

Abstract
Rift Valley fever virus (RVFV), a member of the family Bunyaviridae, genus Phlebovirus, is the causative agent of Rift Valley fever (RVF), a mosquito-borne disease of ruminant animals and humans. The generation of a large sequence database has facilitated studies of the evolution and spread of the virus. Bayesian analyses indicate that currently circulating strains of RVFV are descended from an ancestral species that emerged from a natural reservoir in Africa when large-scale cattle and sheep farming were introduced during the 19th century. Viruses descended from multiple lineages persist in that region, through infection of reservoir animals and vertical transmission in mosquitoes, emerging in years of heavy rainfall to cause epizootics and epidemics. On a number of occasions, viruses from these lineages have been transported outside the enzootic region through the movement of infected animals or mosquitoes, triggering outbreaks in countries such as Egypt, Saudi Arabia, Mauritania and Madagascar, where RVF had not previously been seen. Such viruses could potentially become established in their new environments through infection of wild and domestic ruminants and other animals and vertical transmission in local mosquito species. Despite their extensive geographic dispersion, all strains of RVFV remain closely related at the nucleotide and amino acid level. The high degree of conservation of genes encoding the virion surface glycoproteins suggests that a single vaccine should protect against all currently circulating RVFV strains. Similarly, preservation of the sequence of the RNA-dependent RNA polymerase across viral lineages implies that antiviral drugs targeting the enzyme should be effective against all strains. Researchers should be encouraged to collect additional RVFV isolates and perform whole-genome sequencing and phylogenetic analysis, so as to enhance our understanding of the continuing evolution of this important virus. This review forms part of a series of invited papers in Antiviral Research on the genetic diversity of emerging viruses.
Keywords: Rift Valley fever virus, Bunyavirus, Phlebovirus, Genetic diversity, Phylogenetics
1. Introduction
Rift Valley fever virus (RVFV) is a member of the genus Phlebovirus, one of five genera in the family Bunyaviridae. It was first identified in 1930, during an investigation of an epizootic of abortion of pregnant ewes and death of newborn lambs on a farm in the Rift Valley of Kenya, accompanied by cases of febrile illness in humans (Daubney and Hudson, 1931). Since then, numerous outbreaks of RVF have been reported in many regions of Africa (Figs. 1 and 2). The largest epidemic occurred in Egypt in 1977–1978, when there were an estimated 200,000 human infections, with some 18,000 cases of illness and 600 deaths. In 2000, an outbreak occurred in Saudi Arabia and Yemen, the first detected outside of Africa, raising concern that the disease could spread further into Asia or Europe, or even to the Western Hemisphere (Ahmed et al., 2009).
Fig. 1.
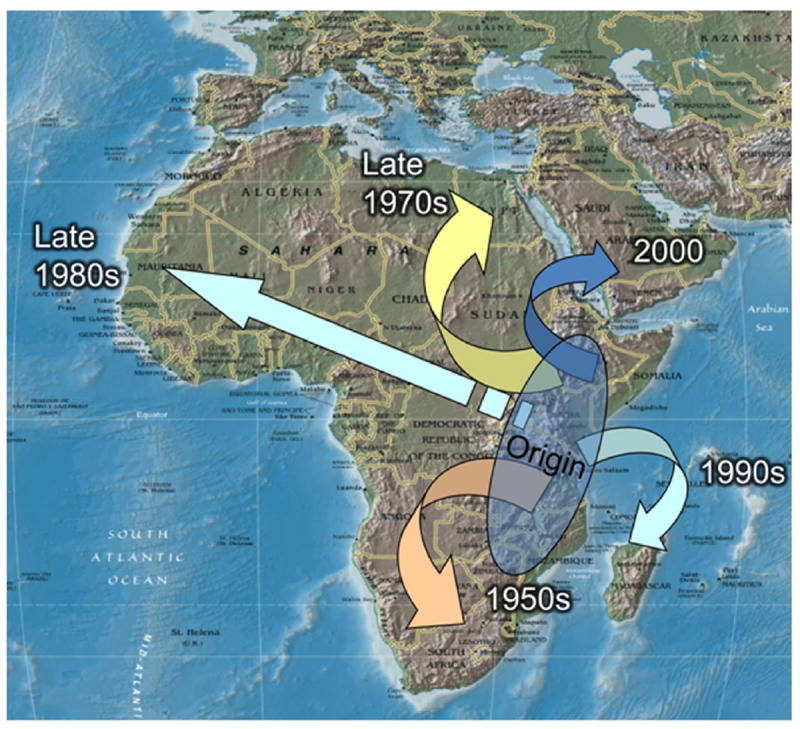
The spread of RVFV outside of sub-Saharan East Africa. Cases were recognized in South Africa in the 1950s, and larger epizootics/epidemics occurred in Egypt in 1978–1979, in Mauritania in the late 1980s, in Madagascar in the 1990s and in Yemen and Saudi Arabia in 2000–2001. Courtesy of Scott Weaver (Weaver and Reisen, 2010).
Fig. 2.
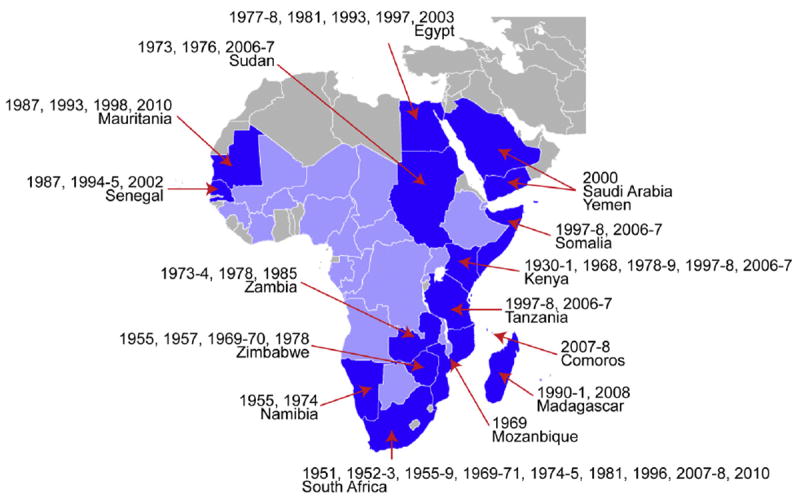
Distribution of RVF as of 2011. Countries that have experienced substantial epizootics and epidemics are shown in dark blue, while those with serologic evidence or virus isolation are marked with light blue. Years of major outbreaks are shown. Based on (Bird et al., 2009; Swanepoel and Coetzer, 2004).
RVFV is classified as a Category A pathogen and an overlap select agent by the US Centers for Disease Control and Prevention and the Department of Agriculture. There is no commercially available vaccine for human or animal use outside endemic countries, including the US and Europe, and effective antiviral drugs have not been identified. There is therefore an urgent need to develop countermeasures against an accidental or deliberate introduction of RVFV into the US or other non-endemic countries. A thorough understanding of the genetic diversity of the virus is needed to develop reliable diagnostic methods, to support the development of antivirals and vaccines and to identify the source of new outbreaks. In this paper, we first present a concise summary of the epidemiology, clinical features, animal models and molecular biology of RVF. We then review the large amount of information that has been obtained during the past two decades on the genetic diversity of RVFV, including phylogenetic analysis of isolates from its “ancestral homeland” in Africa and from epidemics in regions outside the traditional enzootic area. We conclude with an assessment of the impact of RVFV genetic diversity on the development of vaccines, antiviral drugs and rapid diagnostic methods.
2. Rift Valley fever: basic features
2.1. Natural reservoirs and transmission cycles
In its enzootic region in Kenya, Zimbabwe and other countries of central Africa, RVFV persists in the environment through vertical transmission in mosquitoes and horizontal transmission by mosquitoes among animals (presumably native ungulates and rodents, but the principal hosts have not been identified) (Olive et al., 2012). Infected eggs of floodwater Aedes mosquitoes are a principal source of infection, maintaining the virus during dry conditions (Bird et al., 2009; Linthicum et al., 1985). Heavy rainfall increases the hatching of eggs of floodwater Aedes mosquitoes and permanent fresh-water species such as Culex pipiens; the latter act as amplifying vectors, by feeding on viremic ruminants and humans (Pepin et al., 2010). In addition to mosquito transmission, humans become infected through contact with the blood or body fluids of infected animals (Chambers and Swanepoel, 1980; Pepin et al., 2010).
In addition to recurrent disease outbreaks within the traditional RVF enzootic area, the past few decades have seen the spread of RVFV to a number of other countries within and outside of Africa, including Egypt, Madagascar, Mauritania and Saudi Arabia, apparently as the result of the commercial transport of infected animals and/or the windborne movement of infected mosquitoes. Such outbreaks outside the traditional enzootic area might occur in two ways. In the first, imported virus could be introduced directly to cattle, sheep and other livestock, followed by rapid amplification and mosquito-borne spread among animals and humans, causing an epizootic closely followed by an epidemic. If the virus is also introduced to wild reservoir animals and becomes established through vertical transmission among local mosquitoes, it could persist in the environment once the epizootic/epidemic has ended; to date, there is no evidence that this has occurred outside of Africa. Alternatively, the newly introduced virus might first infect local reservoir animals and mosquitoes, persist at low level in the environment, then emerge at later times to cause outbreaks among livestock and humans when heavy rainfall expands the mosquito population (Grobbelaar et al., 2011).
At this point, it is not entirely clear to what extent RVFV has become established in countries outside its “ancestral” enzootic area in Africa. Repeated outbreaks have occurred in Egypt, fortunately on a much smaller scale than the massive 1977 epidemic, suggesting that RVFV is now enzootic in that country, with the extent of disease being limited through livestock vaccination and mosquito control (Ahmed Kamal, 2011). In contrast, a number of outbreaks have also been registered in Madagascar, but evidence to date indicates that they have resulted from repeated introductions of virus from mainland Africa, rather than from the persistence of RVFV in the local environment (Carroll et al., 2011).
2.2. Disease in sheep and cattle
Outbreaks of RVF are often recognized initially as an “abortion storm” in herds of pregnant ruminants (Daubney and Hudson, 1931). Sheep are the most susceptible species, while cattle, goats, and camels are variably susceptible (Findlay, 1931). Forty to 100% of pregnant ewes abort, and the fetus often manifests malformations (Swanepoel and Coetzer, 2004). Newborn lambs suffer peracute disease, characterized by necrotic hepatitis with 95–100% mortality. Some breeds of adult sheep also exhibit hemorrhagic signs, similar to those seen in humans (Olaleye et al., 1996). In cattle, RVFV infection varies greatly in severity. While silent infection can occur, pregnant cattle frequently abort, and the overall mortality rate ranges from 10% to 30% (Swanepoel and Coetzer, 2004).
2.3. Disease in humans
After an incubation period of 4–6 days, RVF typically begins acutely, with chills and fever, malaise, dizziness, weakness, nausea and severe headache and muscle pains. Patients are viremic during the febrile period. Although the initial symptoms may resolve in 3–4 days, they often recur, and complete recovery may take weeks. Neutralizing antibodies sometimes appear as early as the fourth day after the onset of symptoms. A small percentage of patients develop a hemorrhagic fever syndrome, characterized by a macular rash, ecchymoses, bleeding from the gums and melena. Those with hemorrhagic disease often develop diarrhea, low blood pressure, jaundice and/or hepatosplenomegaly, and may die 3–6 days after the onset of symptoms (Ikegami and Makino, 2011). Some patients develop neurological disorders, including hemiparesis, neck rigidity, confusion, visual hallucinations, stupor and coma, beginning weeks to months after the onset of illness. Histopathologically, fatal cases show parenchymal necrosis in the central nervous system (van Velden et al., 1977). Some 1–10% of patients also suffer from partial or complete vision loss, which may begin immediately after the onset of symptoms or up to several months later. Despite chorioretinal scarring, some recovery of vision can occur (Ikegami and Makino, 2011).
2.4. Infection of laboratory animals
When RVFV was first isolated, researchers tested its host range by inoculating a variety of species, and found that a number of non-ruminant animals, such as horses, pigs, rabbits and birds were resistant to infection, while mice and hamsters were highly susceptible (Findlay, 1931). However, RVFV has not been isolated from wild mice or rats in enzootic/endemic areas, suggesting that they are not involved in maintaining the virus in nature (Swanepoel et al., 1978). After experimental inoculation, mice develop acute necrotic hepatitis, which is often fatal; those that survive usually develop encephalitis 1–2 weeks postinfection (Smith et al., 2010). Laboratory rats show a range of susceptibility to RVF. Wistar-Furth (WF) and brown Norway strains usually die within 4 after challenge, whereas Lewis and other strains are resistant to infection (Peters and Anderson, 1981; Peters and Slone, 1982). However, the resistance phenotype varied with the source of animals, suggesting that host factors affect susceptibility (Ritter et al., 2000). Gerbils display an age-dependent susceptibility to RVFV, developing encephalitis with minimal liver injury (Anderson et al., 1988). They have therefore been used as a model of RVF encephalitis.
RVFV also causes disease in certain species of nonhuman primates. Approximately 20% of rhesus macaques develop hemorrhagic fever following intravenous inoculation, while the remainder shows milder illness or subclinical infection (Morrill et al., 1990; Peters et al., 1988). Marmosets are more susceptible, displaying hemorrhagic manifestations with severe liver necrosis and disseminated intravascular coagulation after intravenous or subcutaneous exposure; encephalitis is also observed (Smith et al., 2011).
3. Virion structure, genome, structural and nonstructural proteins
The following sections provide current information on RVFV genetics, with a special emphasis on genetic motifs, structures and functions that are useful for understanding genetic variation among RVFV strains.
3.1. The virion
RVF virions are spherical, consisting of an envelope and a ribonucleocapsid (RNP) (Fig. 3A–D). Virions measure 80–120 nm in diameter. The viral envelope is covered by 122 capsomers, consisting of heterodimers of the Gn and Gc glycoproteins on an icosahedral lattice with T = 12 quasisymmetry. The surface capsomers form 110 cylinder-shaped hexamers and 12 pentamers (Freiberg et al., 2008; Huiskonen et al., 2009; Sherman et al., 2009). Envelope surface projections 9 nm long form distinctive spikes that cover the surface and are embedded in a 7 nm lipid bilayer. The envelope surrounds three RNPs corresponding to the S, M and L genome segments.
Fig. 3.
Morphology of RVFV particles. (A) Negative-stain transmission electron micrograph (TEM) showing pleomorphic RVFV MP-12 particles. (B) Negative-stain TEM micrograph showing spherical RVFV MP-12 particles with a distinct surface structure composed of morphological units with a central cavity. Scale bar: 100 nm. (C) Histogram representing the size distribution of negatively stained RVFV MP-12 particles, with an average diameter of 95 ± 9 nm (n = 84). (D) Surface-shaded representation of RVFV MP-12. Capsomers formed by the surface glycoproteins, Gn and Gc, are indicated in dark brown, and ridges connecting neighboring capsomers are shown in light brown. Figures A–C were adopted and modified with permission from (Freiberg et al., 2008). Figure D was kindly provided by Alex Freiberg (Sherman et al., 2009).
The RNP of bunyaviruses is filamentous, with a length of 200–3000 nm and a width of 10–12 nm. It is reported to be a string-like structure, distinct from the RNPs of other negative-sense RNA viruses that exhibit helical symmetry (Raymond et al., 2010). The ribonucleocapsid is thought to have a pan-handle-like structure, due to complementary sequences at the genome termini (Nichol et al., 2005). The N proteins form ring-like hexamers with an external diameter of 100 Å, and the viral RNA is believed to bind to its cavity (Ferron et al., 2011; Raymond et al., 2010).
3.2. Genome segments and encoded proteins
The RVFV genome consists of three single-stranded RNA segments of negative or ambisense polarity:
the S segment (prototype strain ZH501: 1690 nucleotides (nt));
the M segment (3885 nt);
and the L segment (6404 nt) (Fig. 4) (Schmaljohn and Nichol, 2007).
Fig. 4.
The viral-sense (negative-sense) RNA genome of the prototype ZH501 strain of RVFV. The S segment encodes the N and NSs proteins in an ambi-sense manner. The M segment encodes the NSm, the 78-kDa protein and Gn and Gc, while the L segment encodes the L protein. The 78-kDa and the NSm proteins are synthesized beginning from the first and second AUG of the M mRNA in the preglycoprotein (pre-G) region, which contains five in-frame AUGs.
Because of the lack of a cap-structure at the 5′ ends, no viral proteins are synthesized from viral genomic RNA. All genome segments share identical termini (3′-UGUGUUUC or GAAACACA-5′), and this sequence is largely conserved among viruses in the Phlebovirus genus. The conserved genomic termini form panhandle structures and serve as promoters for genomic RNA synthesis, as well as N encapsidation signals (Schmaljohn and Nichol, 2007). Both N and L are required for the synthesis of viral genomic RNA and mRNA.
As of 2011, the Virus Pathogen Database Resource (ViPR: accessed on April 19, 2012 at http://www.viprbrc.org/brc/home.do?decorator=vipr) lists 158, 106 and 95 full-length sequences of S, M and L segments. The complete genomic sequences of at least 88 strains are currently available. The length of the S segment varies somewhat among the 158 strains; variation occurs by an insertion or deletion at the intergenic region. The 3′-UTR, 5′-UTR and the N and NSs open reading frames (ORFs) do not differ in length, except for the NSs ORF of the C13 strain, in which a large deletion has occurred (Bird et al., 2007c).
The transcription of bunyaviral mRNA utilizes a cap-snatching strategy, by which capped mRNAs of the host are cleaved and the capped 5′ fragments with 10–15 nt are used as primers to synthesize viral mRNA. The resulting viral mRNAs therefore have heterogeneous 5′ end sequences (Lopez et al., 1995; Schmaljohn and Nichol, 2007). The N and L proteins that accumulate during primary transcription initiate subsequent viral RNA replication. A study has suggested that the level of viral RNA accumulation in infected cells is S > M > L (Gauliard et al., 2006). Accumulated viral genomic RNA allows the further amplification of viral mRNA (secondary transcription).
3.2.1. The S segment
The S-segment encodes the ORFs of the N protein (738 nt, 245 amino acids, 25 kDa for prototype strain ZH501) and NSs protein (798 nt, 265 amino acids, 34 kDa) in an ambisense manner (Collett et al., 1985; Struthers et al., 1984). Between the N and NSs ORFs lies an intergenic sequence of 82 nt (Giorgi et al., 1991), which consists of unique poly-C (viral sense) or poly-G (antiviral sense) tracts. The S-segment intergenic region is much more variable among RVFV strains (11%) than the N and NSs ORFs (4%), while the NSs ORF is slightly more variable (4.5%) than the N ORF (3.5%) (Bird et al., 2007c).
The N mRNA is transcribed from the viral-sense portion of the S segment, while NSs mRNA is transcribed from the antiviral-sense portion. The N or NSs mRNA syntheses are terminated at nt 841 or nt 789, respectively, by using 3′-CGUCG-5′ (N mRNA: nt 846–850, NSs mRNA: nt 780–784), in combination with the upstream poly-C or poly-G tracts, respectively (Albarino et al., 2007; Ikegami et al., 2007; Lara et al., 2011). Thus, the overlapped 30–60 nt of 3′ termini of N and NSs mRNA are complementary with the poly-C or poly-G tract sequences.
The N protein encapsidates the viral RNA to form the ribonucleocapsid. Encapsidation is required for transcription and RNA replication (Ikegami et al., 2005a; Lopez et al., 1995). The ribonucleocapsid facilitates the production of virus-like particles (VLP) (Piper et al., 2011), suggesting the role of N proteins in the assembly process.
The NSs protein is indispensable for lethal infection in mice and is considered a major virulence factor of RVFV (Bouloy et al., 2001; Vialat et al., 2000). NSs forms filamentous structure in the nucleus of infected cells (Swanepoel and Blackburn, 1977; Yadani et al., 1999). Alignment of NSs protein sequences indicates that cysteine residues at aa 39, 40, 150, 179 and 195 were highly conserved, suggesting that they are important for three-dimensional structure and filamentous structure formation (Sall et al., 1997). NSs specifically interacts with the p44 subunit protein of TFIIH, which is an essential transcription factor for cellular RNA polymerase I and II, probably interrupting the assembly of the TFIIH complex, resulting in a general transcriptional shutoff (Le May et al., 2004). Although RVFV infection activates transcription factors such as IRF-3, AP-1 and NF-kB, the transcription of IFN-β and other IFN-related genes is not up-regulated (Billecocq et al., 2004). By interacting with Sin3A-associated protein (SAP30), NSs forms a multiprotein complex containing YY1/SAP30/NCoR/Sin3A/HDAC-3 on the IFN-β promoter region, preventing its activation (Le May et al., 2008). NSs also promotes the degradation of two host proteins, double-stranded RNA-dependent protein kinase (PKR) (Habjan et al., 2009; Ikegami et al., 2009) and TFIIH p62 (Kalveram et al., 2011). Degradation probably occurs through the proteasome pathway, although no evidence of ubiquitination of PKR or p62 has been demonstrated.
A recent study showed that 15 or 10 serial passages of RVFV in baby hamster kidney (BHK) 21 cells or Aag2 (Ae. aegypti) cells results in large deletions of the NSs gene, while 30 serial alternating passages between BHK21 cells and Aag2 cells did not result in the deletion of NSs, suggesting that alternating replication in different cell types promotes stability of the NSs gene (Moutailler et al., 2011). It is likely that minor viral populations lacking NSs might adapt to specific cells, and replicate faster than the major population encoding NSs.
3.2.2. The M segment
The M mRNA is synthesized from the negative-sense M segment, and is terminated at nt 3774 by using 3′-CGUCGUCG-5′ in combination with the upstream poly-G tract (Albarino et al., 2007; Ikegami et al., 2007). A precursor polyprotein is co-translationally cleaved into five proteins:
Gn (nt 480–2090, 537 amino acids, mature form: 56 kDa),
Gc (nt 2091–3614, 507 amino acids, mature form: 65 kDa),
NSm (nt 135–479, 115 amino acids, 14 kDa),
the 78-kDa protein (nt 21–2090, 690 amino acids, 78 kDa)
and NSm-Gn (nt 135–2090),
presumably by signal peptidase (Collett et al., 1985; Gerrard et al., 2007; Gerrard and Nichol, 2007; Won et al., 2006). Synthesis of those proteins involves leaky scanning of the ribosome in five initiation codons (nt 21, 135, 174, 411 and 426, with the 5′ end of the initiation codon in the antiviral-sense) located upstream of the Gn coding region. The 78-kDa protein is synthesized from the first AUG and NSm from the second, while the synthesis of Gn most probably occurs from the second, third or fourth AUG (Gerrard and Nichol, 2007; Suzich et al., 1990). Interestingly, all the in-frame AUGs at the upstream of Gn are conserved among 33 RVFV strains, suggesting the functional requirement of those AUGs or NSm (Bird et al., 2007c). The mechanism of Gc synthesis is still largely unknown; two or more precursor proteins might be involved in the process.
The putative signal peptides are located downstream of the first and fifth AUG (Gerrard and Nichol, 2007). Although the synthesis of NSm occurs from the second AUG and the signal peptide is encoded only downstream of the 5th AUG, the precursor protein consisting of the polypeptide ranging from NSm to Gn can enter the secretory pathway, suggesting that the use of a signal peptide downstream of the fifth AUG is functional (Gerrard and Nichol, 2007). The C-terminus of Gn (nt 1776–1919) encodes a Golgi localization signal (Gerrard and Nichol, 2002). When Gc is expressed individually, it localizes to the endoplasmic reticulum, but when co-expressed with Gn, it localizes to the Golgi through the signal sequence (Gerrard and Nichol, 2002). Such co-localization of Gn and Gc in the Golgi should be required for efficient viral assembly and maturation. It is suggested that N-terminus (first 30 amino acids) or the C-terminus (the last 40 amino acids) of a presumable 70-amino-acid-long Gn cytoplasmic tail is required for N or L protein recruitment, respectively (Piper et al., 2011). A coordinated packaging mechanism of S-, M- and L-segment RNA, in which the co-packaging of S- and M-segment can support an efficient co-packaging of L-segment, has been demonstrated (Terasaki et al., 2011). The S-, M- and L-segments encoding the terminal 25 nt of the 5′-UTR were equally competent for genome packaging into VLP, suggesting that the terminal 25 nt encodes a packaging signal (Murakami et al., 2012).
NSm localizes predominantly to the Golgi with some reticular staining, while the 78-kDa proteins localize to the Golgi (Wasmoen et al., 1988). However, a lack of both NSm and 78-kDa protein expression does not alter the localization pattern of Gn and Gc, suggesting that both proteins are dispensable for Gn/Gc synthesis, processing, modification or Golgi retention (Wasmoen et al., 1988). Indeed, recombinant RVFV lacking both 78-kDa and NSm was recovered, and it showed similar replication kinetics to the parental virus, indicating that they are dispensable for replication (Gerrard et al., 2007; Won et al., 2006). However, NSm is probably important for pathogenesis (Bird et al., 2007a). WF rats infected with wild-type RVFV ZH501 strain, which lacks the 78-kDa and NSm proteins, developed neurologic disease, rather than hepatitis (Bird et al., 2007a). Cells infected with RVFV lacking NSm were shown to be more susceptible to apoptosis, suggesting that the protein has an anti-apoptotic function (Won et al., 2007).
Contrary to the expectation that the envelope proteins should be more variable than other RFVF proteins, Gn and Gc are well conserved. No variation in their length has been detected, and all N-linked glycosylation sites are completely conserved among 33 RVFV isolates (Bird et al., 2007c). Antigenic domains in Gn and Gc are well conserved among RVFV strains. However, although mabs raised against a South African virus neutralized strains from other parts of Africa, different binding patters were noted for the attenuated Onderstepoort and the Smithburn neurotropic vaccine viruses, suggesting changes in the ternary structure of their antigenic domains (Besselaar et al., 1991). Similarly, the antigenic differences of neutralizing epitopes II and IV (Keegan and Collett, 1986) of 22 RVFV strains were analyzed by using mabs (Battles and Dalrymple, 1988). Lunyo and 90060 strain (a plaque isolate of ZH501) resisted neutralization by an epitope II-specific mab, while the Smithburn strain showed reduced neutralization by an antibody that recognized epitope IV. On the other hand, none of the strains resisted neutralization by mabs recognizing epitope I and II, suggesting that neutralizing antibodies that react to several different epitopes on Gn and Gc confer protection against most wild-type RVFV strains.
3.2.3. The L segment
The L-segment encodes an RNA-dependent RNA polymerase (L protein: nt 19-6297, 2087 amino acids), which synthesizes both viral mRNA and genomic RNA (Muller et al., 1994). The synthesis of L mRNA is terminated between nt 6364 and 6384 (nt location at antiviral-sense L-segment) by using the stem–loop structure formed around the termination sequence (Ikegami et al., 2007), while it is also proposed that L-segment mRNA synthesis is not terminated and the 3′ end of L mRNA is identical to anti-viral-sense L-segment RNA (Albarino et al., 2007). A more recent study suggested that the L mRNA is terminated at nt 6377, with a presumable role of 5′-CGAUG-3′ for termination (Lara et al., 2011). RVFV L proteins form oligomers by interacting at thir N- and C-termini (Zamoto-Niikura et al., 2009). L is packaged into virus particles (Piper et al., 2011), and synthesizes primary transcripts in infected cells (Ikegami et al., 2005b).
A number of functional motifs have been identified in the RVFV L protein. Regions 1 (aa 60–185) and 2 (aa 650–788) are conserved among bunyaviruses and arenaviruses. Region 3 (aa 895–1206), the “polymerase module,” is conserved among both segmented and non-segmented RNA viruses (Muller et al., 1994). It consists of premotif A and motifs A, B, C and D; its amino acid sequence aligns with the corresponding region of the human immunodeficiency virus (HIV) reverse transcriptase (Muller et al., 1994). All of the L protein functional motifs are conserved among 33 different RVFV strains (Bird et al., 2007c).
4. Genetic diversity of RVFV
A number of analyses of the genetic diversity of RVFV have been published over the past 25 years. Early studies made use of partial gene sequences from a relatively small number of isolates, while more recent work has been based on complete genomic sequences of a much larger number of viruses recovered from many geographic areas. As a consequence of the expanding database, the number of identified viral lineages (distinct genetic groups sharing a common ancestor) of RVFV has increased from 3 in an early analysis (Sall et al., 1997) to 7 in a 2007 study (Bird et al., 2007c) to 15 in the most recent report (mean pairwise distances ≤0.017 within lineages and bootstrap values ≥70%) (Grobbelaar et al., 2011). The latter two studies found that the genetic diversity of RVFV across all lineages is quite limited, with maximum pairwise differences for the S segment and partial M segments of 4% and 5.4% for the nucleotide sequence and 1% and 2.8% for the amino acid sequence. Such limited diversity may in part reflect the evolutionary constraint imposed on arboviruses by their alternating replication in mammalian and arthropod hosts (Coffey et al., 2008; Moutailler et al., 2011). However, the diversity of RVFV is much narrower than that of some other arthropod-borne bunyaviruses, such as Crimean-Congo hemorrhagic fever virus, suggesting that the principal factor responsible for the close phylogenetic relationship among RVFV isolates is the recent emergence of an ancestral virus in Africa and its expansion among introduced sheep and cattle (see below).
4.1. Identification of genetic lineages of RVFV in Africa
In the first report to examine the evolutionary relationships of RVFV viruses, Sall et al. aligned the NSs gene from 18 different strains collected over 38 years from diverse localities in Africa (Sall et al., 1997). The authors chose the NSs gene because it is the most variable among phleboviruses, and is therefore considered to be under weak evolutionary pressure to maintain its sequence; it varied up to 9.6% at the nucleotide level and 9.5% at the amino acid level. Phylogenetic analysis identified two major viral lineages. Cluster I (sub-Saharan) was divided into cluster Ia and Ib, with the latter consisting of West African isolates, while the former contained isolates from different regions in Africa. Cluster II contained viruses isolated in Egypt in 1977 and 1993, which were very similar, suggesting a common origin. Unexpectedly, two distinct lineages corresponding to clusters Ia and Ib were recovered during the Mauritanian outbreak in 1987, and isolates corresponding to clusters Ia and II were identified in Madagascar, suggesting separate introductions of virus onto the island. To further understand RVFV evolution, the authors constructed phylogenetic trees for the Gn and L genes, which showed that viruses in cluster Ia were localized to East/Central Africa, in cluster Ib to West Africa and in cluster II to Egypt (Sall et al., 1999).
The recent availability of high-throughput sequencing has markedly increased the amount of data available for phylogenetic analysis. Bird et al. performed the first study of full-length RVFV sequences, employing 33 viruses isolated from 1944 to 2000 in 10 different African countries and in Saudi Arabia (Bird et al., 2007c). The maximum pairwise differences among the S, M and L segments were 4%, 5% and 4% at the nucleotide level and 1%, 2% and 1% at the amino acid level. The 33 viruses could be separated into seven lineages (Fig. 5):
A (isolates from Zimbabwe in 1974, Egypt in 1977, 1978 and 1979, and Madagascar in 1979);
B (isolates from Kenya in 1983 and 1998, the Central African Republic (CAR) in 1973 and Saudi Arabia in 2000);
C (isolates from Guinea in 1981 and 1984, the CAR in 1969, 1973 and 1974 and Zimbabwe in 1978);
D (isolates from Kenya in 1965, Zimbabwe in 1970, South Africa in 1975, Burkina Faso in 1983 and Mauritania in 1987);
E (isolates from Uganda in 1944 and Zimbabwe in 1974);
F (an isolate from Zimbabwe in 1974); and
G (an isolate from South Africa in 1951).
Fig. 5.
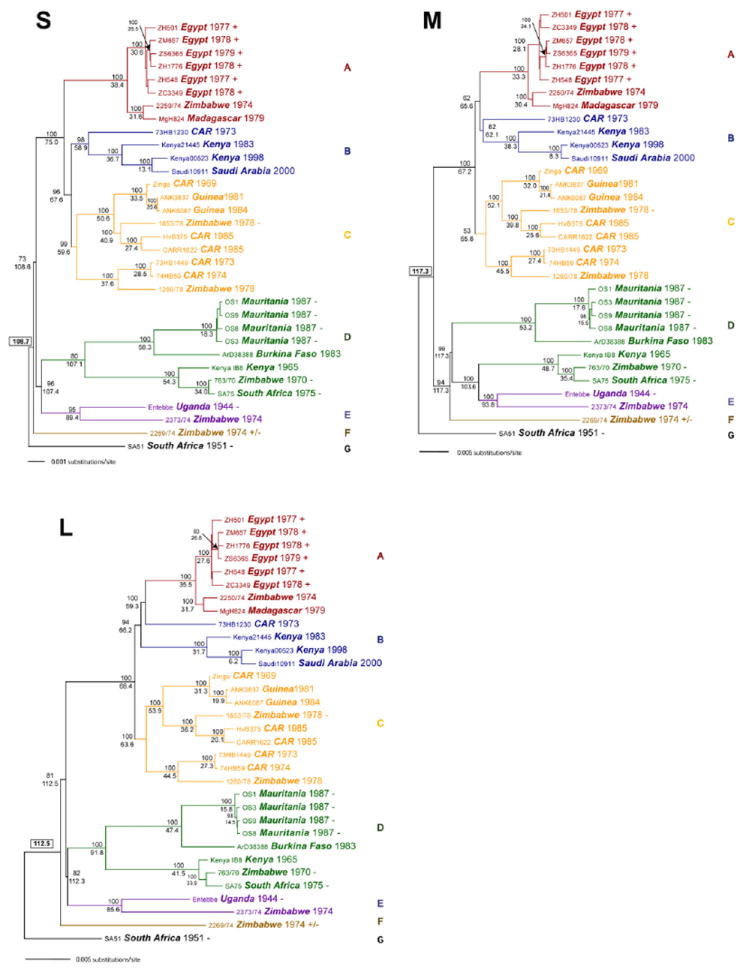
Genetic lineages of 33 RVFV isolates, based on complete sequences of the S segment (A), the M segment (B) and L segment (C) (Bird et al., 2007c). Each taxon name indicates the strain, country of origin, and date of isolation. Virulence in WF rats is indicated by + (an LD50 of approximately 1 PFU), ± (LD50 approximately 2 × 103 PFU), or – (nonlethal). Adopted and modified with permission (Bird et al., 2007c).
The existence of strains from a number of countries within each of lineages A–D is consistent with an epidemiologic picture in which multiple RVFV lineages circulate continuously in central Africa, and individual strains are occasionally transported to regions outside the enzootic zone. Thus, RVFV strains in lineages A, C, D, E and F are found in Zimbabwe, but only viruses in lineage A have been isolated in Egypt, indicating that the 1977–1979 Egyptian outbreak resulted from the point-source introduction of a single genotype from sub-Saharan Africa (Bird et al., 2007c). Limited evidence of differences in virulence among viruses of various lineages is described in section VIII.
In the largest phylogenetic analysis to date, Grobbelaar et al. studied partial M segment sequences from 203 viruses isolated in 16 African countries and Saudi Arabia from 1944 to 2010 (Grobbelaar et al., 2011). The maximum pairwise differences observed were 2.8% at the nucleotide and 5.4% at the amino acid level. As shown in Fig. 6, analysis of the sequences of a large number of viruses permitted the identification of 15 distinct lineages:
A (isolates from Zimbabwe in 1974 and 1978, Egypt in 1977, 1978, 1979 and 1993, and Madagascar in 1979, suggesting that outbreaks in the latter two countries resulted from importations from Zimbabwe);
B (isolate from Kenya in 1972);
C (isolates from Zimbabwe in 1976, 1978, 1979 and 1998, Kenya in 1977, 1983, 1997, 1998 and 2007, Saudi Arabia in 2000, Somalia in 1998, South Africa in 1999, 2008 and 2009, Madagascar in 1991 and 2008, and Mauritania in 2003, suggesting that outbreaks in Saudi Arabia, South Africa, Madagascar and Mauritania all resulted from importations from Zimbabwe or Kenya);
D (isolate from the CAR in 1973);
E (isolates from Zimbabwe in 1975 and 1978, the CAR in 1973 and 1974, and Zambia in 1985);
F (isolate from South Africa in 1981);
G (isolates from the CAR in 1969 and 1985, Guinea in 1981 and 1984, Senegal in 1993 and Zimbabwe in 1978);
H (isolates from South Africa in 2009 and 2010);
I (isolates from South Africa in 1955 and 1956);
J (isolate 2269/74 from Zimbabwe in 1974);
K (isolates from Uganda in 1944, Kenya in 1951, 1962 and 1963, South Africa in 2010, and isolate 2373/74 from Zimbabwe in 1974, plus the Smithfield neurotropic strain (SNS) and the 95EG live vaccine strains);
L (isolates from Egypt in 1995, Kenya in 1963 and 1964, South Africa in 1971, 1974 and 1975 and Zimbabwe in 1969 and 1970);
M (isolates from Uganda in 1955 and South Africa in 1955);
N (isolates from Mauritania in 1987 and 1988, Senegal in 1975, 1983 and 1993 and Burkina Faso in 1983); and (isolate from South Africa in 1951).
Fig. 6.
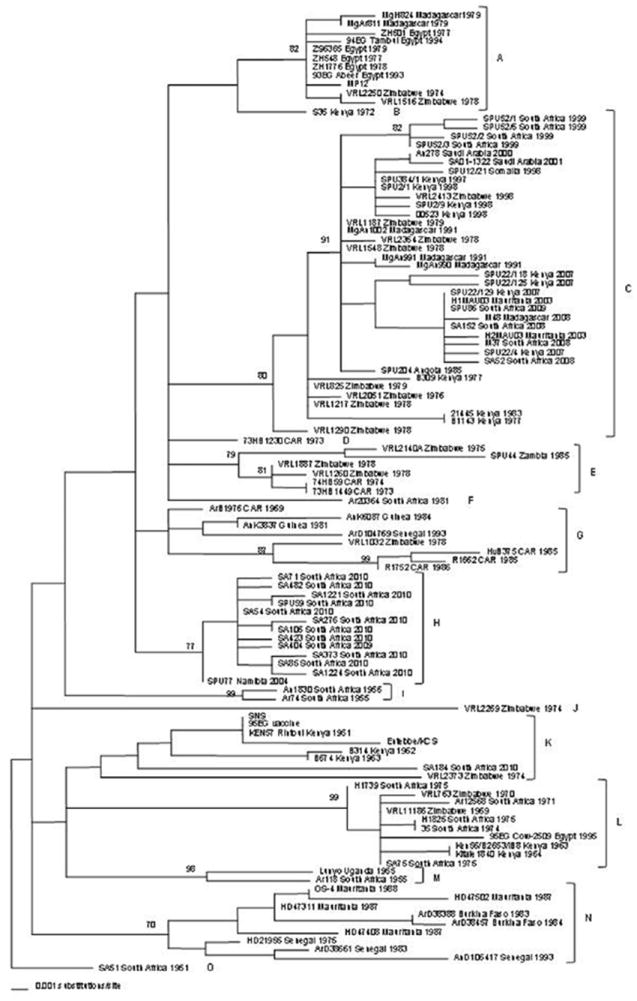
Genetic lineages of 111 RVFV isolates, based on a 490-nt section of the Gn gene. The names, countries of origin and date of isolation are indicated. CAR, Central African Republic; SNS, Smithfield neurotropic strain. From Grobbelaar et al. (2011), with permission.
Compared to the previously cited report by Bird et al., the study created five new lineages (B, F, H, I and M). In addition, lineages B and C were rearranged to create new lineages C, D, E and F, and the former lineage D was rearranged to form lineages L and N. In this new phylogenetic tree, viruses isolated in Zimbabwe were found in lineages A, C, E, G, J, K and L and South African strains were found in lineages C, F, G, H, I, K, L and M. As discussed below, the authors suggested that the increased genetic variability of strains from these two countries could be attributed to use of the live Smithburn vaccine (Grobbelaar et al., 2011).
4.2. Evidence of reassortment
Evidence of genome segment reassortment among RVF viruses was first detected by Sall et al., who found that a virus isolated in 1987 in Mauritania derived its S segment from cluster Ia and its M and L segments from a virus in cluster Ib (Sall et al., 1999). Similarly, a 1984 Senegal strain had an S segment from cluster II and M and L segments from cluster Ib, while a virus isolated in Guinea in 1984 had an S segment from cluster Ib and M and L segments from cluster Ia. Such combinations of genome segments from viruses with different geographic origins clearly indicate the occurrence of reassortment during co-infection of a single host by two virus strains.
In their examination of 33 different RVFV isolates, Bird et al. found no evidence of genetic reassortment (Bird et al., 2007c). On the other hand, a subsequent analysis by the same authors of an outbreak in Kenya in 2006–2007 identified a reassortment event (Bird et al., 2008). The 31 viruses recovered during the epidemic were divided into three discrete lineages (Kenya-1, Kenya-1a and Kenya-2) with up to 1.6% nucleotide divergence. One of them had S and L segments of lineage Kenya-1a, but an M segment from Kenya-2 (Fig. 7). Evidence detected by Grobbelaar et al. of reassortment between the Smithburn vaccine and a wild virus in lineage H, which resulted from the accidental co-infection of a human with both viruses, is discussed below.
Fig. 7.
Evidence of RVFV reassortment in nature. The RVF virus S segment lineages Kenya I (orange), Kenya-1a (blue), and Kenya-2 are extracted from the original figure (Bird et al., 2008). A natural reassortant virus isolated in Kenya in 2007 is shown by arrows. Adopted and modified with permission (Bird et al., 2008).
4.3. Genetic analysis of viruses recovered during RVF outbreaks
Analysis of viruses isolated during epizootics and epidemics has revealed two patterns of RVFV diversity. Outbreaks occurring in the “ancestral homeland” of the virus in east Africa have been caused by viruses from a number of different lineages, indicating that those lineages are maintained continuously in the region through enzootic circulation among mammalian reservoirs by local mosquito species. In contrast, when epizootics/epidemics have taken place in areas such as Egypt, Madagascar and Mauritania, where RVF had not previously been observed, the recovered viruses have been closely related to a central African strain, and have shown very limited genetic diversity. This close similarity indicates that the novel outbreaks resulted from the introduction of a single virus strain from central Africa, followed by mosquitoborne spread among susceptible animals and humans.
4.3.1. South Africa, 1951
After the first reported RVFV outbreak in 1930 in Kenya, the next large epizootic/epidemic to occur was in South Africa in 1951. Because the disease had not been recognized previously in that country, it appeared that the virus had been introduced from other African countries such as East Africa (Daubney and Hudson, 1931) or central Africa (Gear et al., 1951). However, recent phylogenetic analysis shows that the South African SA51 strain, the only strain in lineage O, is quite distinct from other African strains, in particular the earliest isolate from Entebbe, Uganda, which is in lineage K (Fig. 6) (Grobbelaar et al., 2011). It is therefore possible that the SA51 virus is the only isolate to date of an indigenous RVFV strain that circulates at low level in South Africa, or that it represents an as-yet unrecognized strain from within the east African enzootic area.
4.3.2. Zimbabwe, 1970s
Outbreaks of RVF were first reported in cattle and humans in the area around Harare in Zimbabwe (the former Rhodesia) in 1957 (Shone, 1958), and again from 1969 to 1970 (Christie, 1969). The persistence of enzootic activity during 1971–1977 was confirmed by virus isolation and antigen detection (Swanepoel et al., 1979). Another large epidemic occurred in the Harare area in 1978–1979, linked to heavy rainfall. According to recent retrospective analyses, the Zimbabwean isolates belong to several lineages, suggesting the cocirculation of multiple strains in the area around Harare (Bird et al., 2007c; Grobbelaar et al., 2011). Viruses isolated in 1969 and 1970 clustered in lineage L, and were also close to the Smithburn-related 95EG Cow-2509 strain. Among those isolated in 1974, two were in lineage A, near to viruses recovered in Egypt in 1977–1979, while another was close to the Smithburn vaccine strain in lineage K. Strain VRL2140A/75, isolated from a cow in 1975, was in lineage E, related to viruses isolated in CAR and Zambia, while strain VRL2051/76, recovered from a human case in 1976, is in lineage C, close to a virus subsequently isolated in Kenya in 1983 (Grobbelaar et al., 2011). These results show that multiple strains in lineages A, C, J, K and L were already circulating in the Harare area at the time of the large 1978–1979 epidemic. As discussed below, some of these lineages might be the source of outbreaks in Egypt, Madagascar and West Africa.
4.3.3. Egypt, 1977–1978
In July, 1977, an outbreak of RVF began in the Aswan Governorate of southern Egypt, then spread up the Nile Valley, ultimately resulting in more than 200,000 human infections and 600 deaths (Meegan, 1979). It is thought that the virus was introduced into Egypt from Sudan, either by sheep imported to Aswan (Gad et al., 1986) or by wind-borne mosquitoes (Sellers et al., 1982). In 1977, the prototype ZH548 virus was isolated from a 52-year-old man, and strain ZH501 was recovered from a 12-year-old girl who died of a hemorrhagic fever-like illness in Sharqiya (Meegan, 1979). Other Egyptian viruses were isolated from a patient in Gharbiya in 1978, a cow in Asyut in 1978, a sheep in Gharbiya in 1979, and from a mosquito in Sharqiya in 1978 (Anderson and Peters, 1988; Atwa et al., 2011). The study by Bird et al. showed that the maximum pairwise difference among these isolates was 0.33% at the nucleotide level, indicating that the epidemic in Egypt resulted from a single point-source introduction (Bird et al., 2007c).
4.3.4. Egypt, 1993–2003
Since the massive epizootic of 1977–1979, Egypt has made use of livestock vaccination to prevent further outbreaks. Two types of vaccine have been employed: an inactivated preparation of a virus (ZH501) isolated in 1977, and locally produced versions of the live, attenuated Smithburn vaccine from South Africa. Despite these efforts, outbreaks occurred in 1993, 1994, 1997, and 2003, but on a much smaller scale than in 1977 (Arthur et al., 1993; Hanafi et al., 2011). Interestingly, no epizootic preceded the occurrence of human cases in 2003, suggesting that livestock vaccination efforts were effective in limiting the extent of disease.
Sequence analysis has shown that viruses recovered in Egypt in 1977–1979, 1993 and 1994 are all in clade A, and are very closely related. They differ significantly from the Smithburn and Onderstepoort vaccines in clade K, indicating that those live vaccines could not have been the source of the outbreaks (Botros et al., 2006; Grobbelaar et al., 2011). The sequences of viruses isolated during the 2003 outbreak have apparently not been reported. The recurrence of epidemics caused by viruses similar to the causative agent of the massive 1977 outbreak suggests that RVFV has become enzootic among local mammals and mosquitoes in Egypt, from which it can re-emerge under appropriate conditions of rainfall and the availability of susceptible hosts, with the size of such outbreaks being limited through livestock vaccination and vector control. Alternatively, it has been proposed that the reappearance in of viruses closely related to the 1977 epidemic strain might be the result of the repeated importation of infected animals from the same source outside of Egypt, or might reflect the occasional failure of inactivation of the locally produced vaccine, which employs a 1977 virus as the master seed (Ahmed Kamal, 2011; Atwa et al., 2011; Botros et al., 2006). Continued careful surveillance of animals and humans, with sequence analysis of additional isolates, will be required to obtain a better picture of the current epidemiology of RVF in Egypt.
4.3.5. Madagascar, 1979–2009
RVFV was first isolated from mosquitoes and from an accidentally infected laboratory worker in Madagascar in 1979, but no disease in animals or other infections of humans were detected at that time (Swanepoel and Coetzer, 2004). The viruses were closely related to strains isolated during the 1977–1978 Egyptian epidemic (Carroll et al., 2011; Grobbelaar et al., 2011). The first recognized RVF outbreak in humans and livestock occurred in 1991 (Morvan et al., 1991a,b), and epidemics took place again in 2008–2009, during two successive rainy seasons (Andriamandimby et al., 2010). Strains isolated from 1979 to 2008 were closely related to viruses isolated in Zimbabwe in the 1970s (Fig. 8) (Carroll et al., 2011). Phylogenetic analysis of partial S-segment sequences of viruses recovered in Madagascar in 2008 found a maximum nucleotide divergence of 0.96. The 12 sequences all lie within the same clade as viruses isolated in Kenya in 2007, belonging to the Kenya-1 and Kenya-1a lineages (Andriamandimby et al., 2010; Bird et al., 2008). Based on these close similarities, Carroll et al. concluded that all outbreaks in Madagascar have resulted from repeated importations from mainland Africa, rather than from low-level enzootic maintenance (Carroll et al., 2011).
Fig. 8.
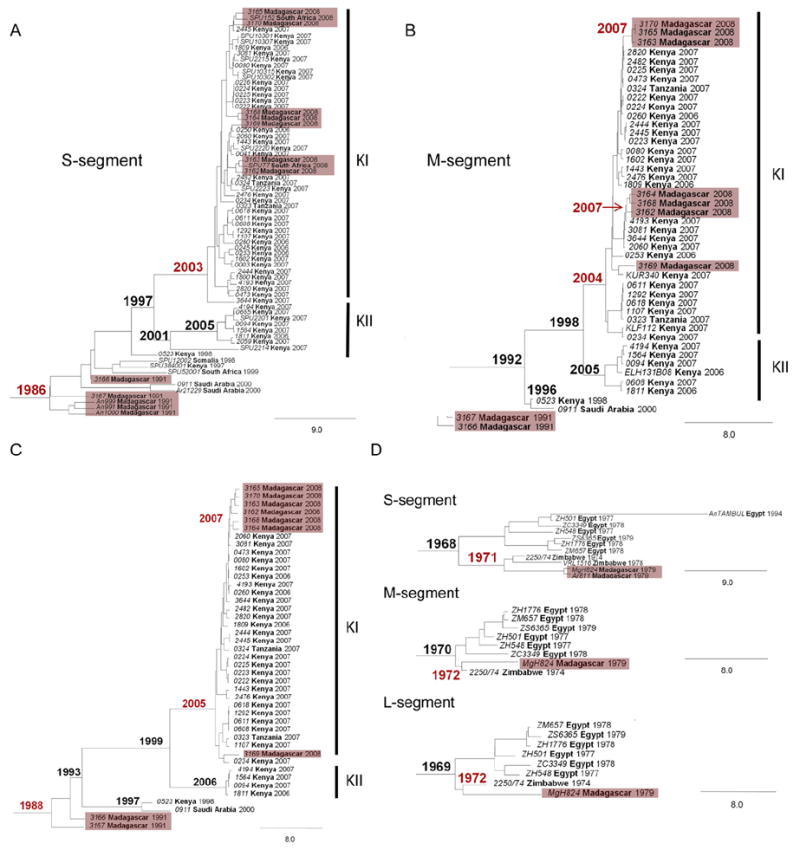
Relationships of viruses isolated in Madagascar to East African (A–C) and Zimbabwean isolates (D). Bayesian coalescent analyses of the S, M and L segments are shown. The maximum clade credibility tree is shown with the year of the most recent common ancestor (MRCA) in boldface at each node. Viruses from Madagascar and nodes containing associated MRCA estimates are shown in red. Lineages from the 2006–2007 East African outbreaks are designated Kenya I (KI) and Kenya II (KII). Adopted and modified with permission (Carroll et al., 2011).
4.3.6. Saudi Arabia and Yemen, 2000–2001
The RVF outbreak in Saudi Arabia and Yemen in 2000–2001 was the first appearance of the disease outside of Africa. Its route of introduction is not known, but it is thought that infected mosquitoes or livestock were transported to the Arabian Peninsula from East Africa (Miller et al., 2002). There is no evidence of viral persistence among mosquito populations in the area. Analysis of the S, M and L segments of viruses recovered from patients and from a pool of mosquitoes showed them to be almost identical to viruses in cluster Ia (East/Central Africa), especially those isolated in Madagascar in 1991 and in Kenya in 1997 (Miller et al., 2002; Shoemaker et al., 2002).
4.3.7. Chad, 2001
In the summer of 2001, an outbreak of febrile illness occurred among French soldiers based in Chad. Genetic analysis of virus recovered from blood samples showed that the troops were infected with a strain of RVFV closely related to cluster Ia from East/Central Africa, which also contains viruses isolated in Kenya, Madagascar and Saudi Arabia (Durand et al., 2003). The authors suggested that the virus was not indigenous to Chad, but had been introduced from outside the country, probably from Kenya.
4.3.8. Mauritania, 2003
The construction of a dam on the Senegal River in Mauritania the late 1980s was soon followed by an outbreak of RVF, and another epidemic occurred in 2003. Analysis of partial S, M and L segments of viruses isolated in 1989 showed that they formed a discrete West African lineage, while strains recovered in 2003 fall within the East/Central African lineage (Faye et al., 2007). In the more recent study by Grobbelaar et al., the 2003 Mauritanian viruses were clustered into lineage C, together with strains isolated in Kenya in 2007, South Africa in 2008 and 2009 and Madagascar in 2008 (Grobbelaar et al., 2011). Lineage C also includes strains isolated in Zimbabwe in 1976–1979 and 1998, suggesting that viruses originating in Zimbabwe might be responsible for outbreaks in Mauritania, Kenya, South Africa and Madagascar (Bird et al., 2007c; Carroll et al., 2011).
4.3.9. Kenya, 2006–2007
Several large epizootics and epidemics of RVF have been reported in Kenya since the first outbreak was recognized in 1930. The virus is maintained through vertical transmission in local Aedes mosquitoes (Linthicum et al., 1985). According to the most recent analysis, Kenyan viruses fall within lineages B, C, K and L, with the majority in lineage C, indicating that multiple lineages circulate in parallel among local reservoir species (Grobbelaar et al., 2011). Little sequence divergence is observed over time: viruses isolated from a cow in 1977 and from Aedes mcintoshi in 1983 are closely related to the strains that were responsible for outbreaks in 1997–1998 and 2006–2007 (Bird et al., 2008; Grobbelaar et al., 2011).
In addition to the indigenous RVFV strains, several viruses isolated in Kenya have clustered within lineage K, suggesting that they originated from the live, mouse-passaged Smithburn vaccine strain (Grobbelaar et al., 2011). These include the Rintoul virus, isolated from a sheep in 1951, and strains B314 and B674, recovered from cattle in 1962 and 1963. In contrast, viruses Ken56/B2653/IB8 and Kitale 1840, isolated from cattle in 1963 and 1964, are in lineage L, close to the Egyptian isolate 95EG Cow-2509. Considering that the Smithburn vaccine was introduced into Kenya in 1951, it seems likely that naturally occurring viruses in lineages K and L are descended from the vaccine (see below) (Grobbelaar et al., 2011).
5. Consequences of the use of live and inactivated livestock vaccines
The widespread use of livestock vaccines derived from wild-type RVFV may add additional diversity to the pool of circulating viruses. When a live, attenuated vaccine is used in the setting of an outbreak, there is the possibility that a viremic animal will be inoculated, providing opportunities for reassortment between wild and vaccine viruses. As noted above, the occurrence of such an event was identified by Grobbelaar et al. in a reassortant virus (SA184/10) recovered from a person who suffered a needle injury while vaccinating sheep with Smithburn vaccine (Grobbelaar et al., 2011). The M segment of the virus localized to lineage K, close to the vaccine sequence, but its S and L segments were clustered with SA54/10 in lineage H, indicating that the individual was infected with the wild-type virus at the time he was accidentally inoculated with the vaccine.
An additional factor complicating the situation is that the multiply mouse-brain-passaged Smithburn vaccine virus retains a certain degree of virulence, and can cause abortions in pregnant females and illness in young calves and lambs. This was demonstrated in an experiment in Egypt in 1995, when European cattle and native buffalo were vaccinated with a locally produced Smithburn vaccine (Botros et al., 2006). Within 72 days post-vaccination, 28 cattle aborted, but no abortion occurred among native buffalo. Interestingly, virus 95EG Cow-2509, isolated from an aborted fetus, had S and L segments corresponding to the Smithburn vaccine sequence in lineage K, but its M segment sequence was closer to viruses in lineage L (Grobbelaar et al., 2011). Because the genetic population of the vaccine has not been characterized, it remains unclear whether the variant occurred through selection of a pre-existing subpopulation or as the result of acquired mutations. This problem could be minimized by employing live, attenuated vaccines that do not produce viremia.
The use of inactivated vaccines also has the potential to increase the diversity of circulating RVFV, if failure to completely inactivate some vaccine batches leads to the re-introduction of live virus to livestock. This possibility is discussed by Ahmed Kamal in a recent review of RVF in Egypt (Ahmed Kamal, 2011). Therefore, it will be important to evaluate the virulence of candidate vaccines and the potential reassortant strains made between the vaccine strain and wt RVFV strain(s).
6. Emergence and evolution of RVFV
Bayesian analyses have indicated that all currently circulating RVFV strains are descended from viruses that emerged in the late 19th century, when European breeds of sheep and cattle were introduced into the Rift Valley region of East Africa on a large scale. The study by Bird et al. indicated that the most recent ancestor of the 33 RVFV strains analyzed existed sometime in the 1880s (Bird et al., 2007c), while the more recent study of partial M-segments of 203 strains dated a most recent common ancestor to 1892 (Grobbelaar et al., 2011). On the other hand, recent study suggests that codon-based substitution model estimates the most common recent ancestor older than that calculated by nucleotide-based substitution model (Wertheim and Kosakovsky Pond, 2011). Therefore, it might also be possible that the most common ancestor could be present earlier than 19th century. Based on the Bayesian analysis by Bird et al., the mean molecular evolutionary rates of the S-, M- and L-segments of RVFV were estimated to be 2.35 × 10−4, 2.42 × 10−4, and 2.78 × 10−4 nucleotide substitution per site per year, respectively, similar to rates that have been calculated for other negative-stranded RNA viruses (Bird et al., 2007c). However, it seems clear that the rate of RVFV evolution may be accelerated when the virus spreads from an area of low-level enzootic maintenance to produce epizootics and epidemics in new geographic regions, vastly increasing the number of mammalian hosts and the turnover of virus. Diversity may also be increased through reassortment among circulating viruses and between wild viruses and live vaccine strains.
7. Quasispecies
No information is currently available on the presence of quasispecies in animals or humans naturally infected with RVFV. However, the isolation of Smithburn strain from vaccinated cattle suggests the presence of viral subpopulations affecting the outcome of disease (Botros et al., 2006). Another study suggested the presence of quasispecies in strains used for laboratory experiments. Thus, when the M segment of ZH501 strain was first sequenced, the nucleotide at position 847 was G (Collett et al., 1985), but it was later identified as A (Battles and Dalrymple, 1988). Morrill et al. demonstrated that strain ZH501 contains two major populations, with either A or G at nt 847 (Morrill et al., 2010). The G-containing recombinant virus is moderately attenuated, causing delayed death in mice, rather than acute, lethal hepatitis (Morrill et al., 2010). It is unknown whether this population was present in the original viral population sampled from a fatal human case, or emerged during repeated passages in suckling mouse brain, fetal rhesus lung and Vero E6 cells (Bird et al., 2007c). A minor variant present in a natural RVFV isolate could have become the major population during replication in the laboratory.
It is now feasible to characterize the entire genetic population of a circulating RVFV strain by performing deep sequencing of individual PCR amplicons. Such information will help to identify genetic markers in specific lineages, so as to understand viral adaptation to specific hosts. In addition, it might be possible to use deep sequencing to identify differences in the genetic signatures of laboratory-derived and wild strains, helping to establish the origin of naturally occurring or deliberately-induced outbreaks.
8. Variation in virulence
Several studies in the 1980s identified major differences in the virulence of RVFV isolates for laboratory rodents. Battles et al. studied 22 different viruses and found that their LD50 values for outbred mice ranged from 2 to more than 106 PFU, and the range was even greater for C57BL6 mice (Battles and Dalrymple, 1988). Anderson and Peters evaluated the virulence of various RVFV isolates, and found that viruses from Egypt were more virulent for WF rats than sub-Saharan viruses, and were less susceptible in vitro to treatment with rat interferon (Anderson and Peters, 1988). More recent analysis of the same data showed that viruses virulent for WF rats are in lineage A, while the less virulent strains belong to lineages C, D and E (Fig. 5) (Bird et al., 2007c). The authors identify a number of amino acid differences in the Gn and Gc proteins that might be responsible for those differences in virulence.
9. Sequence-based detection of RVFV
Detection of RVFV RNA is important for early diagnosis, when infected humans or animals are viremic. Quantitative, sensitive and specific detection of viral genomic RNA is also ideal for testing the effects of antiviral compounds or vaccines. The following sections discuss the advantages and disadvantages of current technologies for sequence-based diagnostics.
9.1. Reverse transcriptase-polymerase chain reaction (RT-PCR)
In the first published study of the use of RT-PCR to detect RVFV, Ibrahim et al. used a Gn-specific primer set to detect RNA in Egyptian Cx. pipiens mosquitoes experimentally infected with strain ZH501 (Ibrahim et al., 1997). The first RT-PCR could detect 50 pfu of virus, while nested RT-PCR could detect 0.5 pfu. The assay could detect RVFV RNA from a single infected mosquito. Jupp et al. also selected the Gn region to amplify RVFV sequences from Ae. aegypti and Eretmapodites quinquevittatus mosquitoes infected with the 845/78 Zimbabwe strain (Jupp et al., 2000). The assay’s sensitivity was superior to infection of Vero cells, but similar to suckling mouse brain inoculation.
RT-PCR was further established as a diagnostic tool to detect NSs sequence in human and animal sera (Sall et al., 2001). The assay was specific and did not amplify other phlebovirus RNAs; its detection limit in human sera was 50 pfu by single and 0.5 pfu by nested RT-PCR. Single RT-PCR was sufficient to detect viremia in infected sheep, while nested RT-PCR was needed to detect viremia in experimentally infected mice. Single RT-PCR also successfully also detected RVFV RNA in samples from patients in Mauritania; a subsequent study showed that the sensitivity was 70.6% and the specificity 97.1%, compared to virus isolation (Sall et al., 2002). The findings suggested that RT-PCR worked better for samples collected early in infection, before IgM was present (Sall et al., 2002).
A generic RT-nested-PCR, designed to detect any phlebovirus, was able to detect RVF, Toscana, sandfly fever, Aguacate, and Punta Toro viruses (Sanchez-Seco et al., 2003).
More recently, a multiplex RT-PCR was developed to detect RVFV, bluetongue virus, rinderpest virus and Peste des petits ruminants virus (Yeh et al., 2011). The assay employs dual-priming oligonucleotides (DPO), which contain two separate priming regions joined by a polydeoxyinosine linker. The assay was highly specific and sensitive, and could detect as little as 101.1 TCID50/ml of RVFV in a blood sample. The primers used for the RT-PCR are shown in Table 1.
Table 1.
The primers and probes used for RT-PCR or real-time RT-PCR for detecting RVFV RNA.
| Primer and probe sequence (5′ to 3′) | Probe label | Target RNA | 5′ position (anti-viral-sense) | References | |
|---|---|---|---|---|---|
| RVF1 | GAC TAC CAG TCA GCT CAT TAC C | M (Gn) | 777 | Ibrahim et al. (1997) | |
| RVF2 | TG TGA ACA ATA GGC ATT GG | M (Gn) | 1327 | ||
| RVF3 | CAG ATG ACA GGT GCT AGC | M (Gn) | 876 | ||
| RVF4 | CT ACC ATG TCC TCC AAT CTT GG | M (Gn) | 1249 | ||
| Primer No. 1 | CCA AAT GAC TAC CAG TCA GC | M (Gn) | 771 | Jupp et al. (2000) | |
| Primer No. 2 | GAC AAA TGA GTC TGG TAG AC | M (Gn) | 1139 | ||
| NSca | CCT TAA CCT CTA ATC AAC | S (NSs) | 850 | Sall et al. (2001) | |
| NSng | TAT CAT GGA TTA CTT TCC | S (NSs) | 1660 | ||
| NS3a | ATG CTG GGA AGT GAT GAG CG | S (NSs) | 962 | ||
| NS2 g | GAT TTG CAG AGT GGT CGT C | S (NSs) | 1629 | ||
| NPhlebo1+ | ATG GAR GGI TTT GTI WSI CII CC | L | 2044 | Sanchez-Seco et al. (2003) | |
| NPhlebo1- | AAR TTR CTI GWI GCY TTI ARI GTI GC | L | 2597 | ||
| Nphlebo2+ | WTI CCI AAI CCI YMS AAR ATG | L | 2071 | ||
| NPhlebo2- | TCY TCY TTR TTY TTR ARR TAR CC | L | 2315 | ||
| F-1 | CCT GGC CTC TTG GAG AAC IIIII CTG GCT TTC TT | S (NSs) | 1322 | Yeh et al. (2011) | |
| F-2 | CCT GGC CTC TTG GAG AAC IIIII CTG GCC TTC TT | S (NSs) | 1322 | ||
| R-1 | GCA CAG GTC AAT CCC TCT GA IIIII GCC TCA GTC | S (NSs) | 1123 | ||
| R-2 | GCA CAG GTC AAT CCC TCT GA IIIII GCC TCG GTC | S (NSs) | 1123 | ||
| NS3m | ATG CTG GGA AGT GAT GAG | S (NSs) | 962 | Garcia et al. (2001) | |
| S432 | ATG ATG ACA TTA GAA GGG A | S (NSs) | 1259 | ||
| CRSSAr | ATT GAC CTG TGC CTG TTG CC | 5′: FAM, 3′: TAMRA | S (NSs) | 1134 | |
| RVS | AAA GGA ACA ATG GAC TCT GGT CA | M (Gn) | 1164 | Drosten et al. (2002) | |
| RVAs | CAC TTC TTA CTA CCA TGT CCT CCA AT | M (Gn) | 1258 | ||
| RVP | AAA GCT TTG ATA TCT CTC AGT GCC CCA A | 5′: FAM, 3′: TAMRA, 3′ end phosphorylation | M (Gn) | 1204 | |
| RVFL-2912fwdGG | TGA AAA TTC CTG AGA CAC ATG G | L | 2912 | Bird et al. (2007b) | |
| RVFL-2981revAC | ACT TCC TTG CAT CAT CTG ATG | L | 3001 | ||
| RVFL-probe-2950 | CAA TGT AAG GGG CCT GTG TGG ACT TGT G | 5′: FAM, 3′: BHQ1 | L | 2950 | |
| RVFV S233F | AAG GCA AAG CAA CTG TGG AG | S (N) | 271 | Naslund et al. (2008) | |
| RVFV S388R | CAG TGA CAG GAA GCC ACT CA | S (N) | 426 | ||
| RVF FP | TGC CAC GAG TYA GAG CCA | S (N) | 234 | Weidmann et al. (2008) | |
| RVF RP | GTG GGT CCG AGA GTY TGC | S (N) | 107 | ||
| RVF P | TCC TTC TCC CAG TCA GCC CCA C | 5′: FAM, 3′: TAMRA | S (N) | 196 | |
| RVF-1F | GAC GCA GCA TTT TGC TCT GCT TAT G | M (Gn) | 1266 | He et al. (2009) | |
| RVF-1R | GTT GTG CAA GGC TCA ACT CTC TGG ATG | M (Gn) | 1462 | ||
| RVF-P | CTT TAT GTG TAG GGT ATG AGA GAG TGG TTG TGA | 5′: HRP | M (Gn) | 1381 |
FAM, 6-carboxyfluorescein; TAMRA, 6-carboxy-tetramethyl-rhodamine; BHQ1, Black Hole Quencher-1; HRP, horseradish peroxidase.
9.2. Real-time RT-PCR
A number of real-time RT-PCR assays have been developed since 2001 for the detection and quantitation of RVFV. The first employed primers and probe specific to the NSs gene, and was able to amplify RVFV strains from all regions of Africa (Garcia et al., 2001). It could detect less than TCID50/ml of MP-12 virus in the supernatant of infected Vero E6 cells. The authors employed the system to demonstrate a reduction in RVFV copy number in cells treated with several antiviral compounds. Another assay, reported by Drosten et al., employed multiple probes to diagnose 8 different types of viral hemorrhagic fever (Drosten et al., 2002). For RVFV, the detection limit was 2.8 × 103 genome equivalents per ml of plasma (8.6–16 RNA copies per reaction).
More recently, a highly sensitive and broadly reactive real-time RT-PCR assay for RVFV has been developed, targeting the L segment (Bird et al., 2007b). To address potential mismatches of previously published primers and probes to different RVFV strains, the authors designed primers and probes specific to highly conserved regions among 40 different RVFV strains. The assay successfully detected viral RNA from 21 distinct strains representing all lineage; its limit of detection was 5 RNA copies per reaction (0.1 pfu of virus ZH501) (Bird et al., 2007b). The assay could detect viral RNA in serum samples at an early stage of infection (days 1–4). The authors found that viral RNA decreased as IgM appeared in serum, and that fatal cases have persistently high levels of circulating RNA.
Another broadly reactive real-time RT-PCR, based on the S segment, with a reported detection limit of 102 RNA copies per reaction, has also been described (Weidmann et al., 2008). The assay can detect 37 different RVFV strains. An inexpensive SYBR Green real-time RT-PCR assay for RVFV has also been developed, with a detection limit of approximately 300 RNA copies (Naslund et al., 2008). A large multiplex PCR assay (“Bio T” RNA assay) has been developed for detecting RVFV and other emerging RNA viruses (He et al., 2009). Its limit of detection is 1.5 × 103 RNA copies per reaction for RVFV. The primers and probes used for the real-time PCR are shown in Table 1.
9.3. Loop-Mediated Isothermal Amplification (LAMP)
Reverse-transcription Loop-Mediated Isothermal Amplification (LAMP) has also been used to detect RVFV (Table 2) (Le Roux et al., 2009; Peyrefitte et al., 2008). The assay uses a set of 6 primers, and is performed at a constant temperature of 60–65 °C without cyclic denaturation of template DNA. Primers bind to the single stranded loop, formed by the unique primer design, which is complementary to the downstream sequence of the primer (Notomi et al., 2000). The reaction is sensitive, specific, and does not require a thermal cycler, which is useful for field diagnostics of pathogens.
Table 2.
The primers for Loop-Mediated Isothermal Amplification for RVFV.
| Primers for LAMP (5′ to 3′) | L gene nucleotides* | References | |
|---|---|---|---|
| F3-RVF (Fwd outer) | TTT CTA AGA TGG GGA GAG C | 6119–6137 | Peyrefitte et al. (2008) |
| B3-RVF (Rev outer) | AAA TCT AAG CCT GAC TTT GC | 6307–6326 | |
| FIP-RVF (Fwd inner: F1c-TTTT-F2) | TGT AGA GTT CTG AAG AGA ACC TTT G TTTT TAG GAA AGT TCT AGA GAC AGG | F1c: 6194–6218, F2: 6144–6164 | |
| BIP-RVF (Rev inner: B1c-TTTT-B2) | GAG ACC AGA AGA GAG CAT TAG GAT TTTT TGG TCT TAG CCT AGC ATG T | B1c: 6219–6242, B2: 6284–6302 | |
| LFP-RVF (Fwd loop) | TCT TTG CTG GAG CAC CGA | 6165–6182 | |
| BLP-RVF (Back loop) | ATC TGG AGT TAT ATG AGG AGA CAG A | 6248–6272 | |
| F3 (Fwd outer) | TGG GGA TCT AGG AAG AAG TT | 4081–4100 | Le Roux et al. (2009) |
| B3 (Rev outer) | GAG GCC ATG ACT TTA CAA ACT | 4269–4289 | |
| F1c-TTTT-F2 (Fwd inner) | AGC ACC TCT GGA TTC TCA TTT ATT TTTT CAG AAA TTG AGA GAC CGT TT | F1c: 4149–4172, F2: 4102–4121 | |
| B1c-TTTT-B2 (Rev inner) | AGA ACA GGC CCA GAA ATA TTG T TTTT GAC AAT GAT GAC ACA ACA C | B1c: 4186–4207, B2: 4238–4256 | |
| LF (Fwd loop) | GTT CAA TCC AGT TCT CTG GTA TGT T | 4123–4147 | |
| LB (Back loop) | CAT TGC AGA GAA AGT CCA TAG CC | 4212–4234 |
Bold TTTT represents the linker between oligoes of F1c and F2, or those of B1c and B2.
represents nucleotide positions at anti-viral-sense.
Two different LAMP assays targeting the L segment have been developed (Le Roux et al., 2009; Peyrefitte et al., 2008). Both can detect low copy numbers of RVFV within 30 min, much faster than real-time RT-PCR. The detection limit is approximately 10 RNA copies per assay. LAMP assays were successful for a wide range of RVFV strains, but were negative for other phleboviruses. In a test of 32 human sera that were positive by virus isolation, all but one were positive by RT-LAMP and TaqMan RT-PCR (Drosten et al., 2002) (Le Roux et al., 2009). Monitoring the accumulation of magnesium pyrophosphate by spectrophotometry allowed quantitative comparison among samples.
10. Conclusion
The strains of RVFV that have caused epizootics and epidemics since the early 20th century appear to be derived from viruses that emerged from a local reservoir when large-scale sheep and cattle farming were introduced into Africa. The comparatively short history of dispersion, combined with the restriction of variation imposed on this arbovirus by its need to replicate both in mammals and mosquitoes, has resulted in comparatively little sequence variation among identified RVFV strains. As a consequence of the high degree of sequence conservation of the M segment genes encoding the virion surface glycoproteins, a single vaccine should be able to elicit protective immunity against all currently circulating viruses. Similarly, the high degree of conservation of the L segment, including segments that encode the functional motifs of the RNA-dependent RNA polymerase, suggests that an antiviral drug targeting the enzyme will be equally effective against all viral strains. The presence of highly conserved regions of the viral genome should also make it possible to develop diagnostic probes that can detect all RVFV strains. At the same time, the existence of a large sequence database will aid efforts to determine the geographic source of the virus in the event of new outbreaks. Continued collection and whole-genome analysis of viral isolates should be actively encouraged, to enhance our understanding of how RVFV circulates within its “ancestral homeland”, how it spreads outside that originally circulating region, and how it is able to establish an enzootic presence in new areas.
Acknowledgments
I thank Mike Bray at NIH for his critical review and communications with author to improve the manuscript; Brian Bird, Serena Carroll and Stuart Nichol at the Centers for Disease Control and Prevention, Atlanta, GA for their permission to generate Figs. 5, 7 and 8 as well as a critical review and comments by Brian Bird; Bob Swanepoel at the National Institute for Communicable Diseases, Sandringham, South Africa, for permission to use Fig. 6; Alex Freiberg at The University of Texas Medical Branch (UTMB) for providing Fig. 3, and Scott Weaver at UTMB for providing Fig. 1 and for critical review of the manuscript. This study has been performed as a part of the SRI project #P19177 (DHS contract#HSHQDC-09-C-00120). T.I. is supported by 5 U54 AI057156 through the Western Regional Center of Excellence, by NIH Grant R01 AI08764301, and by the Sealy Center for Vaccine Development at UTMB. The Virus Pathogen Database and Analysis Resource (ViPR) introduced in the text is wholly funded with federal funds from NIH/NIAID and Department of Health and Human Services under Contract #HHSN272200900041C.
References
- Ahmed J, Bouloy M, Ergonul O, Fooks A, Paweska J, Chevalier V, Drosten C, Moormann R, Tordo N, Vatansever Z, Calistri P, Estrada-Pena A, Mirazimi A, Unger H, Yin H, Seitzer U. International network for capacity building for the control of emerging viral vector-borne zoonotic diseases: ARBO-ZOONET. Euro Surveill. 2009;1419160 [PubMed] [Google Scholar]
- Ahmed Kamal S. Observations on rift valley fever virus and vaccines in Egypt. Virol J. 2011;8:532. doi: 10.1186/1743-422X-8-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albarino CG, Bird BH, Nichol ST. A shared transcription termination signal on negative and ambisense RNA genome segments of Rift Valley fever, sandfly fever Sicilian, and Toscana viruses. J Virol. 2007;81:5246–5256. doi: 10.1128/JVI.02778-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GW, Jr, Peters CJ. Viral determinants of virulence for Rift Valley fever (RVF) in rats. Microb Pathog. 1988;5:241–250. doi: 10.1016/0882-4010(88)90096-4. [DOI] [PubMed] [Google Scholar]
- Anderson GW, Jr, Slone TW, Jr, Peters CJ. The gerbil, Meriones unguiculatus, a model for Rift Valley fever viral encephalitis. Arch Virol. 1988;102:187–196. doi: 10.1007/BF01310824. [DOI] [PubMed] [Google Scholar]
- Andriamandimby SF, Randrianarivo-Solofoniaina AE, Jeanmaire EM, Ravololomanana L, Razafimanantsoa LT, Rakotojoelinandrasana T, Razainirina J, Hoffmann J, Ravalohery JP, Rafisandratantsoa JT, Rollin PE, Reynes JM. Rift Valley fever during rainy seasons, Madagascar, 2008 and 2009. Emerg Infect Dis. 2010;16:963–970. doi: 10.3201/eid1606.091266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur RR, el-Sharkawy MS, Cope SE, Botros BA, Oun S, Morrill JC, Shope RE, Hibbs RG, Darwish MA, Imam IZ. Recurrence of Rift Valley fever in Egypt. Lancet. 1993;342:1149–1150. doi: 10.1016/0140-6736(93)92128-g. [DOI] [PubMed] [Google Scholar]
- Atwa MH, El-Sabagh IM, Amer HM, Saad S, Yousif AA, Shaheen MA. ZH501-VSVRI: is it still the best choice for vaccination against Rift Valley fever in Egypt? Vaccine & Vaccination. 2011;2:1000121. [Google Scholar]
- Battles JK, Dalrymple JM. Genetic variation among geographic isolates of Rift Valley fever virus. Am J Trop Med Hyg. 1988;39:617–631. doi: 10.4269/ajtmh.1988.39.617. [DOI] [PubMed] [Google Scholar]
- Besselaar TG, Blackburn NK, Meenehan GM. Antigenic analysis of Rift Valley fever virus isolates: monoclonal antibodies distinguish between wild-type and neurotropic virus strains. Res Virol. 1991;142:469–474. doi: 10.1016/0923-2516(91)90069-f. [DOI] [PubMed] [Google Scholar]
- Billecocq A, Spiegel M, Vialat P, Kohl A, Weber F, Bouloy M, Haller O. NSs protein of Rift Valley fever virus blocks interferon production by inhibiting host gene transcription. J Virol. 2004;78:9798–9806. doi: 10.1128/JVI.78.18.9798-9806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird BH, Albarino CG, Nichol ST. Rift Valley fever virus lacking NSm proteins retains high virulence in vivo and may provide a model of human delayed onset neurologic disease. Virology. 2007a;362:10–15. doi: 10.1016/j.virol.2007.01.046. [DOI] [PubMed] [Google Scholar]
- Bird BH, Bawiec DA, Ksiazek TG, Shoemaker TR, Nichol ST. Highly sensitive and broadly reactive quantitative reverse transcription-PCR assay for high-throughput detection of Rift Valley fever virus. J Clin Microbiol. 2007b;45:3506–3513. doi: 10.1128/JCM.00936-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird BH, Githinji JW, Macharia JM, Kasiiti JL, Muriithi RM, Gacheru SG, Musaa JO, Towner JS, Reeder SA, Oliver JB, Stevens TL, Erickson BR, Morgan LT, Khristova ML, Hartman AL, Comer JA, Rollin PE, Ksiazek TG, Nichol ST. Multiple virus lineages sharing recent common ancestry were associated with a Large Rift Valley fever outbreak among livestock in Kenya during 2006–2007. J Virol. 2008;82:11152–11166. doi: 10.1128/JVI.01519-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird BH, Khristova ML, Rollin PE, Ksiazek TG, Nichol ST. Complete genome analysis of 33 ecologically and biologically diverse Rift Valley fever virus strains reveals widespread virus movement and low genetic diversity due to recent common ancestry. J Virol. 2007c;81:2805–2816. doi: 10.1128/JVI.02095-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird BH, Ksiazek TG, Nichol ST, Maclachlan NJ. Rift Valley fever virus. J Am Vet Med Assoc. 2009;234:883–893. doi: 10.2460/javma.234.7.883. [DOI] [PubMed] [Google Scholar]
- Botros B, Omar A, Elian K, Mohamed G, Soliman A, Salib A, Salman D, Saad M, Earhart K. Adverse response of non-indigenous cattle of European breeds to live attenuated Smithburn Rift Valley fever vaccine. J Med Virol. 2006;78:787–791. doi: 10.1002/jmv.20624. [DOI] [PubMed] [Google Scholar]
- Bouloy M, Janzen C, Vialat P, Khun H, Pavlovic J, Huerre M, Haller O. Genetic evidence for an interferon-antagonistic function of rift valley fever virus nonstructural protein NSs. J Virol. 2001;75:1371–1377. doi: 10.1128/JVI.75.3.1371-1377.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SA, Reynes JM, Khristova ML, Andriamandimby SF, Rollin PE, Nichol ST. Genetic evidence for Rift Valley fever outbreaks in Madagascar resulting from virus introductions from the East African mainland rather than enzootic maintenance. J Virol. 2011;85:6162–6167. doi: 10.1128/JVI.00335-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers PG, Swanepoel R. Rift valley fever in abattoir workers. Cent Afr J Med. 1980;26:122–126. [PubMed] [Google Scholar]
- Christie GJ. Rift Valley fever. Rhodesia Sci News. 1969;3:238–240. [Google Scholar]
- Coffey LL, Vasilakis N, Brault AC, Powers AM, Tripet F, Weaver SC. Arbovirus evolution in vivo is constrained by host alternation. Proc Natl Acad Sci USA. 2008;105:6970–6975. doi: 10.1073/pnas.0712130105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collett MS, Purchio AF, Keegan K, Frazier S, Hays W, Anderson DK, Parker MD, Schmaljohn C, Schmidt J, Dalrymple JM. Complete nucleotide sequence of the M RNA segment of Rift Valley fever virus. Virology. 1985;144:228–245. doi: 10.1016/0042-6822(85)90320-4. [DOI] [PubMed] [Google Scholar]
- Daubney R, Hudson JR. Enzootic hepatitis or Rift Valley fever: an undescribed virus disease of sheep cattle and man from east Africa. J Path Bact. 1931;34:545–579. [Google Scholar]
- Drosten C, Gottig S, Schilling S, Asper M, Panning M, Schmitz H, Gunther S. Rapid detection and quantification of RNA of Ebola and Marburg viruses, Lassa virus, Crimean-Congo hemorrhagic fever virus, Rift Valley fever virus, dengue virus, and yellow fever virus by real-time reverse transcription-PCR. J Clin Microbiol. 2002;40:2323–2330. doi: 10.1128/JCM.40.7.2323-2330.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand JP, Bouloy M, Richecoeur L, Peyrefitte CN, Tolou H. Rift Valley fever virus infection among French troops in Chad. Emerg Infect Dis. 2003;9:751–752. doi: 10.3201/eid0906.020647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faye O, Diallo M, Diop D, Bezeid OE, Ba H, Niang M, Dia I, Mohamed SA, Ndiaye K, Diallo D, Ly PO, Diallo B, Nabeth P, Simon F, Lo B, Diop OM. Rift Valley fever outbreak with East-Central African virus lineage in Mauritania, 2003. Emerg Infect Dis. 2007;13:1016–1023. doi: 10.3201/eid1307.061487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron F, Li Z, Danek EI, Luo D, Wong Y, Coutard B, Lantez V, Charrel R, Canard B, Walz T, Lescar J. The hexamer structure of Rift Valley fever virus nucleoprotein suggests a mechanism for its assembly into ribonucleoprotein complexes. PLoS Pathog. 2011;7:e1002030. doi: 10.1371/journal.ppat.1002030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay GM. Rift Valley fever on enzootic hepatitis. Trans R Soc Trop Med Hyg. 1931;25:229–262. [Google Scholar]
- Freiberg AN, Sherman MB, Morais MC, Holbrook MR, Watowich SJ. Three-dimensional organization of Rift Valley fever virus revealed by cryoelectron tomography. J Virol. 2008;82:10341–10348. doi: 10.1128/JVI.01191-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gad AM, Feinsod FM, Allam IH, Eisa M, Hassan AN, Soliman BA, el Said S, Saah AJ. A possible route for the introduction of Rift Valley fever virus into Egypt during 1977. J Trop Med Hyg. 1986;89:233–236. [PubMed] [Google Scholar]
- Garcia S, Crance JM, Billecocq A, Peinnequin A, Jouan A, Bouloy M, Garin D. Quantitative real-time PCR detection of Rift Valley fever virus and its application to evaluation of antiviral compounds. J Clin Microbiol. 2001;39:4456–4461. doi: 10.1128/JCM.39.12.4456-4461.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauliard N, Billecocq A, Flick R, Bouloy M. Rift Valley fever virus noncoding regions of L, M and S segments regulate RNA synthesis. Virology. 2006;351:170–179. doi: 10.1016/j.virol.2006.03.018. [DOI] [PubMed] [Google Scholar]
- Gear J, De Meillon B, Measroch V, Davis DH, Harwin H. Rift valley fever in South Africa. 2. The occurrence of human cases in the Orange Free State, the North-Western Cape Province, the Western and Southern Transvaal. B. Field and laboratory investigation. S Afr Med J. 1951;25:908–912. [PubMed] [Google Scholar]
- Gerrard SR, Bird BH, Albarino CG, Nichol ST. The NSm proteins of Rift Valley fever virus are dispensable for maturation, replication and infection. Virology. 2007;359:459–465. doi: 10.1016/j.virol.2006.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrard SR, Nichol ST. Characterization of the Golgi retention motif of Rift Valley fever virus G(N) glycoprotein. J Virol. 2002;76:12200–12210. doi: 10.1128/JVI.76.23.12200-12210.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrard SR, Nichol ST. Synthesis, proteolytic processing and complex formation of N-terminally nested precursor proteins of the Rift Valley fever virus glycoproteins. Virology. 2007;357:124–133. doi: 10.1016/j.virol.2006.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgi C, Accardi L, Nicoletti L, Gro MC, Takehara K, Hilditch C, Morikawa S, Bishop DH. Sequences and coding strategies of the S RNAs of Toscana and Rift Valley fever viruses compared to those of Punta Toro, Sicilian Sandfly fever, and Uukuniemi viruses. Virology. 1991;180:738–753. doi: 10.1016/0042-6822(91)90087-r. [DOI] [PubMed] [Google Scholar]
- Grobbelaar AA, Weyer J, Leman PA, Kemp A, Paweska JT, Swanepoel R. Molecular epidemiology of Rift Valley fever virus. Emerg Infect Dis. 2011;17:2270–2276. doi: 10.3201/eid1712.111035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habjan M, Pichlmair A, Elliott RM, Overby AK, Glatter T, Gstaiger M, Superti-Furga G, Unger H, Weber F. NSs protein of rift valley fever virus induces the specific degradation of the double-stranded RNA-dependent protein kinase. J Virol. 2009;83:4365–4375. doi: 10.1128/JVI.02148-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanafi HA, Fryauff DJ, Saad MD, Soliman AK, Mohareb EW, Medhat I, Zayed AB, Szumlas DE, Earhart KC. Virus isolations and high population density implicate Culex antennatus (Becker) (Diptera: Culicidae) as a vector of Rift Valley Fever virus during an outbreak in the Nile Delta of Egypt. Acta Trop. 2011;119:119–124. doi: 10.1016/j.actatropica.2011.04.018. [DOI] [PubMed] [Google Scholar]
- He J, Kraft AJ, Fan J, Van Dyke M, Wang L, Bose ME, Khanna M, Metallo JA, Henrickson KJ. Simultaneous Detection of CDC Category “A” DNA and RNA Bioterrorism Agents by Use of Multiplex PCR & RT-PCR Enzyme Hybridization Assays. Viruses. 2009;1:441–459. doi: 10.3390/v1030441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huiskonen JT, Overby AK, Weber F, Grunewald K. Electron cryomicroscopy and single-particle averaging of Rift Valley fever virus: evidence for GN-GC glycoprotein heterodimers. J Virol. 2009;83:3762–3769. doi: 10.1128/JVI.02483-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim MS, Turell MJ, Knauert FK, Lofts RS. Detection of Rift Valley fever virus in mosquitoes by RT-PCR. Mol Cell Probes. 1997;11:49–53. doi: 10.1006/mcpr.1996.0075. [DOI] [PubMed] [Google Scholar]
- Ikegami T, Makino S. The pathogenesis of Rift Valley fever. Viruses. 2011;3:493–519. doi: 10.3390/v3050493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami T, Narayanan K, Won S, Kamitani W, Peters CJ, Makino S. Rift Valley fever virus NSs protein promotes post-transcriptional downregulation of protein kinase PKR and inhibits eIF2alpha phosphorylation. PLoS Pathog. 2009;5:e1000287. doi: 10.1371/journal.ppat.1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami T, Peters CJ, Makino S. Rift valley fever virus nonstructural protein NSs promotes viral RNA replication and transcription in a minigenome system. J Virol. 2005a;79:5606–5615. doi: 10.1128/JVI.79.9.5606-5615.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami T, Won S, Peters CJ, Makino S. Rift Valley fever virus NSs mRNA is transcribed from an incoming anti-viral-sense S RNA segment. J Virol. 2005b;79:12106–12111. doi: 10.1128/JVI.79.18.12106-12111.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami T, Won S, Peters CJ, Makino S. Characterization of Rift Valley fever virus transcriptional terminations. J Virol. 2007;81:8421–8438. doi: 10.1128/JVI.02641-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jupp PG, Grobbelaar AA, Leman PA, Kemp A, Dunton RF, Burkot TR, Ksiazek TG, Swanepoel R. Experimental detection of Rift Valley fever virus by reverse transcription-polymerase chain reaction assay in large samples of mosquitoes. J Med Entomol. 2000;37:467–471. doi: 10.1093/jmedent/37.3.467. [DOI] [PubMed] [Google Scholar]
- Kalveram B, Lihoradova O, Ikegami T. NSs Protein of Rift Valley Fever Virus Promotes Post-Translational Downregulation of the TFIIH Subunit p62. J Virol. 2011;85:6234–6243. doi: 10.1128/JVI.02255-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan K, Collett MS. Use of bacterial expression cloning to define the amino acid sequences of antigenic determinants on the G2 glycoprotein of Rift Valley fever virus. J Virol. 1986;58:263–270. doi: 10.1128/jvi.58.2.263-270.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara E, Billecocq A, Leger P, Bouloy M. Characterization of wild-type and alternate transcription termination signals in the Rift Valley fever virus genome. J Virol. 2011;85:12134–12145. doi: 10.1128/JVI.05322-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le May N, Dubaele S, Proietti De Santis L, Billecocq A, Bouloy M, Egly JM. TFIIH transcription factor, a target for the Rift Valley hemorrhagic fever virus. Cell. 2004;116:541–550. doi: 10.1016/s0092-8674(04)00132-1. [DOI] [PubMed] [Google Scholar]
- Le May N, Mansuroglu Z, Leger P, Josse T, Blot G, Billecocq A, Flick R, Jacob Y, Bonnefoy E, Bouloy M. A SAP30 complex inhibits IFN-beta expression in Rift Valley fever virus infected cells. PLoS Pathog. 2008;4:e13. doi: 10.1371/journal.ppat.0040013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roux CA, Kubo T, Grobbelaar AA, van Vuren PJ, Weyer J, Nel LH, Swanepoel R, Morita K, Paweska JT. Development and evaluation of a real-time reverse transcription-loop-mediated isothermal amplification assay for rapid detection of Rift Valley fever virus in clinical specimens. J Clin Microbiol. 2009;47:645–651. doi: 10.1128/JCM.01412-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linthicum KJ, Davies FG, Kairo A, Bailey CL. Rift Valley fever virus (family Bunyaviridae, genus Phlebovirus). Isolations from Diptera collected during an inter-epizootic period in Kenya. J Hyg (Lond) 1985;95:197–209. doi: 10.1017/s0022172400062434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez N, Muller R, Prehaud C, Bouloy M. The L protein of Rift Valley fever virus can rescue viral ribonucleoproteins and transcribe synthetic genome-like RNA molecules. J Virol. 1995;69:3972–3979. doi: 10.1128/jvi.69.7.3972-3979.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meegan JM. The Rift Valley fever epizootic in Egypt 1977–78. 1. Description of the epizzotic and virological studies. Trans R Soc Trop Med Hyg. 1979;73:618–623. doi: 10.1016/0035-9203(79)90004-x. [DOI] [PubMed] [Google Scholar]
- Miller BR, Godsey MS, Crabtree MB, Savage HM, Al-Mazrao Y, Al-Jeffri MH, Abdoon AM, Al-Seghayer SM, Al-Shahrani AM, Ksiazek TG. Isolation and genetic characterization of Rift Valley fever virus from Aedes vexans arabiensis, Kingdom of Saudi Arabia. Emerg Infect Dis. 2002;8:1492–1494. doi: 10.3201/eid0812.020194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrill JC, Ikegami T, Yoshikawa-Iwata N, Lokugamage N, Won S, Terasaki K, Zamoto-Niikura A, Peters CJ, Makino S. Rapid accumulation of virulent rift valley Fever virus in mice from an attenuated virus carrying a single nucleotide substitution in the m RNA. PLoS ONE. 2010;5:e9986. doi: 10.1371/journal.pone.0009986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrill JC, Jennings GB, Johnson AJ, Cosgriff TM, Gibbs PH, Peters CJ. Pathogenesis of Rift Valley fever in rhesus monkeys: role of interferon response. Arch Virol. 1990;110:195–212. doi: 10.1007/BF01311288. [DOI] [PubMed] [Google Scholar]
- Morvan J, Fontenille D, Saluzzo JF, Coulanges P. Possible Rift Valley fever outbreak in man and cattle in Madagascar. Trans R Soc Trop Med Hyg. 1991a;85:108. doi: 10.1016/0035-9203(91)90178-2. [DOI] [PubMed] [Google Scholar]
- Morvan J, Saluzzo JF, Fontenille D, Rollin PE, Coulanges P. Rift Valley fever on the east coast of Madagascar. Res Virol. 1991b;142:475–482. doi: 10.1016/0923-2516(91)90070-j. [DOI] [PubMed] [Google Scholar]
- Moutailler S, Roche B, Thiberge JM, Caro V, Rougeon F, Failloux AB. Host alternation is necessary to maintain the genome stability of rift valley fever virus. PLoS Negl Trop Dis. 2011;5:e1156. doi: 10.1371/journal.pntd.0001156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller R, Poch O, Delarue M, Bishop DH, Bouloy M. Rift Valley fever virus L segment: correction of the sequence and possible functional role of newly identified regions conserved in RNA-dependent polymerases. J Gen Virol. 1994;75:1345–1352. doi: 10.1099/0022-1317-75-6-1345. [DOI] [PubMed] [Google Scholar]
- Murakami S, Terasaki K, Narayanan K, Makino S. Roles of the coding and noncoding regions of rift valley Fever virus RNA genome segments in viral RNA packaging. J Virol. 2012;86:4034–4039. doi: 10.1128/JVI.06700-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslund J, Lagerqvist N, Lundkvist A, Evander M, Ahlm C, Bucht G. Kinetics of Rift Valley Fever Virus in experimentally infected mice using quantitative real-time RT-PCR. J Virol Methods. 2008;151:277–282. doi: 10.1016/j.jviromet.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Nichol ST, Beaty BJ, Elliott RM, Goldbach RW, Plyusnin A, Tesh RB. The Bunyaviridae. VIIIth Report of the International Committee on Taxonomy of Viruses. 2005:695–716. [Google Scholar]
- Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N, Hase T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28:E63. doi: 10.1093/nar/28.12.e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olaleye OD, Tomori O, Fajimi JL, Schmitz H. Experimental infection of three Nigerian breeds of sheep with the Zinga strain of the Rift Valley Fever virus. Rev Elev Med Vet Pays Trop. 1996;49:6–16. [PubMed] [Google Scholar]
- Olive MM, Goodman SM, Reynes JM. The role of wild mammals in the maintenance of rift valley Fever virus. J Wildl Dis. 2012;48:241–266. doi: 10.7589/0090-3558-48.2.241. [DOI] [PubMed] [Google Scholar]
- Pepin M, Bouloy M, Bird BH, Kemp A, Paweska J. Rift Valley fever virus (Bunyaviridae: Phlebovirus): an update on pathogenesis, molecular epidemiology, vectors, diagnostics and prevention. Vet Res. 2010;41:61. doi: 10.1051/vetres/2010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters CJ, Anderson GWJ. Pathogenesis of Rift Valley Fever. In: Swartz TA, Klingberg MA, Goldblum N, Papier CM, editors. Rift Valley Fever S Karger AG Contr Epidem Biostatist. Vol. 3. Vol. 1981. Switzerland: 1981. pp. 21–41. [Google Scholar]
- Peters CJ, Jones D, Trotter R, Donaldson J, White J, Stephen E, Slone TW., Jr Experimental Rift Valley fever in rhesus macaques. Arch Virol. 1988;99:31–44. doi: 10.1007/BF01311021. [DOI] [PubMed] [Google Scholar]
- Peters CJ, Slone TW. Inbred rat strains mimic the disparate human response to Rift Valley fever virus infection. J Med Virol. 1982;10:45–54. doi: 10.1002/jmv.1890100107. [DOI] [PubMed] [Google Scholar]
- Peyrefitte CN, Boubis L, Coudrier D, Bouloy M, Grandadam M, Tolou HJ, Plumet S. Real-time reverse-transcription loop-mediated isothermal amplification for rapid detection of rift valley Fever virus. J Clin Microbiol. 2008;46:3653–3659. doi: 10.1128/JCM.01188-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper ME, Sorenson DR, Gerrard SR. Efficient cellular release of Rift Valley fever virus requires genomic RNA. PLoS ONE. 2011;6:e18070. doi: 10.1371/journal.pone.0018070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond DD, Piper ME, Gerrard SR, Smith JL. Structure of the Rift Valley fever virus nucleocapsid protein reveals another architecture for RNA encapsidation. Proc Natl Acad Sci USA. 2010;107:11769–11774. doi: 10.1073/pnas.1001760107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter M, Bouloy M, Vialat P, Janzen C, Haller O, Frese M. Resistance to Rift Valley fever virus in Rattus norvegicus: genetic variability within certain ‘inbred’ strains. J Gen Virol. 2000;81:2683–2688. doi: 10.1099/0022-1317-81-11-2683. [DOI] [PubMed] [Google Scholar]
- Sall AA, de AZ PM, Zeller HG, Digoutte JP, Thiongane Y, Bouloy M. Variability of the NS(S) protein among Rift Valley fever virus isolates. J Gen Virol. 1997;78:2853–2858. doi: 10.1099/0022-1317-78-11-2853. [DOI] [PubMed] [Google Scholar]
- Sall AA, Macondo EA, Sene OK, Diagne M, Sylla R, Mondo M, Girault L, Marrama L, Spiegel A, Diallo M, Bouloy M, Mathiot C. Use of reverse transcriptase PCR in early diagnosis of Rift Valley fever. Clin Diagn Lab Immunol. 2002;9:713–715. doi: 10.1128/CDLI.9.3.713-715.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sall AA, Thonnon J, Sene OK, Fall A, Ndiaye M, Baudez B, Mathiot C, Bouloy M. Single-tube and nested reverse transcriptase-polymerase chain reaction for detection of Rift Valley fever virus in human and animal sera. J Virol Methods. 2001;91:85–92. doi: 10.1016/s0166-0934(00)00252-4. [DOI] [PubMed] [Google Scholar]
- Sall AA, Zanotto PM, Sene OK, Zeller HG, Digoutte JP, Thiongane Y, Bouloy M. Genetic reassortment of Rift Valley fever virus in nature. J Virol. 1999;73:8196–8200. doi: 10.1128/jvi.73.10.8196-8200.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Seco MP, Echevarria JM, Hernandez L, Estevez D, Navarro-Mari JM, Tenorio A. Detection and identification of Toscana and other phleboviruses by RT-nested-PCR assays with degenerated primers. J Med Virol. 2003;71:140–149. doi: 10.1002/jmv.10465. [DOI] [PubMed] [Google Scholar]
- Schmaljohn C, Nichol ST. Bunyaviridae. In: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE, editors. Fields Virology. fifth ed. Lippincott, Williams & Wilkins; Philadelphia, PA: 2007. pp. 1741–1789. [Google Scholar]
- Sellers RF, Pedgley DE, Tucker MR. Rift Valley fever, Egypt 1977: disease spread by windborne insect vectors? Vet Rec. 1982;110:73–77. doi: 10.1136/vr.110.4.73. [DOI] [PubMed] [Google Scholar]
- Sherman MB, Freiberg AN, Holbrook MR, Watowich SJ. Single-particle cryo-electron microscopy of Rift Valley fever virus. Virology. 2009;387:11–15. doi: 10.1016/j.virol.2009.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker T, Boulianne C, Vincent MJ, Pezzanite L, Al-Qahtani MM, Al-Mazrou Y, Khan AS, Rollin PE, Swanepoel R, Ksiazek TG, Nichol ST. Genetic analysis of viruses associated with emergence of Rift Valley fever in Saudi Arabia and Yemen, 2000–01. Emerg Infect Dis. 2002;8:1415–1420. doi: 10.3201/eid0812.020195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shone DK. Rift Valley fever in Southern Rhodesia. Cent Afr J Med. 1958;4:284–286. [PubMed] [Google Scholar]
- Smith DR, Bird BH, Lewis B, Johnston SC, McCarthy S, Keeney A, Botto M, Donnelly G, Shamblin J, Albarino CG, Nichol ST, Hensley LE. Development of a Novel Non-human Primate Model for Rift Valley Fever. J Virol. 2011;86:2109–2120. doi: 10.1128/JVI.06190-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DR, Steele KE, Shamblin J, Honko A, Johnson J, Reed C, Kennedy M, Chapman JL, Hensley LE. The pathogenesis of Rift Valley fever virus in the mouse model. Virology. 2010;407:256–267. doi: 10.1016/j.virol.2010.08.016. [DOI] [PubMed] [Google Scholar]
- Struthers JK, Swanepoel R, Shepherd SP. Protein synthesis in Rift Valley fever virus-infected cells. Virology. 1984;134:118–124. doi: 10.1016/0042-6822(84)90277-0. [DOI] [PubMed] [Google Scholar]
- Suzich JA, Kakach LT, Collett MS. Expression strategy of a phlebovirus: biogenesis of proteins from the Rift Valley fever virus M segment. J Virol. 1990;64:1549–1555. doi: 10.1128/jvi.64.4.1549-1555.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanepoel R, Blackburn NK. Demonstration of nuclear immunofluo rescence in Rift Valley fever infected cells. J Gen Virol. 1977;34:557–561. doi: 10.1099/0022-1317-34-3-557. [DOI] [PubMed] [Google Scholar]
- Swanepoel R, Blackburn NK, Efstratiou S, Condy JB. Studies on Rift Valley fever in some African murids (Rodentia: Muridae) J Hyg (Lond) 1978;80:183–196. doi: 10.1017/s0022172400053535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanepoel R, Coetzer JAW. Rift Valley fever. In: Coetzer JAW, Thompson GR, Tustin RD, editors. Infectious Diseases of Livestock with Special Reference to Southern Africa. second ed. Oxford University Press; Cape Town, South Africa: 2004. pp. 1037–1070. [Google Scholar]
- Swanepoel R, Manning B, Watt JA. Fatal Rift Valley fever of man in Rhodesia. Cent Afr J Med. 1979;25:1–8. [PubMed] [Google Scholar]
- Terasaki K, Murakami S, Lokugamage KG, Makino S. Mechanism of tripartite RNA genome packaging in Rift Valley fever virus. Proc Natl Acad Sci USA. 2011;108:804–809. doi: 10.1073/pnas.1013155108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Velden DJ, Meyer JD, Olivier J, Gear JH, McIntosh B. Rift Valley fever affecting humans in South Africa: a clinicopathological study. S Afr Med J. 1977;51:867–871. [PubMed] [Google Scholar]
- Vialat P, Billecocq A, Kohl A, Bouloy M. The S segment of rift valley fever phlebovirus (Bunyaviridae) carries determinants for attenuation and virulence in mice. J Virol. 2000;74:1538–1543. doi: 10.1128/jvi.74.3.1538-1543.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasmoen TL, Kakach LT, Collett MS. Rift Valley fever virus M segment: cellular localization of M segment-encoded proteins. Virology. 1988;166:275–280. doi: 10.1016/0042-6822(88)90174-2. [DOI] [PubMed] [Google Scholar]
- Weaver SC, Reisen WK. Present and future arboviral threats. Antiviral Res. 2010;85:328–345. doi: 10.1016/j.antiviral.2009.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidmann M, Sanchez-Seco MP, Sall AA, Ly PO, Thiongane Y, Lo MM, Schley H, Hufert FT. Rapid detection of important human pathogenic Phleboviruses. J Clin Virol. 2008;41:138–142. doi: 10.1016/j.jcv.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Wertheim JO, Kosakovsky Pond SL. Purifying selection can obscure the ancient age of viral lineages. Mol Biol Evol. 2011;28:3355–3365. doi: 10.1093/molbev/msr170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won S, Ikegami T, Peters CJ, Makino S. NSm and 78-kilodalton proteins of Rift Valley fever virus are nonessential for viral replication in cell culture. J Virol. 2006;80:8274–8278. doi: 10.1128/JVI.00476-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won S, Ikegami T, Peters CJ, Makino S. NSm protein of Rift Valley fever virus suppresses virus-induced apoptosis. J Virol. 2007;81:13335–13345. doi: 10.1128/JVI.01238-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadani FZ, Kohl A, Prehaud C, Billecocq A, Bouloy M. The carboxy-terminal acidic domain of Rift Valley Fever virus NSs protein is essential for the formation of filamentous structures but not for the nuclear localization of the protein. J Virol. 1999;73:5018–5025. doi: 10.1128/jvi.73.6.5018-5025.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh JY, Lee JH, Seo HJ, Park JY, Moon JS, Cho IS, Choi IS, Park SY, Song CS, Lee JB. Simultaneous Detection of Rift Valley Fever, Bluetongue, Rinderpest, and Peste des Petits Ruminants Viruses by a Single-Tube Multiplex Reverse Transcriptase-PCR Assay Using a Dual Priming Oligonucleotide System. J Clin Microbiol. 2011;49:1389–1394. doi: 10.1128/JCM.00710-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamoto-Niikura A, Terasaki K, Ikegami T, Peters CJ, Makino S. Rift Valley fever virus L protein forms a biologically active oligomer. J Virol. 2009;83:12779–12789. doi: 10.1128/JVI.01310-09. [DOI] [PMC free article] [PubMed] [Google Scholar]