

Abstract
We investigated the effect of delayed, prolonged systemic inflammation on stroke outcomes and progesterone (P4) neuroprotection in middle-aged rats. After transient middle cerebral artery occlusion/reperfusion (MCAO) surgery, rats received P4 (8 or 16 mg/kg) or vehicle injections at 2h, 6h and every 24h until day 7 post-occlusion. At 24h post-injury systemic inflammation was induced by giving 3 doses of lipopolysaccharide (LPS; 50 mg/kg, at 4h intervals) to model post-stroke infections. We measured serum brain-derived neurotrophic factor (BDNF), pro-inflammatory cytokines, and behavioral parameters at multiple times. Serum BDNF levels decreased more in the vehicle+LPS group compared to vehicle-alone at 3 and 7 days post-injury (P<0.05). Vehicle-alone showed a significant increase in IL-1β, IL-6, and TNFα levels at different times following stroke and these levels were further elevated in the vehicle+LPS group. P4 at both doses produced a significant (P<0.05) decline in cytokine levels compared to vehicle and vehicle+LPS. P4 restored BDNF levels at 3 and 7 days post-stroke (P<0.05). Behavioral assessment (rotarod, grip strength, sensory neglect and locomotor activity tests) at 3, 5 and 7 days post-stroke revealed that the vehicle group had significant (P<0.05) deficits in all tests compared to intact controls, and performance was worse in the vehicle+LPS group. P4 at both doses produced significant functional improvement on all tests. Systemic inflammation did not show an additive effect on infarct volume but P4 at both doses showed significant infarct reduction. We suggest that post-stroke infection exacerbates stroke outcomes and P4 exerts neuroprotective/modulatory effects through its systemic anti-inflammatory and BDNF regulatory actions.
Keywords: BDNF, Immunodepression, LPS, Post-stroke infection, Progesterone
1. INTRODUCTION
Stroke is a complex systemic disease causing severe long-term disability and death worldwide (Mathers et al., 2009). Stroke prognosis is highly variable, depending not only on stroke severity and the patient’s age, but also on stroke-associated complications. Post-stroke infection (such as pneumonia and urinary tract infections) is one of the major complications during the acute phase and accounts for one-third of all in-hospital deaths and the worsening of long-term outcomes in ischemic stroke patients (Grau et al., 1999; Emsley and Hopkins, 2008; Koennecke et al., 2011). Preventive antibiotic treatments to reduce infection rate and improve stroke outcome have not been very successful. According to a recent meta-analysis, prophylactic antibiotic treatment in acute-stroke patients is effective in preventing infections but does not reduce mortality (Westendorp et al., 2012). Importantly, none of the clinical trials in this meta-analysis were sufficiently powered to support an analysis of the impact of a preventive antibiotic treatment on stroke outcome. Moreover, a major problem with preventive antibacterial therapy is that it can promote antibiotic resistance in common bacteria with very serious consequences for the patient’s well-being and recovery (Meisel et al., 2012).
Stroke disturbs the normally well-balanced interplay between the nervous and immune systems by compromising sympathetic and parasympathetic neural connections with lymphoid organs including the hypothalamo-pituitary-adrenal (HPA) axis (Woiciechowsky et al., 1998; Meisel et al., 2005). The sympathetic nervous system (SNS) has been reported to play a key role in impaired anti-bacterial immune response following stroke in a mouse model of focal cerebral ischemia (Prass et al., 2003, 2006). Clinical studies suggest that stress-mediated immunodepression driven by the SNS and HPA axis is an essential facilitating factor in the onset of post-stroke infections (Chamorro et al., 2007; Harms et al., 2008, 2011). In this scenario, immediately after stroke onset, massive release of pro-inflammatory cytokines activates the HPA axis and parasympathetic and sympathetic nervous systems. The activated HPA axis and SNS release noradrenaline, glucocorticoids and acetylcholine, mounting a “systemic anti-inflammatory response” that negatively affects the function of the innate and adaptive immune systems and contributes to morbidity, infection and death in stroke patients (Emsley et al., 2003; Smith et al., 2004; Prass et al. 2003; Meisel et al., 2005). Some investigators have proposed that selective immunomodulation to prevent post-stroke infections may be an alternative to preventive antibiotic treatment (Meisel and Meisel, 2011).
In the past two decades, only one drug has been shown to be clinically effective for stroke treatment, with genetically engineered tissue plasminogen activators (tPAs) still the only FDA-approved agents to treat stroke. Given the high risk-to-benefit ratio and very narrow therapeutic time window for tPA, development of an agent which has no negative side effects, and can efficiently prevent secondary ischemic brain damage, inhibit HPA axis-associated immunodepression, and enhance neuronal repair would be a major breakthrough. We propose that the neurosteroid progesterone (P4) might be a suitable candidate. P4 is a pleotropic drug targeting several perpetrators of stroke injury. P4’s safety and efficacy have already been demonstrated in two independent phase II clinical trials for traumatic brain injury (TBI) (Wright et al., 2007; Xiao et al., 2008). Two independent Phase III, NIH- and industry-sponsored, multi-center trials of P4 treatment for moderate to severe TBI are now underway and in interim analysis (http://clinicaltrials.gov/ct2/show/record/NCT00822900 and http://www.synapse-trial.com/). If successful, these trials could stimulate further interest in testing P4 in patients with adult ischemic stroke (Stein, 2012). Our laboratory has recently published a number of studies demonstrating substantial neuroprotection by P4 in several animal stroke models (Sayeed et al., 2006; Sayeed et al., 2007; Sayeed and Stein, 2009; Ishrat et al., 2009, 2010, 2012). P4 treatment not only improves functional/behavioral outcomes in several injury models, but also reduces inflammatory cytokines, brain tissue necrosis, apoptosis, and cerebral edema (Stein, 2008).
In this study, we tested the hypothesis that P4, as a potent anti-inflammatory agent, would inhibit post-stroke systemic infection and inflammation-induced cellular damage, and improve functional recovery. To mimic the systemic effect of post-stroke infections, we administered multiple doses of lipopolysaccharide (LPS) to maintain prolonged systemic inflammation (Langdon et al., 2010). Serum pro-inflammatory cytokines (interleukin-1β (IL-1β), interleukin-6 (IL-6) and tumor necrosis factor α (TNFα)) and brain-derived neurotrophic factor (BDNF) were measured at multiple time points. We also investigated whether the systemic inflammation increased brain infarction and worsened functional outcomes, and whether P4 treatment would reduce infarct volume and improve functional recovery.
2. EXPERIMENTAL PROCEDURES
All behavioral, biochemical and histological assays were performed independently and were double-blinded.
2.1. Animals, treatment regimen and induced systemic inflammation
Middle-aged male Sprague-Dawley rats (450–500 grams; 13 months of age at the beginning of the experiments; Charles River Laboratories, Wilmington, MA) were used according to procedures approved by the Institutional Animal Care and Use Committee, Emory University (Protocol # 200–1517). The rats were quarantined for 7 days before the experiment and housed in individual cages in a room maintained at 21–25° C, 45–50% humidity, a 12-h light/dark cycle and free access to food and water. Rats were randomized to the treatment conditions and then the identity of the groups was coded to avoid experimenter bias. The animals were formed into 5 groups (n = 6/group): (1) Sham+vehicle (Sham); (2) MCAO+vehicle (Veh); (3) MCAO+vehicle+LPS (Veh+LPS); (4) MCAO+LPS+P4 8 mg/kg (LPS+P1); (5) MCAO+LPS+P4 16 mg/kg (LPS+P2). P4 (P-0130; Sigma-Aldrich Co., St. Louis, MO) was dissolved in 22.5% 2-hydroxypropyl-β cyclodextrin and administered by intraperitoneal (i.p.) injection 2 h post-occlusion, and then subcutaneously (s.c.) at 6 h, and then every 24 h until day 7 post-occlusion. To model the systemic effect of post-stroke infections, LPS (50 μg/kg; i.p; 3 separate doses at 4-h intervals) were given at 24, 28 and 32 h post-occlusion (Langdon et al., 2010). On day 7, animals were killed and their brains removed after transcardial perfusion. For each outcome measure, we calculated the starting sample sizes and power needed to reject the null hypothesis with a p-value of 0.05. The number of rats per group at these criteria was determined to be 4 to reject the null hypothesis (H0) at P < 0.05 with a power of 0.80 (effect size f = 0.25).
2.2. Transient middle cerebral artery occlusion (MCAO)
Prior to MCAO surgery, isoflurane anesthesia was induced at 5% and then maintained at 1.5–2% during surgery in 2:1 nitrous oxide and oxygen. Focal cerebral ischemia was induced by occlusion of the right middle cerebral artery as previously described (Longa et al., 1989). A midline incision was made on the ventral surface of the neck, and the right common carotid arteries were isolated and ligated with 6.0 silk suture. The internal carotid artery and the pterygopalatine artery were temporarily occluded with a microvascular clip. A 4-0 Doccol filament (Doccol Corporation, Redlands, CA) was introduced into the internal carotid artery through the incision in the external carotid artery. The filament was advanced approximately 20 mm distal to the carotid bifurcation. Relative cerebral blood flow (CBF) was monitored by laser Doppler (LD) for the entire 2 h of occlusion. Drug treatment was randomly assigned 5 min before onset of reperfusion. After 2 h of MCAO, the occluding filament was withdrawn back into the common carotid artery to allow for reperfusion. Relative CBF was then monitored for 5 min before the wound was sutured and the rats were then permitted to recover from anesthesia. Pulse oximetry (SurgiVet™ V3304; Waukesha, WI) was used to maintain heart rate at approximately 350 bpm, with blood oxygen saturation (SpO2) levels N95%. Anesthesia duration was the same for all groups.
2.3. Temperature monitoring
Body temperature was monitored throughout surgery (by rectal probe) and maintained at ~37.0° C using an automated heat lamp (Harvard Apparatus, South Natick, MA). We monitored post-ischemic core body temperature hourly from 1–4 h post-injury (before LPS injections) and then 24–35 h post-injury (after LPS injections).
2.4. Serum cytokines and BDNF assay
For serum isolation, blood was collected from the tail vein by the tail nick method at 6, 24, 48, 72 and 168 h post-injury and allowed to coagulate for 30 min at room temperature followed by centrifugation for 5 min at 1000×g. Using sandwich ELISA kits, serum IL-1β (BMS630, eBioscience, San Diego, CA), IL-6 (BMS625, eBioscience) and TNFα (BMS622, eBioscience) were measured at 6, 24, 48, and 72 h post-stroke. Total BDNF (CYT306, Millipore, Billerica, MA) levels were measured at 1, 3 and 7 days post-stroke according to the manufacturer’s instructions.
2.5. Behavioral testing
2.5.1. Motor Coordination--Accelerating Rotarod
Motor impairment was assessed with the accelerating rotarod (Ishrat et al., 2009). Rats were given 3 training sessions 5 min apart before surgery. The animals were habituated to the stationary rod, and then placed on the rotating rod. The rod was started at 2 rpm and then accelerated linearly to 20 rpm within 300 sec. Latency to fall off the rotarod was determined before ischemia (pre-surgery) and at 3, 5 and 7 days post-surgery.
2.5.2. Grip strength
Forelimb grip strength was measured pre-surgery and again at 3, 5 and 7 days post-surgery with a grip-strength meter (Columbus Instruments, Columbus, OH). A digital reading (in Newtons) of 3 successive trials was obtained for each rat, and then averaged for analysis.
2.5.3. Sticky-tape removal test
To assess somatosensory dysfunction after tMCAO, we used a modified adhesive removal (sticky-tape) test. A removable sticky label was placed on the underside of the animal’s paw contralateral to the stroke, and the time taken for the rats to sense (contact latency) and remove (removal latency) the label was recorded during a 180-sec observation period. Two trials per animal were averaged for analysis before ischemia (pre-surgery) and at 3, 5 and 7 days post-surgery.
2.5.4. Spontaneous locomotor activity
DigiscanTM activity-monitoring boxes were used to assay spontaneous motor activity pre-surgery and then at 3, 5, and 7 days post-surgery. Each session lasted 5 min and was conducted under red-light conditions.
2.6. Analysis of infarct volume
Cerebral infarct size was evaluated using previously applied methods (Ishrat et al., 2010). On post-ischemia day 7, animals were deeply anesthetized using isoflurane. After transcardial perfusion with cold saline followed by 10% buffered formalin, brains were extracted, fixed in gradient sucrose solution and cut coronally into 20-μm sections for histological analysis. Brain sections were stained in 0.1% cresyl violet solution for 10 min at 45° C, and then rinsed in distilled water. Stained sections were fixed by serial dehydration in alcohol and xylene and mounted with xylene-based cytoseal. Fixed sections were coded to hide group identity and then scanned. The infarct areas, defined as areas showing reduced Nissl staining under light microscopy, were traced and quantified with an image-analysis system. The infarct area was measured on each brain section using NIH imaging software (Image-J, version 1.38, NIH, Rockville, MD). Infarct size was then calculated by multiplying the infarct area on each section by the distance between sections and represented as a percentage of the size of the contralateral hemispheric side±SEM.
2.7. Spleen and thymic atrophy
On the day of euthanasia (day 7 post-stroke), spleen and thymus were isolated from different groups to determine the systemic effect of stroke on the peripheral immune system and modulatory effects of P4. We compared the organ weights in different groups as a marker of post-stroke atrophy.
2.8. Statistical analysis of data
With sample sizes determined by power analysis, repeated measures one-way analysis of variance (RM-ANOVA) was used for behavioral experiments followed by LSD and Tukey’s tests for independent comparisons. Significance was set at P<0.05. Data are presented as mean ± standard error of the mean (SEM). For brain infarction data, we used unpaired t-tests (two-tailed).
3. RESULTS
3.1. Cerebral blood flow measurements and body temperature
Cortical CBF was reduced by at least 70% of pre-ischemic values in all rats subjected to MCAO. There were no significant differences in cortical CBF after occlusion among rats given vehicle, P4, LPS or combination treatment. There were no significant differences in the increase in relative CBF in drug-treated compared to vehicle-treated rats after reperfusion (data not shown). We recorded each animal’s post-ischemic core temperature from 1–4 h post-injury (before LPS injections) and then 24–35 h post-injury (after LPS injections). We did not observe any significant increase in core body temperature on the day of surgery in any group. Administration of LPS at 24, 28, and 32 h post-injury slightly increased core temperature (by ~ 0.4°C) for only a 5-h interval (28–33 h post-injury) in all the rats subjected to MCAO surgery (data not shown).
3.2. Effect of P4 on spleen and thymic atrophy
We compared the organ weight of spleen (Fig. 1A) and thymus (Fig. 1B) in the different experimental groups. One-way ANOVA revealed a significant group effect in spleen (F (4, 25) = 3.433; P<0.023) and thymus (F (4, 25) = 4.281; P<0.009) weights. A significant (P<0.05) reduction in spleen (P<0.006) and thymus weights (P<0.001) was observed in the Veh+LPS group compared to their sham counterparts. The Veh-alone group showed no difference in spleen weight but had a significant (P<0.01) reduction in thymus weight compared to shams. P4 at both 8 and 16 mg doses showed a significant recovery in spleen weight (P<0.01; P<0.009 respectively) and thymus weight (P<0.02; P<0.03 respectively) compared to the Veh+LPS group.
Fig. 1.


Effect of P4 on stroke-induced spleen (A) and thymic (B) atrophy 7 days post-injury. Values are expressed as means ± SEM. Significant difference #P<0.05 compared to sham; *P<0.05 compared to vehicle+LPS (Veh+LPS).
3.3. P4 modulates inflammatory cytokine levels
3.3.1. Interleukin-1β (IL-1β)
Repeated measures ANOVA revealed a significant group effect (F(4,25) = 153.60; P<0.001). Post-hoc analysis showed that serum IL-1β levels were significantly (P<0.05) higher in the Veh group at all time points compared to sham values (Fig. 2A). Delayed multiple LPS injections starting 24 h post-injury significantly (P<0.05) enhanced IL-1β release at 48 and 72 h compared to the Veh group. P4 at both doses significantly decreased IL-1β levels at all time points compared to the Veh and Veh+LPS groups. There was no significant difference between the two doses of P4.
Fig. 2.


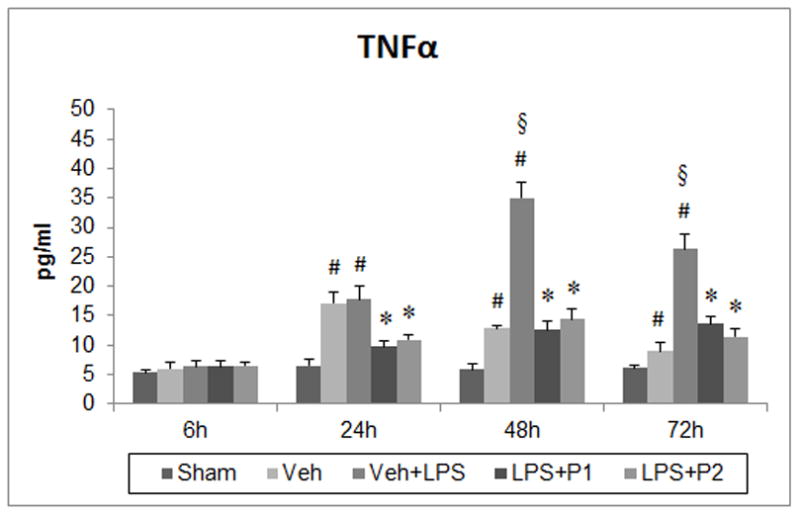
Effect of post-stroke systemic inflammation on inflammatory markers (A) IL-1β, (B) IL-6 and (C) TNFα at 6, 24, 48 and 72 h post-injury and modulation by P4. Values are expressed as means ± SEM. Significant difference #P<0.05 compared to sham; *P<0.05 compared to vehicle (Veh) and vehicle+LPS (Veh+LPS); §P<0.05 compared to Veh.
3.3.2. Interleukin-6 (IL-6)
Repeated measures ANOVA revealed a significant group effect (F(4,25) = 179.56; P<0.001). Post-hoc analysis showed that serum IL-6 levels were significantly (P<0.05) higher in the Veh group at all time points compared to sham values (Fig. 2B). Delayed multiple LPS injections (starting 24 h post-injury) significantly (P<0.05) enhanced IL-6 release at 48 and 72 h in the Veh+LPS group compared to Veh values. P4 at both doses significantly decreased IL-6 levels at all time points compared to the Veh or Veh+LPS groups. There was no significant difference between the two doses of P4.
3.3.3. Tumor necrosis factor-α (TNF-α)
Repeated measures ANOVA revealed a significant group effect (F(4,25) = 58.94; P<0.001). Post-hoc analysis showed that serum TNF-α levels were significantly (P<0.05) higher in the Veh and Veh+LPS groups at 24, 48 and 72 h post-injury compared to sham values (Fig. 2C). Delayed multiple LPS injections (starting 24 h post-injury) significantly (P<0.05) enhanced TNF-α release at 48 and 72 h compared to the Veh group. P4 at both doses significantly decreased TNF-α levels at 24, 48 and 72 h post-injury compared to Veh or Veh+LPS groups. There was no significant difference between the two doses of P4.
3.4. P4 provides trophic support to ischemic brain by modulating BDNF
Repeated measures ANOVA revealed a significant group effect (F(4,25) = 24.838; P<0.001). Post-hoc analysis showed that serum BDNF levels were significantly (P<0.001) decreased in the Veh and Veh+LPS groups at days 3 and 7 post-injury compared to sham-operated rats (Fig. 3). This effect was more pronounced in the Veh+LPS group at days 3 and 7 post-injury (P<0.01 and P<0.001 respectively) compared to Veh. At day 1 post-injury, no difference in serum BDNF levels was observed in any groups. P4 treatment at both doses showed a significant increase (P<0.001) in BDNF levels compared to the Veh and Veh+LPS groups at 3 and 7 days post-injury. The highest levels of BDNF were observed at day 7 post-injury in P4-treated groups, suggesting direct involvement of BDNF in P4’s neurotrophic action. There was no difference in the two doses of P4 on BDNF release.
Fig. 3.

Effect of P4 on serum BDNF levels at 1, 3, and 7 days post-injury. Values are expressed as means ± SEM. Significant difference #P<0.05 compared to sham; *P<0.05 compared to vehicle (Veh) and vehicle+LPS (Veh+LPS); §P<0.05 compared to Veh.
3.5. Delayed, systemic inflammation exacerbates post-stroke functional outcomes and P4 treatment reduces the ensuing behavioral deficits
3.5.1. Rotarod
Repeated measures ANOVA revealed a significant group effect (F(4, 26) = 33.059, P<0.001) in rotarod activity. Vehicle-treated rats showed a significantly (P<0.001) impaired ability to remain on the moving rotarod compared to shams at days 3, 5 and 7 post-injury (Fig 4A). Delayed LPS exposure in vehicle-+-LPS-treated rats significantly (P<0.01, 0.04) worsened rotarod performance compared to the Veh group at days 3 and 7 post-injury respectively. This detrimental effect of LPS was highly significant (P<0.001) compared to sham values. Post-hoc analyses showed that P4 treatment at both 8 and 16 mg/kg significantly (P<0.001) reduced this motor deficit at days 3, 5 and 7 post-stroke compared to the Veh+LPS group. On day 7, there were no significant differences in motor recovery between the two doses of P4.
Fig. 4.
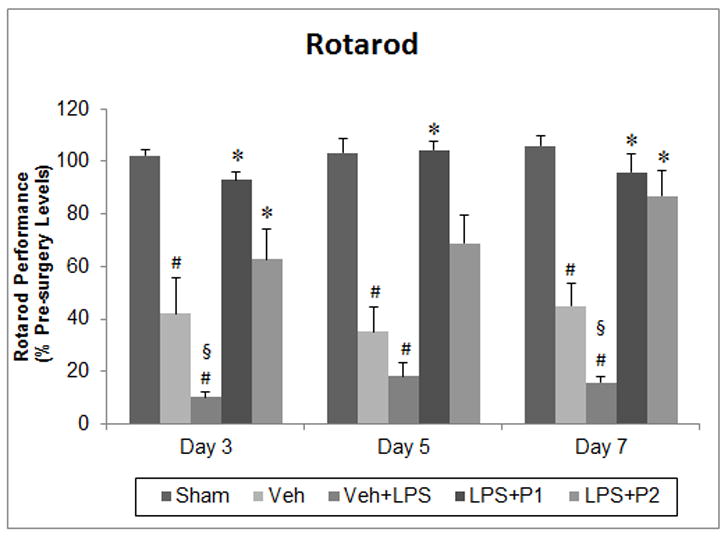



Effect of post-stroke systemic inflammation on functional outcomes at 3, 5 and 7 days post-injury using rotarod (A), grip strength (B), sticky tape removal (C), and locomotor activity (D) tests. Values are expressed as means ± SEM. Significant difference #P<0.05 compared to sham; *P<0.05 compared to vehicle (Veh) and vehicle+LPS (Veh+ LPS); §P<0.05 compared to Veh. CL, contact latency; RL, removal latency.
3.5.2. Grip strength
A significant group effect (F(4, 26) = 42.263, P<0.001) was shown by repeated measures ANOVA (Fig 4B). A significant (P<0.001) reduction in muscle strength in the Veh animals was observed at 3, 5 and 7 days post-injury compared to shams. Post-hoc analyses showed a significant (P<0.01, 0.05, 0.01) decrease in grip strength in the Veh+LPS group compared to the Veh alone group. P4 at both doses significantly (P<0.001) improved grip strength at 3, 5 and 7 days post-stroke compared to the Veh+LPS group. There was no significant difference in motor recovery between the two doses of P4 at day 7 post-injury.
3.5.3. Sticky-tape removal
Repeated measures ANOVA showed significant group effects in the sticky-tape contact (F(4, 26) = 65.973, P<0.001) and removal (F(4, 26) = 150.712, P<0.001) latencies. Post-hoc analyses showed a significant (P<0.001) increase in contact and removal latency to remove the sticker from the contralateral forepaw in the Veh group compared to sham values at 3, 5 and 7 days post-injury (Fig. 4C). Systemic inflammation significantly increased contact latency (P<0.001, 0.01 at days 3 and 5 respectively) and removal latency (P<0.002, 0.01, 0.001 at days 3, 5 and 7 respectively) in the Veh+LPS group compared to Veh animals. P4 treatment showed a highly significant (P<0.001) decrease in both contact and removal latencies at all time points compared to the Veh+LPS group. No significant differences in contact and removal latency were observed between the two doses of P4 at day 7 post-injury.
3.5.4. Locomotor Activity
Repeated measures ANOVA showed significant group effects (F(4, 25) = 63.423, P<0.001). Post-hoc analyses showed a significant (P<0.05) decrease in distance travelled in the Veh group at all time points compared to shams and this effect was worse in the Veh+LPS animals (Fig. 4D). P4 treatment at both doses equally produced a significant (P<0.001) recovery in locomotor activity at days 3, 5 and 7 compared to the Veh and Veh+LPS groups.
3.5. Infarct volume: effect of systemic inflammation and P4 treatment
On day 7 post-injury, infarct volume in the Veh group was not significantly different from that in the Veh+LPS group (Fig. 5). However, P4 at both 8 and 16 mg significantly (P<0.05) reduced infarct volume compared to either of the vehicle groups.
Fig. 5.

Effect of post-stroke systemic inflammation and P4 treatment on brain infarction. Representative photographs of cresyl violet-stained sections are shown. Values are expressed as means ± SEM. Significant difference *P<0.05 compared to Veh and Veh+LPS groups. Data were analyzed using a two-tailed unpaired t-test. Delayed systemic inflammation did not increase infarct size. P4 showed a similar reduction in infarct size at both doses.
4. DISCUSSION
Our findings demonstrate that: (1) delayed and prolonged systemic inflammation exacerbates stroke outcome, and (2) P4 treatment enhances behavioral recovery and reduces stroke infarct volume in middle-aged rats with an induced systemic infection. We also show that P4 treatment inhibits the inflammatory response to ischemic injury and enhances neurotrophic BDNF levels with BDNF having been shown to play a critical role in neuronal recovery/repair (Greenberg et al., 2009; Meyer et al., 2012; Su et al., 2012).
4.1. Progesterone and the systemic inflammatory response to ischemic stroke
Both CNS-specific and systemic inflammation can play a critical role in the outcome of brain injury, but the systemic component of the disease is infrequently given attention in animal experiments on brain injury and its molecular and functional consequences (see Stein, 2012 for review). However, a few laboratories have demonstrated the effects of acute systemic inflammation induced by a single bolus dose of LPS either immediately before or after ischemia to model the systemic effects of an infection (Spencer et al., 2007; McColl et al., 2007, 2008; Marsh et al., 2009). In our project, we adopted a delayed prolonged systemic inflammation approach to post-stroke infection because we think that model is more clinically relevant (Langdon et al., 2010). Since delayed low doses of LPS do not produce the acute inflammation or febrile response that may occur at higher doses (Spencer et al., 2007), our model produced more prolonged inflammatory conditions to mimic what might occur in human stroke patients.
Infection is the most common cause of fever in stroke victims (Kumar et al., 2010). In the acute stage of the disease, a post-stroke rise in body temperature has been independently associated with an increased risk of poor outcome in patients (Polderman, 2008), so we believe it is clinically important to differentiate whether the injury and associated functional deficits result from the stroke itself or from post-stroke pyrexia. In our study we recorded each animal’s post-ischemic core temperature at several time points over 48 h. Post-stroke injection of LPS at 24, 28, and 32 h slightly increased core temperature (by ~0.40° C) only over a 5-h period (28 to 33 h post-injury), an increase not sufficient to worsen functional deficits or brain infarction, given the finding that a post-ischemic brain temperature of 39° C showed no effects on stroke outcomes (Kim et al., 1996). Unlike our study with its prolonged post-injury time points, previous reports on hyperthermia and stroke observed brain temperature only during or shortly after stroke (MacLellan et al., 2006). Further, the comparatively low doses of LPS may not have been sufficient to significantly raise body temperature. This finding is consistent with others (Langdon, 2010). Taking all the data together we propose that the observed deficits in short-term stroke outcomes are due in mainly to systemic inflammation induced by LPS.
Stroke is a multi-organ systemic disease which induces a biphasic effect on the peripheral immune system characterized by an early activation of peripheral leukocytes followed by a delayed severe immunosuppression and atrophy of the spleen and thymus (Dziennis et al., 2011). Offner et al. (2006) reported the drastic effects of stroke on the gross morphology and cell numbers in spleen and thymus, and on splenocyte function and cellular distribution by 96 h after stroke. Our findings are in agreement with these reports. We observed a reduction in spleen and thymus weights in the Veh+LPS group which was significantly restored after P4 treatment. These findings suggest that P4 not only provides neuroprotection to the ischemic brain but may also modulate post-stroke immunodepression. Since spleen and thymus are the major organs which regulate peripheral immune cells under both healthy and pathological conditions, it would be interesting to explore the specific cellular/molecular effects of P4 on splenic/thymic atrophy following stroke. We are currently investigating these mechanisms in another series of experiments.
We observed that MCAO significantly increases serum levels of pro-inflammatory cytokines IL-1β and IL-6 starting at 6 h and for up to 72 h post-injury. Serum TNFα level was significantly higher at 24, 48 and 72 h, but no significant difference was observed at 6 h among the treatment groups. It is possible that some TNFα derives from activated astrocytes and that may be why we did not observe higher serum/systemic TNFα levels at 6 h. Although TNFα has been reported to be up-regulated in ischemic brain within 6 h post-injury (Jiang et al., 2011), serum cytokines may have a different response from those of brain cytokines. Our findings are in agreement with Langdon et al. (2010), who also observed a delayed response for TNFα and fast response for IL-1β and IL-6. These cytokine levels were further elevated by LPS injections administered at 24 h post-injury. The cause of this increase could be that microglia and macrophages recognize LPS and respond by secreting pro-inflammatory cytokines such as TNFa and IL-6 (Boje and Arora, 1992; Bhat et al., 1998). Our findings are in agreement with previous reports also showing elevated levels of serum IL-1β, IL-6 and TNFα after prolonged LPS injection at various time points after injury (Langdon et al., 2010). A single low dose of LPS given immediately after global ischemia has been reported to elevate IL-6 and TNFα (Spencer et al., 2007). McColl et al. (2007) for the first time demonstrated that acute systemic inflammation induced by LPS injection is detrimental to stroke outcome, and that the resulting brain damage and neurological deficits were IL-1 dependent in stroke in mice. Systemic inflammation has been reported to impair stroke outcome by increasing brain inflammation, blood-brain barrier injury and brain edema formation in mice after experimental stroke (Denes et al., 2011). We observed that P4 treatment at both 8 and 16 mg doses significantly reduced serum levels of IL-1β, IL-6 and TNFα compared to the Veh+LPS group. There are other studies showing that P4 is a potential anti-inflammatory agent following acute stroke or TBI (Gibson et al., 2005; Cekic et al., 2011; Hua et al., 2011; Wang et al., 2011). P4 is also known to reduce LPS and IFN-γ-stimulated nitrite production in a dose-dependent manner (Miller et al., 1996; Robert and Spitzer, 1997).
4.2. Progesterone and BDNF: the neurotrophic response to injury
P4 not only protects ischemic brain from inflammatory damage, it also provides trophic support to the injured brain by up-regulating neurotrophic factors that promote functional recovery. Neurotrophic factors are known to exert beneficial effects on cellular and behavioral recovery following brain injury (Sofroniew et al., 2001; Althaus et al., 2008; Su et al., 2012; Morali et al., 2012). The best-understood trophic factors in the contexts of TBI and stroke are NGF and BDNF. BDNF is mainly synthesized by neurons and found in substantial amounts in brain (Hu and Russek, 2008; Meyer et al., 2012), where it plays a crucial role in neuronal survival and plasticity through the p75NTR and TrkB receptors (Chen et al., 2005; Greenberg et al., 2009). BDNF is also present in the blood, where it is at a higher concentration in serum than in plasma (Fujimura et al., 2002). Although the cellular source of the BDNF present in plasma is not known, there are several reports suggesting that BDNF may be secreted into plasma by endothelial or circulating immune cells (Kerschensteiner et al., 1999; Nakahashi et al., 2000; Bayas et al., 2002; Wang et al., 2006). The brain has been hypothesized to be an additional source of the BDNF present in plasma based on the evidence of parallel changes in serum and cortical brain BDNF during postnatal development in rats (Karege et al., 2002). BDNF is one of the neurotrophic factors whose expression is affected by cerebral ischemia (Abe and Hayashi, 1997) and appears to protect against cerebral ischemia-induced neuronal loss (Mattson et al., 2004). Direct intracerebral, intraventricular, and intravenous administration of BDNF has been shown to reduce infarct volume in several stroke models (Mizuno et al., 2000; Shi et al., 2009).
We measured serum BDNF levels at 1, 3 and 7 days post-injury to determine whether BDNF plays a role in P4’s beneficial effects in post-stroke systemic inflammation and functional recovery. We found that ischemic injury induces a significant decrease in serum BDNF levels at 3 and 7 days but not at 24 h post-injury. We also observed a significant increase in serum BDNF levels in our P4-treated group at 3 and 7 days post-injury. BDNF expression is thought to be modulated by P4 in several brain injury models (Coughlan et al., 2009; Jodha et al., 2009) and is involved in P4’s mechanisms of action in the CNS (González et al., 2004; Kaur et al., 2007; Swiatek-De Lange et al., 2007). We previously reported that brain BDNF levels remained unchanged at 24 h post ischemia in a permanent rat model of stroke but decreased significantly at 3 and 14 days post-stroke. P4 treatment increased BDNF levels and was associated with better functional outcomes (Ishrat et al., 2012).
4.2.1. Do serum and brain levels of BDNF have to match?
It remains unclear whether serum BDNF levels match brain BDNF levels following stroke. There is both positive and negative evidence. Some researchers report that both serum and brain BDNF levels complement each other in response to stroke (Di Lazzaro et al., 2007; Jimenez et al., 2009; Yang et al., 2011; Zhou et al., 2011). In contrast, others argue that changes in regional brain BDNF levels are not associated with changes in plasma or serum (Elfving et al., 2010; Luo et al., 2010). One recent study suggests that circulating BDNF levels do not mirror brain BDNF levels after permanent stroke, and severe stroke is associated with high plasma BDNF in the very acute stage (Bejot et al., 2011). However, this study has the limitation that the stroke model used lacked reperfusion after the initial blockage, so the data may apply only to patients or experimental subjects lacking reperfusion. These findings suggest that reperfusion may be associated with the passage of BDNF from the brain into the blood. In the present study, we did not measure and compare brain BDNF with serum BDNF levels at different time points, but it is an intriguing question needing further investigation to confirm whether brain BDNF levels mirror systemic BDNF levels following stroke, and whether the BDNF expression profile is time-dependent and varies in different stroke models. Further studies are also needed to explore whether systemic BDNF over-expression is simply a consequence of improved health of the P4-treated animals or a mechanism underlying P4 neuroprotection].
4.2.2. How BDNF and inflammation are related
In addition to providing trophic support, BDNF also plays a critical role in modulating the inflammatory process under different pathologic conditions. It inhibits microglial apoptosis and promotes microglial proliferation and phagocytic activity in vitro (Zhang et al., 2003). BDNF has also been reported to decrease TNFα expression while up-regulating the expression of IL-10, an anti-inflammatory cytokine, in a model of multiple sclerosis (Makar et al., 2009). BDNF activates NFκB and protects cells from serum starvation and glutamate toxicity in vitro (Yeiser et al., 2004; Kajiya et al., 2009). Activated NFκB not only inhibits apoptosis but also promotes neuronal survival, whereas its inhibition aggravates ischemic injury (Hill et al., 2001; Valerio et al., 2009). In an experimental stroke model, intranasal BDNF has been reported to modulate local inflammation and protects against ischemic brain by up-regulating anti-inflammatory cytokine IL-10, down-regulating the pro-inflammatory cytokine TNFα, and increasing the DNA-binding activity of NFκB (Jiang et al., 2011). These findings support our data on systemic BDNF levels and cytokine expression. We speculate that high systemic BDNF levels may contribute to the observed anti-inflammatory effects of P4 and provide trophic support for the repair of damaged neurons in the injured brain. If true, this is one of the reasons P4 can be understood as a pleiotropic hormone (Stein, 2008; 2012).
4.3. Does LPS make a stroke infarct worse? It depends
We observed that animals with MCAO treated with either vehicle or multiple doses of LPS developed similar infarct size by day 7. Langdon et al. (2010) reported that young male rats subjected to 24-h delayed LPS treatment showed larger infarct volumes than animals treated with vehicle. Conversely, we observed no effect of LPS treatment on infarct volume in middle-aged rats. The difference may be due to the age factor. It has been reported that because of their attenuated inflammatory response to stroke, aged animals tend to develop smaller infarcts than their younger counterparts (Sieber et al., 2011). We speculate that because middle-aged rats have smaller infarcts, the low LPS dose we used may not have been sufficient to affect infarct volume. We observed that P4 treatment at both 8 mg and 16 mg/kg reduced infarct volume by 43.25% and 48.19% respectively compared to the Veh+LPS group. We and others have previously reported beneficial effects of P4 in different stroke models (Gibson and Murphy, 2004; Gibson et al., 2005; Sayeed et al., 2006, 2007; Ishrat et al., 2009).
In our study ischemia/reperfusion injury significantly impaired locomotion, grip strength, sensory neglect and motor activity in middle-aged rats over 7 days post-injury. Prolonged, systemic inflammation induced by LPS injections beginning 24 h after the stroke worsened the functional deficits compared to vehicle-treated animals. Systemic inflammation has been reported to impair behavioral functions in rats, and our findings are in agreement with those studies (McColl et al., 2007; Langdon et al., 2010). Treatment with P4 at both doses attenuated functional deficits on various behavioral tests (rotarod, grip strength, locomotion and sensory neglect) at different time points over the 7 days of post-injury treatment. These findings are consistent with previous reports (Gibson and Murphy, 2004; Gibson et al., 2005; Sayeed et al., 2006, 2007; Ishrat et al., 2009; Gibson et al., 2011).
5.0. CONCLUSION
In this study, we investigated the short-term effects of post-stroke infection and the neuroprotective effects of the hormone P4 in middle-aged rats. Our findings indicate that post-stroke systemic inflammation worsens stroke outcomes as assessed by multiple outcome measures including expression of pro-inflammatory cytokines, infarct volume and behavioral testing. P4 treatment at both 8 and 16 mg/kg showed equally beneficial effects. Our data can be interpreted to indicate that BDNF can play a key role in modulating the detrimental effects of post-stroke systemic inflammation and providing trophic support essential for brain repair. At this point, we do not know all of the exact mechanisms or signaling pathways by which P4 modulates post-stroke systemic inflammation—this needs to be investigated in future studies. We will also need to consider the long-term and systemic effects of post-stroke infection on functional outcomes and the role of P4 treatment in keeping infection at bay. In most animal studies of stroke and TBI, mechanisms (and treatments) that can affect and alter systemic shock and sepsis are rarely given consideration in determining the biomarkers of injury and repair. This area of investigation needs much more attention if we are to understand and treat the complex factors that contribute to stroke and brain injury. Overall, our preliminary findings suggest that P4 treatment may be beneficial not only in the treatment of ischemic stroke itself but also in fighting post-stroke infections.
HIGHLIGHTS.
Prolonged systemic post-stroke inflammation worsens stroke outcomes in middle-aged rats.
Systemic inflammation worsens behavioral deficits in stroke animals.
Progesterone treatment reduces infarct volume after transient stroke.
P4 reduced functional deficits by modulating pro-inflammatory cytokines and BDNF.
Acknowledgments
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke grants U01 NSO 62676 to DGS and philanthropic gifts from Allen & Company. The authors would like to thank Leslie McCann for her invaluable editorial assistance.
ABBREVIATIONS
- ANOVA
analysis of variance
- BDNF
brain-derived neurotrophic factor
- CNS
central nervous system
- FDA
food and drug administration
- HPA
hypothalamo-pituitary adrenal axis
- IL-1β
interleukin-1β
- IL-6
interleukin-6
- LPS
lipopolysaccharide
- MCAO
middle cerebral artery occlusion
- P4
progesterone
- SEM
standard error of the mean
- SNS
sympathetic nervous system
- TBI
traumatic brain injury
- TNFα
tumor necrosis factor alpha
- tPA
tissue plasminogen activator
Footnotes
DISCLOSURE/CONFLICT OF INTEREST
D.G.S. is entitled to royalty payment (~6.5%) from BHR Pharmaceuticals related to research on progesterone and brain injury. His future financial interests may be affected by the outcome of this research. The terms of this arrangement have been reviewed and approved by Emory University, which receives the largest share of any benefits (~60.0%) in accordance with its business practices and conflict of interest policies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe K, Hayashi T. Expression of the glial cell line-derived neurotrophic factor gene in rat brain after transient MCA occlusion. Brain Res. 1997;21:230–234. doi: 10.1016/s0006-8993(97)01041-x. [DOI] [PubMed] [Google Scholar]
- Althaus HH, Kloppner S, Klopfleisch S, Schmitz M. Oligodendroglial cells and neurotrophins: a polyphonic cantata in major and minor. J Mol Neurosci. 2008;35:65–79. doi: 10.1007/s12031-008-9053-y. [DOI] [PubMed] [Google Scholar]
- Bayas A, Hummel V, Kallmann BA, Karch C, Toyka KV, et al. Human cerebral endothelial cells are a potential source for bioactive BDNF. Cytokine. 2002;19:55–58. doi: 10.1006/cyto.2002.0892. [DOI] [PubMed] [Google Scholar]
- Béjot Y, Mossiat C, Giroud M, Prigent-Tessier A, Marie C. Circulating and brain BDNF levels in stroke rats. Relevance to clinical studies. PLoS One. 2011;6:e29405. doi: 10.1371/journal.pone.0029405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat NR, Zhang P, Lee JC, Hogan EL. Extracellular signal-regulated kinase and p38 subgroups of mitogen activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-alpha gene expression in endotoxin-stimulated primary glial cultures. J Neurosci. 1998;18:1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boje KM, Arora PK. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992;587:250–256. doi: 10.1016/0006-8993(92)91004-x. [DOI] [PubMed] [Google Scholar]
- Cekic M, Cutler SM, VanLandingham JW, Stein DG. Vitamin D deficiency reduces the benefits of progesterone treatment after brain injury in aged rats. Neurobiol Aging. 2011;32:864–874. doi: 10.1016/j.neurobiolaging.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamorro A, Amaro S, Vargas M, Obach V, Cervera A, Gomez-Choco M, Torres F, Planas AM. Catecholamines, infection, and death in acute ischemic stroke. J Neurol Sci. 2007;252:29–35. doi: 10.1016/j.jns.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Chen J, Zhang C, Jiang H, Li Y, Zhang L, Robin A, Katakowski M, Lu M, Chopp M. Atorvastatin induction of VEGF and BDNF promotes brain plasticity after stroke in mice. J Cereb Blood Flow Metab. 2005;25:281–290. doi: 10.1038/sj.jcbfm.9600034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlan T, Gibson C, Murphy S. Progesterone, BDNF and neuroprotection in the injured CNS. Int J Neurosci. 2009;119:1718–40. doi: 10.1080/00207450903116430. [DOI] [PubMed] [Google Scholar]
- Dénes A, Ferenczi S, Kovács KJ. Systemic inflammatory challenges compromise survival after experimental stroke via augmenting brain inflammation, blood- brain barrier damage and brain oedema independently of infarct size. J Neuroinflammation. 2011;8:164. doi: 10.1186/1742-2094-8-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lazzaro V, Profice P, Pilato F, Dileone M, Florio L, et al. BDNF plasma levels in acute stroke. Neurosci Lett. 2007;422:128–130. doi: 10.1016/j.neulet.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Dziennis S, Mader S, Akiyoshi K, Ren X, Ayala P, Burrows GG, Vandenbark AA, Herson PS, Hurn PD, Offner HA. Therapy with recombinant T-cell receptor ligand reduces infarct size and infiltrating inflammatory cells in brain after middle cerebral artery occlusion in mice. Metab Brain Dis. 2011;26:123–133. doi: 10.1007/s11011-011-9241-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley HCA, Smith CJ, Gavin CM, Georgiou RF, Vail A, Barberan EM, Hallenbeck JM, del Zoppo GJ, Rothwell NJ, Tyrrell PJ, Hopkins SJ. An early and sustained peripheral inflammatory response in acute ischaemic stroke: relationships with infection and atherosclerosis. J Neuroimmunol. 2003;139:93–101. doi: 10.1016/s0165-5728(03)00134-6. [DOI] [PubMed] [Google Scholar]
- Emsley HC, Hopkins SJ. Acute ischemic stroke and infection: recent and emerging concepts. Lancet Neurol. 2008;7:341–353. doi: 10.1016/S1474-4422(08)70061-9. [DOI] [PubMed] [Google Scholar]
- Elfving B, Plougmann PH, Müller HK, Mathé AA, Rosenberg R, et al. Inverse correlation of brain and blood BDNF levels in a genetic rat model of depression. Int J Neuropsychopharmacol. 2010;13:563–572. doi: 10.1017/S1461145709990721. [DOI] [PubMed] [Google Scholar]
- Fujimura H, Altar CA, Chen R, Nakamura T, Nakahashi T, et al. Brain derived neurotrophic factor is stored in human platelets and released by agonist stimulation. Thromb Haemost. 2002;87:728–734. [PubMed] [Google Scholar]
- Gibson CL, Murphy SP. Progesterone enhances functional recovery after middle cerebral artery occlusion in male mice. J Cereb Blood Flow Metab. 2004;24:805–813. doi: 10.1097/01.WCB.0000125365.83980.00. [DOI] [PubMed] [Google Scholar]
- Gibson CL, Constantin D, Prior MJ, Bath PM, Murphy SP. Progesterone suppresses the inflammatory response and nitric oxide synthase-2 expression following cerebral ischemia. Exp Neurol. 2005;193:522–530. doi: 10.1016/j.expneurol.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Gibson CL, Coomber B, Murphy SP. Progesterone is neuroprotective following cerebral ischaemia in reproductively ageing female mice. Brain. 2011;134:2125–2133. doi: 10.1093/brain/awr132. [DOI] [PubMed] [Google Scholar]
- González SL, González Deniselle F, Labombarda MC, Guennoun R, Schumacher M, De Nicola AF. Progesterone up-regulates neuronal brain-derived neurotrophic factor expression in the injured spinal cord. Neuroscience. 2004;125:605–614. doi: 10.1016/j.neuroscience.2004.02.024. [DOI] [PubMed] [Google Scholar]
- Grau AJ, Buggle F, Schnitzler P, Spiel M, Lichy C, Hacke W. Fever and infection early after ischemic stroke. J Neurol Sci. 1999;171:115–120. doi: 10.1016/s0022-510x(99)00261-0. [DOI] [PubMed] [Google Scholar]
- Greenberg ME, Xu B, Lu B, Hempstead BL. New insights in the biology of BDNF synthesis and release: implications in CNS function. J Neurosci. 2009;29:12764–12767. doi: 10.1523/JNEUROSCI.3566-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms H, Prass K, Meisel C, Klehmet J, Rogge W, Drenckhahn C, Gohler J, Bereswill S, Gobel U, Wernecke KD, Wolf T, Arnold G, Halle E, Volk HD, Dirnagl U, Meisel A. Preventive antibacterial therapy in acute ischemic stroke: a randomized controlled trial. PLoS One. 2008;3:e2158. doi: 10.1371/journal.pone.0002158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms H, Reimnitz P, Bohner G, Werich T, Klingebiel R, Meisel C, Meisel A. Influence of stroke localization on autonomic activation, immunodepression, and post-stroke infection. Cerebrovasc Dis. 2011;32:552–560. doi: 10.1159/000331922. [DOI] [PubMed] [Google Scholar]
- Hill WD, Hess DC, Carroll JE, Wakade CG, Howard EF, Chen Q, Cheng C, Martin-Studdard A, Waller JL, Beswick RA. The NF-kappaB inhibitor diethyldithiocarbamate (DDTC) increases brain cell death in a transient middle cerebral artery occlusion model of ischemia. Brain Res Bull. 2001;55:375–386. doi: 10.1016/s0361-9230(01)00503-2. [DOI] [PubMed] [Google Scholar]
- Hu Y, Russek SJ. BDNF and the diseases nervous system: a delicate balance between adaptative and pathological process of gene regulation. J Neurochem. 2008;105:1–17. doi: 10.1111/j.1471-4159.2008.05237.x. [DOI] [PubMed] [Google Scholar]
- Hua F, Wang J, Ishrat T, Wei W, Atif F, Sayeed I, Stein DG. Genomic profile of Toll-like receptor pathways in traumatically brain-injured mice: effect of exogenous progesterone. J Neuroinflammation. 2011;8:42. doi: 10.1186/1742-2094-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishrat T, Sayeed I, Atif F, Hua F, Stein DG. Progesterone is neuroprotective against ischemic brain injury through its effects on the phosphoinositide 3-kinase/protein kinase B signaling pathway. Neuroscience. 2012;210:442–450. doi: 10.1016/j.neuroscience.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishrat T, Sayeed I, Atif F, Hua F, Stein DG. Progesterone and allopregnanolone attenuate blood-brain barrier dysfunction following permanent focal ischemia by regulating the expression of matrix metalloproteinases. Exp Neurol. 2010;226:183–190. doi: 10.1016/j.expneurol.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishrat T, Sayeed I, Atif F, Stein DG. Effects of progesterone administration on infarct volume and functional deficits following permanent focal cerebral ischemia in rats. Brain Res. 2009;1257:94–101. doi: 10.1016/j.brainres.2008.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Wei N, Lu T, Zhu J, Xu G, Liu X. Intranasal brain-derived neurotrophic factor protects brain from ischemic insult via modulating local inflammation in rats. Neurosci. 2011;172:398–405. doi: 10.1016/j.neuroscience.2010.10.054. [DOI] [PubMed] [Google Scholar]
- Jimenez I, Sobrino T, Rodrigez-Yanez M, Pouso M, Cristobo M, et al. High serum levels of leptin are associated with post-stroke depression? Psychological Med. 2009;39:1201–1209. doi: 10.1017/S0033291709005637. [DOI] [PubMed] [Google Scholar]
- Jodha PK, Kaur P, Underwood W, Lydon JP, Singh M. The differences in neuroprotective efficacy of progesterone and medroxyprogesterone acetate correlate with their effects on brain-derived neurotrophic factor expression. Endocrinology. 2009;150:3162–3168. doi: 10.1210/en.2008-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur P, Jodhka PK, Underwood WA, Bowles CA, de Fiebre NC, de Fiebre CM, Singh M. Progesterone increases brain-derived neurotrophic factor expression and protects against glutamate toxicity in a mitogen-activated protein kinase- and phosphoinositide-3 kinase-dependent manner in cerebral cortical explants. J Neurosci Res. 2007;85:2441–2449. doi: 10.1002/jnr.21370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajiya M, Shiba H, Fujita T, Takeda K, Uchida Y, Kawaguchi H, Kitagawa M, Takata T, Kurihara H. Brain-derived neurotrophic factor protects cementoblasts from serum starvation-induced cell death. J Cell Physiol. 2009;221:696–706. doi: 10.1002/jcp.21909. [DOI] [PubMed] [Google Scholar]
- Karege F, Schwald M, Cisse M. Postnatal developmental profile of brain derived neurotrophic factor in rat brain and platelets. Neurosci Lett. 2002;328:261–264. doi: 10.1016/s0304-3940(02)00529-3. [DOI] [PubMed] [Google Scholar]
- Kerschensteiner M, Gallmeier E, Behrens L, Leal VV, Misgeld T, et al. Activated human T cells, B cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lesions: a neuroprotective role of inflammation? J Exp Med. 1999;189:865–870. doi: 10.1084/jem.189.5.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Busto R, Dietrich WD, Kraydieh S, Ginsberg MD. Delayed postischemic hyperthermia in awake rats worsens the histopathological outcome of transient focal cerebral ischemia. Stroke. 1996;27:2274–2280. doi: 10.1161/01.str.27.12.2274. [DOI] [PubMed] [Google Scholar]
- Koennecke HC, Belz W, Berfelde D, Endres M, Fitzek S, Hamilton F, Kreitsch P, Mackert BM, Nabavi DG, Nolte CH, Pohls W, Schmehl I, Schmitz B, von Brevern M, Walter G, Heuschmann PU. Factors influencing inhospital mortality and morbidity in patients treated on a stroke unit. Neurology. 2011;77:965–972. doi: 10.1212/WNL.0b013e31822dc795. [DOI] [PubMed] [Google Scholar]
- Kumar S, Selim MH, Caplan LR. Medical complications after stroke. Lancet Neurol. 2010;9:105–118. doi: 10.1016/S1474-4422(09)70266-2. [DOI] [PubMed] [Google Scholar]
- Langdon KD, Maclellan CL, Corbett D. Prolonged, 24-h delayed peripheral inflammation increases short- and long-term functional impairment and histopathological damage after focal ischemia in the rat. J Cereb Blood Flow Metab. 2010;30:1450–1459. doi: 10.1038/jcbfm.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Luo KR, Hong CJ, Liou YJ, Hou SJ, Huang YH, et al. Differential regulation of neurotrophin S100B and BDNF in two rat models of depression. Prog Neuro-Psychopharmacol Biol Psychiatry. 2010;34:1433–1439. doi: 10.1016/j.pnpbp.2010.07.033. [DOI] [PubMed] [Google Scholar]
- MacLellan CL, Davies LM, Fingas MS, Colbourne F. The influence of hypothermia on outcome after intracerebral hemorrhage in rats. Stroke. 2006;37:1266–1270. doi: 10.1161/01.STR.0000217268.81963.78. [DOI] [PubMed] [Google Scholar]
- Makar TK, Bever CT, Singh IS, Royal W, Sahu SN, Sura TP, Sultana S, Sura KT, Patel N, Dhib-Jalbut S, Trisler D. Brain-derived neurotrophic factor gene delivery in an animal model of multiple sclerosis using bone marrow stem cells as a vehicle. J Neuroimmunol. 2009;210:40–51. doi: 10.1016/j.jneuroim.2009.02.017. [DOI] [PubMed] [Google Scholar]
- Marsh B, Stevens SL, Packard AE, Gopalan B, Hunter B, Leung PY, Harrington CA, Stenzel-Poore MP. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J Neurosci. 2009;29:9839–9849. doi: 10.1523/JNEUROSCI.2496-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathers CD, Boerma T, Ma Fat D. Global and regional causes of death. Br Med Bull. 2009;92:7–32. doi: 10.1093/bmb/ldp028. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Maudsley S, Martin B. BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci. 2004;27:589–594. doi: 10.1016/j.tins.2004.08.001. [DOI] [PubMed] [Google Scholar]
- McColl BW, Rothwell NJ, Allan SM. Systemic inflammatory stimulus potentiates the acute phase and CXC chemokine responses to experimental stroke and exacerbates brain damage via interleukin-1- and neutrophil-dependent mechanisms. J Neurosci. 2007;27:4403–4412. doi: 10.1523/JNEUROSCI.5376-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl BW, Rothwell NJ, Allan SM. Systemic inflammation alters the kinetics of cerebrovascular tight junction disruption after experimental stroke in mice. J Neurosci. 2008;28:9451–9462. doi: 10.1523/JNEUROSCI.2674-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. 2005;6:775–786. doi: 10.1038/nrn1765. [DOI] [PubMed] [Google Scholar]
- Meisel C, Meisel A. Suppressing immunosuppression after stroke. N Engl J Med. 2011;365:2134–2136. doi: 10.1056/NEJMcibr1112454. [DOI] [PubMed] [Google Scholar]
- Meisel A, Meisel C, Harms H, Hartmann O, Ulm L. Predicting post-stroke infections and outcome with blood-based immune and stress markers. Cerebrovasc Dis. 2012;33:580–588. doi: 10.1159/000338080. [DOI] [PubMed] [Google Scholar]
- Meyer M, Gonzalez Deniselle MC, Gargiulo-Monachelli G, Garay LI, Schumacher M, Guennoun R, De Nicola AF. Progesterone effects on neuronal brain-derived neurotrophic factor and glial cells during progression of Wobbler mouse neurodegeneration. Neurosci. 2012;201:267–279. doi: 10.1016/j.neuroscience.2011.11.034. [DOI] [PubMed] [Google Scholar]
- Miller L, Alley EW, Murphy WJ, Russell SW, Hunt JS. Progesterone inhibits inducible nitric oxide synthase gene expression and nitric oxide production in murine macrophages. J Leukoc Biol. 1996;59:442–450. doi: 10.1002/jlb.59.3.442. [DOI] [PubMed] [Google Scholar]
- Mizuno M, Yamada K, Olariu A, Nawa H, Nabeshima T. Involvement of brain-derived neurotrophic factor in spatial memory formation and maintenance in a radial arm maze test in rats. J Neurosci. 2000;20:7116–7121. doi: 10.1523/JNEUROSCI.20-18-07116.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moralí G, Montes P, González-Burgos I, Velázquez-Zamora DA, Cervantes M. Cytoarchitectural characteristics of hippocampal CA1 pyramidal neurons of rats, four months after global cerebral ischemia and progesterone treatment. Restor Neurol Neurosci. 2012;30:1–8. doi: 10.3233/RNN-2011-0605. [DOI] [PubMed] [Google Scholar]
- Nakahashi T, Fujimura H, Altar CA, Li J, Kambayashi J, et al. Vascular endothelial cells synthesize and secrete brain-derived neurotrophic factor. FEBS Lett. 2000;470:113–117. doi: 10.1016/s0014-5793(00)01302-8. [DOI] [PubMed] [Google Scholar]
- Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A, Vandenbark AA, Hurn PD. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol. 2006;176:6523–6531. doi: 10.4049/jimmunol.176.11.6523. [DOI] [PubMed] [Google Scholar]
- Polderman KH. Induced hypothermia and fever control for prevention and treatment of neurological injuries. Lancet. 2008;371:1955–1969. doi: 10.1016/S0140-6736(08)60837-5. [DOI] [PubMed] [Google Scholar]
- Prass KC, Meisel CH, Braun J, Halle E, Wolf T, Ruscher K, Victorov IV, Priller J, Dirnagl U, et al. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J Exp Med. 2003;198:725–736. doi: 10.1084/jem.20021098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prass K, Braun JS, Dirnagl U, Meisel C, Meisel A. Stroke propagates bacterial aspiration to pneumonia in a model of cerebral ischemia. Stroke. 2006;37:2607–2612. doi: 10.1161/01.STR.0000240409.68739.2b. [DOI] [PubMed] [Google Scholar]
- Robert R, Spitzer JA. Effects of female hormones (17beta-estradiol and progesterone) on nitric oxide production by alveolar macrophages in rats. Nitric Oxide. 1997;1:453–462. doi: 10.1006/niox.1997.0157. [DOI] [PubMed] [Google Scholar]
- Sayeed I, Guo Q, Hoffman SW, Stein DG. Allopregnanolone, a progesterone metabolite, is more effective than progesterone in reducing cortical infarct volume after transient middle cerebral artery occlusion. Ann Emerg Med. 2006;47:381–389. doi: 10.1016/j.annemergmed.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Sayeed I, Stein DG. Progesterone as a neuroprotective factor in traumatic and ischemic brain injury. Prog Brain Res. 2009;175:219–237. doi: 10.1016/S0079-6123(09)17515-5. [DOI] [PubMed] [Google Scholar]
- Sayeed I, Wali B, Stein DG. Progesterone inhibits ischemic brain injury in a rat model of permanent middle cerebral artery occlusion. Restor Neurol Neurosci. 2007;25:151–159. [PubMed] [Google Scholar]
- Shi Q, Zhang P, Zhang J, Chen X, Lu H, Tian Y, Parker TL, Liu Y. Adenovirus-mediated brain-derived neurotrophic factor expression regulated by hypoxia response element protects brain from injury of transient middle cerebral artery occlusion in mice. Neurosci Lett. 2009;465:220–225. doi: 10.1016/j.neulet.2009.08.049. [DOI] [PubMed] [Google Scholar]
- Sieber MW, Claus RA, Witte OW, Frahm C. Attenuated inflammatory response in aged mice brains following stroke. PLoS One. 2011;6:e26288. doi: 10.1371/journal.pone.0026288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ, Emsley HC, Gavin CM, Georgiou RF, Vail A, Barberan EM, del Zoppo GJ, Hallenbeck JM, Rothwell NJ, Hopkins SJ, Tyrrell PJ. Peak plasma interleukin-6 and other peripheral markers of inflammation in the first week of ischaemic stroke correlate with brain infarct volume, stroke severity and long-term outcome. BMC Neurol. 2004;4:2. doi: 10.1186/1471-2377-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Howe CL, Mobley WC. Nerve growth factor signaling, neuroprotection, and neural repair. Annu Rev Neurosci. 2001;24:1217–1281. doi: 10.1146/annurev.neuro.24.1.1217. [DOI] [PubMed] [Google Scholar]
- Spencer SJ, Mouihate A, Pittman QJ. Peripheral inflammation exacerbates damage after global ischemia independently of temperature and acute brain inflammation. Stroke. 2007;38:1570–1577. doi: 10.1161/STROKEAHA.106.476507. [DOI] [PubMed] [Google Scholar]
- Stein DG. Progesterone exerts neuroprotective effects after brain injury. Brain Res Rev. 2008;57:386–397. doi: 10.1016/j.brainresrev.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein DG. A clinical/translational perspective: Can a developmental hormone play a role in the treatment of traumatic brain injury? Horm Behav. 2012 May 14; doi: 10.1016/j.yhbeh.2012.05.004. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Su C, Cunningham RL, Rybalchenko N, Singh M. Progesterone Increases the Release of Brain-Derived Neurotrophic Factor from Glia via Progesterone Receptor Membrane Component 1 (Pgrmc1)-Dependent ERK5 Signaling. Endocrinol. 2012;153:4389–4400. doi: 10.1210/en.2011-2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiatek-De Lange M, Stampfl A, Hauck SM, Zischka H, Gloeckner CJ, Deeg CA, Ueffing M. Membrane-initiated effects of progesterone on calcium dependent signaling and activation of VEGF gene expression in retinal glial cells. Glia. 2007;55:1061–1073. doi: 10.1002/glia.20523. [DOI] [PubMed] [Google Scholar]
- Valerio A, Dossena M, Bertolotti P, Boroni F, Sarnico I, Faraco G, Chiarugi A, Frontini A, Giordano A, Liou HC, De Simoni MG, Spano P, Carruba MO, Pizzi M, Nisoli E. Leptin is induced in the ischemic cerebral cortex and exerts neuroprotection through NFkappaB/c-Rel-dependent transcription. Stroke. 2009;40:610–617. doi: 10.1161/STROKEAHA.108.528588. [DOI] [PubMed] [Google Scholar]
- Wang H, Ward N, Boswell M, Katz DM. Secretion of brain-derived neurotrophic factor from brain microvascular endothelial cells. Eur J Neurosci. 2006;23:1665–1670. doi: 10.1111/j.1460-9568.2006.04682.x. [DOI] [PubMed] [Google Scholar]
- Wang J, Zhao Y, Liu C, Jiang C, Zhao C, Zhu Z. Progesterone inhibits inflammatory response pathways after permanent middle cerebral artery occlusion in rats. Mol Med Report. 2011;4:319–324. doi: 10.3892/mmr.2011.418. [DOI] [PubMed] [Google Scholar]
- Westendorp WF, Vermeij JD, Vermeij F, Den Hertog HM, Dippel DW, van de Beek D, Nederkoorn PJ. Antibiotic therapy for preventing infections in patients with acute stroke. Cochrane Database Syst Rev. 2012;1:CD008530. doi: 10.1002/14651858.CD008530.pub2. [DOI] [PubMed] [Google Scholar]
- Woiciechowsky C, Asadullah K, Nestler D, Eberhardt B, Platzer C, Schoning B, Glockner F, Lanksch WR, Volk HD, Docke WD. Sympathetic activation triggers systemic interleukin-10 release in immunodepression induced by brain injury. Nat Med. 1998;4:808–813. doi: 10.1038/nm0798-808. [DOI] [PubMed] [Google Scholar]
- Wright DW, Kellermann AL, Hertzberg VS, Clark PL, Frankel M, Goldstein FC, Salomone JP, Dent LL, Harris OA, Ander DS, Lowery DW, Patel MM, Denson DD, Gordon AB, Wald MM, Gupta S, Hoffman SW, Stein DG. ProTECT: a randomized clinical trial of progesterone for acute traumatic brain injury. Ann Emerg Med. 2007;49:391–402. doi: 10.1016/j.annemergmed.2006.07.932. [DOI] [PubMed] [Google Scholar]
- Xiao G, Wei J, Yan W, Wang W, Lu Z. Improved outcomes from the administration of progesterone for patients with acute severe traumatic brain injury: a randomized controlled trial. Crit Care. 2008;12:R61. doi: 10.1186/cc6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Zhang Z, Sun D, Xu Z, Yuan Y, et al. Low serum BDNF may indicate the development of PSD in patients with acute ischemic stroke. Int J Geriatr Psychiatry. 2011;26:495–502. doi: 10.1002/gps.2552. [DOI] [PubMed] [Google Scholar]
- Yeiser EC, Rutkoski NJ, Naito A, Inoue J, Carter BD. Neurotrophin signaling through the p75 receptor is deficient in traf6−/− mice. J Neurosci. 2004;24:10521–10529. doi: 10.1523/JNEUROSCI.1390-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Geula C, Lu C, Koziel H, Hatcher LM, Roisen FJ. Neurotrophins regulate proliferation and survival of two microglial cell lines in vitro. Exp Neurol. 2003;183:469–481. doi: 10.1016/s0014-4886(03)00222-x. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Lu T, Yue X, Zhu W, Ma M, et al. Decreased serum-brain derived neurotrophic factor (BDNF) is associated with post-stroke depression but not with BDNF gene Val66met polymorphism. Clin Chem Lab Med. 2011;49:185–189. doi: 10.1515/CCLM.2011.039. [DOI] [PubMed] [Google Scholar]