG, Mayeux R, Goate AM. Rare variants in APP, PSEN1 and PSEN2 increase risk for AD in late-onset Alzheimer’s disease families. PloS one. 2012;7(2):e31039. doi: 10.1371/journal.pone.0031039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Theuns J, Van Broeckhoven C. Locus-specific mutation databases for neurodegenerative brain diseases. Human mutation (Journal Article) 2012 doi: 10.1002/humu.22117; 10.1002/humu.22117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome. 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D. The structure of haplotype blocks in the human genome. Science. 2002;296(5576):2225–9. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- Goedert M, Ghetti B, Spillantini MG. Frontotemporal dementia: implications for understanding Alzheimer disease. Cold Spring Harbor perspectives in medicine. 2012;2(2):a006254. doi: 10.1101/cshperspect.a006254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heutink P. Untangling tau-related dementia. Human molecular genetics. 2000;9(6):979–86. doi: 10.1093/hmg/9.6.979. [DOI] [PubMed] [Google Scholar]
- Hodges J. Familial frontotemporal dementia and amyotrophic lateral sclerosis associated with the C9ORF72 hexanucleotide repeat. Brain: a journal of neurology. 2012;135(Pt 3):652–5. doi: 10.1093/brain/aws033. [DOI] [PubMed] [Google Scholar]
- Ishiura H, Takahashi Y, Mitsui J, Yoshida S, Kihira T, Kokubo Y, Kuzuhara S, Ranum LP, Tamaoki T, Ichikawa Y, Date H, Goto J, Tsuji S. C9ORF72 Repeat Expansion in Amyotrophic Lateral Sclerosis in the Kii Peninsula of JapanC9ORF72 Repeat Expansion in ALS. Archives of Neurology (Journal Article) 2012:1–5. doi: 10.1001/archneurol.2012.1219. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, Habu T, Liu W, Okuda H, Koizumi A. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. American Journal of Human Genetics. 2011;89(1):121–30. doi: 10.1016/j.ajhg.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luty AA, Kwok JB, Thompson EM, Blumbergs P, Brooks WS, Loy CT, Dobson-Stone C, Panegyres PK, Hecker J, Nicholson GA, Halliday GM, Schofield PR. Pedigree with frontotemporal lobar degeneration--motor neuron disease and Tar DNA binding protein-43 positive neuropathology: genetic linkage to chromosome 9. BMC neurology 8(Journal Article) 2008:32. doi: 10.1186/1471-2377-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney CJ, Beck J, Rohrer JD, Lashley T, Mok K, Shakespeare T, Yeatman T, Warrington EK, Schott JM, Fox NC, Rossor MN, Hardy J, Collinge J, Revesz T, Mead S, Warren JD. Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features. Brain: a journal of neurology. 2012;135(Pt 3):736–50. doi: 10.1093/brain/awr361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majounie E, Abramzon Y, Renton AE, Perry R, Bassett SS, Pletnikova O, Troncoso JC, Hardy J, Singleton AB, Traynor BJ. Repeat expansion in C9ORF72 in Alzheimer’s disease. The New England journal of medicine. 2012a;366(3):283–4. doi: 10.1056/NEJMc1113592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chio A, Restagno G, Nicolaou N, Simon-Sanchez J, van Swieten JC, Abramzon Y, Johnson JO, Sendtner M, Pamphlett R, Orrell RW, Mead S, Sidle KC, Houlden H, Rohrer JD, Morrison KE, Pall H, Talbot K, Ansorge O, Chromosome ALSFTDC, Consortium I, Hernandez DG, Arepalli S, Sabatelli M, Mora G, Corbo M, Giannini F, Calvo A, Englund E, Borghero G, Floris GL, Remes AM, Laaksovirta H, McCluskey L, Trojanowski JQ, Van Deerlin VM, Schellenberg GD, Nalls MA, Drory VE, Lu CS, Yeh TH, Ishiura H, Takahashi Y, Tsuji S, Le Ber I, Brice A, Drepper C, Williams N, Kirby J, Shaw P, Hardy J, Tienari PJ, Heutink P, Morris HR, Pickering-Brown S, Traynor BJ French research network on FFA. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet neurology. 2012b;11(4):323–30. doi: 10.1016/s1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34(7):939–44. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Mok K, Traynor BJ, Schymick J, Tienari PJ, Laaksovirta H, Peuralinna T, Myllykangas L, Chio A, Shatunov A, Boeve BF, Boxer AL, DeJesus-Hernandez M, Mackenzie IR, Waite A, Williams N, Morris HR, Simon-Sanchez J, van Swieten JC, Heutink P, Restagno G, Mora G, Morrison KE, Shaw PJ, Rollinson PS, Al-Chalabi A, Rademakers R, Pickering-Brown S, Orrell RW, Nalls MA, Hardy J. Chromosome 9 ALS and FTD locus is probably derived from a single founder. Neurobiology of aging. 2012;33(1):209.e3–e8. doi: 10.1016/j.neurobiolaging.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Al-Chalabi A, Andersen PM, Hosler B, Sapp P, Englund E, Mitchell JE, Habgood JJ, de Belleroche J, Xi J, Jongjaroenprasert W, Horvitz HR, Gunnarsson LG, Brown RH., Jr A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology. 2006;66(6):839–44. doi: 10.1212/01.wnl.0000200048.53766.b4. [DOI] [PubMed] [Google Scholar]
- Murray ME, DeJesus-Hernandez M, Rutherford NJ, Baker M, Duara R, Graff-Radford NR, Wszolek ZK, Ferman TJ, Josephs KA, Boylan KB, Rademakers R, Dickson DW. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta Neuropathologica. 2011;122(6):673–90. doi: 10.1007/s00401-011-0907-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj AC, Beecham GW, Martin ER, Gallins PJ, Powell EH, Konidari I, Whitehead PL, Cai G, Haroutunian V, Scott WK, Vance JM, Slifer MA, Gwirtsman HE, Gilbert JR, Haines JL, Buxbaum JD, Pericak-Vance MA. Dementia revealed: novel chromosome 6 locus for late-onset Alzheimer disease provides genetic evidence for folate-pathway abnormalities. PLoS genetics. 2010;6(9):e1001130. doi: 10.1371/journal.pgen.1001130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, St George-Hyslop P, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, DeCarli C, DeKosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Parisi JE, Perl DP, Peskind E, Petersen RC, Poon WW, Quinn JF, Rajbhandary RA, Raskind M, Reisberg B, Ringman JM, Roberson ED, Rosenberg RN, Sano M, Schneider LS, Seeley W, Shelanski ML, Slifer MA, Smith CD, Sonnen JA, Spina S, Stern RA, Tanzi RE, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Williamson J, Woltjer RL, Cantwell LB, Dombroski BA, Beekly D, Lunetta KL, Martin ER, Kamboh MI, Saykin AJ, Reiman EM, Bennett DA, Morris JC, Montine TJ, Goate AM, Blacker D, Tsuang DW, Hakonarson H, Kukull WA, Foroud TM, Haines JL, Mayeux R, Pericak-Vance MA, Farrer LA, Schellenberg GD. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nature genetics. 2011;43(5):436–41. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademakers R, Dermaut B, Peeters K, Cruts M, Heutink P, Goate A, Van Broeckhoven C. Tau (MAPT) mutation Arg406Trp presenting clinically with Alzheimer disease does not share a common founder in Western Europe. Human mutation. 2003;22(5):409–11. doi: 10.1002/humu.10269. [DOI] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M, Consortium I, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–68. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollinson S, Halliwell N, Young K, Callister JB, Toulson G, Gibbons L, Davidson YS, Robinson AC, Gerhard A, Richardson A, Neary D, Snowden J, Mann DM, Pickering-Brown SM. Analysis of the hexanucleotide repeat in C9ORF72 in Alzheimer’s disease. Neurobiology of aging (Journal Article) 2012 doi: 10.1016/j.neurobiolaging.2012.01.109. [DOI] [PubMed] [Google Scholar]
- Simon-Sanchez J, Dopper EG, Cohn-Hokke PE, Hukema RK, Nicolaou N, Seelaar H, de Graaf JR, de Koning I, van Schoor NM, Deeg DJ, Smits M, Raaphorst J, van den Berg LH, Schelhaas HJ, De Die-Smulders CE, Majoor-Krakauer D, Rozemuller AJ, Willemsen R, Pijnenburg YA, Heutink P, van Swieten JC. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain: a journal of neurology. 2012;135(Pt 3):723–35. doi: 10.1093/brain/awr353. [DOI] [PubMed] [Google Scholar]
- Snowden JS, Rollinson S, Thompson JC, Harris JM, Stopford CL, Richardson AM, Jones M, Gerhard A, Davidson YS, Robinson A, Gibbons L, Hu Q, DuPlessis D, Neary D, Mann DM, Pickering-Brown SM. Distinct clinical and pathological characteristics of frontotemporal dementia associated with C9ORF72 mutations. Brain: a journal of neurology. 2012;135(Pt 3):693–708. doi: 10.1093/brain/awr355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP, Hardy J, Fischbeck KH. Toxic proteins in neurodegenerative disease. Science (New York, NY) 2002;296(5575):1991–5. doi: 10.1126/science.1067122. [DOI] [PubMed] [Google Scholar]
- Vance C, Al-Chalabi A, Ruddy D, Smith BN, Hu X, Sreedharan J, Siddique T, Schelhaas HJ, Kusters B, Troost D, Baas F, de Jong V, Shaw CE. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain: a journal of neurology. 2006;129(Pt 4):868–76. doi: 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.