

Abstract
DNA transposon-based vectors have emerged as new potential delivery tools in therapeutic gene transfer. Such vectors are now showing promise in hematopoietic stem cells and primary human T cells, and clinical trials with transposon-engineered cells are on the way. However, the use of plasmid DNA as a carrier of the vector raises safety concerns due to the undesirable administration of bacterial sequences. To optimize vectors based on the Sleeping Beauty (SB) DNA transposon for clinical use, we examine here SB transposition from DNA minicircles (MCs) devoid of the bacterial plasmid backbone. Potent DNA transposition, directed by the hyperactive SB100X transposase, is demonstrated from MC donors, and the stable transfection rate is significantly enhanced by expressing the SB100X transposase from MCs. The stable transfection rate is inversely related to the size of circular donor, suggesting that a MC-based SB transposition system benefits primarily from an increased cellular uptake and/or enhanced expression which can be observed with DNA MCs. DNA transposon and transposase MCs are easily produced, are favorable in size, do not carry irrelevant DNA, and are robust substrates for DNA transposition. In accordance, DNA MCs should become a standard source of DNA transposons not only in therapeutic settings but also in the daily use of the SB system.
Keywords: antibiotic resistance marker, DNA transposition, minicircle, plasmid backbone, Sleeping Beauty
Introduction
The Sleeping Beauty (SB) DNA transposon is a promising nonviral integrating vector system for therapeutic gene transfer. The combination of the ability to stably integrate transgene cassettes into the genome of mammalian cells and an easy, cost-effective production, characteristic for a plasmid-based vector system, has made the SB transposon system an attractive and widely studied gene delivery tool for gene therapy applications (reviewed in ref. 1,2).
Since its reconstruction in 1997 from ancient Tc1/mariner elements resident in the salmonid fish genome,3 the SB vector has been subjected to several modifications aimed at improving the efficacy and safety of the system. Modifications of the inverted repeat sequences in the transposon have resulted in an improved SB transposon, with a more than threefold increased transposition efficiency compared with the original transposon.4 Mutational analyses of the SB transposase sequence have led to several hyperactive transposase mutants with increased transposition activities,5,6,7 and a high-throughput polymerase chain reaction (PCR)-based DNA-shuffling strategy facilitated the development of the hyperactive transposase SB100X, which has an estimated 100-fold enhanced efficiency compared with the original SB10 transposase.8 To remove the risk of inadvertent integration of the transposase gene into the genome and remobilization of inserted transposons, transposition experiments using RNA as a source of SB transposase have been carried out successfully both in vitro and in vivo.9,10,11,12 In addition, attempts to direct transposon integration into “safe” chromosomal regions have led to the analysis of DNA-binding fusion proteins designed to tether the SB transposase to specific sequences in the genome,13 and proof-of-principle of SB-mediated targeted chromosomal integration in human cells has been achieved.14 As of today, long-term gene expression after SB-mediated delivery has been observed for a wide range of primary cells (including CD34+,8,15,16,17 primary T,11,18,19,20 and human embryonic stem cells12,21) and tissues (including liver,22,23,24 lung,25,26,27,28,29 and neuronal tissue30,31). Recently, the first clinical trial using SB-directed gene insertion has been initiated for treatment of patients with B-lymphoid malignancies with adoptive immunotherapy.32
One of the desirable properties of the SB vector system is consistent excision from donor DNA and genomic integration of defined transposon sequences, leaving out the plasmid bacterial backbone. Before the transposition event can take place, however, plasmid DNA has to be delivered to the nucleus of treated cells, implying that undesirable bacterial sequences, such as origin of replication and antibiotic resistance gene, are administered into patient cells. In patients, horizontal transfer of antibiotic resistance genes to pathogenic bacteria would be possible in theory, raising some safety concerns related to clinical use of plasmid DNA.33 In addition, plasmid bacterial sequences contain unmethylated CpG motifs, which may trigger the activation of inflammatory responses upon plasmid administration,34,35 making bacterial backbone sequences a possible safety concern for plasmid-based vector systems. Adding to these potential drawbacks, antibiotic resistance genes are relatively large and are likely, therefore, to have a negative effect on the plasmid transfection rate.
Minicircle (MC) DNA vectors are small, supercoiled molecules devoid of plasmid bacterial sequences. They are believed to be superior to standard plasmid DNA as gene delivery vehicles due to observations of more persistent and increased transgene expression both in vitro and in vivo.36,37,38,39,40,41,42 MC DNA molecules can be obtained through a phage integrase ΦC31-mediated site-specific recombination between attB and attP sites, leading to excision of the intervening bacterial backbone sequence, which is then digested by the I-SceI endonuclease and degraded. Production of MC DNA has previously involved costly and labor-intensive techniques, but with the construction, by Chen et al., of a genetically modified E. coli strain that stably expresses a set of inducible ΦC31 integrase and I-SceI endonuclease enzymes, generation of MC DNA is now comparable with plasmid production protocols.43
To improve the safety profile and the efficacy of the SB system, we combined in this study the SB transposon vector technology with the MC DNA technology. We created a MC-based SB transposon donor vector, and measured efficient transposition from the MC donor in a range of different cell lines in vitro. In HeLa cells, stable transfection rates obtained by mobilizing transposons from the MC donor at restricted transposase dosages were improved relative to the standard plasmid vector. To analyze SB-mediated transposition by a MC-based system by which both the transposon and the transposase expression cassette were delivered in the context of MC DNA, we also constructed a MC-based SB100X transposase vector, and found increased stable transfection rates in transfections with the MC SB100X vector compared to the corresponding plasmid vector. Our findings suggest that MC-based SB transposon vectors represent an efficient alternative to plasmid DNA as donors of SB transposons and as carriers of SB transposase expression cassettes.
Results
Construction and production of SB transposon MCs
To study SB DNA transposition from MC DNA, we generated the plasmids pMC.SBT/PGK-puro and pMC.SBT/CMV-eGFP, which contain a PGK-puro and a CMV-eGFP cassette, respectively, situated in a SB transposon flanked by attB and attP recombination sites (Figure 1a). To allow the left inverted repeat and right inverted repeat to be separated in the MC context, we inserted a 193-bp fragment, taken from the eGFP gene, after the right inverted repeat of the SBT/PGK-puro transposon and a 225-bp fragment, taken from the β-gal gene, after the right inverted repeat of the SBT/CMV-eGFP transposon. We aimed at an outer distance of approximately 300 bp between the transposon ends in the transposon MC constructs, as this distance has previously been shown to be favorable for mobilizing the SB transposon from circular DNA substrates.5 The pMC.SBT/PGK-puro and pMC.SBT/CMV-eGFP plasmids were then transformed into the E. coli strain ZYCY10P3S2T, and MC DNA was subsequently produced by overnight growth of a transformed colony, followed by incubation with a MC induction mix and purification on a plasmid purification column. This procedure resulted in the production of MC DNA without plasmid contamination as verified by restriction digestion analysis (Figure 1b).
Figure 1.
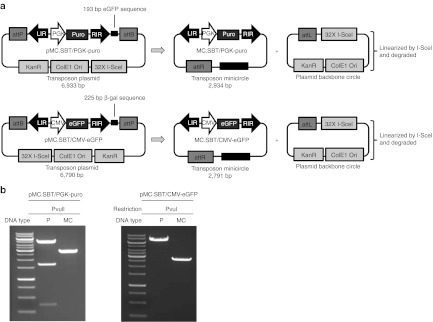
Construction of SB transposon MC vectors. (a) Schematic representation of MC transposon construction. The MC-producing plasmids, pMC.SBT/PGK-puro and pMC.SBT/CMV-eGFP, contain a Sleeping Beauty DNA transposon flanked by attB and attP recombination sites. Upon ΦC31-mediated recombination, plasmid backbone sequences are excised leading to formation of a transposon MC and a plasmid backbone circle. The backbone circle is subsequently linearized by I-SceI restriction and degraded. (b) Restriction digestion analysis of plasmid and MC DNA. Plasmid or MC DNA of 1 µg was digested with PvuII or PvuI endonuclease and analyzed by gel electrophoresis on a 1% agarose gel. attB, bacterial attachment site; attL, left hybrid sequence; attP, phage attachment site; attR, right hybrid sequence; β-gal, β-galactosidase; CMV, cytomegalovirus promoter; ColE1 Ori, E. coli origin of replication; eGFP, enhanced green fluorescent protein; I-SceI, I-SceI restriction site; KanR, kanamycin resistance gene; LIR, left inverted repeat; MC, minicircle; P, plasmid; PGK, phosphoglycerate kinase promoter; puro, puromycin resistance gene; RIR, right inverted repeat.
Efficient SB-mediated transposition from MC vectors
To examine the rate of stably transfecting cells using the MC transposon donor, we made a colony-forming transposition assay in HeLa cells that have been used conventionally in studies of SB efficiency. MC DNA of 380 ng or pMC.SBT/PGK-puro plasmid DNA of 900 ng (corresponding to equal molar amounts) were cotransfected into HeLa cells together with 100 ng of a helper plasmid expressing either the inactive transposase mSB, the original transposase SB10, or the hyperactive transposase SB100X. Transposition efficiencies were determined based on number of resistant colonies obtained after puromycin selection. Efficient transposition from the MC donor was observed with both the SB10 and SB100X transposase (Figure 2a). Although there was no statistical difference between plasmid and MC transposition, a tendency of increased transposition from the MC donor was observed for SB10 (P = 0.05). The SB100X gave rise to an approximately fivefold increase in transposition compared with SB10, which is in agreement with results from previous HeLa transfection experiments using relatively high transposon and transposase amounts.8 Southern blot analysis of the puromycin-resistant clones showed that both types of transposon donors created a mean of approximately seven insertions per clone (Supplementary Figure S1). For the plasmid donor, we identified from 2 to 13 insertions per clone (total of 8 clones), whereas the number of insertions per clone ranged from 3 to 12 with the MC donor (total of 13 clones). Notably, the far majority of these insertions were transposon insertions, although random insertion of plasmid and MC DNA could also be detected in some of the clones.
Figure 2.

Efficient transposition of SB transposon from MC donor. (a) Transposition activity of plasmid and MC donor in HeLa cells. MC of 380 ng or pMC.SBT/PGK-puro plasmid DNA of 900 ng was cotransfected together with 100 ng of pCMV-mSB, pCMV-SB10, or pCMV-SB100X plasmid into HeLa cells. The pcDNA3.1D/V5.TOPO plasmid was included as irrelevant DNA to obtain a total amount of 1,000 ng of DNA in each transfection. Transfected cells were selected with puromycin for 10 days, and resistant colonies were counted as a measure of transposition activity. Mean ± SEM values are shown (N = 3). (b) Detection of MC excision products. MC of 380 ng or pMC.SBT/PGK-puro plasmid DNA of 900 ng was cotransfected together with 100 ng of pCMV-mSB or pCMV-SB100X plasmid into HeLa cells. Irrelevant DNA (pcDNA3.1D/V5.TOPO) was included to obtain a total amount of 1,000 ng of DNA in each transfection. Low-molecular weight DNA was extracted 2 days after transfection, and 50 ng of DNA from each transfection was used in a PCR analysis with a primer set specific for MC excision products (top) or a primer set specific for MC and plasmid excision products (bottom). (c) Transposition activity of plasmid and MC donor in RPE, F9, and HaCaT cells. MC of 210 ng or pMC.SBT/PGK-puro plasmid DNA of 500 ng was cotransfected together with 50 ng of pCMV-mSB or pCMV-SB100X plasmid into RPE, F9, and HaCaT cells. The pcDNA3.1D/V5.TOPO plasmid was included as irrelevant DNA, and a total amount of 1,000 ng of DNA was used in each transfection. Colony numbers were obtained as described in Figure 2a. Mean ± SEM values are shown (N = 3). attR, right hybrid sequence; CMV, cytomegalovirus promoter; MC, minicircle; PCR, polymerase chain reaction; PGK, phosphoglycerate kinase promoter; RPE, retina pigment epithelium; SB, Sleeping Beauty.
We also evaluated transposition activities by detecting excision circles, which are plasmid, or MC, backbone DNA molecules formed after transposon excision and cellular repair of the excision site. As the outer length between the transposon inverted repeats was much smaller in the MC donor compared with the plasmid donor (4,325 bp compared with 327 bp, respectively), we were interested in studying if excision circles were also formed after transposon excision from MC DNA. MC or the parental pMC.SBT/PGK-puro plasmid was cotransfected with helper plasmids pCMV-mSB or pCMV-SB100X into HeLa cells, and low-molecular weight DNA was extracted 2 days after transfection. A PCR specific for the repaired excision site in MC excision circles resulted in a distinct 259-bp PCR band that was detected in the DNA sample originating from MC and pCMV-SB100X cotransfections (Figure 2b, top). This showed that excision site repair leading to robust formation and purification of a 327-bp circle took place after transposition from the MC donor. In addition, we used a primer set that would amplify excision sites from both plasmid and MC donors. The expected 128-bp PCR band was observed for both types of substrates (Figure 2b, bottom). Approximately, equal band intensities suggested that transposon excision frequencies from plasmid and MC DNA were in the same range, although potential differences in plasmid and MC excision circle stability and repaired circle extraction could have some impact on a direct comparison.
To measure the stable transfection rate of the transposon MC vector in other cell types, we performed colony-forming assays in retina pigment epithelium (RPE) cells, embryonic carcinoma cells (F9), and human keratinocytes (HaCaT). Efficient transposition from the transposon MC vector was observed in all three cell types, although statistical difference between plasmid and MC transposition could not be detected (Figure 2c). These data collectively demonstrated that MC transposon donors under the experimental conditions used mediated levels of DNA transposition that were comparable with those of conventional plasmid donors.
Increased transposition efficiencies from MC DNA with low transposase dosages in HeLa cells
The transposon-to-transposase plasmid ratio is known to affect SB transposition efficiency.5 Too high transposase concentrations, for instance, may lead to overproduction inhibition that reduces the transposition activity through unknown mechanisms. To investigate transposition from the MC donor at low transposase amounts and at different transposon-to-transposase ratios, we lowered the amount of SB100X-encoding plasmid to 20 ng and measured the stable transfection rate in HeLa cells transfected with increasing amounts of transposon donor DNA (ranging from 50 to 1,500 ng plasmid DNA and from 21 to 635 ng MC DNA). Irrelevant plasmid DNA was included in each transfection to ensure that the cells were transfected with equal amounts of DNA. As shown in Figure 3a, reducing the amount of SB100X-encoding plasmid resulted in a considerable relative enhancement of transposition from the MC donor compared with the plasmid vector (P < 0.05 for the four lowest transposon concentrations; P = 0.09 for transfections with 1,500/635 ng donor DNA). These findings demonstrated improved performance of MC DNA as a transposon donor in cells with restricted levels of transposase.
Figure 3.

Transposition activity of different SB transposon donors in HeLa cells. Mean ± SEM values are shown (N = 3) (a) Increased transposition activity of MC donor compared with plasmid donor at different transposon DNA dosages. pCMV-SB100X plasmid of 20 ng was cotransfected together with five different dosages of MC or pMC.SBT/PGK-puro plasmid DNA into HeLa cells. The pcDNA3.1D/V5.TOPO plasmid was included as irrelevant DNA, and a total amount of 2,000 ng of DNA was used in each transfection. Colony numbers were obtained as described in Figure 2a. (b) Increased transposition activity with decreased transposon vector size. pLV.SBT/PGK-puro plasmid of 132 ng, pMC.SBT/PGK-puro plasmid of 100 ng, pSBT/PGK-puro plasmid of 76 ng or MC.SBT/PGK-puro DNA of 42 ng (corresponding to equal molar amounts) were cotransfected with 20 ng of pCMV-SB100X and pcDNA3.1D/V5.TOPO irrelevant DNA (to a total amount of 1,000 ng) into HeLa cells. Transposition activities are shown as relative numbers, with the transposition activity of the pLV.SBT/PGK-puro plasmid corresponding to 4,167 ± 475 puromycin resistant colonies. (c) Transient expression levels from plasmid and MC DNA in HeLa cells. MC of 411 ng or pMC.SBT/CMV-eGFP plasmid DNA of 1,000 ng was cotransfected together with 1,000 ng pAAV2-siRNA plasmid (encoding the AsRed marker gene) into HeLa cells. The pcDNA3.1D/V5.TOPO plasmid was included as irrelevant DNA to obtain equal DNA amounts in each transfection. eGFP and AsRed expression was measured 2 days after transfection by flow cytometry. Mean ± SEM values are shown (N = 5). (d) Transposition activity of plasmid and MC donor in HeLa cells measured after cell-sorting of transfected cells. MC of 411 ng or pMC.SBT/CMV-eGFP plasmid DNA of 1,000 ng was cotransfected together with 20 ng of pCMV-mSB or pCMV-SB100X plasmid into HeLa cells. The pcDNA3.1D/V5.TOPO plasmid was included as irrelevant DNA to obtain a total amount of 1,500 ng of DNA in each transfection. Two days after transfection, eGFP-positive cells were sorted and cultivated. Eighteen days after transfection, cells were harvested and analyzed by flow cytometry to detect eGFP expression. Mean ± SEM values are shown (N = 3). *Significant difference between plasmid and MC donor (Student's t test, P < 0.05). CMV, cytomegalovirus promoter; eGFP, enhanced green fluorescent protein; MC, minicircle; MFI, median fluorescence intensity; PGK, phosphoglycerate kinase promoter; SB, Sleeping Beauty.
Stable transfection rate is inversely related to size of transposon donor
We hypothesized that the difference in transposition rates from the MC and pMC.SBT/PGK-puro donors was a reflection of the different sizes of these donors. To test this possibility and to ensure that the enhanced transposition rate obtained with the MC donor did not simply reflect that the pMC.SBT/PGK-puro for other reasons was suboptimal as a DNA transposon donor, we compared donors of different sizes, all carrying the SB/PGK-puro transposon. In addition to MC and pMC.SBT/PGK-puro, we included the plasmids, pSBT/PGK-puro (5,282 bp) and pLV.SBT/PGK-puro (9,135 bp), which have previously been shown to serve as efficient transposon donors.44,45 Colony-forming assays showed a significant increase in the stable transfection rate when the SBT/PGK-puro transposon was mobilized from the MC donor compared with all three plasmid donors (P < 0.01; Figure 3b). In addition, an inverse linear relationship between transposition activity and transposon vector size was observed. These findings indicated that the vector efficiency was improved by the enhanced transfection of smaller DNA molecules although it cannot be excluded that higher levels of transposition could result from the smaller outer distance between the transposon ends. Together, our findings indicate that the MC transposon vector is superior to plasmid transposon vector at conditions where cellular uptake of the DNA substrate and the amount of transposase are limiting factors.
The mobilization of transposons from MC DNA benefits from increased MC DNA transfection efficiency
Our initial findings lent support to the notion that MC DNA is not necessarily a better substrate for transposition but benefits primarily from increased rates of transfection due to the lack of irrelevant DNA sequences. To address this aspect further, equal molar amounts of MC.SBT/CMV-eGFP and the pMC.SBT/CMV-eGFP plasmid were transfected into HeLa cells together with a plasmid encoding the AsRed reporter as a transfection control. Levels of AsRed and eGFP expression were measured 2 days after transfection by flow cytometry. Whereas the AsRed level and the percentage of AsRed positive cells were equal in all transfections, a fivefold increase in eGFP median fluorescence intensity (P < 0.0001; Figure 3c, left) and a 1.4-fold increase in percentage of eGFP-positive cells (P < 0.0004; Figure 3c, right) was observed for the MC construct compared with the plasmid construct. Although we cannot formally exclude that MC DNA supported an enhanced level of transgene expression relative to plasmid DNA, this result most likely indicated that the transfection efficiency of MC DNA was increased relative to that of its plasmid counterpart.
To investigate SB transposition from MC DNA under nonselective conditions, we performed a DNA transposition assay based on the mobilization of an eGFP-expressing DNA transposon. Equal molar amounts of pMC.SBT/CMV-eGFP plasmid and MC.SBT/CMV-eGFP (Figure 1a) were transfected into HeLa cells together with pCMV-SB100X or pCMV-mSB. Two days after transfection, eGFP-positive cells were sorted for each transfection by flow cytometry and further passaged for 16 days to allow dilution of episomal DNA. As shown in Figure 3d (left panel), a significant higher percentage of eGFP-positive cells (P < 0.05) was seen with the MC donor compared with the plasmid donor at 2 days after transfection. After sorting and passaging of the cells for 18 days, we measured a marked increase in the percentage of eGFP-positive cells for both plasmid and MC donors in the presence of SB100X. With this setup, the stable transfection rate, as determined by the percentage of eGFP-positive cells, was similar for the two types of donors (P = 0.69; Figure 3d, right). In the presence of mSB, a significant increase in eGFP-positive cells (P < 0.01) was observed for the MC donor compared with the plasmid donor, suggesting that MC episomes were more stable or more stably supported expression of the eGFP gene relative to episomal plasmid DNA. Alternatively, the MC transfections gave rise to random insertion events more frequently than the plasmid transfections, but this remains questionable as the far majority of such insertions would not result in eGFP expression due to expected damage of the CMV-eGFP transgene cassette in the case of random MC integration. Together, our data suggest that the high transposition activity observed with the MC donor at restricted transposase amounts is a result of enhanced transfection of MC DNA due to the smaller size compared with plasmid DNA and that this difference does not necessarily reflect an increased rate of transposition per se from the MC donor.
MC DNA is superior to plasmid DNA as transposon donor in comparisons of transfections performed with equal weight units of the two donors
Conventional plasmid transfections suffer from the fact that the bacterial plasmid backbone, all in all a large amount of irrelevant DNA, is loaded into the cells. By excluding irrelevant segments of DNA, it is possible, within a fixed amount of DNA, to increase copy numbers of the transgene expression cassette. To illustrate this advantage, we analyzed the stable transfection rate at restricted transposase amounts in RPE, F9, and HaCaT cells. First, we transfected these cell types with equal molar amounts of pMC.SBT/PGK-puro plasmid (100 ng) or MC.SBT/PGK-puro (42 ng) in the presence of SB100X expressed from 20 ng pCMV-SB100X. A slightly higher number of resistant colonies was obtained with MC DNA compared with plasmid DNA in all three cell types (Figure 4a), although this increase in transposition efficiency was not statistically significant. However, when we transfected equal weight units of MC and plasmid DNA, the MC donor led to a significantly higher transposition activity than the plasmid donor (P < 0.01), with a 1.9-fold, 1.8-fold, and 2.0-fold increase in activity for MC transposon versus plasmid transposon in RPE, F9, and HaCaT cells, respectively (Figure 4b). These data show that at fixed DNA amounts, higher transposition efficiencies can be obtained with MC transposon donors compared with plasmid transposon donors.
Figure 4.

MC transposon donor facilitates enhanced transposition activity compared with plasmid donor at equal weight units of DNA. *Significant difference between plasmid and MC donor (Student's t test, P < 0.05). Mean ± SEM values are shown (N = 3). (a) Transposition activity of plasmid and MC transposon donor in RPE, F9, and HaCaT cells at low transposase dosage. MC of 42 ng or pMC.SBT/PGK-puro plasmid DNA of 100 ng was cotransfected together with 20 ng of pCMV-mSB or pCMV-SB100X plasmid into RPE, F9, and HaCaT cells. The pcDNA3.1D/V5.TOPO plasmid was included as irrelevant DNA to obtain a total amount of 1,000 ng of DNA in each transfection. Colony numbers were obtained as described in Figure 2a. (b) Transposition activity with equal weight units of MC and plasmid DNA in RPE, F9, and HaCaT cells. MC of 100 ng or pMC.SBT/PGK-puro plasmid DNA was cotransfected together with 20 ng of pCMV-mSB or pCMV-SB100X plasmid into RPE, F9, and HaCaT cells. Colony numbers were obtained as described in Figure 2a. CMV, cytomegalovirus promoter; MC, minicircle; PGK, phosphoglycerate kinase promoter; RPE, retina pigment epithelium; SB, Sleeping Beauty
Expression of SB100X from MC DNA leads to enhanced stable transfection rates
Recent examples of successful use of mRNA as a source of SB transposase represent one approach to avoid delivery of irrelevant nucleic acids, such as bacterial backbone DNA sequences, to cells treated by DNA transposition vectors.9,10,11 Expression of the SB transposase from a MC vector would also allow for a SB transposition system that is devoid of bacterial backbone sequences. To obtain an SB vector system in which both transposon donor and transposase helper was contained in MC DNA, we constructed the pMC.CMV-SB100X plasmid, containing a CMV-SB100X cassette (Figure 5a), and produced MC DNA as previously described. Pure MC DNA without plasmid contamination was produced as verified by restriction digestion analysis (Figure 5b). To measure transposition mediated by the SB100X MC vector, we performed a colony-forming assay in HeLa cells using 450 ng of MC.SBT/PGK-puro MC transposon donor and 50 ng pMC.CMV-SB100X plasmid or 17 ng MC.CMV-SB100X MC DNA (corresponding to equal molar amounts). As shown in Figure 5c, more than twofold increase in the number of resistant colonies was obtained with the SB100X MC vector compared with the SB100X plasmid vector (P < 0.05). This finding indicates that a lower amount of transposase vector DNA (one third of the DNA amount) can be used to obtain a high transposition efficiency when using the SB100X MC transposase vector compared with the plasmid vector.
Figure 5.

Enhanced transposition by MC-encoded SB100X in HeLa cells. (a) Schematic representation of the MC-producing plasmid pMC.CMV-SB100X and the resulting SB100X MC transposase vector. (b) Restriction digestion analysis of SB100X plasmid and MC DNA. Plasmid or MC DNA of 1 µg was digested with PstI endonuclease and analyzed by gel electrophoresis on a 1% agarose gel. (c) Increased stable transfection rate with SB100X MC DNA compared with SB100X plasmid DNA in HeLa cells. MC.SBT/PGK-puro MC DNA of 450 ng was cotransfected together with 50 ng of pCMV-mSB, 50 ng of pMC.CMV-SB100X plasmid, or 17 ng of MC.CMV-SB100X MC DNA into HeLa cells. The pcDNA3.1D/V5.TOPO plasmid was included as irrelevant DNA to obtain a total amount of 1,000 ng in each transfection. Colony numbers were obtained as described in Figure 2a. *Significant difference between SB100X plasmid and MC (Student's t test, P < 0.05). Mean ± SEM values are shown (N = 3). attP, phage attachment site; attB, bacterial attachment site; attR, right hybrid sequence; ColE1 Ori, E. coli origin of replication; CMV, cytomegalovirus promoter; I-SceI, I-SceI restriction site; KanR, kanamycin resistance gene; MC, minicircle; P, plasmid; PGK, phosphoglycerate kinase promoter; SB, Sleeping Beauty; SB100X, SB100X transposase gene.
Discussion
Bacterial backbone sequences, situated in plasmid DNA, contain elements that can cause serious safety problems when plasmid-based vectors are used for gene therapy. One such element is the antibiotic resistance gene necessary for bacterial selection. Due to risks such as (i) expression of antibiotic resistance genes in patient cells leading to unintended immune responses and (ii) transmission of antibiotic resistance genes to host bacteria via horizontal gene transfer, regulatory agencies recommend avoiding the use of antibiotic resistance markers.33,46 Furthermore, unmethylated CpG motifs, frequently present in bacterial DNA sequences, have been observed to activate the innate immune system of the host by binding to the Toll-like receptor 9 (TLR9) of antigen-presenting cells.47 The SB DNA transposon delivery system typically consists of two plasmids, a transposon donor and a transposase helper plasmid, respectively. In this study, we have shown that MC DNA devoid of plasmid bacterial backbone sequences can serve as a donor substrate for SB-mediated transposition and as an expression vector for the SB100X transposase. We detected efficient transposition in transfected HeLa, RPE, F9, and HaCaT cells with the MC donor showing a transposition activity that was equal to or higher than the activity of the standard plasmid donor at same molar amounts. We also observed a correlation between transposition efficiency and transposon vector size, suggesting that reducing the size of the transposon vector, as in the MC donor, leads to better overall transposition activities. In continuation hereof, we observed higher transgene expression levels with the MC donor compared with the plasmid donor, shortly after transfection, suggesting that reduced vector size gives rise to increased transfection efficiency, at least in lipid-based transfections. When transfecting the same amount of MC donor and plasmid donor (equal weight units), we saw significant higher transposition efficiencies with the MC donor compared with plasmid donor, reflecting that an increased amount of transposon units per weight of DNA can be obtained with the MC donor. Finally, we observed an increased stable transfection rate when both transposon donor and transposase helper was situated in MC DNA, indicating that robust transposition with low DNA transposase amounts is possible when using an SB100X MC expression vector. In conclusion, we propose that MC-based SB transposon vectors constitute an efficient, bacterial backbone free alternative to plasmid-based transposon vectors for gene therapy applications. On the basis of our findings, we recommend that MC DNA is considered a primary donor for transposon delivery for future clinical use of DNA transposons and for the daily use of the SB system with particular relevance in hard-to-transfect cell types and preclinical animal models.
Materials and Methods
Plasmid construction. The MC producer plasmid pMC.BESPX was kindly provided by Z.-Y. Chen and M. A. Kay (Stanford University School of Medicine, Stanford, CA) and was previously described in ref. 43. The pMC.SBT/PGK-puro plasmid was constructed by ligation of a SBT/PGK-puro.eGFP PCR fragment into SalI-digested pMC.BESPX. The SBT/PGK-puro.eGFP PCR fragment was generated by overlap extension PCR of an 193-bp eGFP PCR fragment amplified from peGFP.N1 (Clontech, Mountain View, CA) and a SBT/PGK-puro PCR fragment amplified from pSBT/PGK-puro (previously referred to as pT/puro).44 pMC.SBT/CMV-eGFP was made by insertion of a PCR-amplified SBT/PGK-eGFP.β-gal cassette into SalI-digested pMC.BESPX followed by a replacement of the PGK promoter with a CMV promoter amplified from peGFP.N1 (AgeI/PacI digestion). The SBT/PGK-eGFP.Δβ-gal cassette was constructed by overlap extension PCR of a 225-bp β-gal PCR fragment amplified from pSBT/β-gal.PGK-puro. β-gal48 and a SBT/PGK-eGFP PCR fragment amplified from pSBT/PGK-eGFP (B. Moldt and J.G. Mikkelsen, unpublished data). To construct pCMV-SB100X, the SB100X gene was PCR-amplified from pCMV(CAT)T7-SB100X8 and inserted into SacII-digested pCMV-SB3 replacing the SB10 gene. The pMC.CMV-SB100X plasmid was made by ligation of a CMV-SB100X PCR fragment, amplified from pCMV-SB100X, into SpeI/SalI-digested pMC.BESPX followed by downstream insertion of an SV40 polyA PCR fragment amplified from pCMV-SB100X. The plasmids pCMV-SB, pCMV-mSB and pLV.SBT/PGK-puro (previously referred to as pLV/puro-PGK-SBT) have been described previously.3,45,49 All produced DNA constructs were verified by restriction digestion and DNA sequencing.
MC production. MC DNA was produced as described by Chen et al.43 Briefly, pMC.SBT/PGK-puro or pMC.SBT/CMV-eGFP plasmid was transformed into the E. coli strain ZYCY10P3S2T, which contains 10 copies of the ΦC31 integrase gene and 3 copies of the I-SceI endonuclease gene, all regulated by the L-arabinose–inducible araCBAD system. Bacteria were grown overnight at 37 °C in TB medium supplemented with 50 µg/ml kanamycin. A MC induction mix comprising 1 volume of LB medium, 0.04 volume of 1 N NaOH, and 0.02% L-arabinose was added, and the bacteria were grown for a further 24 hours at 32 °C. MC DNA was purified, hereafter, on a plasmid purification column (Macherey Nagel, Düren, Germany). Bacterial genomic DNA contamination was removed by treatment with Plasmid-Safe ATP-Dependent DNase (Epicentre Biotechnologies, Madison, WI) according to manufacturer's instructions.
Cell culture and transposition assays. HeLa (human cervical cancer), HaCaT (human keratinocyte), and F9 (murine embryonal teratocarcinoma) cells were maintained in Dulbecco's modified Eagle's medium (Lonza, Basel, Switzerland) supplemented with 10% fetal bovine serum (Lonza), 0.26 mg/ml glutamine, 54 ng/ml penicillin, and 36 ug/ml streptomycin. The F9 cells were grown on gelatin-coated material. Human retinal pigment epithelium (RPE) cells were maintained in culture medium containing 50% Ham's F-12 Nutrient Mixture (Invitrogen, Carlsbad, CA) and 50% Dulbecco's modified Eagle's medium with serum, glutamine, penicillin, and streptomycin, as described above. To measure rates of stable transfection, cells were plated at 1.5 × 105 cells/well in six-well dishes 1 day before cotransfection with (i) a MC or plasmid transposon donor and (ii) a helper plasmid encoding either SB10, SB100X, or mSB. In analysis of the SB100X MC vector, cotransfections were performed with (i) the MC transposon donor MC.SBT/PGK-puro and (ii) the helper plasmid pMC.CMV-SB100X or the helper MC vector MC.CMV-SB100X. The pcDNA3.1D/V5.TOPO plasmid (Invitrogen) was used as irrelevant DNA to obtain equal DNA amounts in each transfection. Transfections were carried out using FuGene-6 (Roche, Basel, Switzerland) according to manufacturer's instructions using 3 µl of reagent per 1 µg of DNA. One day after transfection, cells were split in varying densities and plated in 10-cm dishes. Two days after transfection, selection medium containing 1 µg/ml puromycin (Invitrogen) was added to the cells. After 10 days of selection, colonies of cells were stained with 0.6% methylene blue, air-dried, and counted. Southern blot analysis was performed as previously described.50
Transposon excision assay. HeLa cells were seeded in six-well dishes (1.5 × 105 cells/well) and transfected with 380 ng of MC DNA or 900 ng of pMC.SBT/PGK-puro plasmid DNA together with 100 ng of pCMV-SB100X or pCMV-mSB. The pcDNA3.1D/V5.TOPO plasmid was used as stuffer DNA. Transfections were carried out using FuGene-6 (Roche) according to manufacturer's instructions using 3 µl of reagent per 1 µg of DNA. Low-molecular weight DNA was extracted 2 days after transfection using the QIAprep miniprep kit (CA 91355; Qiagen, Valencia, Spain) according to the manufacturer's instructions, except for a 1-hour incubation period at 55 °C (0.6% sodium dodecyl sulfate and 0.08 mg/ml proteinase K together with buffer P1) instead of the lysis step with buffer P2 after resuspension of the cell pellet. Extracted DNA of 50 ng was used as template for a MC specific excision PCR with primers 5′-GGATCACTCTCGGCATGGAC-3′ and 5′-ACTGGGTGCTCAGGTAGTGGT-3′ and for an excision PCR with primers 5′-GCCCCCATGGGTCGACGTTTA-3′ and 5′-ACTGGGTGCTCAGGTAGTGGT-3′. Thirty cycles (95 °C 30 seconds, 65 °C 30 seconds, and 72 °C 5 seconds) were used in both PCRs.
Transfection efficiency analysis by flow cytometry. HeLa cells were seeded in six-well dishes (1.5 × 105 cells/well) and cotransfected with 411 ng of MC DNA or 1,000 ng of pMC.SBT/CMV-eGFP plasmid DNA together with 1,000 ng of pAAV2-siRNA (Applied Viromics, Fremont, CA,) encoding the red fluorescent marker AsRed. The pcDNA3.1D/V5.TOPO plasmid was used as irrelevant DNA to obtain equal DNA amounts in each transfection. Transfections were carried out using FuGene-6 according to manufacturer's instructions using 3 µl of reagent per 1 µg of DNA. Cells were harvested 2 days after transfection and analyzed by flow cytometry on a BD FACSAria III cell sorter to detect eGFP and AsRed expression.
Transposition assay with cell sorting. HeLa cells were seeded in six-well dishes (1.5 × 105 cells/well) and transfected with 411 ng of MC DNA or 1,000 ng of pMC.SBT/CMV-eGFP plasmid DNA together with 20 ng of pCMV-SB100X or pCMV-mSB. The pcDNA3.1D/V5.TOPO plasmid was used as irrelevant DNA to obtain equal DNA amounts in each transfection. Transfections were carried out using FuGene-6 according to manufacturer's instructions using 3 µl of reagent per 1 µg of DNA. Two days after transfection green fluorescent protein positive cells were sorted by FACS using a BD FACSAria III cell sorter and subsequently cultivated. Cells were harvested 18 days after transfection and analyzed by flow cytometry to detect eGFP expression.
SUPPLEMENTARY MATERIAL Figure S1. Copy number detection by Southern blot analysis of puromycin-resistant HeLa colonies.
Acknowledgments
The authors thank Zhi-Ying Chen and Mark A. Kay for providing the MC production system, and Lajos Mátés, Zoltán Ivics, and Zsuzanna Izsvák for providing the plasmid encoding the SB100X transposase. This work was made possible through support by the Danish National Advanced Technology Foundation the Lundbeck Foundation, the Novo Nordisk Foundation, Aase og Ejnar Danielsens Fond, Agnes og Poul Friis Fond, Grosserer A. V. Lykfeldt og Hustrus Legat, Else og Mogens Wedell-Wedellsborgs Fond, Fonden af 17-12-1981, Kong Christian den Tiendes Fond, and Frits, Georg og Marie Cecilie Gluds Legat. J.G.M. is a member of the Aarhus Research Center for Innate Immunology (ARCII) established through funding by the AU-Ideas program at Aarhus University. The authors declared no conflict of interest.
Supplementary Material
Copy number detection by Southern blot analysis of puromycin-resistant HeLa colonies.
References
- Izsvák Z, Hackett PB, Cooper LJ., and, Ivics Z. Translating Sleeping Beauty transposition into cellular therapies: victories and challenges. Bioessays. 2010;32:756–767. doi: 10.1002/bies.201000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronovich EL, McIvor RS., and, Hackett PB. The Sleeping Beauty transposon system: a non-viral vector for gene therapy. Hum Mol Genet. 2011;20 R1:R14–R20. doi: 10.1093/hmg/ddr140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivics Z, Hackett PB, Plasterk RH., and, Izsvák Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–510. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- Cui Z, Geurts AM, Liu G, Kaufman CD., and, Hackett PB. Structure-function analysis of the inverted terminal repeats of the sleeping beauty transposon. J Mol Biol. 2002;318:1221–1235. doi: 10.1016/s0022-2836(02)00237-1. [DOI] [PubMed] [Google Scholar]
- Zayed H, Izsvák Z, Walisko O., and, Ivics Z. Development of hyperactive sleeping beauty transposon vectors by mutational analysis. Mol Ther. 2004;9:292–304. doi: 10.1016/j.ymthe.2003.11.024. [DOI] [PubMed] [Google Scholar]
- Yant SR, Park J, Huang Y, Mikkelsen JG., and, Kay MA. Mutational analysis of the N-terminal DNA-binding domain of sleeping beauty transposase: critical residues for DNA binding and hyperactivity in mammalian cells. Mol Cell Biol. 2004;24:9239–9247. doi: 10.1128/MCB.24.20.9239-9247.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baus J, Liu L, Heggestad AD, Sanz S., and, Fletcher BS. Hyperactive transposase mutants of the Sleeping Beauty transposon. Mol Ther. 2005;12:1148–1156. doi: 10.1016/j.ymthe.2005.06.484. [DOI] [PubMed] [Google Scholar]
- Mátés L, Chuah MK, Belay E, Jerchow B, Manoj N, Acosta-Sanchez A.et al. (2009Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates Nat Genet 41753–761. [DOI] [PubMed] [Google Scholar]
- Wilber A, Frandsen JL, Geurts JL, Largaespada DA, Hackett PB., and, McIvor RS. RNA as a source of transposase for Sleeping Beauty-mediated gene insertion and expression in somatic cells and tissues. Mol Ther. 2006;13:625–630. doi: 10.1016/j.ymthe.2005.10.014. [DOI] [PubMed] [Google Scholar]
- Wilber A, Wangensteen KJ, Chen Y, Zhuo L, Frandsen JL, Bell JB.et al. (2007Messenger RNA as a source of transposase for sleeping beauty transposon-mediated correction of hereditary tyrosinemia type I Mol Ther 151280–1287. [DOI] [PubMed] [Google Scholar]
- Jin Z, Maiti S, Huls H, Singh H, Olivares S, Mátés L.et al. (2011The hyperactive Sleeping Beauty transposase SB100X improves the genetic modification of T cells to express a chimeric antigen receptor Gene Ther 18849–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilber A, Linehan JL, Tian X, Woll PS, Morris JK, Belur LR.et al. (2007Efficient and stable transgene expression in human embryonic stem cells using transposon-mediated gene transfer Stem Cells 252919–2927. [DOI] [PubMed] [Google Scholar]
- Yant SR, Huang Y, Akache B., and, Kay MA. Site-directed transposon integration in human cells. Nucleic Acids Res. 2007;35:e50. doi: 10.1093/nar/gkm089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivics Z, Katzer A, Stüwe EE, Fiedler D, Knespel S., and, Izsvák Z. Targeted Sleeping Beauty transposition in human cells. Mol Ther. 2007;15:1137–1144. doi: 10.1038/sj.mt.6300169. [DOI] [PubMed] [Google Scholar]
- Hollis RP, Nightingale SJ, Wang X, Pepper KA, Yu XJ, Barsky L.et al. (2006Stable gene transfer to human CD34(+) hematopoietic cells using the Sleeping Beauty transposon Exp Hematol 341333–1343. [DOI] [PubMed] [Google Scholar]
- Sumiyoshi T, Holt NG, Hollis RP, Ge S, Cannon PM, Crooks GM.et al. (2009Stable transgene expression in primitive human CD34+ hematopoietic stem/progenitor cells, using the Sleeping Beauty transposon system Hum Gene Ther 201607–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, Huang X, Nodland SE, Mátés L, Ma L, Izsvák Z.et al. (2009Stable gene transfer and expression in cord blood-derived CD34+ hematopoietic stem and progenitor cells by a hyperactive Sleeping Beauty transposon system Blood 1141319–1330. [DOI] [PubMed] [Google Scholar]
- Singh H, Manuri PR, Olivares S, Dara N, Dawson MJ, Huls H.et al. (2008Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system Cancer Res 682961–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Guo H, Kang J, Choi S, Zhou TC, Tammana S.et al. (2008Sleeping Beauty transposon-mediated engineering of human primary T cells for therapy of CD19+ lymphoid malignancies Mol Ther 16580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Wilber A, McIvor RS., and, Zhou X. DNA transposons for modification of human primary T lymphocytes. Methods Mol Biol. 2009;506:115–126. doi: 10.1007/978-1-59745-409-4_9. [DOI] [PubMed] [Google Scholar]
- Orbán TI, Apáti A, Németh A, Varga N, Krizsik V, Schamberger A.et al. (2009Applying a “double-feature” promoter to identify cardiomyocytes differentiated from human embryonic stem cells following transposon-based gene delivery Stem Cells 271077–1087. [DOI] [PubMed] [Google Scholar]
- Ohlfest JR, Frandsen JL, Fritz S, Lobitz PD, Perkinson SG, Clark KJ.et al. (2005Phenotypic correction and long-term expression of factor VIII in hemophilic mice by immunotolerization and nonviral gene transfer using the Sleeping Beauty transposon system Blood 1052691–2698. [DOI] [PubMed] [Google Scholar]
- Liu L, Mah C., and, Fletcher BS. Sustained FVIII expression and phenotypic correction of hemophilia A in neonatal mice using an endothelial-targeted sleeping beauty transposon. Mol Ther. 2006;13:1006–1015. doi: 10.1016/j.ymthe.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Kren BT, Unger GM, Sjeklocha L, Trossen AA, Korman V, Diethelm-Okita BM.et al. (2009Nanocapsule-delivered Sleeping Beauty mediates therapeutic Factor VIII expression in liver sinusoidal endothelial cells of hemophilia A mice J Clin Invest 1192086–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belur LR, Frandsen JL, Dupuy AJ, Ingbar DH, Largaespada DA, Hackett PB.et al. (2003Gene insertion and long-term expression in lung mediated by the Sleeping Beauty transposon system Mol Ther 8501–507. [DOI] [PubMed] [Google Scholar]
- Liu L, Sanz S, Heggestad AD, Antharam V, Notterpek L., and, Fletcher BS. Endothelial targeting of the Sleeping Beauty transposon within lung. Mol Ther. 2004;10:97–105. doi: 10.1016/j.ymthe.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Liu H, Liu L, Fletcher BS., and, Visner GA. Sleeping Beauty-based gene therapy with indoleamine 2,3-dioxygenase inhibits lung allograft fibrosis. FASEB J. 2006;20:2384–2386. doi: 10.1096/fj.06-6228fje. [DOI] [PubMed] [Google Scholar]
- Belur LR, Podetz-Pedersen K, Frandsen J., and, McIvor RS. Lung-directed gene therapy in mice using the nonviral Sleeping Beauty transposon system. Nat Protoc. 2007;2:3146–3152. doi: 10.1038/nprot.2007.460. [DOI] [PubMed] [Google Scholar]
- Lin EH, Keramidas M, Rome C, Chiu WT, Wu CW, Coll JL.et al. (2011Lifelong reporter gene imaging in the lungs of mice following polyethyleneimine-mediated sleeping-beauty transposon delivery Biomaterials 321978–1985. [DOI] [PubMed] [Google Scholar]
- Wu A, Oh S, Ericson K, Demorest ZL, Vengco I, Gharagozlou S.et al. (2007Transposon-based interferon gamma gene transfer overcomes limitations of episomal plasmid for immunogene therapy of glioblastoma Cancer Gene Ther 14550–560. [DOI] [PubMed] [Google Scholar]
- Ohlfest JR, Lobitz PD, Perkinson SG., and, Largaespada DA. Integration and long-term expression in xenografted human glioblastoma cells using a plasmid-based transposon system. Mol Ther. 2004;10:260–268. doi: 10.1016/j.ymthe.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Williams DA. Sleeping beauty vector system moves toward human trials in the United States. Mol Ther. 2008;16:1515–1516. doi: 10.1038/mt.2008.169. [DOI] [PubMed] [Google Scholar]
- Vandermeulen G, Marie C, Scherman D., and, Préat V. New generation of plasmid backbones devoid of antibiotic resistance marker for gene therapy trials. Mol Ther. 2011;19:1942–1949. doi: 10.1038/mt.2011.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Li S, Pitt BR., and, Huang L. The inhibitory role of CpG immunostimulatory motifs in cationic lipid vector-mediated transgene expression in vivo. Hum Gene Ther. 1999;10:2153–2161. doi: 10.1089/10430349950017149. [DOI] [PubMed] [Google Scholar]
- Yew NS., and, Cheng SH. Reducing the immunostimulatory activity of CpG-containing plasmid DNA vectors for non-viral gene therapy. Expert Opin Drug Deliv. 2004;1:115–125. doi: 10.1517/17425247.1.1.115. [DOI] [PubMed] [Google Scholar]
- Darquet AM, Rangara R, Kreiss P, Schwartz B, Naimi S, Delaère P.et al. (1999Minicircle: an improved DNA molecule for in vitro and in vivo gene transfer Gene Ther 6209–218. [DOI] [PubMed] [Google Scholar]
- Chen ZY, He CY, Ehrhardt A., and, Kay MA. Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo. Mol Ther. 2003;8:495–500. doi: 10.1016/s1525-0016(03)00168-0. [DOI] [PubMed] [Google Scholar]
- Chang CW, Christensen LV, Lee M., and, Kim SW. Efficient expression of vascular endothelial growth factor using minicircle DNA for angiogenic gene therapy. J Control Release. 2008;125:155–163. doi: 10.1016/j.jconrel.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Chen Z, Hu S, Jia F, Li Z, Hoyt G.et al. (2009Novel minicircle vector for gene therapy in murine myocardial infarction Circulation 12011 SupplS230–S237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn MJ, McElmurry RT, Lees CJ, DeFeo AP, Chen ZY, Kay MA.et al. (2011Minicircle DNA-based gene therapy coupled with immune modulation permits long-term expression of a-L-iduronidase in mice with mucopolysaccharidosis type I Mol Ther 19450–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Xiao X, Zhao P, Xue G, Zhu Y, Zhu X.et al. (2006Minicircle-IFNgamma induces antiproliferative and antitumoral effects in human nasopharyngeal carcinoma Clin Cancer Res 124702–4713. [DOI] [PubMed] [Google Scholar]
- Stenler S, Andersson A, Simonson OE, Lundin KE, Chen ZY, Kay MA.et al. (2009Gene transfer to mouse heart and skeletal muscles using a minicircle expressing human vascular endothelial growth factor J Cardiovasc Pharmacol 5318–23. [DOI] [PubMed] [Google Scholar]
- Kay MA, He CY., and, Chen ZY. A robust system for production of minicircle DNA vectors. Nat Biotechnol. 2010;28:1287–1289. doi: 10.1038/nbt.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant SR., and, Kay MA. Nonhomologous-end-joining factors regulate DNA repair fidelity during Sleeping Beauty element transposition in mammalian cells. Mol Cell Biol. 2003;23:8505–8518. doi: 10.1128/MCB.23.23.8505-8518.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldt B, Miskey C, Staunstrup NH, Gogol-Döring A, Bak RO, Sharma N.et al. (2011Comparative genomic integration profiling of Sleeping Beauty transposons mobilized with high efficacy from integrase-defective lentiviral vectors in primary human cells Mol Ther 191499–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayrhofer P, Schleef M., and, Jechlinger W. Use of minicircle plasmids for gene therapy. Methods Mol Biol. 2009;542:87–104. doi: 10.1007/978-1-59745-561-9_4. [DOI] [PubMed] [Google Scholar]
- Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R.et al. (1995CpG motifs in bacterial DNA trigger direct B-cell activation Nature 374546–549. [DOI] [PubMed] [Google Scholar]
- Dalsgaard T, Moldt B, Sharma N, Wolf G, Schmitz A, Pedersen FS.et al. (2009Shielding of sleeping beauty DNA transposon-delivered transgene cassettes by heterologous insulators in early embryonal cells Mol Ther 17121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant SR, Meuse L, Chiu W, Ivics Z, Izsvak Z., and, Kay MA. Somatic integration and long-term transgene expression in normal and haemophilic mice using a DNA transposon system. Nat Genet. 2000;25:35–41. doi: 10.1038/75568. [DOI] [PubMed] [Google Scholar]
- Sharma N, Hollensen AK, Bak RO, Staunstrup NH, Schrøder LD., and, Mikkelsen JG. The impact of cHS4 insulators on DNA transposon vector mobilization and silencing in retinal pigment epithelium cells. PLoS ONE. 2012;7:e48421. doi: 10.1371/journal.pone.0048421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Copy number detection by Southern blot analysis of puromycin-resistant HeLa colonies.