

Abstract
Glioma and medulloblastoma represent the most commonly occurring malignant brain tumors in adults and in children respectively. Recent genomic and transcriptional approaches present a complex group of diseases, and delineate a number of molecular subgroups within tumors that share a common histopathology. Differences in cells of origin, regional niches, developmental timing and genetic events all contribute to this heterogeneity. In an attempt to recapitulate the diversity of brain tumors, an increasing array of genetically engineered mouse models (GEMMs) has been developed. These models often utilize promoters and genetic drivers from normal brain development, and can provide insight into specific cells from which these tumors originate. GEMMs show promise in both developmental biology and developmental therapeutics. This review describes numerous murine brain tumor models in the context of normal brain development, and the potential for these animals to impact brain tumor research.
Keywords: development, medulloblastoma, glioma, murine model, transgene
1. Introduction
Human brain tumors comprise a multitude of various different tumors that can be distinguished at both a histological and a molecular level. This review summarizes use of genetically engineered mice to model glioma and the embryonal tumors medulloblastoma (MB) and primitive neuroectodermal tumors (PNETs), focused on developmental aspects and molecular pathways.
2. Normal Brain Development and Brain Tumor Development
The brain is formed during embryogenesis and continues to develop after birth. The central nervous system (CNS) arises from the neural plate on day 19 in human embryos. As the plate folds along its anteroposterior axis (primary neurulation), it develops into a tube with vesicles that are the anlagen of fore- mid- and hindbrain [1]. More posteriorly the narrow tube develops into the spinal cord, which forms both from primary and secondary neurulation, the latter process referring to tube formation arising through canalization of a solid cylindrical neural structure. Neural tube closure begins at Embryonic Day (E) 8.5 and finishes at E10.5 in mice. Proper neurulation of the CNS relies on the inhibition of bone morphogenetic protein (BMP) signaling [2].
2.1 Developing forebrain and cellular origin of gliomas
Cortical neurogenesis begins around E9-E10 in the mouse where Fibroblast Growth Factor (FGF)-driven neuroepithelial cells line the forebrain ventricles and spinal canal giving rise to radial glial cells [3]. Radial glial cells are primary progenitor cells that eventually give rise to mature neurons as well as oligodendrocytes, astrocytes and ependymal cells [4] (Figure 1). The differentiation of these mature cells starts from radial glia-derived progenitors and are driven by specific developmental morphogens which promote brain patterning and give rise to the complex brain structure [1]. Radial glia or neural stem cells (NSCs) are bipolar, and divide asymmetrically to enable both self-renewal, and to generate progenitors and more differentiated brain cells that form the adult brain. In humans (and to a lesser degree in rodents), a group of unipolar radial glia-like cells arising from the outer subventricular zone (so-called ORG cells) have been shown recently to contribute to both neural migration and the expanded numbers of neurons in human cortex [5].
Figure 1. Cells involved in normal brain development from embryo to adult.

The schematic figure shows regions and cells of origin for glioma (in forebrain, above) and MB (in cerebellum, below). Processes of the radial glia (RG) serve as guides for migrating more immature neurons [196,197]. RG born from neuroepithelial stem cells could give rise to basically all other cell types including ependymal cells that line the ventricles. However, they first give rise to more restricted intermediate progenitors that determine the path for astrocytes (protoplasmic (in GM) or fibrous (in WM)), oligodendrocytes and neurons. Radial glia cells form neural stem cells, specifically called type B cells (SVZ astrocytes) that reside in the subventricular zone (SVZ) even in the adult forebrain [12]. Similar adult neural stem cells also exist in the dentate gyrus granule cell layer in the hippocampus [13]. Radial glia form neural stem cells that can give rise to multiple cerebellar cell types, and also more directly (around birth) convert into Bergmann glia that keep their extended processes even in adult brain. In cerebellum, granule precursor cells are also formed and divide in the external germinal layer (EGL) from the boost of SHH-producing large Purkinje neurons. Before the cerebellar EGL layer disappears (around three weeks of age in mice) the GPCs that constitute EGL migrate down the Bergmann glial processes and pass Purkinje neurons before they settle down as more differentiated granule neurons in the IGL. GM: Grey matter; WM: White matter; NSC: Neuroepithelial stem cell; ORG: Outer SVZ radial glia-like cells; VRG: ventricular epithelium radial glia; OPC: Oligodendrocyte precursor cell; SVZ: Subventricular zone; SGZ: Subgranular zone; VZ; Ventricular zone at 4th ventricle; RL: Rhombic lip; EGL: External germinal layer; GPC: Granule cell progenitor; ML: Molecular layer; PL: Purkinje cell layer; IGL: Internal granular layer. Examples of markers that are expressed, or not expressed, in the various cell types are shown followed by + or −, respectively.
Progenitors, including oligodendrocyte precursor cells (OPCs) and ependymal cells arise from radial glia and NSCs. These cells become regionally restricted depending on intrinsic maturation, and on cues from developmental organizing pathways like Sonic Hedgehog (SHH), Wingless proteins (WNTs), BMPs and FGFs. Brain lipid-binding protein (BLBP) is expressed by radial glia cells in brain and spinal cord [6–8]. When the Blbp promoter was used to drive Cre/loxP to follow cellular fate using the Rosa26 reporter, it was evident that neurons in almost all brain regions were labeled [9] suggesting they all originate from these BLBP-positive cells. OPCs are presumably also derived from the radial glia. OPCs express NG2 and continue to produce myelin-producing oligodendrocytes [10]. Adult OPCs are also born and are presumably derived from NSCs [11] that reside in the adult forebrain in lateral regions of the lateral ventricles and in the dentate gyrus in the hippocampus. Such NSCs are termed “type B” cells, slowly dividing subventricular zone (SVZ) astrocytes or subgranular zone (SGZ) astrocytes [12,13]. During neurogenesis, type B NSCs give rise to transit amplifying progenitors (type C cells) that later give rise to immature neuroblasts (type A cells). Type A cells in rodents migrate to the olfactory bulb where they differentiate into olfactory neurons [14]. Ependymal cells also originate from radial glia, are born in the embryonic and early postnatal brain and do not divide after differentiation [15]. As discussed in detail below, cells of origin for glioma are probably heterogeneous, as both NSCs and OPCs that are both derived from a neuroepithelial cell could generate brain tumors in mouse models (Figure 1).
2.2. Developing hindbrain and cellular origin of MB
Segmentation plays a prominent role in the developing hindbrain. One area of segmentation, the isthmic region, lies at the midbrain-hindbrain boundary of the neural tube. During E9.5 in mice the so-called isthmic organizer secretes FGFs and WNTs, which provide regional identity and pattern proliferation along the anterior/posterior axis [16]. Abrogation of Otx2 and Gbx2 in mice indicated these proteins were essential for regionalization of the midbrain and rostral hindbrain [17]. The region where transcription factors like En1, Pax2, and Otx2 are expressed acquires midbrain identity, while FGF8 also expressed in the isthmus, produces a strong local signal associated to cerebellum fate determination [18].
The cerebellum is in control of balance control and motor coordination but also shares with the cerebrum some role in cognitive functions, speech and spatial memory [19], functions that may be affected in children with brain tumors, or after surgical resection [20]. Two distinct germinal zones give rise to the cells of the cerebellum [21] that arise from rhombomere 1. After E10 in mice, the ventricular zone (VZ), the primary germinal zone, forms along the fourth ventricle. The VZ of the cerebellar anlage contains primitive cells like neuroepithelial cells and radial glia. These PTF1A-positive progenitors exit the cell cycle, migrate radially into the cerebellum and give rise to all GABAergic cells [22], Purkinje neurons and interneurons including Basket and Stellate cells. Interestingly, deleting Ptf1a leads to failure of VZ cells to generate GABAergic neuronal cell types. [23]. This GABAergic linage could be traced through E18 when these embryos died. Some radial glias from VZ convert into Bergmann glial cells [24]. Precursors of Bergmann glia are found after E15, and they keep their processes once made by their radial predecessors [25].
The second cerebellar germinal zone forms along the anterior aspect of the rhombic lip (RL) structure and give rise to glutamatergic neurons [26] including cerebellar granule neurons [27,28], the most abundant cell type in the entire CNS. Cells exiting the anterior RL migrate over the cerebellar anlage forming the external germinal layer (EGL). The EGL consists of MATH1-positive granule cell progenitors (GCPs) that continue to proliferate. The peak of this proliferation occurs at P7 [29] in response to SHH produced by Purkinje neurons [30,31]. However, SHH that prevents differentiation and promotes proliferation of granule neuron precursors is also present in the embryonic cerebrospinal fluid, that circulates within the cerebellar VZ by E12.5-E15 [32].
Whether the RL is the sole source of SHH-dependent granule cells remains unclear. When Math1 is depleted there is a failure to produce granule cells but the RL still forms [28]. Fate mapping has suggested that Math1-positive cells are not the definite RL stem cells. Instead, another yet undefined RL stem cell population may actually give rise to Math1-positive cells [33,34]. Since the RL and EGL cells are both derived from GFAP-positive cells [35] both populations could conceivably give rise to SHH-driven MB [36,37]. This suggests GFAP-positive VZ cells are certainly a major contributor to both these germinal zones. For example, if the VZ marker PTF1A is deleted, some mutant VZ progenitor cells aberrantly migrate to express RL markers like MATH1 and ZIC1/2 [38].
The cerebellum is structurally complete by 3 weeks of age in mice and at ~2 years of age in human [39]. The fully developed cerebellar cortex consists of molecular, Purkinje cell, and internal granule cell layers. The development of VZ and RL are regulated by NOTCH activity in the VZ, and BMPs that signal from the roof plate. Early, during embryonic cerebellar development BMPs stimulate production of RL-derived neurons and expression of MATH1 [40–42]. Later on, during postnatal periods BMPs instead inhibit MATH1, SHH-induced GCP proliferation and MB development [43,44]. Loss of Notch1 in the cerebellar primordium leads to severe hypoplasia and an increased RL neurogenesis [41,45] while overexpression of Notch2 activity postnatally inhibits GCP differentiation [46]. Thus NOTCH signaling like SHH signaling acts to arrest the differentiation state of cerebellar precursors at different developmental stages.
As for glioma, the cells of origin for MB are not clearly identified, and it is likely that more than one type of cell can generate these tumors (Figure 1). In fact, neural stem cells, granule precursor cells and progenitor cells from the lower RL of the developing dorsal spinal cord all give rise to MB in mouse models, as further discussed below.
3 Glioma
3.1 Classification and prognosis
Gliomas are the most common tumors in the central nervous system (CNS). They are classified histopathologically as astrocytomas, oligodendrogliomas, mixed oligo-astrocytomas and ependymomas, and graded with regard to malignancy as I- IV according to the World Health Organization (WHO) classification system. Grade I and II tumors are slowly growing, while high-grade gliomas (of grade III and IV) are malignant tumors with a high proliferation rate. In addition, grade IV gliomas, also called glioblastoma multiform (GBM), show areas of necrotic pathology and of vascular proliferation.
The vast majority of grade IV gliomas present de novo as primary GBM, however rarely, secondary GBM progresses from lower grade gliomas. Although the primary and secondary GBM display the same histopathologic features, they have different underlying genetic mutations and molecular characteristics. The prognosis for GBM is unfortunately dismal, with grade rather than stage typically the main prognostic stratifier. The alkylating agent temozolomide in combination with radiation and chemotherapy results in a 5-year overall survival of less than 10% after diagnosis [47]. Lower-grade astrocytomas, and oligodendroglioma patients have a better prognosis where oligodendroglioma with a combination of LOH (loss of heterozygosity) on Chromosome 1p/19q often respond better to chemotherapy [48].
3.2 Molecular characterization
There has been great progress in the characterization of gliomas at both genomic and transcriptional levels. Several large-scale genomic efforts, including the Cancer Genome Atlas Research network have focused on GBM and in fact, the TCGA database now includes over 500 GBMs (http://cancergenome.nih.gov) [49]. Three key signaling pathways commonly disrupted in a majority of GBMs include the receptor tyrosine kinase (RTK) signaling pathways as well as the tumor suppressor pathways of RB and p53 (Figure 2). In addition to the identification of mutations affecting these pathways, new genes have been implicated as key players in gliomagenesis, and new subgroups of GBM have been identified on the basis of these genetic aberrations and gene expression profiles.
Figure 2. Common molecular pathways of glioma.
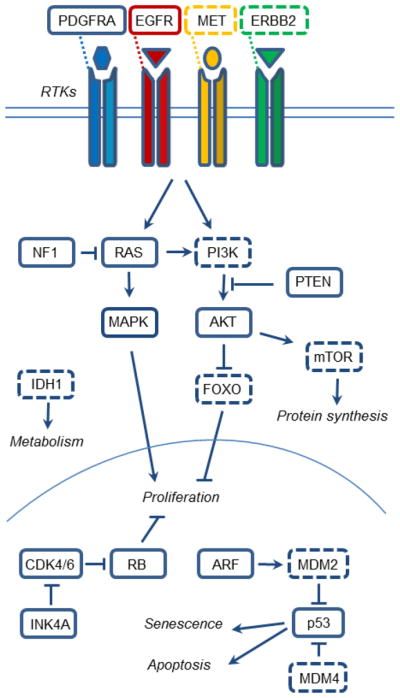
Major molecular pathways altered in glioma, starting with receptor tyrosine kinases (RTKs) on the cell surface, cytosolic proteins and nuclear proteins/transcription factors. Shown are also the relationship between these brain cancer proteins and their mechanisms of action. Proteins surrounded by broken lines identify glioma pathways that are not yet modeled in mice or described in this review.
Analysis of high-grade glioma (WHO grade III and IV) gene expression patterns initially identified three subgroups, named Proneural, Mesenchymal and Proliferative types. These groups displayed features of neuronal progenitors, mesenchymal cells and proliferating cells, respectively [50]. A larger set of GBM patient samples from the TCGA were later analyzed to identify four separate groups; Classical, Mesenchymal, Proneural and Neural [51]. The tumors of the Classical group are characterized by EGFR amplification and loss of PTEN and CDKN2A. Mutations in TP53 are rarely found in this group. Activation of SHH and Notch are relatively common. The Mesenchymal group of GBM is characterized by mutation or loss of NF1, PTEN and TP53. They display an angiogenic or mesenchymal gene expression profile with activated TNF and NFkB pathways. The Neural group of GBM often shows overexpression and amplification of EGFR, and displays a gene expression profile resembling normal brain.
The Proneural subtype of GBM often shows PDGFRA amplifications and loss of TP53, CDKN2A and PTEN. The Proneural tumors also commonly have mutations in IDH1 and in genes encoding PI3K. The gene expression profiles of Proneural tumors resemble neuronal and oligodendrocyte progenitor cells. The genetic aberrations of secondary GBMs mostly resemble the Proneural subtype, with TP53 and IDH mutations and overexpression of PDGFRA. These genetic aberrations are emerging early in the tumor progression from low- to high grade astrocytic glioma. Analysis of IDH1 mutation status in human glioma samples showed an association of mutated IDH1 with frontal lobe location, Proneural gene expression signature, altered methylation pattern and a better prognosis [52].
Oligodendrogliomas are grade II-III gliomas with oligodendroglia-like histology, often displaying LOH on chromosome 1p and 19q as well as loss of IDH1 and overexpression of EGFR and PDGFRA. Pilocytic astrocytomas are benign (Grade I) tumors often carrying activating mutations in BRAF [53,54], and activation of the MAPK signaling pathway. Roughly 15% of Neurofibromatosis Type 1 (NF1) patients also have pilocytic astrocytomas, particularly in the optic pathway [55].
GBM is also a lethal brain tumor in children where DNA copy number and gene expression signatures indicate some differences. Interestingly, mutations in regulatory Histone 3.3 and chromatin remodeling genes are highly prevalent in pediatric GBMs but also in adult GBMs [56].
Ependymomas are glial tumors with many different genetic aberrations, including loss of CDKN2A, PTEN and overexpression of NOTCH1 and EPHB2. Ependymomas are a heterogeneous group of tumors that probably consists of several subgroups, arising in different locations in the CNS [57].
3.3 Mouse models of gliomas
Genetically engineered mouse models (GEMMs) have been extensively used to model human gliomas. Mutations and genetic aberrations found in human gliomas have been introduced into both germ-line cells and somatic cells by various methods (Table I). The use of inducible gene-expression constructs and tissue-specific promoters allows for the targeting of a specific cell type at a specific time point or developmental stage. GEMMs have provided means to study both the mechanisms of tumor initiation and progression as well as to find new potential targets for treatment.
Table I.
Mouse models of glioma divided after classification and WHO grade with driving cancer gene, putative targeted cell of origin (Targeted cell) and the mechanism of the model as indicated.
| Cancer genes | Targeted cell | Mechanism | Reference |
|---|---|---|---|
| Ependymoma models | |||
| EphB2/Ink4a/Arf−/− | NSCs | Transplant | [57] |
| Pilocytic astrocytoma models (I) | |||
| BRAFV600E | Nestin | RCAS | [91] |
| Astrocytoma models (II-III) | |||
| v-src | GFAP | Transgenic | [97] |
| Arf−/− | - | Transgenic | [98] |
| c-myc | GFAP | Transgenic | [99] |
| K-ras | GFAP | Cre | [100] |
| H-Ras | GFAP | Transgenic | [101] |
| H-Ras/Ptenloxp/loxp | GFAP | Cre | [102] |
| T121/Pten+/− | GFAP | Cre | [103] |
| EGFR/Ink4a/Arf−/− | Nestin | RCAS | [60] |
| Oligodendroglioma models (II-III) | |||
| H-ras | GFAP | Transgenic | [79] |
| H-ras/EGFRvIII | GFAP | Transgenic | [62] |
| v-ErbB/Ink4a/Arf+/− | S100β | Transgenic | [61] |
| v-ErbB/Trp53−/− | S100β | Transgenic | [63] |
| PDGFB/Ink4a/Arf−/− | - | Retroviral | [104] |
| PDGFB/Ink4a/Arf−/− | Nestin | RCAS | [72] |
| PDGFB/Akt | Nestin | RCAS | [105] |
| PDGFB | CNP | RCAS | [73] |
| PDGFB | Embryo | Retroviral | [69] |
| PDGFAL | GFAP | Transgenic | [77] |
| Glioblastoma (GBM) models (IV) | |||
| Nf1+/−/Trp53+/− | - | Transgenic | [83] |
| PDGFB | - | Retroviral | [70] |
| PDGFB | Nestin | RCAS | [106] |
| PDGFB/Trp53−/− | GFAP | Transgenic | [76] |
| K-Ras/Akt | Nestin | RCAS | [80] |
| K-Ras/Ink4a/Arf−/− | GFAP, Nestin | RCAS | [81] |
| K-Ras/Akt/Ptenloxp/loxp | Nestin | RCAS/Cre | [107] |
| Nf1loxp/+/Trp53+/− | GFAP | Cre | [85] |
| Ptenloxp/+/Trp53loxp/loxp | GFAP | Cre | [108] |
| Ptenloxp/+/Nf1loxp/+/Trp53loxp/− | GFAP | Cre | [87] |
| p53−/− | Embryo | Mutagen | [109] |
| Ptenloxp/+/Nf1loxp/+/Trp53loxp/loxp | SVZ, Nestin | CreER | [88] |
| Akt/H-Ras/Trp53+/− | SVZ, HC, GFAP | Lentiviral/Cre | [82] |
| Ptenloxp/loxp/Trp53loxp/loxp/Rbloxp/loxp | SVZ, GFAP | Adenoviral/Cre | [89] |
| Nf1+/−/Trp53loxp/loxp | GFAP | Transgenic/Cre | [84] |
| PDGFB/Ptenloxp/loxp/Trp53loxp/loxp | Subcortical WM | Retroviral/Cre | [78] |
| Ptenloxp/loxp/Trp53loxp/loxp/Rb1loxp/loxp | Astrocyte (GFAP) OPC (NG2), NSCs (GFAP/Nestin) | CreER | [90] |
| Nf1−/−/Trp53−/− | MADM/Cre | [94] | |
| EGFRvIII/Ink4a/Arf−/− | NSC, Astrocyte | Transplant | [64] |
| Pten−/−/EGFRvIII/ Ink4a/Arf−/− | Striatum | Adenoviral/Cre | [65] |
| TGFα/EGFRwt/Ink4a/Arf−/− | Striatum | Lentiviral/Cre | [66] |
NSCs: neural stem cells; SVZ: Subventricular zone ;OPC: Oligodendrocyte precursor cell; WM: White matter. HC: Hippocampus.
3.4 Models using RTKs or growth factors
Excessive growth factor signaling is one crucial component in the glioma development. Genetic analysis of high-grade tumors reveals a high frequency of aberrations affecting the RTK/RAS/PI3K signaling pathways. The most commonly mutated RTK is EGFR, which is found mutated or amplified in 45% of GBMs. Activating mutations or amplifications are also found in PDGFRA (13%), ERBB2 (8%) and MET (4%) [49].
The most commonly constitutively activated form of EGFR in glioma is EGFRvIII that has lost parts of its extracellular domain and signals independent of ligand binding [58,59]. Constitutively activated forms of EGFR have been used in combination with other genetic aberrations such as loss of tumor suppressors Ink4a/Arf, p53 or Pten, to induce both low- and high grade gliomas in mice [60-63]. The Classical subgroup of GBM is characterized by EGFR signaling together with loss of CDKN2A. GEMMs with this combination of genetic aberrations have been generated in several ways.
Low-grade gliomas formed when an active form of EGFR was expressed in Nestin-positive progenitor cells of newborn Ink4a/Arf−/− mice. Active EGFR was introduced into Nestin-expressing cells or GFAP-expressing cells by the avian retroviral RCAS/tv-a system. In this model, tumors/glioma-like lesions formed more frequently from Nestin-positive cells, than GFAP-positive cells [60]. In a transplantation mouse model system using newborn EGFRvIII overexpressing Ink4a/Arf−/− primary cells, both NSCs and mature astrocytes were shown to induce high grade gliomas when orthotopically transplanted into recipient mouse brain [64].
Excessive EGFR signaling together with loss of Ink4a/Arf has also been targeted in cells of oligodendroglial lineage. Transgenic expression of active EGFR in the form of the transforming avian allele v-erbB, in S100β-positive oligodendrocytes of mice resulted in low grade oligodendrogliomas [61]. When combined with either loss of p53 or loss of Ink4a/Arf, misexpression of v-erbB gave high grade oligodendrogliomas with a reduced latency. This model was used to demonstrate that tumors originated from an expanded pool of oligodendrocyte precursor cells [63], an observation also demonstrated in human oligodendroglioma
GBMs have been generated in mice by combining EGFRvIII with loss of both Pten and Ink4a/Arf. The expression of EGFRvIII was induced in adult mouse striatum by adenoviral Cre [65]. Recently, another inducible mouse model showed that overexpression of wild-type EGFR together with the ligand transforming growth factor alpha (TGFα), could also induce GBM. In this model, lentiviruses were used to introduce TGFα together with Cre to the striatum of Ink4a/Arf−/− mice resulting in oncogene dependent, high grade gliomas [66].
Analysis of human glioma tissue show that overexpression of PDGFRA is found in both low-and high-grade tumors [67]. The activating ligands (PDGF-A, -B, -C and -D) are also expressed in human gliomas, and the expression patterns of PDGF ligands and receptors PDGFRA and PDGFRB in glioma tissue suggest the presence of autocrine and paracrine loops stimulating tumor growth [68]. Ectopic expression of the PDGF ligands, and PDGFB in particular, has been used extensively to model different glioma subtypes in mice. Retroviral expression of PDGFB has been shown to cause malignant oligodendroglial tumors, when injected into embryonic [69] or newborn mouse brain [70]. Experiments on newborn rat brains show that retroviral PDGFB can cause a shift in the differentiation of NSCs, producing more PDGFRA/NG2/OLIG2-expressing OPCs [71].
A targeted approach using the RCAS/tv-a system, induced expression of PDGFB specifically in GFAP-, Nestin- or CNP-expressing cells, resulting in oligodendrogliomas and some mixed oligoastrocytomas [72,73]. The gliomas induced by PDGFB with the RCAS/tv-a system were altered to a more malignant tumor phenotype when combined with a second genetic aberration such as loss of p53 or Ink4a/Arf [74,75].
In contrast to the retroviral models, transgenic expression of PDGFB from the GFAP promoter only resulted in gliomas when combined with loss of p53. Mice over-expressing PDGFB on a Trp53 null background developed GBM-like tumors with high expression of PDGFRA on tumor cells and PDGFRB on tumor vessels [76]. In comparison, the same human GFAP promoter was also used to overexpress the long form of PDGFA. In contrast to the high-grade astrocytic gliomas formed by PDGFB in Trp53 null mice, transgenic expression of PDGFAL instead led to rapid expansion of oligodendrocyte progenitor cells and oligoastrocytomas in a Trp53 wild-type background [77].
Retroviral expression of PDGFB together with Cre has been used to generate high-grade astrocytic tumors in subcortical white matter regions of adult Pten−/− as well as Pten−/−Trp53−/− mice. Gene expression profiling of the resulting tumors revealed a strong similarity to the human Proneural GBM subtype [78].
3.5 Models using other oncogenes
Several glioma models target the downstream signaling pathways activated by growth factor signaling. Activated RTKs signal via many pathways, including the RAS/MAPK and PI3K/AKT pathways, to promote cell proliferation and survival. Activation of different components in these pathways promotes glioma formation in mice (Figure 2).
Transgenic mice overexpressing a constitutively active form of RAS (V12Ha-ras) under the GFAP promoter develop astrocytic gliomas. Increasing the expression levels of the transgene resulted in progression of the tumors from low- to high-grade gliomas [79]. Adding overexpression of activated EGFRvIII to the model turned the gliomas to a more oligodendrocytic pathology [62]. On the other hand, when the RCAS/tv-a system was used to somatically introduce an activated form of K-Ras into newborn mice, transduction of activated Akt was also needed for tumor formation. High grade gliomas formed only when the activated Akt and K-Ras genes were introduced together into Nestin-expressing neural progenitors, and tumors did not form from GFAP-expressing cells [80]. The same activated variant of K-Ras in combination with loss of the tumor suppressor locus Ink4a/Arf resulted in glioma formation from both GFAP- and Nestin-positive cells, some with a sarcoma-like phenotype [81].
In adult mouse brain, lentiviral vectors carrying H-Ras and Akt induced high-grade astrocytomas when introduced to GFAP-expressing cells of both SVZ and hippocampus. On a Trp53 null background, the developing gliomas became more malignant, and cortical tumors also formed in addition to the SVZ and hippocampal tumors [82].
3.6 Models using tumor suppressor genes
The signaling activity of Ras is regulated by NF1, which has been found to be mutated in 18% of human GBM [49]. Loss of Nf1 has been used to model gliomas in mice in combination with loss of other tumor suppressors such as Trp53, Rb or Ink4a/Arf.
The tumor suppressor genes Trp53 and Nf1 are located on the same chromosome, allowing for an easy way for targeting the two genes at once. Mice heterozygous for both Trp53 and Nf1 genes develop low and high grade astrocytomas displaying loss of heterozygosity (LOH) for the remaining Trp53 and Nf1 allele [83]. More restricted approaches where Trp53 and Nf1 were mutated or deleted only in GFAP-expressing cells also resulted in high grade astrocytomas [84,85]. Gliomas formed only when Trp53 was deleted before or simultaneously as Nf1, indicating that loss of Trp53 is crucial for the initiation of gliomagenesis [85]. Loss of p53 is indeed an early event in human gliomagenesis, and mutations in TP53, and LOH at chromosomal region 17p containing the TP53 gene are commonly found in both low- and high grade gliomas [86].
A tumor suppressor gene commonly found inactivated in human high-grade gliomas, but not in low-grade tumors is PTEN. Loss of PTEN leads to hyperactivation of AKT and increased proliferation and migration. Adding heterozygosity for Pten to a mouse model with deleted Nf1 and Trp53 in GFAP-expressing cells resulted in a more malignant tumor phenotype, and frequent LOH of the remaining allele of Pten in progressed grade IV tumors [87]. Conditional mouse models have further been utilized to study the tumor suppressing roles of p53, NF1 and PTEN in different cell types and in different areas of the brain. Conditional loss of Nf1 and Trp53 in Nestin-expressing cells resulted in high grade astrocytomas when targeting both newborn and adult SVZ. However, no tumors were formed by targeting cortical Nestin-expressing cells with the same genetic aberrations [88]. Another study showed that combined loss of Trp53 and Pten in GFAP-expressing cells of adult SVZ resulted in high grade astrocytic gliomas, while loss of Trp53 and Pten in GFAP-expressing cells in other regions failed to generate tumors. Mice carrying conditional alleles of Trp53, Pten and Rb were also generated. Mice with loss of all three genes developed high grade tumors, with reduced latency, and in some cases a PNET-like phenotype [89]. Gliomas induced by the combined loss of Trp53 and Pten or Trp53, Pten and Rb showed gene expression profiles similar to human GBM, including Proneural, Mesenchymal and Proliferative subgroups [90]. The RB signaling pathway is one of the three critical pathways often mutated in human GBM, [49]. RB signaling was found altered in 78% of primary GBMs. The most common aberrations were deletions and mutations in CDKN2A and CDKN2B [49]. Many genetically engineered mouse models of glioma include loss of the Cdkn2a or Ink4a/Arf locus, coding for both Ink4a and Arf. Targeted deletion of this locus thus affects both p53 and RB pathways.
3.7 Models for other glioma subgroups
Pediatric gliomas are histologically similar to adult gliomas, although they arise in different anatomic locations and show differences in signaling. Grade I pilocytic astrocytomas are more common in children than adults. These benign tumors are characterized by an active MAPK signaling pathway defined by gene fusions involving BRAF [54] that results in constitutive activation of BRAF kinase activity. Pilocytic astrocytoma has recently been modeled in mice by introducing a mutated active form (V600E) of BRAF into Nestin-expressing cells using the RCAS/tv-a system [91].
Pediatric gliomas of grade II-IV are often located in the brain stem. These brain stem gliomas or DIPGs (diffuse intrinsic pontine gliomas) are often driven by excessive PDGF signaling. The RCAS/tv-a system was used to overexpress PDGFB specifically in Nestin-positive cells of newborn mouse brain stem, located adjacent to the fourth ventricle. PDGFB expression in these cells resulted in low-grade brain stem gliomas, and when PDGFB was introduced into Ink4a/Arf−/− mice, more malignant, high grade brain stem gliomas developed [92]. Low-grade, diffusely growing brain stem gliomas were also occasionally found in the transgenic PDGFB/Trp53−/− model, where GFAP was used to direct transgene expression [76].
Transgenic overexpression of PDGFB in GFAP-expressing cells has also been used to model spinal glioma with a mixed oligoastrocytic phenotype. Over-expressing PDGFB on a Trp53 heterozygous background reduced the tumor latency in this model [93].
3.8 Cells of origin for glioma
Mouse models for glioma are commonly generated by introducing genetic aberrations found in human tumors into mouse brain. The genes have been targeted in putative cells of origin at different developmental stages with the help of specific promoters. Inducible systems have revealed early steps in tumorigenesis in adult mouse brain; however a paucity of specific markers has made it difficult to identify cells of origin. Sophisticated methods to target genes somatically in specific brain areas have reduced the number of manipulated cells in the mouse brain, narrowing the search for the origins of glioma.
Early steps of tumorigenesis have been studied in mouse models, to provide insights about the origin of tumors. In transgenic mice with activated alleles of EGFR (v-erbB), under control of the human S100β promoter, analysis of tumor location by MRI, followed by identification of dividing cells in the same area, revealed NG2+ OPCs as cells of origin for oligodendroglial tumors [63]. Retroviral transfer of PDGFB to myelinating OPCs expressing Cnp using the RCAS/tv-a system also suggested OPCs a cell of origin for PDGF-driven oligodendrogliomas [73]. Early steps of tumor formation have also been analyzed in a mouse model for astrocytic GBM. Here, constitutive transgenic expression of GFAP-Cre was used to knock-out Trp53 in Nf1 heterozygous mice. Tumor progenitor cells with mutated p53 were tracked during early tumor formation and were found to migrate from SVZ to other brain regions along the corpus callosum, a large white matter structure that connects the two hemispheres [84]. A mosaic mouse model, also with targeted deletions of Nf1 and Trp53, was used recently to show that, although the combined genetic aberrations were induced in the NSC compartment of SVZ, the actual tumorigenesis occurred in the developing OPCs [94].
Another strategy to pin point origin has been to reduce the number of targeted cells. The promoters used to direct gene expression are typically active in many different cell types at several developmental stages. Local injections of virally expressed Cre have been used in combination with specific promoters to target cells selectively in a small area. Comparison of tumor forming abilities in different brain areas showed that both Nestin-positive and GFAP-positive cells residing in neurogenic niches were more easily transformed than cells in differentiated areas [82,88]. On the other hand, results from RCAS/tv-a glioma models indicate no difference in the tumor forming ability of cells residing in different brain areas [95]. These studies illustrate the necessity for the right cellular context in modeling tumor formation, as the same genetic alterations may give completely different outcomes depending on cell type.
An alternative approach to elucidate cell of origin has been to compare the gene expression profiles of human tumors with the expression profiles of different normal cell types in the brain. This has proven to be very successful in the case of ependymomas, where gene expression profiling proved the cell of origin of ependymomas to be radial glia [96]. The same type of comparison of gene expression profiles was then further used to identify putative cell types to target when developing relevant mouse models for different subtypes of ependymoma [57].
4 Embryonal brain tumors
4.1 Classification and prognosis
Medulloblastoma (MB) is the most common malignant brain tumor of childhood accounting for 20% of pediatric CNS tumors [110]. Surgical resection, radiation and chemotherapy have led to an improvement in overall survival of 70–80% in MB patients [111]. However, the quality of life for survivors is often compromised by treatment-related side effects. MB are also the largest group under embryonal brain tumors followed by primitive neuroectodermal tumors (PNETs), pineal gland tumors, ependymoblastomas and atypical teratoid/rhabdoid tumors (ATRTs) [112]. Embryonal tumors like MB are thought to originate from cerebellar or even spinal cord structures while PNETs form in other structures of the CNS most commonly in the forebrain, cerebrum. A rare and highly lethal embryonal tumor with abundant neuropil and true rosettes (ETANTR), has also been described recently as a variety of PNET [113].
4.2 Molecular characterization of MB
An international consensus panel has now agreed on dividing human MB into four major molecular subgroups [114], WNT, SHH, Group 3 and Group 4 (Figure 3). This followed after a number of large sample gene expression studies of human MB confirmed very different expression patterns in these tumors [115–118]. The first two groups are characterized for expressing SHH and WNT pathway markers while Group 3 shows a photoreceptor/GABAergic profile and Group 4 presents a neuronal/glutamatergic profile [116]. Further, Group 3 MB has a subset of MYC-amplified tumors and SHH, WNT and group 4 MB all have subsets of MYCN-amplified tumors [114,119].
Figure 3. Molecular pathways in four MB subgroups.
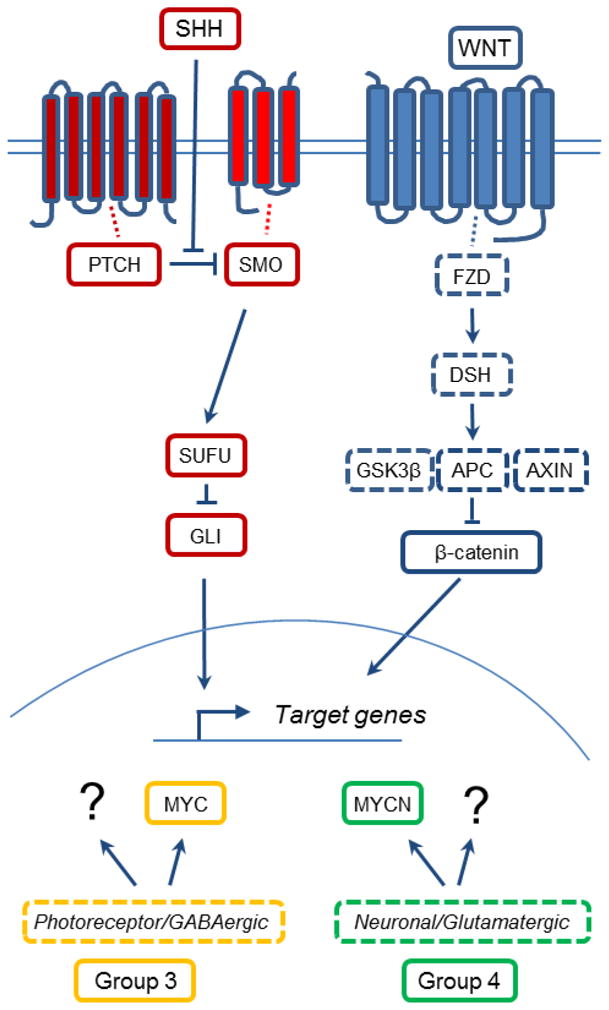
The four major molecular pathways altered in MB. Shown are two simplified versions of the SHH and the WNT pathway that represent two of the MB subgroups. MB of Group 3 and Group 4 present some degree of MYC and MYCN alterations, respectively. Group 3 and Group 4 further present a profile of not yet identified Photoreceptor/GABAergic and Neuronal/Glutamatergic pathways, respectively. Proteins surrounded by broken lines identify MB pathways that are not yet modeled in mice or described in this review. ?: unknown pathway/mechanism.
4.3 Mouse models of MB
Mouse models for MB can be divided into developmental pathways that are important in cerebellar development. While early reports focused on the SHH pathway (SHH models), recent reports describe non-SHH models that depend on WNT signaling, as well as models for Group 3 MB and Group 4 MB, that are not so clearly correlated with any given developmental pathway (Table II).
Table II.
Mouse models of MB with suggested molecular subtype (MB profile), dissemination/metastasis (Mets), mechanism of the model and putative cell of origin.
| Genes/pathway | MB profile; Mets | Mechanism; Putative origin | Reference |
|---|---|---|---|
| SHH models | |||
| Ptch+/− | SHH; - | KO; GPC | [120] |
| Ptch+/−; | SHH; - | KO, irradiation; GPC | [121] |
| Ptch+/−; Trp53−/− | SHH; - | KO; GPC | [122] |
| Ptch+/−; aInk4c+/- | SHH; - | KO; GPC | [123] |
| Ptch+/−; Ptc2+/- | SHH; - | KO; GPC | [124] |
| Ptch+/−; aKip1+/− | SHH; - | KO; GPC | [125] |
| Ptch+/−; Hic1−/− | SHH; - | KO; GPC | [126] |
| Math1-CreE; Ptchc/c | SHH; - | CKO; GPC | [37] |
| GFAP-Cre; Ptchc/c | SHH; - | CKO; GFAP-positive cell | [37] |
| GFAP-Cre; Rbloxp/loxp; aTrp53loxp/loxp | SHH; - | CKO; GFAP-positive cell | [127] |
| GFAP-Cre; Rbloxp/loxp; aTrp53loxp/loxp | SHH; - | OM; GFAP-positive cell | [128] |
| Nestin-Cre: Ptchc/c | SHH; - | CKO; Nestin-positive cell | [129] |
| ND2-SmoAhemizygous | SHH; - | CTG; GPC | [130] |
| ND2-SmoA/SmoA | SHH; Mets | CTG; GPC | [131] |
| SmoM2 | SHH; - | CTG; GPC | [132] |
| GFAP-Cre or Math1-Cre or Olig2-Cre or Tlx-Cre; SmoM2; | SHH; - | CTG; NSC, GPC or Olig2- positive cell | [36] |
| Sufu+/−; Trp53−/− | SHH; - | KO; GPC | [133] |
| Shh | SHH; - | Ectopic; GPC | [134] |
| Nestin-tva-Shh | SHH; - | RCAS; Nestin-positive cell | [135] |
| Nestin-tva-Shh; c-myc; Akt; IGF2 | SHH; - | RCAS; Nestin-positive cell | [136] |
| Nestin-tva-Shh; Mycn | SHH; - | RCAS; Nestin-positive cell | [137] |
| Nestin-tva-Shh; Bcl-2 | SHH; - | RCAS; Nestin-positive cell | [138] |
| Nestin-Cre;Brca2 | SHH; - | CTG; Nestin-positive cell | [139] |
| Nestin-Cre;Lig4 | SHH; - | CTG; Nestin-positive cell | [140] |
| Nestin-Cre;Xrcc2 | SHH; - | CTG; Nestin-positive cell | [140] |
| Nestin-Cre;Xrcc4 | SHH; - | CTG; Nestin-positive cell | [141] |
| Parp1−/−; Trp53−/− | SHH; - | KO; GPC | [142] |
| Lig4−/−; Trp53−/− | SHH; - | KO; GPC | [143] |
| SB11; Ptch+/− | SHH; Mets | SB/KO; GPC | [144] |
| SB11; Trp53mut | SHH; Mets | SB/KO; GPC | [144] |
| Non-SHH models | |||
| bProm1+Lin -sorted; cMyc; DNp53−/− | Group 3 | OM/RV/KO; NSC | [145] |
| Atoh1-GFP-sorted; Myc or Mycn; dTrp53−/−; | Group 3 or SHH | OM/RV/KO; NSC or GPC | [146] |
| Glt1-tTA; TRE-MYCN | Group 3/4; Mets | TG; Glt1-positive cell | [147] |
| GFAP-tva-MycnT58A from P0 or E16 NSCs | Group 4 or SHH | OM/RCAS; GFAP-postive NSC | [148] |
| Blbp-Cre;Ctnnb1;Trp53loxp/loxp | WNT | CTG; LRL precursor | [149] |
or −/−;
or Math1-GFP-sorted;
WT or T58A mutant;
with or without Cdkn2c−/−;
KO: knock-out; CKO: conditional knock-out; CTG: conditional transgenic model; GPC: granule cell precursor; NSC: neural stem cell; OM: orthotopic model; RV: retroviral; SB: Sleeping Beauty; DN: dominant-negative.
4.4 MB models for SHH pathway
The Hedgehog gene was first identified in Drosophila melanogaster [150] and is highly conserved between invertebrates and mammalians. Sonic Hedgehog (SHH) promotes neurogenesis of granule cell precursors in the developing cerebellum [30,31]. Purkinje neurons secrete this mitogen, promoting proliferation of GCPs. The gradient of SHH also drives migration of GCPs from external germinal layer to the internal granule layer [151].
SHH acts as a ligand for its receptor Patched, the 12-pass transmembrane protein (PTCH). PTCH in the absence of ligand, suppresses Smoothened (SMO), a 7-pass transmembrane protein. When SHH binds PTCH, SMO is derepressed, leading to the activation of GLI family transcription factors (Figure 3). The activation of GLI is in part regulated by a complex inhibitory protein complex that includes suppressor of fused (SUFU).
Germline mutations in PTCH lead to Gorlin’s syndrome, characterized by increasing incidence of basal cell carcinoma, MB and rhabdomyosarcoma [152]. The first transgenic model for this syndrome and for SHH-driven MB was the Ptch knock-out mouse, described more than 15 years ago [120]. When knocked out constitutively, the model is embryonic lethal, however mice heterozygous for Ptch+/− develop MB at 14% incidence after 5 weeks. The incidence of SHH-driven tumors is dramatically increased with radiation [121] or when p53 is additionally mutated [122]. Constitutively active Smo, SmoA, can drive MB at a high incidence (48%) from the NeuroD2 promoter in GCPs [130]. Homozygous SmoA (Smo/Smo) further increase incidence and also promote leptomeningeal spread [131]. Another model drives another active Smo, SmoM2, ubiquitously using either sporadic leakage or global induction after birth giving rise to MB [132] and also other types of tumors like those described in Gorlin’s syndrome above.
The primary cilium, a non-motile single organelle on the cell membrane, often referred to as the cells antenna, is essential for the SHH pathway [153] as SMO and PTCH are both localized to this structure [154,155]. During SHH stimulation SMO is recruited to replace PTCH at this structure leading to downstream activation of GLI proteins. It is therefore not surprising that primary cilia are required for SHH-driven MB formation, as shown when Kif3a null or Ift88 null mice, which lack primary cilia [156] were crossed with the SmoM2 model.
Shh itself drives MB using in utero injection of a Shh-containing retrovirus [134] or when it is restricted to Nestin-expressing progenitors using the RCAS/tv-a system [135]. Further, different combinations of SHH with oncogenes like c-myc, IGF or AKT increases MB penetrance in the latter system [136]. Similarly, N-myc or Bcl-2 increases MB penetrance together with Shh [137]. N-MYC is a downstream target of the SHH pathway and actually essential for driving SHH-driven tumors. In normal GPCs [157] as well as in MBs driven conditionally from Math1-directed Ptch loss [158], pre-tumorigenic GPCs required N-myc in order to generate tumors. Interestingly, downstream SHH-pathway members like the transcription factor GLI1 cannot alone (or together with oncogenes like N-myc) give rise to MB, at least not in nestin-expressing cells [159,137]. By contrast, SHH is tumorigenic without GLI1 expression, as SHH-driven MB were formed also in Gli1 null mutants [134]. However, SHH still required MATH1 for its tumorigenic capacity [160].
The negative regulator Sufu induces SHH-like MB, but only when p53 is also depleted [133]. Interestingly, as p53 mutations are prevalent in human SHH MB [161] it is not surprising to find Trp53 null mice driving SHH-driven MB in different combinations. Besides driving more tumors in Ptch mutants [122] Trp53 loss promotes tumorigenesis when combined with Rb loss in GCPs [127]. Similarly, when critical components of the DNA repair machinery are suppressed, p53 further drives SHH-positive MB. This is true when Trp53 null mice are crossed with mice that lost the DNA-repair gene Poly-(ADP-ribose) polymerase (Parp) [142] or when crossed with mice depleted in critical non-homogenous end joining proteins like LIG4 [143], XRCC2 [140] or XRCC4 [141].
4.5 MB models for WNT pathway
Wnt was initially discovered in forward genomic screens in the early eighties when Nusse and Varmus observed that mouse mammary tumor virus integrations in a novel Int target gene could promote breast tumor development in mice [162]. Fly homologues with a wingless (Wg) phenotype were identified subsequently, with WNT representing a hybrid between Wg and Int. In the activated canonical WNT pathway WNT proteins bind to cell-surface receptors of the Frizzled family (FZD) that cause the receptors to activate Dishevelled family proteins (DSH) which results in an increased amount of β-catenin that reaches the nucleus (Figure 3). WNT proteins are like SHH proteins morphogens important for brain patterning. About 10–15% of MBs are associated with aberrant WNT signaling and of generally good prognosis with mutations in the β-catenin gene, CTNNB1 often found in this subgroup [163]. Further, mutations in adenomatous polyposis coli (APC) a scaffolding protein critical to this pathway, are often identified in patients with Turcot’s syndrome, with APC mutant pedigrees occasionally generating MB. The origin of these tumors was recently directed to a region of the lower RL structure in BLBP-positive, Ctnnb1 mutated, Trp53 defective mice [149]. Here, cells of origin were targeted embryonically, where CTNNB1 was transduced via electroporation into OLIG3/ZIC1-positive cells of origin. Such tumors, formed in the brain stem, could recapitulate human WNT MB that also localize to this structure by MRI.
4.6 Models for Group 3 and Group 4 MB
Group 3 and Group 4 constitute more than 60% of human MB and are also the groups that correlate strongest with worst prognosis [119]. MYC is amplified in a subset of Group 3 tumors [114], whereas MYCN is amplified in a subset of Group 4 MB [164]. A transgenic mouse was generated driving human MYCN in hindbrain NSCs, the GTML model [147]. This mouse used a Tet-Off model to drive classic or LC/A tumors from the Glutamate transporter 1 (Glt1) promoter, and is regulatable, where expression of MYCN could be turned off using doxycycline, leading to tumor regression through senescence. This is today the only transgenic model that can be used to follow non-SHH MB formation and also model disseminated tumors via leptomeningeal spread.
More other recent models used wild-type or mutationally stabilized T58A versions of MYC or MYCN to generate non-SHH tumors. Two different labs used MYC in combination with p53 loss to drive anaplastic MB from P7 cerebellar stem cells, following orthotopic transplantation [145,146]. Expression profiles from these tumors correlated with of human Group 3 MB and expressed the Group 3 signature marker, NPR3 [118,114]. Interestingly, while MYCN drove GCPs to become SHH tumors, MYC apparently reprogrammed these same cells to a SHH-independent lineage [146]. While these data suggest that MYC and MYCN sub serve distinct functions in transformation and reprogramming, the basis for these differences remains unexplained.
Another model investigated MYCN biology in a time and space-dependent matter by driving it from distinct populations of NSCs. Here, prenatal NSCs generated SHH-driven MBs in response to N-MYC expression. In contrast, N-MYC generated non-SHH MB from postnatal cerebellar stem cells [148]. These postnatal tumors expressed markers for Group 4 MB, like KCNA1 [118,114]. These MYC/MYCN models described are the first that could be used to recapitulate MYC and MYCN-amplified Group 3 and Group 4 MB, respectively; brain tumors that have a very poor prognosis [119]. These non-SHH models, including the majority of the GTML tumors, showed insensitivity to SMO inhibitors, like cyclopamine, that inhibits the SHH-pathway. Interestingly, inhibitors of PI3K/mTOR proved effective in MYC-generated MB [145] suggesting a role for this pathway in maintenance.
4.7 Models for metastatic MB
MB can disseminate through the cerebrospinal fluid into the leptomenigeal space. Metastases mark poor prognosis, are found in up to 30–40% of children at diagnosis, and are frequently observed in recurrent MB [165,166]. All molecular subgroups of human MB except the WNT group present this feature. Mouse models that show leptomeningeal spread include the Smo/Smo model [131] and the GTML model [147]. The mechanisms of dissemination have recently been studied in SHH-driven Ptch1 heterozygous and p53 mutant MB models, that were combined with a Sleeping Beauty system [144] for functional cancer gene discovery. Interestingly, metastases in MB from mice and patients lost MYCN amplification (but not MYC amplification) when these were found in the primary tumor [144]. Interestingly, metastases from the SB screen present higher expression of PDGFRα both in primary tumors and metastases [144], (metastases in human MBs also show high levels of PDGFRα [167].) suggesting a therapeutic target.
4.8 Other embryonal brain tumor models
Retrovirus-mediated transfer of SV40 virus large T antigen generates a large percentage of PNETs in animals [168–170]. Further, RCAS/tv-a viruses containing c-myc generated PNETs from GFAP-expressing forebrain cells in p53-deficient mice [171]. Beta-catenin activation enhanced tumor progression in this model. Malignant glioma with PNET characteristics (MG-PNET) is a rare forebrain tumor described with MYCN amplified in 40% of human cases [172]. Such tumors were modeled using N-MYC overexpressed in GFAP-positive NSCs isolated from forebrain VZ regions followed by orthotopic transplantation [148]. While these first brain tumor models used perinatal NSCs, brain tumors could also be generated from adult NSCs in another model that followed PNET formation after orthotopic transplantation.
Recombination of PTEN/p53 in adult NSCs gave rise to gliomas, whereas deletion of Rb/p53 or Rb/p53/PTEN generated PNETs, indicating an important role of an early Rb loss in driving the PNET phenotype [89]. Germ line mutations of INI1 (SNF5) have been identified in 78% of children with atypic theratoid/rhabdoid tumors (ATRTs) [173], indicating that INI1 is the mayor tumor suppressor in this disease. INI1 loss has recently been found to promote an aberrant activation of the SHH/GLI pathway [174]. Mouse models that recapitulate ATRTs are still missing. While homozygous deletion of Ini1/Snf5 results in early embryonic lethality heterozygotes develop primitive sarcomas but not ATRTs [175,176]. Finally, adult MB comprises three molecular variants [177] with SHH tumors being the most common and with Group 3 MB missing. Whether mouse models better recapitulate these adult or childhood MB is not clear, especially as the timing of tumor formation in mice cannot be compared to that of human tumors.
4.9 Cell of origin for embryonal brain tumor development
Many labs have suggested a putative cell of origin for their MB model. Importantly, as MB at the transcriptome level is not just one but at least four different types of tumors [114], one might suggest different types of originating cell for these molecularly distinct brain tumors. The most studied candidate for MB is the granule neuron precursor that has been shown to give rise to MB in SHH-models [36,37]. The SmoM2 model described above was used to carefully study cell of origin for MB when it was further refined to give rise to MB only [36]. This was achieved by using specific promoters expressed in distinct cell types including the Tlx3 promoter for GPCs in the EGL of Lobes VI-IX and the Math1 promoter for all types of GCPs. The authors also used the Olig2 promoter and the human GFAP promoter for cells expressed in various parts of the RL, two promoters that drive expression of SmoM2 in cells that later give rise to GPCs as well as cerebellar astrocytes and interneurons. Similarly, Ptch loss could be driven conditionally and restrict Cre using promoters like Math1 and GFAP to target the cerebellar cell types we just described [37]. Importantly, these two models confirm that SHH-driven MB could be generated from primitive NSCs as well as more restricted GCPs, with MBs developing spatially and coupled to the cell of origin that expressed the specific promoter.
Surprisingly, WNT-driven MBs were recently shown to emanate from Olig3-positive lower RL progenitors in the developing dorsal brain stem [149]. Other types of brain stem cells, particularly GCPs of the cochlear nuclei, a derivative of the auditory lower RL, have recently been shown to generate SHH-driven tumors [178]. Math1-postive GCPs do not generate MYC-driven MB. It is rather Math1-negative NSCs that generate such tumors that correlate with molecular profiles of human Group 3 MB [146,145]. Interestingly, the age at transformation might determine the type of tumor generated. While GFAP-positive NSCs isolated from embryonic cerebellum give rise to SHH MB following N-MYC transduction, GFAP-positive NSCs isolated at postnatal ages generates a KCNA1-positive N-MYC-driven MB characteristic of a human Group 4 MB lacking SHH-dependence [148]. This clearly demonstrates the importance of intrinsic differences in distinct NSC populations. However, a detailed analysis of cells of origin in MYC/MYCN-driven tumors in models in vivo would reveal what specific cells that really respond to transformation and produce these Group 3 and Group 4 tumors. Moreover, what types of cells generate those subgroups of Group 3 and Group 4 MB that do not express MYC proteins, or that show MYC/MYCN amplification?
5. Improved mouse models for human brain tumors
5.1 Current models recapitulate human brain tumor histology
A prominent feature of glioma is the ability to diffusely invade into surrounding normal parenchyma which makes complete removal difficult upon resection. Grade III gliomas (anaplastic astrocytoma, anaplastic oligodendroglioma and anaplastic oligoastrocytoma) are characterized by increased cellularity, nuclear atypia, and proliferative activity. Grade IV GBMs also contain areas of microvascular proliferation and necrosis often surrounded by pseudopalisading cells [179].
MB typically presents with poorly differentiated cells. MB has histologically been classified into five variants: classic, desmoplastic/nodular, MB with extensive nodularity, large cell MB and anaplastic MB [180].
PNETs occur predominantly in children and adolescents and show aggressive clinical behavior. They may be phenotypically undifferentiated, or present differentiation along neuronal, astrocytic and ependymal lines. CNS PNETs include supratentorial PNETs but also similar tumors located in the brain stem and spinal cord [112].
Many of the models described in this review present histological characteristics and close pathological similarities/hallmarks of the human brain tumor they model. Three examples of transgenic models that present such features are presented in Figure 4.
Figure 4. Transgenic brain tumor models that recapitulate human brain tumors.
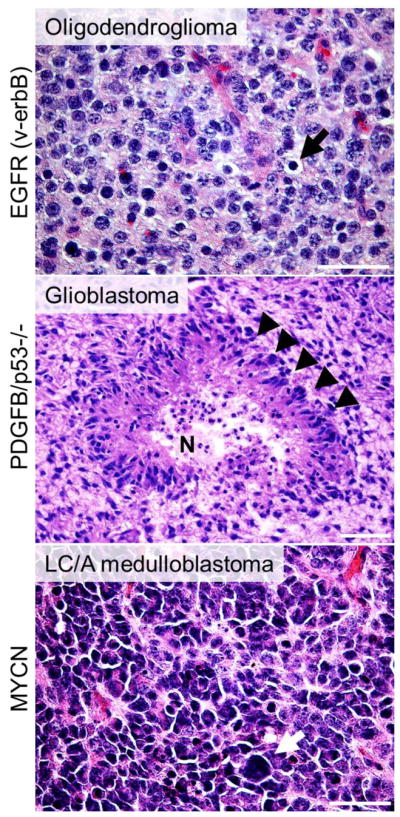
Three examples of models where different types of brain tumors are generated from specific promoters that targets a distinct population of cells in the brain with three clinically relevant cancer genes, v-erbB (activated EGFR), PDGFB (that activates PDGFRα), Trp53 as well as MYCN. A) Tumor cells from an S100β-v-erbB-driven oligodendroglioma with typical round nuclei and perinuclear cytoplasmic retraction (black arrows). [61] B) A GFAP-PDGFB-driven Trp53 null GBM with characteristic pseudopalisadation (black arrowheads) surrounding a necrotic area (N) [76]. C) A Glt1-MYCN-driven LC/A MB with atypical large cells (white arrow) [147].
5.2 New models targeting different brain tumor subtypes
There is a need for new models to recapitulate those cancer genes frequently altered in brain tumors. This is evident when studying the identified cancer genes in high-throughput expression data (as described above) but also from identified gene alterations in recent whole genome sequencing efforts for brain tumors [181,182,161]. As sequencing technology improves and gets cheaper, it is likely that whole genome sequencing will be the method to use to find important mutations that drive or promote these brain tumors. Large scale sequencing of each major cancer is ongoing through the International Cancer Genome Consortium [183]. Of course, mutations found in these screens needs to be validated functionally, like using GEMMs. Valuable features for existing brain tumor models are listed in Figure 5. Also included are needs for new models to recapitulate all currently understood subtypes and clinically relevant pathways.
Figure 5. New animal models for human brain tumors.

Useful features for great brain tumor models and models that are still missing or ongoing.
5.3 Forward genetics in mice: Retroviral and Sleeping Beauty strategies in brain tumors
Retroviral tagging as an instrument to find novel tumor-initiating genes has been used in murine glioma models driven by a Moloney Murine Leukemia Virus containing human PDGFB [184–186]. Here, over 60 common insertion sites (CIS) were identified that contained genes that could collaborate in PDGF-driven brain tumor formation. Several candidate genes in these CIS have been functionally evaluated as having a clear role in gliomagenesis [187–191] suggesting such retroviral screens are useful tools in understanding and identifying brain tumor initiating events. Several sleeping beauty transposon-based techniques have also been used to drive glioma [192–194] and MB [195] in mice and identified genetic drivers in the genesis of these brain tumors. Forward genetics using sleeping beauty has successfully been able to drive SHH-driven MB to disseminate along the leptomenigeal space to form metastases [144]. Additional Sleeping Beauty systems promise to identify novel cancer genes and to identify and validate candidate drivers for brain tumors.
6. Conclusion and future perspectives
Numerous mouse models exist that have been helpful in recapitulating diverse brain tumors that occur in humans, especially glioma and MB. A further refinement of mouse-to-human transcriptome/genome correlation will clarify how well these models recapitulate clinically relevant subtypes. The origin of brain tumors is almost certainly diverse. After several decades of brain tumor modeling in animals, a view of how to manipulate mouse brain tumors is now beginning to emerge. A challenge for future research is to build better parallels with human brain and brain tumor development. A major need is for markers that are better and more specific markers than the broadly expressed markers commonly used in the models described in this review.
Many of the models employ the most clinically relevant (most frequently altered) genes found in human brain tumors. Some of these gene alterations function solely in initiation while others are specifically involved in progression or dissemination of the disease. GEMMs have been useful for, and could further clarify how particular cancer genes contribute to the biology of brain tumors. GEMMs could further be used to identify those cancer proteins that are essential for maintenance of brain tumors; proteins that represent promising therapeutic targets. Hopefully, existing and future GEMMs for human brain tumor subtypes can help us find cures for diverse brain tumors with dismal prognoses like GBM, and can guide us to select more effective and more precise compounds that could replace the damaging and long-lasting effects of radiation and chemotherapy, especially in children with MB.
Acknowledgments
We acknowledge the Swedish Childhood Cancer Foundation, the Swedish Cancer Society, the Pediatric Brain Tumor Foundation, the Swedish Research Council, Åke Wibergs stiftelse, Lions Cancerforskningsfond and Stiftelsen Lars Hiertas Minne, NIH grants CA133091, NS055750, CA102321, CA128583, CA148699, CA163155, CA081403, Burroughs Wellcome Fund, Alex’s Lemonade Stand, Katie Dougherty, Pediatric Brain Tumor, Samuel G. Waxman and V Foundations. We apologize to authors whose work we did not cite, due to space restrictions in this review. Authors further declare no conflict of interest.
References
- 1.Lumsden A, Krumlauf R. Patterning the vertebrate neuraxis. Science. 1996;274(5290):1109–1115. doi: 10.1126/science.274.5290.1109. [DOI] [PubMed] [Google Scholar]
- 2.Liu A, Niswander LA. Bone morphogenetic protein signalling and vertebrate nervous system development. Nat Rev Neurosci. 2005;6(12):945–954. doi: 10.1038/nrn1805. [DOI] [PubMed] [Google Scholar]
- 3.Rowitch DH, Kriegstein AR. Developmental genetics of vertebrate glial-cell specification. Nature. 2010;468(7321):214–222. doi: 10.1038/nature09611. [DOI] [PubMed] [Google Scholar]
- 4.Malatesta P, Hartfuss E, Gotz M. Isolation of radial glial cells by fluorescent-activated cell sorting reveals a neuronal lineage. Development. 2000;127(24):5253–5263. doi: 10.1242/dev.127.24.5253. [DOI] [PubMed] [Google Scholar]
- 5.Hansen DV, Lui JH, Parker PR, Kriegstein AR. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature. 2010;464(7288):554–561. doi: 10.1038/nature08845. [DOI] [PubMed] [Google Scholar]
- 6.Feng L, Hatten ME, Heintz N. Brain lipid-binding protein (BLBP): a novel signaling system in the developing mammalian CNS. Neuron. 1994;12(4):895–908. doi: 10.1016/0896-6273(94)90341-7. [DOI] [PubMed] [Google Scholar]
- 7.Hartfuss E, Galli R, Heins N, Gotz M. Characterization of CNS precursor subtypes and radial glia. Dev Biol. 2001;229(1):15–30. doi: 10.1006/dbio.2000.9962. [DOI] [PubMed] [Google Scholar]
- 8.Kurtz A, Zimmer A, Schnutgen F, Bruning G, Spener F, Muller T. The expression pattern of a novel gene encoding brain-fatty acid binding protein correlates with neuronal and glial cell development. Development. 1994;120(9):2637–2649. doi: 10.1242/dev.120.9.2637. [DOI] [PubMed] [Google Scholar]
- 9.Anthony TE, Klein C, Fishell G, Heintz N. Radial glia serve as neuronal progenitors in all regions of the central nervous system. Neuron. 2004;41(6):881–890. doi: 10.1016/s0896-6273(04)00140-0. [DOI] [PubMed] [Google Scholar]
- 10.Noble M. Precursor cell transitions in oligodendrocyte development. J Cell Biol. 2000;148(5):839–842. doi: 10.1083/jcb.148.5.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Menn B, Garcia-Verdugo JM, Yaschine C, Gonzalez-Perez O, Rowitch D, Alvarez-Buylla A. Origin of oligodendrocytes in the subventricular zone of the adult brain. J Neurosci. 2006;26(30):7907–7918. doi: 10.1523/JNEUROSCI.1299-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Doetsch F, Caille I, Lim DA, Garcia-Verdugo JM, Alvarez-Buylla A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell. 1999;97(6):703–716. doi: 10.1016/s0092-8674(00)80783-7. [DOI] [PubMed] [Google Scholar]
- 13.Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, et al. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4(11):1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- 14.Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spassky N, Merkle FT, Flames N, Tramontin AD, Garcia-Verdugo JM, Alvarez-Buylla A. Adult ependymal cells are postmitotic and are derived from radial glial cells during embryogenesis. J Neurosci. 2005;25(1):10–18. doi: 10.1523/JNEUROSCI.1108-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sgaier SK, Millet S, Villanueva MP, Berenshteyn F, Song C, Joyner AL. Morphogenetic and cellular movements that shape the mouse cerebellum; insights from genetic fate mapping. Neuron. 2005;45(1):27–40. doi: 10.1016/j.neuron.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 17.Simeone A. Positioning the isthmic organizer where Otx2 and Gbx2meet. Trends Genet. 2000;16(6):237–240. doi: 10.1016/s0168-9525(00)02000-x. [DOI] [PubMed] [Google Scholar]
- 18.Joyner AL, Liu A, Millet S. Otx2, Gbx2 and Fgf8 interact to position and maintain a mid-hindbrain organizer. Curr Opin Cell Biol. 2000;12(6):736–741. doi: 10.1016/s0955-0674(00)00161-7. [DOI] [PubMed] [Google Scholar]
- 19.Ito M. Control of mental activities by internal models in the cerebellum. Nat Rev Neurosci. 2008;9(4):304–313. doi: 10.1038/nrn2332. [DOI] [PubMed] [Google Scholar]
- 20.Levisohn L, Cronin-Golomb A, Schmahmann JD. Neuropsychological consequences of cerebellar tumour resection in children: cerebellar cognitive affective syndrome in a paediatric population. Brain. 2000;123(Pt 5):1041–1050. doi: 10.1093/brain/123.5.1041. [DOI] [PubMed] [Google Scholar]
- 21.Hatten ME, Heintz N. Mechanisms of neural patterning and specification in the developing cerebellum. Annu Rev Neurosci. 1995;18:385–408. doi: 10.1146/annurev.ne.18.030195.002125. [DOI] [PubMed] [Google Scholar]
- 22.Hoshino M, Nakamura S, Mori K, Kawauchi T, Terao M, Nishimura YV, et al. Ptf1a, a bHLH transcriptional gene, defines GABAergic neuronal fates in cerebellum. Neuron. 2005;47(2):201–213. doi: 10.1016/j.neuron.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 23.Pascual M, Abasolo I, Mingorance-Le Meur A, Martinez A, Del Rio JA, Wright CV, et al. Cerebellar GABAergic progenitors adopt an external granule cell-like phenotype in the absence of Ptf1a transcription factor expression. Proc Natl Acad Sci U S A. 2007;104(12):5193–5198. doi: 10.1073/pnas.0605699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edwards MA, Yamamoto M, Caviness VS., Jr Organization of radial glia and related cells in the developing murine CNS. An analysis based upon a new monoclonal antibody marker. Neuroscience. 1990;36(1):121–144. doi: 10.1016/0306-4522(90)90356-9. [DOI] [PubMed] [Google Scholar]
- 25.Morales D, Hatten ME. Molecular markers of neuronal progenitors in the embryonic cerebellar anlage. J Neurosci. 2006;26(47):12226–12236. doi: 10.1523/JNEUROSCI.3493-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hevner RF, Hodge RD, Daza RA, Englund C. Transcription factors in glutamatergic neurogenesis: conserved programs in neocortex, cerebellum, and adult hippocampus. Neurosci Res. 2006;55(3):223–233. doi: 10.1016/j.neures.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 27.Akazawa C, Ishibashi M, Shimizu C, Nakanishi S, Kageyama R. A mammalian helix-loop-helix factor structurally related to the product of Drosophila proneural gene atonal is a positive transcriptional regulator expressed in the developing nervous system. J Biol Chem. 1995;270(15):8730–8738. doi: 10.1074/jbc.270.15.8730. [DOI] [PubMed] [Google Scholar]
- 28.Ben-Arie N, Bellen HJ, Armstrong DL, McCall AE, Gordadze PR, Guo Q, et al. Math1 is essential for genesis of cerebellar granule neurons. Nature. 1997;390(6656):169–172. doi: 10.1038/36579. [DOI] [PubMed] [Google Scholar]
- 29.Altman J, Bayer SA. Development of the cerebellar system: In Relation to its Evolution, Structure and Functions. Boca Raton, FL: CRC Press; 1997. [Google Scholar]
- 30.Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22(1):103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- 31.Dahmane N, Ruiz i Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126(14):3089–3100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- 32.Huang X, Liu J, Ketova T, Fleming JT, Grover VK, Cooper MK, et al. Transventricular delivery of Sonic hedgehog is essential to cerebellar ventricular zone development. Proc Natl Acad Sci U S A. 2010;107(18):8422–8427. doi: 10.1073/pnas.0911838107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Machold R, Fishell G. Math1 is expressed in temporally discrete pools of cerebellar rhombic-lip neural progenitors. Neuron. 2005;48(1):17–24. doi: 10.1016/j.neuron.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 34.Wang VY, Rose MF, Zoghbi HY. Math1 expression redefines the rhombic lip derivatives and reveals novel lineages within the brainstem and cerebellum. Neuron. 2005;48(1):31–43. doi: 10.1016/j.neuron.2005.08.024. [DOI] [PubMed] [Google Scholar]
- 35.Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31(2):85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]
- 36.Schuller U, Heine VM, Mao J, Kho AT, Dillon AK, Han YG, et al. Acquisition of granule neuron precursor identity is a critical determinant of progenitor cell competence to form Shh-induced medulloblastoma. Cancer Cell. 2008;14(2):123–134. doi: 10.1016/j.ccr.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, et al. Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell. 2008;14(2):135–145. doi: 10.1016/j.ccr.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aldinger KA, Elsen GE. Ptf1a is a molecular determinant for both glutamatergic and GABAergic neurons in the hindbrain. J Neurosci. 2008;28(2):338–339. doi: 10.1523/JNEUROSCI.5139-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raaf J, Kernohan J. A study of the external granular layer in the cerebellum. Am J Anat. 1944;(75):151–172. [Google Scholar]
- 40.Chizhikov VV, Lindgren AG, Currle DS, Rose MF, Monuki ES, Millen KJ. The roof plate regulates cerebellar cell-type specification and proliferation. Development. 2006;133(15):2793–2804. doi: 10.1242/dev.02441. [DOI] [PubMed] [Google Scholar]
- 41.Machold RP, Kittell DJ, Fishell GJ. Antagonism between Notch and bone morphogenetic protein receptor signaling regulates neurogenesis in the cerebellar rhombic lip. Neural Dev. 2007;2:5. doi: 10.1186/1749-8104-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alder J, Lee KJ, Jessell TM, Hatten ME. Generation of cerebellar granule neurons in vivo by transplantation of BMP-treated neural progenitor cells. Nat Neurosci. 1999;2(6):535–540. doi: 10.1038/9189. [DOI] [PubMed] [Google Scholar]
- 43.Rios I, Alvarez-Rodriguez R, Marti E, Pons S. Bmp2 antagonizes sonic hedgehog-mediated proliferation of cerebellar granule neurones through Smad5 signalling. Development. 2004;131(13):3159–3168. doi: 10.1242/dev.01188. [DOI] [PubMed] [Google Scholar]
- 44.Zhao H, Ayrault O, Zindy F, Kim JH, Roussel MF. Post-transcriptional down-regulation of Atoh1/Math1 by bone morphogenic proteins suppresses medulloblastoma development. Genes Dev. 2008;22(6):722–727. doi: 10.1101/gad.1636408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lutolf S, Radtke F, Aguet M, Suter U, Taylor V. Notch1 is required for neuronal and glial differentiation in the cerebellum. Development. 2002;129(2):373–385. doi: 10.1242/dev.129.2.373. [DOI] [PubMed] [Google Scholar]
- 46.Solecki DJ, Liu XL, Tomoda T, Fang Y, Hatten ME. Activated Notch2 signaling inhibits differentiation of cerebellar granule neuron precursors by maintaining proliferation. Neuron. 2001;31(4):557–568. doi: 10.1016/s0896-6273(01)00395-6. [DOI] [PubMed] [Google Scholar]
- 47.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 48.Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DM, Hammond RR, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst. 1998;90(19):1473–1479. doi: 10.1093/jnci/90.19.1473. [DOI] [PubMed] [Google Scholar]
- 49.Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9(3):157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 51.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lai A, Kharbanda S, Pope WB, Tran A, Solis OE, Peale F, et al. Evidence for sequenced molecular evolution of IDH1 mutant glioblastoma from a distinct cell of origin. J Clin Oncol. 2011;29(34):4482–4490. doi: 10.1200/JCO.2010.33.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest. 2008;118(5):1739–1749. doi: 10.1172/JCI33656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68(21):8673–8677. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lewis RA, Gerson LP, Axelson KA, Riccardi VM, Whitford RP. von Recklinghausen neurofibromatosis. II. Incidence of optic gliomata. Ophthalmology. 1984;91(8):929–935. doi: 10.1016/s0161-6420(84)34217-8. [DOI] [PubMed] [Google Scholar]
- 56.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 57.Johnson RA, Wright KD, Poppleton H, Mohankumar KM, Finkelstein D, Pounds SB, et al. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature. 2010;466(7306):632–636. doi: 10.1038/nature09173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wong AJ, Ruppert JM, Bigner SH, Grzeschik CH, Humphrey PA, Bigner DS, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci U S A. 1992;89(7):2965–2969. doi: 10.1073/pnas.89.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ekstrand AJ, Longo N, Hamid ML, Olson JJ, Liu L, Collins VP, et al. Functional characterization of an EGF receptor with a truncated extracellular domain expressed in glioblastomas with EGFR gene amplification. Oncogene. 1994;9(8):2313–2320. [PubMed] [Google Scholar]
- 60.Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998;12(23):3675–3685. doi: 10.1101/gad.12.23.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weiss WA, Burns MJ, Hackett C, Aldape K, Hill JR, Kuriyama H, et al. Genetic determinants of malignancy in a mouse model for oligodendroglioma. Cancer Res. 2003;63(7):1589–1595. [PubMed] [Google Scholar]
- 62.Ding H, Shannon P, Lau N, Wu X, Roncari L, Baldwin RL, et al. Oligodendrogliomas result from the expression of an activated mutant epidermal growth factor receptor in a RAS transgenic mouse astrocytoma model. Cancer Res. 2003;63(5):1106–1113. [PubMed] [Google Scholar]
- 63.Persson AI, Petritsch C, Swartling FJ, Itsara M, Sim FJ, Auvergne R, et al. Non-stem cell origin for oligodendroglioma. Cancer Cell. 2010;18(6):669–682. doi: 10.1016/j.ccr.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bachoo RM, Maher EA, Ligon KL, Sharpless NE, Chan SS, You MJ, et al. Epidermal growth factor receptor and Ink4a/Arf: convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell. 2002;1(3):269–277. doi: 10.1016/s1535-6108(02)00046-6. [DOI] [PubMed] [Google Scholar]
- 65.Zhu H, Acquaviva J, Ramachandran P, Boskovitz A, Woolfenden S, Pfannl R, et al. Oncogenic EGFR signaling cooperates with loss of tumor suppressor gene functions in gliomagenesis. Proc Natl Acad Sci U S A. 2009;106(8):2712–2716. doi: 10.1073/pnas.0813314106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Acquaviva J, Jun HJ, Lessard J, Ruiz R, Zhu H, Donovan M, et al. Chronic activation of wild-type epidermal growth factor receptor and loss of Cdkn2a cause mouse glioblastoma formation. Cancer Res. 2011;71(23):7198–7206. doi: 10.1158/0008-5472.CAN-11-1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fleming TP, Saxena A, Clark WC, Robertson JT, Oldfield EH, Aaronson SA, et al. Amplification and/or overexpression of platelet-derived growth factor receptors and epidermal growth factor receptor in human glial tumors. Cancer Res. 1992;52(16):4550–4553. [PubMed] [Google Scholar]
- 68.Hermanson M, Funa K, Hartman M, Claesson-Welsh L, Heldin CH, Westermark B, et al. Platelet-derived growth factor and its receptors in human glioma tissue: expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992;52(11):3213–3219. [PubMed] [Google Scholar]
- 69.Calzolari F, Appolloni I, Tutucci E, Caviglia S, Terrile M, Corte G, et al. Tumor progression and oncogene addiction in a PDGF-B-induced model of gliomagenesis. Neoplasia. 2008;10(12):1373–1382. doi: 10.1593/neo.08814. following 1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Uhrbom L, Hesselager G, Nister M, Westermark B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 1998;58(23):5275–5279. [PubMed] [Google Scholar]
- 71.Assanah MC, Bruce JN, Suzuki SO, Chen A, Goldman JE, Canoll P. PDGF stimulates the massive expansion of glial progenitors in the neonatal forebrain. Glia. 2009;57(16):1835–1847. doi: 10.1002/glia.20895. [DOI] [PubMed] [Google Scholar]
- 72.Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes Dev. 2001;15(15):1913–1925. doi: 10.1101/gad.903001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lindberg N, Kastemar M, Olofsson T, Smits A, Uhrbom L. Oligodendrocyte progenitor cells can act as cell of origin for experimental glioma. Oncogene. 2009;28(23):2266–2275. doi: 10.1038/onc.2009.76. [DOI] [PubMed] [Google Scholar]
- 74.Hesselager G, Uhrbom L, Westermark B, Nister M. Complementary effects of platelet-derived growth factor autocrine stimulation and p53 or Ink4a-Arf deletion in a mouse glioma model. Cancer Res. 2003;63(15):4305–4309. [PubMed] [Google Scholar]
- 75.Tchougounova E, Kastemar M, Brasater D, Holland EC, Westermark B, Uhrbom L. Loss of Arf causes tumor progression of PDGFB-induced oligodendroglioma. Oncogene. 2007;26(43):6289–6296. doi: 10.1038/sj.onc.1210455. [DOI] [PubMed] [Google Scholar]
- 76.Hede SM, Hansson I, Afink GB, Eriksson A, Nazarenko I, Andrae J, et al. GFAP promoter driven transgenic expression of PDGFB in the mouse brain leads to glioblastoma in a Trp53 null background. Glia. 2009;57(11):1143–1153. doi: 10.1002/glia.20837. [DOI] [PubMed] [Google Scholar]
- 77.Nazarenko I, Hedren A, Sjodin H, Orrego A, Andrae J, Afink GB, et al. Brain abnormalities and glioma-like lesions in mice overexpressing the long isoform of PDGF-A in astrocytic cells. PLoS One. 2011;6(4):e18303. doi: 10.1371/journal.pone.0018303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lei L, Sonabend AM, Guarnieri P, Soderquist C, Ludwig T, Rosenfeld S, et al. Glioblastoma models reveal the connection between adult glial progenitors and the proneural phenotype. PLoS One. 2011;6(5):e20041. doi: 10.1371/journal.pone.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ding H, Roncari L, Shannon P, Wu X, Lau N, Karaskova J, et al. Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res. 2001;61(9):3826–3836. [PubMed] [Google Scholar]
- 80.Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25(1):55–57. doi: 10.1038/75596. [DOI] [PubMed] [Google Scholar]
- 81.Uhrbom L, Dai C, Celestino JC, Rosenblum MK, Fuller GN, Holland EC. Ink4a-Arf loss cooperates with KRas activation in astrocytes and neural progenitors to generate glioblastomas of various morphologies depending on activated Akt. Cancer Res. 2002;62(19):5551–5558. [PubMed] [Google Scholar]
- 82.Marumoto T, Tashiro A, Friedmann-Morvinski D, Scadeng M, Soda Y, Gage FH, et al. Development of a novel mouse glioma model using lentiviral vectors. Nat Med. 2009;15(1):110–116. doi: 10.1038/nm.1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reilly KM, Loisel DA, Bronson RT, McLaughlin ME, Jacks T. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000;26(1):109–113. doi: 10.1038/79075. [DOI] [PubMed] [Google Scholar]
- 84.Wang Y, Yang J, Zheng H, Tomasek GJ, Zhang P, McKeever PE, et al. Expression of mutant p53 proteins implicates a lineage relationship between neural stem cells and malignant astrocytic glioma in a murine model. Cancer Cell. 2009;15(6):514–526. doi: 10.1016/j.ccr.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, et al. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005;8(2):119–130. doi: 10.1016/j.ccr.2005.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.von Deimling A, Eibl RH, Ohgaki H, Louis DN, von Ammon K, Petersen I, et al. p53 mutations are associated with 17p allelic loss in grade II and grade III astrocytoma. Cancer Res. 1992;52(10):2987–2990. [PubMed] [Google Scholar]
- 87.Kwon CH, Zhao D, Chen J, Alcantara S, Li Y, Burns DK, et al. Pten haploinsufficiency accelerates formation of high-grade astrocytomas. Cancer Res. 2008;68(9):3286–3294. doi: 10.1158/0008-5472.CAN-07-6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, et al. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell. 2009;15(1):45–56. doi: 10.1016/j.ccr.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jacques TS, Swales A, Brzozowski MJ, Henriquez NV, Linehan JM, Mirzadeh Z, et al. Combinations of genetic mutations in the adult neural stem cell compartment determine brain tumour phenotypes. EMBO J. 2010;29(1):222–235. doi: 10.1038/emboj.2009.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chow LM, Endersby R, Zhu X, Rankin S, Qu C, Zhang J, et al. Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell. 2011;19(3):305–316. doi: 10.1016/j.ccr.2011.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gronych J, Korshunov A, Bageritz J, Milde T, Jugold M, Hambardzumyan D, et al. An activated mutant BRAF kinase domain is sufficient to induce pilocytic astrocytoma in mice. J Clin Invest. 2011;121(4):1344–1348. doi: 10.1172/JCI44656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Becher OJ, Hambardzumyan D, Walker TR, Helmy K, Nazarian J, Albrecht S, et al. Preclinical evaluation of radiation and perifosine in a genetically and histologically accurate model of brainstem glioma. Cancer Res. 2010;70(6):2548–2557. doi: 10.1158/0008-5472.CAN-09-2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hitoshi Y, Harris BT, Liu H, Popko B, Israel MA. Spinal glioma: platelet-derived growth factor B-mediated oncogenesis in the spinal cord. Cancer Res. 2008;68(20):8507–8515. doi: 10.1158/0008-5472.CAN-08-1063. [DOI] [PubMed] [Google Scholar]
- 94.Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011;146(2):209–221. doi: 10.1016/j.cell.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hambardzumyan D, Amankulor NM, Helmy KY, Becher OJ, Holland EC. Modeling Adult Gliomas Using RCAS/t-va Technology. Transl Oncol. 2009;2(2):89–95. doi: 10.1593/tlo.09100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005;8(4):323–335. doi: 10.1016/j.ccr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 97.Weissenberger J, Steinbach JP, Malin G, Spada S, Rulicke T, Aguzzi A. Development and malignant progression of astrocytomas in GFAP-v-src transgenic mice. Oncogene. 1997;14(17):2005–2013. doi: 10.1038/sj.onc.1201168. [DOI] [PubMed] [Google Scholar]
- 98.Kamijo T, Bodner S, van de Kamp E, Randle DH, Sherr CJ. Tumor spectrum in ARF-deficient mice. Cancer Res. 1999;59(9):2217–2222. [PubMed] [Google Scholar]
- 99.Jensen NA, Pedersen KM, Lihme F, Rask L, Nielsen JV, Rasmussen TE, et al. Astroglial c-Myc overexpression predisposes mice to primary malignant gliomas. J Biol Chem. 2003;278(10):8300–8308. doi: 10.1074/jbc.M211195200. [DOI] [PubMed] [Google Scholar]
- 100.Abel TW, Clark C, Bierie B, Chytil A, Aakre M, Gorska A, et al. GFAP-Cre-mediated activation of oncogenic K-ras results in expansion of the subventricular zone and infiltrating glioma. Mol Cancer Res. 2009;7(5):645–653. doi: 10.1158/1541-7786.MCR-08-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shannon P, Sabha N, Lau N, Kamnasaran D, Gutmann DH, Guha A. Pathological and molecular progression of astrocytomas in a GFAP:12 V-Ha-Ras mouse astrocytoma model. Am J Pathol. 2005;167(3):859–867. doi: 10.1016/S0002-9440(10)62057-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wei Q, Clarke L, Scheidenhelm DK, Qian B, Tong A, Sabha N, et al. High-grade glioma formation results from postnatal pten loss or mutant epidermal growth factor receptor expression in a transgenic mouse glioma model. Cancer Res. 2006;66(15):7429–7437. doi: 10.1158/0008-5472.CAN-06-0712. [DOI] [PubMed] [Google Scholar]
- 103.Xiao A, Wu H, Pandolfi PP, Louis DN, Van Dyke T. Astrocyte inactivation of the pRb pathway predisposes mice to malignant astrocytoma development that is accelerated by PTEN mutation. Cancer Cell. 2002;1(2):157–168. doi: 10.1016/s1535-6108(02)00029-6. [DOI] [PubMed] [Google Scholar]
- 104.Uhrbom L, Hesselager G, Ostman A, Nister M, Westermark B. Dependence of autocrine growth factor stimulation in platelet-derived growth factor-B-induced mouse brain tumor cells. Int J Cancer. 2000;85(3):398–406. doi: 10.1002/(sici)1097-0215(20000201)85:3<398::aid-ijc17>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 105.Dai C, Lyustikman Y, Shih A, Hu X, Fuller GN, Rosenblum M, et al. The characteristics of astrocytomas and oligodendrogliomas are caused by two distinct and interchangeable signaling formats. Neoplasia. 2005;7(4):397–406. doi: 10.1593/neo.04691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shih AH, Dai C, Hu X, Rosenblum MK, Koutcher JA, Holland EC. Dose-dependent effects of platelet-derived growth factor-B on glial tumorigenesis. Cancer Res. 2004;64(14):4783–4789. doi: 10.1158/0008-5472.CAN-03-3831. [DOI] [PubMed] [Google Scholar]
- 107.Hu X, Pandolfi PP, Li Y, Koutcher JA, Rosenblum M, Holland EC. mTOR promotes survival and astrocytic characteristics induced by Pten/AKT signaling in glioblastoma. Neoplasia. 2005;7(4):356–368. doi: 10.1593/neo.04595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature. 2008;455(7216):1129–1133. doi: 10.1038/nature07443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gil-Perotin S, Marin-Husstege M, Li J, Soriano-Navarro M, Zindy F, Roussel MF, et al. Loss of p53 induces changes in the behavior of subventricular zone cells: implication for the genesis of glial tumors. J Neurosci. 2006;26(4):1107–1116. doi: 10.1523/JNEUROSCI.3970-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Saran A. Medulloblastoma: role of developmental pathways, DNA repair signaling, and other players. Curr Mol Med. 2009;9(9):1046–1057. doi: 10.2174/156652409789839080. [DOI] [PubMed] [Google Scholar]
- 111.Schmidt AL, Brunetto AL, Schwartsmann G, Roesler R, Abujamra AL. Recent therapeutic advances for treating medulloblastoma: focus on new molecular targets. CNS Neurol Disord Drug Targets. 2010;9(3):335–348. doi: 10.2174/187152710791292602. [DOI] [PubMed] [Google Scholar]
- 112.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007;114(2):97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Eberhart CG, Brat DJ, Cohen KJ, Burger PC. Pediatric neuroblastic brain tumors containing abundant neuropil and true rosettes. Pediatr Dev Pathol. 2000;3(4):346–352. doi: 10.1007/s100249910049. [DOI] [PubMed] [Google Scholar]
- 114.Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2011 doi: 10.1007/s00401-011-0922-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24(12):1924–1931. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- 116.Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, van Sluis P, et al. Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One. 2008;3(8):e3088. doi: 10.1371/journal.pone.0003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cho YJ, Tsherniak A, Tamayo P, Santagata S, Ligon A, Greulich H, et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol. 2011;29(11):1424–1430. doi: 10.1200/JCO.2010.28.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, et al. Medulloblastoma comprises four distinct molecular variants. J Clin Oncol. 2011;29(11):1408–1414. doi: 10.1200/JCO.2009.27.4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012;123(4):473–484. doi: 10.1007/s00401-012-0958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277(5329):1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- 121.Pazzaglia S, Mancuso M, Atkinson MJ, Tanori M, Rebessi S, Majo VD, et al. High incidence of medulloblastoma following X-ray-irradiation of newborn Ptc1 heterozygous mice. Oncogene. 2002;21(49):7580–7584. doi: 10.1038/sj.onc.1205973. [DOI] [PubMed] [Google Scholar]
- 122.Wetmore C, Eberhart DE, Curran T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res. 2001;61(2):513–516. [PubMed] [Google Scholar]
- 123.Uziel T, Zindy F, Xie S, Lee Y, Forget A, Magdaleno S, et al. The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes Dev. 2005;19(22):2656–2667. doi: 10.1101/gad.1368605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lee Y, Miller HL, Russell HR, Boyd K, Curran T, McKinnon PJ. Patched2 modulates tumorigenesis in patched1 heterozygous mice. Cancer Res. 2006;66(14):6964–6971. doi: 10.1158/0008-5472.CAN-06-0505. [DOI] [PubMed] [Google Scholar]
- 125.Ayrault O, Zindy F, Rehg J, Sherr CJ, Roussel MF. Two tumor suppressors, p27Kip1 and patched-1, collaborate to prevent medulloblastoma. Mol Cancer Res. 2009;7(1):33–40. doi: 10.1158/1541-7786.MCR-08-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Briggs KJ, Corcoran-Schwartz IM, Zhang W, Harcke T, Devereux WL, Baylin SB, et al. Cooperation between the Hic1 and Ptch1 tumor suppressors in medulloblastoma. Genes Dev. 2008;22(6):770–785. doi: 10.1101/gad.1640908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14(8):994–1004. [PMC free article] [PubMed] [Google Scholar]
- 128.Sutter R, Shakhova O, Bhagat H, Behesti H, Sutter C, Penkar S, et al. Cerebellar stem cells act as medulloblastoma-initiating cells in a mouse model and a neural stem cell signature characterizes a subset of human medulloblastomas. Oncogene. 2010;29(12):1845–1856. doi: 10.1038/onc.2009.472. [DOI] [PubMed] [Google Scholar]
- 129.Hatton BA, Villavicencio EH, Pritchard J, LeBlanc M, Hansen S, Ulrich M, et al. Notch signaling is not essential in sonic hedgehog-activated medulloblastoma. Oncogene. 2010;29(26):3865–3872. doi: 10.1038/onc.2010.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res. 2004;64(21):7794–7800. doi: 10.1158/0008-5472.CAN-04-1813. [DOI] [PubMed] [Google Scholar]
- 131.Hatton BA, Villavicencio EH, Tsuchiya KD, Pritchard JI, Ditzler S, Pullar B, et al. The Smo/Smo model: hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res. 2008;68(6):1768–1776. doi: 10.1158/0008-5472.CAN-07-5092. [DOI] [PubMed] [Google Scholar]
- 132.Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D, et al. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res. 2006;66(20):10171–10178. doi: 10.1158/0008-5472.CAN-06-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lee Y, Kawagoe R, Sasai K, Li Y, Russell HR, Curran T, et al. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene. 2007;26(44):6442–6447. doi: 10.1038/sj.onc.1210467. [DOI] [PubMed] [Google Scholar]
- 134.Weiner HL, Bakst R, Hurlbert MS, Ruggiero J, Ahn E, Lee WS, et al. Induction of medulloblastomas in mice by sonic hedgehog, independent of Gli1. Cancer Res. 2002;62(22):6385–6389. [PubMed] [Google Scholar]
- 135.Rao G, Pedone CA, Coffin CM, Holland EC, Fults DW. c-Myc enhances sonic hedgehog-induced medulloblastoma formation from nestin-expressing neural progenitors in mice. Neoplasia. 2003;5(3):198–204. doi: 10.1016/S1476-5586(03)80052-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rao G, Pedone CA, Del Valle L, Reiss K, Holland EC, Fults DW. Sonic hedgehog and insulin-like growth factor signaling synergize to induce medulloblastoma formation from nestin-expressing neural progenitors in mice. Oncogene. 2004;23(36):6156–6162. doi: 10.1038/sj.onc.1207818. [DOI] [PubMed] [Google Scholar]
- 137.Browd SR, Kenney AM, Gottfried ON, Yoon JW, Walterhouse D, Pedone CA, et al. N-myc can substitute for insulin-like growth factor signaling in a mouse model of sonic hedgehog-induced medulloblastoma. Cancer Res. 2006;66(5):2666–2672. doi: 10.1158/0008-5472.CAN-05-2198. [DOI] [PubMed] [Google Scholar]
- 138.McCall TD, Pedone CA, Fults DW. Apoptosis suppression by somatic cell transfer of Bcl-2 promotes Sonic hedgehog-dependent medulloblastoma formation in mice. Cancer Res. 2007;67(11):5179–5185. doi: 10.1158/0008-5472.CAN-06-4177. [DOI] [PubMed] [Google Scholar]
- 139.Frappart PO, Lee Y, Lamont J, McKinnon PJ. BRCA2 is required for neurogenesis and suppression of medulloblastoma. EMBO J. 2007;26(11):2732–2742. doi: 10.1038/sj.emboj.7601703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Frappart PO, Lee Y, Russell HR, Chalhoub N, Wang YD, Orii KE, et al. Recurrent genomic alterations characterize medulloblastoma arising from DNA double-strand break repair deficiency. Proc Natl Acad Sci U S A. 2009;106(6):1880–1885. doi: 10.1073/pnas.0806882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yan CT, Kaushal D, Murphy M, Zhang Y, Datta A, Chen C, et al. XRCC4 suppresses medulloblastomas with recurrent translocations in p53-deficient mice. Proc Natl Acad Sci U S A. 2006;103(19):7378–7383. doi: 10.1073/pnas.0601938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tong WM, Ohgaki H, Huang H, Granier C, Kleihues P, Wang ZQ. Null mutation of DNA strand break-binding molecule poly(ADP-ribose) polymerase causes medulloblastomas in p53(−/−) mice. Am J Pathol. 2003;162(1):343–352. doi: 10.1016/S0002-9440(10)63825-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lee Y, McKinnon PJ. DNA ligase IV suppresses medulloblastoma formation. Cancer Res. 2002;62(22):6395–6399. [PubMed] [Google Scholar]
- 144.Wu X, Northcott PA, Dubuc A, Dupuy AJ, Shih DJ, Witt H, et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature. 2012;482(7386):529–533. doi: 10.1038/nature10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pei Y, Moore CE, Wang J, Tewari AK, Eroshkin A, Cho YJ, et al. An Animal Model of MYC-Driven Medulloblastoma. Cancer Cell. 2012;21(2):155–167. doi: 10.1016/j.ccr.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kawauchi D, Robinson G, Uziel T, Gibson P, Rehg J, Gao C, et al. A mouse model of the most aggressive subgroup of human medulloblastoma. Cancer Cell. 2012;21(2):168–180. doi: 10.1016/j.ccr.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Swartling FJ, Grimmer MR, Hackett CS, Northcott PA, Fan QW, Goldenberg DD, et al. Pleiotropic role for MYCN in medulloblastoma. Genes Dev. 2010;24(10):1059–1072. doi: 10.1101/gad.1907510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Swartling FJ, Savov V, Persson AI, Chen J, Hackett CS, Grimmer MR, et al. Distinct Neural Stem Cell Populations Give Rise to Disparate Brain Tumors in Response to N-MYC. Cancer Cell. 2012;25(5):601–613. doi: 10.1016/j.ccr.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468(7327):1095–1099. doi: 10.1038/nature09587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287(5785):795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- 151.Espinosa JS, Luo L. Timing neurogenesis and differentiation: insights from quantitative clonal analyses of cerebellar granule cells. J Neurosci. 2008;28(10):2301–2312. doi: 10.1523/JNEUROSCI.5157-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85(6):841–851. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 153.Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci U S A. 2005;102(32):11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317(5836):372–376. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- 155.Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437(7061):1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 156.Han YG, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez-Buylla A. Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med. 2009;15(9):1062–1065. doi: 10.1038/nm.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. 2003;130(1):15–28. doi: 10.1242/dev.00182. [DOI] [PubMed] [Google Scholar]
- 158.Kessler JD, Hasegawa H, Brun SN, Yang ZJ, Dutton JW, Wang F, et al. N-myc alters the fate of preneoplastic cells in a mouse model of medulloblastoma. Genes Dev. 2009;23(2):157–170. doi: 10.1101/gad.1759909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Stecca B, Ruiz i Altaba A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009;28(6):663–676. doi: 10.1038/emboj.2009.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Flora A, Klisch TJ, Schuster G, Zoghbi HY. Deletion of Atoh1 disrupts Sonic Hedgehog signaling in the developing cerebellum and prevents medulloblastoma. Science. 2009;326(5958):1424–1427. doi: 10.1126/science.1181453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rausch T, Jones DT, Zapatka M, Stutz AM, Zichner T, Weischenfeldt J, et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell. 2012;148(1–2):59–71. doi: 10.1016/j.cell.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Nusse R, van Ooyen A, Cox D, Fung YK, Varmus H. Mode of proviral activation of a putative mammary oncogene (int-1) on mouse chromosome 15. Nature. 1984;307(5947):131–136. doi: 10.1038/307131a0. [DOI] [PubMed] [Google Scholar]
- 163.Ellison DW, Dalton J, Kocak M, Nicholson SL, Fraga C, Neale G, et al. Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol. 2011;121(3):381–396. doi: 10.1007/s00401-011-0800-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Korshunov A, Remke M, Kool M, Hielscher T, Northcott PA, Williamson D, et al. Biological and clinical heterogeneity of MYCN-amplified medulloblastoma. Acta Neuropathol. 2011 doi: 10.1007/s00401-011-0918-8. [DOI] [PubMed] [Google Scholar]
- 165.Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7(10):813–820. doi: 10.1016/S1470-2045(06)70867-1. [DOI] [PubMed] [Google Scholar]
- 166.von Hoff K, Hinkes B, Gerber NU, Deinlein F, Mittler U, Urban C, et al. Long-term outcome and clinical prognostic factors in children with medulloblastoma treated in the prospective randomised multicentre trial HIT'91. Eur J Cancer. 2009;45(7):1209–1217. doi: 10.1016/j.ejca.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 167.MacDonald TJ, Brown KM, LaFleur B, Peterson K, Lawlor C, Chen Y, et al. Expression profiling of medulloblastoma: PDGFRA and the RAS/MAPK pathway as therapeutic targets for metastatic disease. Nat Genet. 2001;29(2):143–152. doi: 10.1038/ng731. [DOI] [PubMed] [Google Scholar]
- 168.Eibl RH, Kleihues P, Jat PS, Wiestler OD. A model for primitive neuroectodermal tumors in transgenic neural transplants harboring the SV40 large T antigen. Am J Pathol. 1994;144(3):556–564. [PMC free article] [PubMed] [Google Scholar]
- 169.Theuring F, Gotz W, Balling R, Korf HW, Schulze F, Herken R, et al. Tumorigenesis and eye abnormalities in transgenic mice expressing MSV-SV40 large T-antigen. Oncogene. 1990;5(2):225–232. [PubMed] [Google Scholar]
- 170.al-Ubaidi MR, Font RL, Quiambao AB, Keener MJ, Liou GI, Overbeek PA, et al. Bilateral retinal and brain tumors in transgenic mice expressing simian virus 40 large T antigen under control of the human interphotoreceptor retinoid-binding protein promoter. J Cell Biol. 1992;119(6):1681–1687. doi: 10.1083/jcb.119.6.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Momota H, Shih AH, Edgar MA, Holland EC. c-Myc and beta-catenin cooperate with loss of p53 to generate multiple members of the primitive neuroectodermal tumor family in mice. Oncogene. 2008;27(32):4392–4401. doi: 10.1038/onc.2008.81. [DOI] [PubMed] [Google Scholar]
- 172.Perry A, Miller CR, Gujrati M, Scheithauer BW, Zambrano SC, Jost SC, et al. Malignant gliomas with primitive neuroectodermal tumor-like components: a clinicopathologic and genetic study of 53 cases. Brain Pathol. 2009;19(1):81–90. doi: 10.1111/j.1750-3639.2008.00167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Biegel JA, Tan L, Zhang F, Wainwright L, Russo P, Rorke LB. Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res. 2002;8(11):3461–3467. [PubMed] [Google Scholar]
- 174.Jagani Z, Mora-Blanco EL, Sansam CG, McKenna ES, Wilson B, Chen D, et al. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat Med. 2010;16(12):1429–1433. doi: 10.1038/nm.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Guidi CJ, Sands AT, Zambrowicz BP, Turner TK, Demers DA, Webster W, et al. Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol Cell Biol. 2001;21(10):3598–3603. doi: 10.1128/MCB.21.10.3598-3603.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Roberts CW, Galusha SA, McMenamin ME, Fletcher CD, Orkin SH. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc Natl Acad Sci U S A. 2000;97(25):13796–13800. doi: 10.1073/pnas.250492697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Remke M, Hielscher T, Northcott PA, Witt H, Ryzhova M, Wittmann A, et al. Adult medulloblastoma comprises three major molecular variants. J Clin Oncol. 2011;29(19):2717–2723. doi: 10.1200/JCO.2011.34.9373. [DOI] [PubMed] [Google Scholar]
- 178.Grammel D, Warmuth-Metz M, von Bueren AO, Kool M, Pietsch T, Kretzschmar HA, et al. Sonic hedgehog-associated medulloblastoma arising from the cochlear nuclei of the brainstem. Acta Neuropathol. 2012;123(4):601–614. doi: 10.1007/s00401-012-0961-0. [DOI] [PubMed] [Google Scholar]
- 179.Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359(5):492–507. doi: 10.1056/NEJMra0708126. [DOI] [PubMed] [Google Scholar]
- 180.Gilbertson RJ, Ellison DW. The origins of medulloblastoma subtypes. Annu Rev Pathol. 2008;3:341–365. doi: 10.1146/annurev.pathmechdis.3.121806.151518. [DOI] [PubMed] [Google Scholar]
- 181.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331(6016):435–439. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hudson TJ, Anderson W, Artez A, Barker AD, Bell C, Bernabe RR, et al. International network of cancer genome projects. Nature. 2010;464(7291):993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Johansson FK, Brodd J, Eklof C, Ferletta M, Hesselager G, Tiger CF, et al. Identification of candidate cancer-causing genes in mouse brain tumors by retroviral tagging. Proc Natl Acad Sci U S A. 2004;101(31):11334–11337. doi: 10.1073/pnas.0402716101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Johansson FK, Goransson H, Westermark B. Expression analysis of genes involved in brain tumor progression driven by retroviral insertional mutagenesis in mice. Oncogene. 2005;24(24):3896–3905. doi: 10.1038/sj.onc.1208553. [DOI] [PubMed] [Google Scholar]
- 186.Johansson Swartling F. Identifying candidate genes involved in brain tumor formation. Ups J Med Sci. 2008;113(1):1–38. doi: 10.3109/2000-1967-215. [DOI] [PubMed] [Google Scholar]
- 187.Swartling FJ, Ferletta M, Kastemar M, Weiss WA, Westermark B. Cyclic GMP-dependent protein kinase II inhibits cell proliferation, Sox9 expression and Akt phosphorylation in human glioma cell lines. Oncogene. 2009;28(35):3121–3131. doi: 10.1038/onc.2009.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Phillips JJ, Huillard E, Robinson AE, Ward A, Lum DH, Polley MY, et al. Heparan sulfate sulfatase SULF2 regulates PDGFRalpha signaling and growth in human and mouse malignant glioma. J Clin Invest. 2012;122(3):911–922. doi: 10.1172/JCI58215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Tchougounova E, Jiang Y, Brasater D, Lindberg N, Kastemar M, Asplund A, et al. Sox5 can suppress platelet-derived growth factor B-induced glioma development in Ink4a-deficient mice through induction of acute cellular senescence. Oncogene. 2009;28(12):1537–1548. doi: 10.1038/onc.2009.9. [DOI] [PubMed] [Google Scholar]
- 190.Ferletta M, Uhrbom L, Olofsson T, Ponten F, Westermark B. Sox10 has a broad expression pattern in gliomas and enhances platelet-derived growth factor-B--induced gliomagenesis. Mol Cancer Res. 2007;5(9):891–897. doi: 10.1158/1541-7786.MCR-07-0113. [DOI] [PubMed] [Google Scholar]
- 191.Wolf RM, Draghi N, Liang X, Dai C, Uhrbom L, Eklof C, et al. p190RhoGAP can act to inhibit PDGF-induced gliomas in mice: a putative tumor suppressor encoded on human chromosome 19q13.3. Genes Dev. 2003;17(4):476–487. doi: 10.1101/gad.1040003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Wiesner SM, Decker SA, Larson JD, Ericson K, Forster C, Gallardo JL, et al. De novo induction of genetically engineered brain tumors in mice using plasmid DNA. Cancer Res. 2009;69(2):431–439. doi: 10.1158/0008-5472.CAN-08-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Collier LS, Adams DJ, Hackett CS, Bendzick LE, Akagi K, Davies MN, et al. Whole-body sleeping beauty mutagenesis can cause penetrant leukemia/lymphoma and rare high-grade glioma without associated embryonic lethality. Cancer Res. 2009;69(21):8429–8437. doi: 10.1158/0008-5472.CAN-09-1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Bender AM, Collier LS, Rodriguez FJ, Tieu C, Larson JD, Halder C, et al. Sleeping beauty-mediated somatic mutagenesis implicates CSF1 in the formation of high-grade astrocytomas. Cancer Res. 2010;70(9):3557–3565. doi: 10.1158/0008-5472.CAN-09-4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Dupuy AJ, Akagi K, Largaespada DA, Copeland NG, Jenkins NA. Mammalian mutagenesis using a highly mobile somatic Sleeping Beauty transposon system. Nature. 2005;436(7048):221–226. doi: 10.1038/nature03691. [DOI] [PubMed] [Google Scholar]
- 196.Rakic P. Mode of cell migration to the superficial layers of fetal monkey neocortex. J Comp Neurol. 1972;145(1):61–83. doi: 10.1002/cne.901450105. [DOI] [PubMed] [Google Scholar]
- 197.Hatten ME. Riding the glial monorail: a common mechanism for glial-guided neuronal migration in different regions of the developing mammalian brain. Trends Neurosci. 1990;13(5):179–184. doi: 10.1016/0166-2236(90)90044-b. [DOI] [PubMed] [Google Scholar]