

Abstract
Intramolecular photoinduced cyclizations are investigated in photoprecursors assembled in a modular fashion via a Diels-Alder reaction of acetylenic dienophiles with subsequent Michael additions of aromatic ketones to install a chromophore capable of initiating Paternò-Büchi cycloadditions or radical cyclization cascades. The protolytic oxametathesis in these systems allows for rapid access to novel polycyclic scaffolds decorated by formyl groups and carboxylates suitable for subsequent modifications. In conformationally constrained photoprecursors a radical rearrangement takes place resulting in intramolecular 1,3-diradical cyclopentanation of the double bond.
Introduction
Photoinduced reactions provide expeditious access to prohibitively strained polycyclic targets which are difficult or impossible to synthesize using ground state chemistry. Often the photogenerated semiproducts are introduced into secondary transformations to take advantage of the strain installed at the photochemical step. For example, Rawal's “general strategy for increasing molecular complexity” 1 utilizes the intramolecular Paternò-Büchi reaction in acyl norbonenes2 to access extremely strained polycyclic oxetanes, which are subsequently treated with Cohen's LDBB3 to trigger a radical anion fragmentation cascade yielding di- and triquinanes in a total of two simple steps. We demonstrated that under acidic conditions photogenerated polycyclic oxetanes undergo several spectacular cationic transformations.4 Such oxetanes can also be reverted pyrolytically,5 or via an electron transfer-induced reaction,6 to an alternative pair of an alkene and an aldehyde, Scheme 1, in a manner resembling the metathesis reaction, for which Jones5 suggested the term “carbonyl-olefin metathesis.”
Scheme 1.
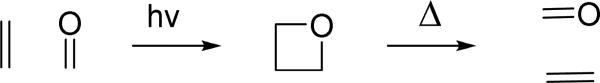
Such oxametathesis reactions have considerable synthetic potential. We have recently shown that polycyclic alkenes outfitted with photoactive benzoyl or heterobenzoyl groups yield highly strained oxetanes which undergo protolytic oxametathetic cycloreversion to offer access to novel polycyclic scaffolds under very mild conditions, ambient temperature and catalytic amounts of HCl, as shown in Scheme 2 (top).7 Unlike with the pyrolytic oxametathesis, most functional groups are expected to tolerate these mild conditions, which enhances the preparative value of the method.
Scheme 2.

In this paper we examine topological and stereochemical effects on the outcomes of the intramolecular Paternò-Büchi reaction and subsequent protolytic cycloreversion in polycycloalkenes tethered to the benzoyl chromophore via a more flexible tether containing an extra methylene group. We will show that, unlike the case of aroyl norbornenes or bicyclo[2.2.2]octenes, i.e. the photoprecursors where benzoyls are directly attached to the polycyclic scaffold, the overall reaction course is highly dependent on the stereochemistry of the starting keto-alkene, allowing for superb reaction control and at the same time greater diversity of the products. The impetus behind this research was to evaluate in the context of diversity-oriented synthesis the prospects of modular design of photoprecursors for photoprotolytic oxametathesis, and to probe how subtle variations in the structure of the starting materials could allow for considerable changes in the reaction's regio- and stereochemical outcomes or, alternatively, turn off the Paternò-Büchi channel altogether, switching to a completely different photoreactivity.
Results and Discussion
We took inventory of efficient coupling reactions and chose Diels-Alder additions of reactive dienophiles, dimethyl acetylene-dicarboxylate (DMAD) and propiolates with cyclic dienes,8 with subsequent Michael additions of benzoyl-containing chromophores to the electrophilic double bond. This formally allows for three diversity inputs in the synthesis of photoprecursors. The Diels-Alder step with acetylenes produces only one type homo-conjugated diene, for example, norbornadiene in the case of cyclopentadiene. However, the Michael addition step potentially produces several diastereomers of which we isolated and purified the photoactive endo-phenacyl isomers and utilized them to probe the effects of steric congestion in the photoprecursors on the outcome of photoprotolytic oxametathesis.
Moderate endo-selectivity of the enolate addition is achieved with spirocyclopropanonorbornadienes 3, 4 and bicyclo[2.2.2]octadienes 5, 6. However, even in the 7-unsubstituted norbornadienes 1, 2 the addition of substituted enolates (R≠H) occurs with modest endo bias. The endo-selectivity is improved with bulkier R groups in the enolate: diphenylethanone (R=Ph) exhibits better than 95% of endo-selectivity.
Scheme 2 illustrates that Paternò-Büchi reaction in the endo-aroyl precursors A, described in ref7, yields only one type oxetane B (γ-oxetane) which upon treatment with catalytic amounts of acid cycloreverts into bicyclo[n.2.1]alkenes C possessing a formyl group. The modular approach which we explore in this work gives phenacyl photoprecursors 7 in which the chromophore is connected to the bicyclic core via a more flexible tether, so that the initial Paternò-Büchi step can potentially yield both regioisomeric oxetanes, 8 and 10. The resulting cycloreversion products in these systems possess either [3.n.1] (9) or [n.3.0] (11) bicyclic topology.
The first example, shown in Scheme 3, demonstrates the regiochemical outcome of the intramolecular Paternò-Büchi cycloaddition in the simplest case of a bicyclo[2.2.2]octane system in which the second substituent, methoxycarbonyl group, is exo-trans to the photoactive phenacyl pendant. In this case the aroyl group is free to rotate unobstructed, and the regiochemistry of the [2+2] photocycloaddition presumably reflects some intrinsic properties of the system. Judging by the subsequent examples, which involve hydrogen-abstraction reactions successfully competing with Paternò-Büchi cycloadditions, it is most likely that this photochemistry originates from the triplet state of aroyl pendants. The initial 1,4-diradicals leading to oxetanes 8a and 10a can be classified as exo-trig and endo-trig.9 We will refer to the products of exo-trig cyclization as γ-oxetanes and the endo-trig – as δ (with α being the attachment point of the phenacyl pendant to the bicyclic core).
Scheme 3.

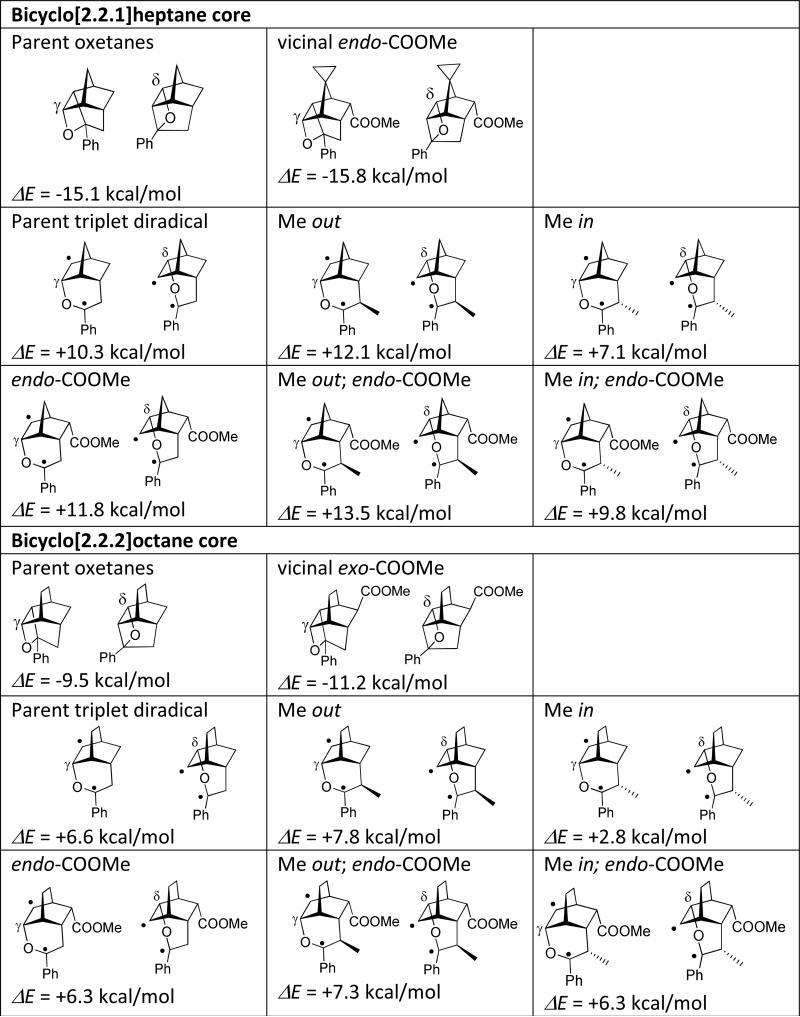
Oxetanes 8a and 10a form in a nearly 1:1 ratio indicating that in these systems there is no intrinsic bias for cyclization associated with the tether. The γ-oxetane is more strained than the δ-oxetane: DFT calculations gave 11.2 kcal/mol difference (Table 1, B3LYP/6-311+G(d,p), Gaussian 09, rev. A.02; see SI for full reference and computational details). Generally, both γ- and δ-oxetanes described in this study are not stable on silica gel during column chromatography. In this particular case only δ-oxetane could be purified (44% isolated yield) and fully characterized by NMR and xray crystallography. The γ-oxetane 8a was characterized in a mixture with 10a by 1H NMR and introduced into subsequent acid-catalyzed cycloreversion without additional purification.
Table 1.
DFT, B3LYP/6-311+G(d,p), relative energies of oxetanes and triplet 1,4-diradicals EDFT-δ-EDFT-γ)
|
Both oxetanes undergo cycloreversion, when subjected to mild acidic conditions, completing the oxametathesis cycle. The mechanism for formation of the presumed initial products of oxametathesis, aldehydes 9a and 11a, is shown in Scheme 4.
Scheme 4.

Expectedly, under slightly acidic reaction conditions aldehydes 9a and 11a are subject to epimerization via enolization, which brings their formyl groups into the exo-position. This is why the preferred experimental procedure for protolytic oxametathesis involved trapping the aldehydes in a form of cyclic acetals, 1,3-dioxolanes 12a and 13a, by running the acid-catalyzed oxetane cycloreversion in the presence of ethylene glycol to partially prevent such epimerization. Ultimately, the one-pot procedure was developed in which the Paternò-Büchi step was carried out in dichloromethane containing HCl and ethylene glycol, as shown in Scheme 5.
Scheme 5.
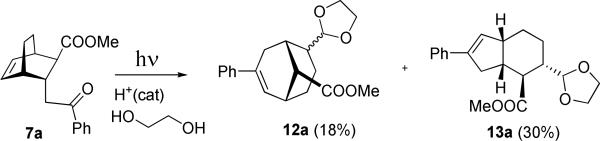
The isolated yields of dioxolanes 12a and 13a are modest, but this is a straightforward one-pot synthetic photochemical procedure starting from the readily available 7a.
Addition of the enolate of propiophenone to Michael acceptor 5 yields several diastereomers. We isolated and purified the trans-diastereomer 7b (Z=CH2CH2, Y=H, exo-COOMe), possessing endo-phenacyl and exo-COOMe groups, which is Paternò-Büchi-confident. Under the conditions of protolytic oxametathesis this endo-diastereomer showed very high regioselectivity. According to the 1H NMR of reaction mixture, irradiation of 7b produced exclusively δ-oxetane 10b (>95%), which was sufficiently stable on the silica gel column to allow for its isolation in 65% yield. Since the length of the tether is identical in both cases, the regiochemical outcome of this reaction is likely determined by the conformational bias introduced by the methyl group which defines the initial conformation of the photoactive pendant with respect to the double bond of the bicyclic core, Scheme 6.
Scheme 6.
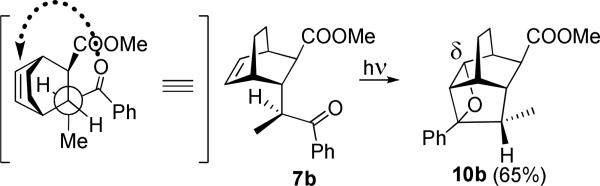
Provided that in the low energy conformation the benzoyl pendant and the methyl group are “out” (i.e., hydrogen is “in”, under the bicyclic core) as shown in Scheme 6, the formation of oxetane 10b occurs with the least motion of the excited carbonyl.
Energetically, in the parent bicyclo[2.2.2]octane system, the γ-diradical of type DR-8a (Scheme 3) is approximately 7 kcal/mol more stable than the δ-diradical of type DR-10a. The fact that oxetanes 8a and 10a are formed in nearly equal amounts implies that while the γ-diradical may be forming to a greater extent, it fails to close the oxetane ring after the intersystem crossing into the singlet manifold because the γ-oxetane is prohibitively (>11 kcal/mol) more strained than the δ-oxetane. The relative orientation of singly occupied orbitals in both γ- and δ-diradicals is similar; there is no reason to believe that the spin-orbit coupling matrix element is considerably greater for one of them.10 We suggest that due to the unfavorable energetics of the reaction channel leading to the γ-oxetane, the γ-diradicals after intersystem crossing from the triplet to singlet potential energy surface partition into the Grob fragmentation channel more frequently than the δ-diradicals (which readily cyclize to form more stable δ-oxetanes). For 7a these two trends cancel out leading to nearly equal amounts of 8a and 10a. In contrast, the methyl substitution in the tether of 7b changes the energetics: the calculated energy difference for triplet γ- and δ-diradicals is 7.8 kcal/mol for the epimer of 7b which produces the diradical with Me out (or exo-). However, for 7b itself (Me is in or endo- in the diradical) this energy difference decreases to only 2.8 kcal/mol @ B3LYP/6-311+G(d,p). Given that the resulting δ-oxetane 10b is still more than 12 kcal/mol more stable than the γ-oxetane, the regiospecific outcome in Scheme 6 becomes readily predictable. It is also possible, but unlikely, that the Paternò-Büchi reaction in these bicyclic systems occurs via a concerted mechanism, i.e. via a singlet four-membered transition state. We failed to computationally locate singlet transition states on the reaction hypersurface.
We found that the protolytic transformation of (a relatively more stable, and therefore purifiable) 10b into 13b requires elevated temperature of 70°C. At ambient temperature elimination to form 14 occurs. The structure of this elimination product, alkene 14, was characterized with xray crystallography. Remarkably, all these diverse modes of reactivity are realized with very high degree of selectivity.
Michael additions to dicarboxylates 2 and 4 produced diastereomeric trans-dicarboxylates which provided an opportunity to probe the photochemistry in the systems where the vicinal endo-carboxylate group severely constrains the conformational mobility of the aroyl chromophore. Generally low yielding Michael additions to bicyclo[2.2.2]octane 6 possessing two methoxycarbonyl groups rendered it not practical for generating endo photoprecursors. This is why we focused mostly on the bicyclic[2.2.1]heptane cores assembled with DMAD. The Diels-Alder product of DMAD and spiroheptadiene also improved the endo-selectivity of Michael additions of enolates of acetophenone, propiophenone, valerophenone, and 3-phenylpropiophenone.
As judged by the 1H NMR of the reaction mixture, acetophenone adduct 7c formed γ-oxetane 8c exclusively, which in the presence of catalytic acid smoothly transforms into the product of oxametathesis 9c and subsequently into its acetal 12c. This is in keeping with the least motion hypothesis, as the counter clockwise rotation is blocked by the endo-COOMe.
Our calculations show that in the parent bicyclo[2.2.1]heptane system the DFT energy difference between γ- and δ-oxetane, 15.1 kcal/mol, is even greater than in the bicyclo[2.2.2]octane framework. The endo-COOMe group in the spirocyclopropano-norbornene-based oxetanes slightly increases this energy difference to 15.8 kcal/mol. However, the γ-diradical leading to γ-oxetane is 10.3 kcal/mol more stable than the δ-diradical, so the δ-channel is no longer competitive.
As we later discovered, further increase in steric congestion via the introduction of the methyl group in the tether could shut down the Paternò-Büchi channel altogether in the photoprecursors already outfitted with the endo-COOMe brake.
Michael addition of propiophenone enolate to bis-carboxylate 2 produced a single endo-diastereomer 7d. The DFT structure of its most stable conformer shows that introduction of the methyl group in the α-position locks the phenyl group in the anti-conformation to this methyl (its xray structure is in keeping with this computational prediction). As shown, even upon clockwise rotation, the phenyl group is predicted to encounter a steric clash with the endo-COOMe before the excited carbonyl reaches the double bond. This suppresses the Paternò-Büchi channel almost completely, as we detect less than 5% of the γ-oxetane in this series. The major product 15 (75%, Scheme 9) is a result of an unprecedented cascade photoreaction. The stereochemistry of this transformation is proved unambiguously, as the structures of both the photoprecursor 7d and the product 15 are determined by xray analysis.
Scheme 9.

The tricycle[3.3.0.03,7]octane (bisnoradamantane11 or stellane in Gleiter's12 terminology) core of 15 is often accessed via intramolecular Paternò-Büchi reactions in endo-acyl norbornenes as extensively studied by Sauers.13 In contrast, the reaction shown in Scheme 9 produces the stellane core as a result of a radical cyclization in which two C-C bonds are formed. Two plausible mechanistic rationales are shown in Scheme 9. Both mechanisms involve the initial β-hydrogen abstraction (from the Me-group) by the excited benzoyl. This 1,3-diradical can recombine to form the cyclopropane ring as in the Yang cyclization.14 This is followed by secondary photoinduced cyclopropane ring opening to form a more stable 1,3-diradical, which subsequently adds to the double bond (top mechanism). An alternative, mechanism involves a 1,2-radical shift of the bicyclic core facilitated by the enol formation, followed by the keto-enol equilibrium, second excitation with H-abstraction, and an interrupted Yang cyclization (bottom).
In both cases the secondary 1,3-diradical undergoes cycloaddition to the readily accessible double bond, which constitutes radical cyclopentanation of alkenes. The minor product 16 (15%) is considerably more strained, possessing a cyclobutane moiety conceivably resulting from a formal [2+2] cycloaddition of the enol form of the photoprecursor.
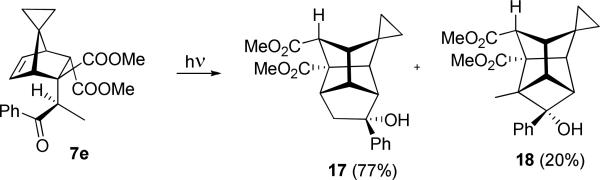
The spirocyclopropanonorbornadiene series with propiophenone gave the same result – the major photoproduct is derived from cyclopentanation of the double bond in 7e with the secondary 1,3-diradical, Scheme 11. As in the case above, both the structure of the photoprecursor 7e and the product 17 is determined by xray crystallographic analysis.
Scheme 11.
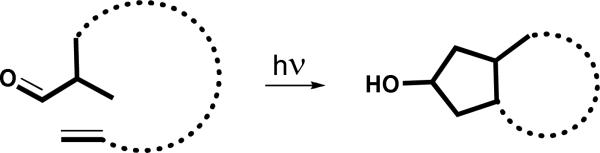
The general topology of this novel photorearrangement/cyclopentanation of double bonds is shown in Scheme 12, which constitutes a one pot photoinduced synthesis of fused cyclopentanols – a useful addition to the toolbox of photoassisted synthetic chemistry.
Scheme 12.
In these transformations we did not observe the products of cyclopentanation by the initially photogenerated 1,3-diradical. Presumably it is too short-lived (it has one primary radical center) to successfully cyclopentanate the double bond. Although, from the point of relative stability of the products, the 1,3-cycloaddition of the primary 1,3-diradical ostensibly produces less strained polycyclic structure. In order to probe whether such alternative reactivity is possible, we simply stabilized the second radical center by employing β-phenylpropiophenone at the Michael addition step. We were able to separate chromatographically both diastereomers of the endo-photoprecursors (7f and 7f’), which are expected to produce much more stable doubly benzylic initial 1,3-diradical. The structure of one of these photoprecursors, 7f, was unambiguously determined by xray crystallography, and the structure of 7f’ was inferred from that. However, even with this additional stabilization the initial 1,3-diradical failed to endo-cyclopentanate the norbornene double bond. This is somewhat in keeping with the fact that we did not find any literature reports on 1,3- or 1,4- Yang cyclizations (or Norrish Type II fragmentations) interrupted by diradical cycloadditions to a suitably positioned double bond.
Diastereomeric photoprecursors 7f and 7f’ exhibited very different reactivity. As it is shown in Scheme 13, diastereomer 7f forms the major photoproduct 19, which is similar to the formation of 15 described in Scheme 10, and the minor cyclobutane 20.
Scheme 13.

Scheme 10.

In contrast, irradiation of diastereomer 7f’ was not a clean reaction. We isolated the product of benzyl radical addition 21 in poor yield of 20%. As shown in Scheme 14, our mechanistic rationale for its formation involves the initial hydrogen abstraction by the excited carbonyl to give a stable 1,3-diradical, which lives long enough for its benzylic terminus to attack the double bond. However, instead of recombination to form the cyclopentyl ring, the ketyl radical simply delivers hydrogen to terminate the second radical center, restoring the benzoyl group.
Scheme 14.

As diastereomer 7f can only form a 1,3-diradical in which the ketyl – not benzylic – radical center can reach the double bond, one draws a conclusion that the ketyl radical is not capable of initiating radical addition to the double bond or such reaction is too slow and cannot compete with the rearrangement leading to the secondary 1,3-diradical and, eventually, to the product of cyclopentanation 19.
The diastereomeric spirocyclopropanes 7g and 7g’ behave in a similar manner with 7g yielding the rearranged cyclopentanation product 22 as the major product and its homobenzylic epimer 7g’ producing polycycle 24, presumably via the same mechanism shown in Scheme 14.
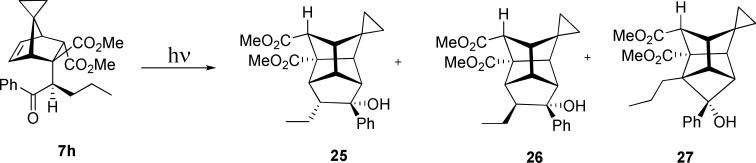
Valerophenone-based photoprecursor 7h also offers stabilization of the second radical center, although in this case we isolated only one endo-diastereomer. Irradiation of 7h yielded two diastereomers of the rearranged cyclopentanation products 25,26 plus the minor product of a formal [2+2] cycloaddition 27. We did not detect any radical addition products similar to 21 and 24 above. It is unlikely that we misassigned any of these, as the benzoyl's carbonyl group is easily detectable by 13C NMR. The plausible explanation for these results is that just like the primary radical derived from 7d in Scheme 10, the ethyl-stabilized secondary radical does not live long enough for attacking the double bond. Instead, the rearrangement and subsequent endo-cyclopentanation to form 25, 26 occurs. The cyclopentanation is not stereospecific in this case as both epimers at the ethyl bearing center are observed. The major product 25 was not entirely separable from the minor [2+2] product 27, and were characterized in a mixture by 1H NMR by analogy with 19 and 22.
Photoprecursors 7d-7h are not Paternò-Büchi competent and therefore they utilize a different reaction pathway available for them, which is initiated by intramolecular hydrogen abstraction. To probe how severe the Paternò-Büchi channel is impeded in these sterically congested systems we attempted to prepare the Michael adducts of dimethyl norbornadiene-2,3-dicarboxylate with cyclic aromatic ketones, indanone, 3-methyl indanone, and 3-phenylindanone, in which the carbonyl is held out and therefore not capable of β-hydrogen abstraction. Regrettably, we were able to isolate and purify only the exo, i.e. photo-inactive, Michael products 7l through 7p (see experimental).
We also synthesized the 1:1 Diels-Alder adduct 2815 (obtained with small amounts of 2:1 adducts1629 and 30) of furan with DMAD. However, under the conditions of the subsequent Michael addition step 7-oxanorbornadiene 28 fragmented yielding pyranone 31 instead of the desired endo-phenacyl bicyclic photoprecursor (two plausible mechanisms are shown in Scheme 17).
Scheme 17.

Summary
In summary, rapid modular assembly of bicyclic scaffolds outfitted with conformationally flexible aroylmethyl chromophores provides access to Paternò-Büchi confident photoprecursors capable of protolytic oxametathesis transformation leading to novel polycyclic systems decorated with functional groups useful for subsequent transformations. At the same time, conformationally-constrained photoprecursors can be readily obtained in a similar fashion. Irradiation of these starting materials initiates hydrogen abstraction and is accompanied by a cascade radical rearrangement/1,3-diradical endo-cycloaddition to the double bond, which amounts to intramolecular diradical cyclopentanation. This expeditious growth of complexity in just a few synthetically simple steps attests to the versatility of these new transformations and offers additional useful tool for the synthetic chemistry tool chest.
Experimental Section
General procedure for preparation of the Diels-Alder adducts (1-6)
(A): A solution of dimethyl acetylenedicarboxylate in 10 mL of furan (used as a solvent and reagent) was stirred at room temperature for 24 h (longer reaction time causes the formation of 1a and 1b). After the reaction was completed, the solvent was removed on a high vacuum pump. The crude reaction mixture was purified on a flash silica gel column using a mixture of hexane and EtOAc as an eluent.
(B): A solution of dimethyl acetylenedicarboxylate (1.0 eq) in 10 mL of DCM and freshly distilled 1,3-cyclopentadiene (1.0-2.0 eq) was stirred at room temperature for 24 h. After the reaction was completed, the solvent was removed on a high vacuum pump. The resulting product was used without further purification unless noted otherwise.
(C): Acetylenedicarboxylic acid monopotassium salt (1.0 eq) and HCl (4.0 M solution in dioxane, 1.0 eq) were stirred in MeOH for 10 min. After that freshly distilled 1,3-cyclopentadiene (2.0 eq) was added and the resulting mixture was stirred at room temperature for 48-72 h. When the reaction was completed, MeOH was removed, the crude residue was dissolved in DCM, washed with Na2CO3 (sat.), water and brine. The organic layer was dried over anhydrous Na2SO4. The organic solvent was dried and evaporated to get pure product.
(D): A solution of propiolic acid methyl ester (1.0 eq) and 1,3-cyclohexadiene (or freshly distilled 1,3-cyclopentadiene) (1.0-5.0 eq) in 10 mL of 1,2-dichlorobenzene or toluene was heated in a pressure vessel at 70-75°C overnight. After the reaction was cooled to room temperature, the solvent was removed on a high vacuum pump. The crude reaction mixture was purified on a silica gel column using a mixture of hexane and EtOAc as an eluent.
Methyl bicyclo[2.2.1]hepta-2,5-diene-2-carboxylate (1):8a
(procedure B) from 1.0 mL of propiolic acid methyl ester (11.2 mmol) and 3.0 mL of 1,3-cyclopentadiene (35.6 mmol): 0.98 g (58 %). 1H NMR (500 MHz, CDCl3) δ = 7.65 (d, J = 3.2 Hz, 1H), 6.91 (dd, J = 5.1, 3.1 Hz, 1H), 6.73 (dd, J = 5.1, 3.1 Hz, 1H), 3.90 (m, 1H), 3.73 (s, 3H), 3.72 (m, 1H), 2.15 (ddd, J = 6.5, 1.6, 1.6 Hz, 1H), 2.12 (d, J = 6.5 Hz, 1H).
Dimethyl bicyclo[2.2.1]hepta-2,5-diene-2,3-dicarboxylate (2):8b
(procedure B) from 0.7 mL of dimethyl acetylenedicarboxylate (5.69 mmol) and 0.5 mL of 1,3-cyclopentadiene (5.95 mmol): 1.13 g (95 %). (Procedure C) from 6.0 g of acetylenedicarboxylic acid monopotassium salt (39.4 mmol) and 0.7 mL of 1,3-cyclopentadiene (83.2 mmol): 7.95 g (97 %). 1H NMR (500 MHz, CDCl3) δ = 6.92 (dd, J = 1.9, 1.9 Hz, 2H), 3.94 (m, 2H), 3.79 (s, 6H), 2.28 (ddd, J = 6.8, 1.6, 1.6 Hz, 1H), 2.10 (ddd, J = 6.8, 1.5, 1.5 Hz, 1H). 13C NMR (500 MHz, CDCl3) δ = 165.5, 152.5, 142.4, 73.0, 53.5, 52.1.
Methyl spiro[bicyclo[2.2.1]hepta-2,5-diene-2-carboxylate-7,1′-cyclopropane] (3):8c
(procedure D) from 1.5 mL of propiolic acid methyl ester (16.9 mmol) and 1.25 mL of spiro[4.2]hepta-4,6-diene (12.5 mmol): 0.78 g (35 %). 1H NMR (500 MHz, CDCl3) δ = 7.71 (dd, J = 3.3, 1.1 Hz, 1H), 6.98 (ddd, J = 5.3, 3.2, 0.7 Hz, 1H), 6.81 (ddd, J = 5.3, 3.2, 0.8 Hz, 1H), 3.76 (s, 3H), 3.40 (m, 1H), 3.20 (m, 1H), 0.59-0.52 (m, 4H).
Dimethyl spiro[bicyclo[2.2.1]hepta-2,5-diene-2,3-dicarboxylate-7,1′-cyclopropane] (4):8d
(procedure B) from 0.6 mL of dimethyl acetylenedicarboxylate (4.88 mmol) and 0.5 mL of spiro[4.2]hepta-4,6-diene (4.99 mmol) at -15°C → 20°C, gradient (hexane/EtOAc gradient 30:1 → 20:1): 0.77 g (68 %). 1H NMR (500 MHz, CDCl3) δ = 6.96 (dd, J = 2.0, 2.0 Hz, 2H), 3.79 (s, 6H), 3.43 (dd, J = 2.0, 2.0 Hz, 2H), 0.68-0.64 (m, 2H), 0.59-0.55 (m, 2H). 13C NMR (500 MHz, CDCl3) δ = 165.4, 152.0, 141.8, 69.3, 58.5, 52.0, 9.4, 9.3.
Methyl bicyclo[2.2.2]octa-2,5-diene-2-carboxylate (5):8e
(procedure D) from 3.0 mL of propiolic acid methyl ester (33.6 mmol) and 7.5 mL of 1,3-cyclohexadiene (78.7 mmol): 5.17 g (88%). 1H NMR (500 MHz, CDCl3) δ = 7.29 (dd, J = 6.4, 1.9 Hz, 1H), 6.38 (ddd, J = 7.4, 6.1, 1.5 Hz, 1H), 6.27 (ddd, J = 7.3, 5.9, 1.5 Hz, 1H), 4.20 (m, 1H), 3.76 (m, 1H), 3.73 (s, 3H), 1.38-1.29 (m, 4H).
Dimethyl bicyclo[2.2.2]octa-2,5-diene-2,3-dicarboxylate (6):8f
(procedure B) from 0.5 mL of dimethyl acetylenedicarboxylate (4.07 mmol) and 0.6 mL of 1,3-cyclohexadiene (6.30 mmol) for 120 h (hexane/EtOAc gradient 50:1 → 10:1): 0.20 g (22 %). 1H NMR (500 MHz, CDCl3) δ = 6.38 (dd, J = 4.4, 3.2 Hz, 2H), 4.04 (m, 2H), 3.77 (s, 6H), 1.51-1.45 (m, 2H), 1.44-1.39 (m, 2H). 13C NMR (500 MHz, CDCl3) δ = 166.5, 142.3, 133.6, 52.1, 38.9, 24.5.
Preparation of the Michael adducts. General procedure
(A): Diisopropylamine (1.4 eq) was dissolved in THF at -10°C, then n-BuLi (1.6 M solution in hexanes 1.2 eq) was added and the resulting mixture was stirred for 20 min at -10°C (in some cases -78°C) to 0°C gradient. After that, carbonyl compound was added (1.0 eq) and the mixture was stirred at room temperature for additional 30-40 min. After the anion was generated, the Diels-Alder adduct (1-7) was added (1.0 eq) at 20°C and the resulting mixture was stirred overnight. After the reaction was completed DCM was added, the resulting mixture was quenched with NH4Cl, washed with water and brine. The organic layer was dried over anhydrous Na2SO4 and removed under vacuum. The crude mixture was purified on a silica gel column using a mixture of hexane and EtOAc as an eluent.
(B): Fresh LDA (1.8 M solution, 1.3 eq) was added under nitrogen atmosphere to the solution of a carbonyl compound (1.0 eq) in THF at -5°C to 0°C. After the anion was generated (in 20-30 min), the Diels-Alder adduct (1-6) was added (1.0 eq) at room temperature and the resulting mixture was stirred for 10-20 h. After the reaction was completed DCM was added, the resulting mixture was quenched with NH4Cl, washed with water and brine. The organic layer was dried over anhydrous Na2SO4 and removed under vacuum. The crude mixture was purified on a silica gel column using a mixture of hexane and EtOAc as an eluent.
Methyl endo-5-phenacylbicyclo[2.2.2]oct-2-ene-exo-6-carboxylate (7a)
(procedure B) from 10.7 mL of LDA (19.18 mmol), 1.75 mL of acetophenone (15.06 mmol) and 2.25 g of 5 (13.70 mmol) (hexane/EtOAc gradient 50:1 → 10:1): 1.40 g (37 %). 1H NMR (500 MHz, CDCl3) δ = 7.95 (d, J = 8.1 Hz, 2H), 7.57 (t, J = 7.4 Hz, 1H), 7.47 (t, J = 7.8 Hz, 2H), 6.40 (ddd, J = 8.0, 6.8, 1.1 Hz, 1H), 6.25 (t, J = 7.1 Hz, 1H), 3.74 (s, 3H), 2.91 (dd, J = 16.3, 7.6 Hz, 1H), 2.86-2.80 (m, 2H), 2.77 (dddd, J = 12.7, 5.4, 1.8, 1.8 Hz, 1H), 2.54 (m, 1H), 2.07 (dt, J = 5.4, 2.3 Hz, 1H), 1.73-1.62 (m, 2H), 1.34-1.27 (m, 1H), 1.17-1.10 (m, 1H). 13C NMR (500 MHz, CDCl3) δ = 198.4, 175.0, 137.0, 134.3, 133.0, 132.9, 128.5, 128.1, 51.8, 50.1, 46.0, 36.8, 34.3, 33.0, 25.2, 20.1. HRMS (ESI/TOF) calcd for C18H20NaO3+ (MNa+) 307.1310, found 307.1292.
Methyl endo-5-(1-methylphenacyl)bicyclo[2.2.2]oct-2-ene-exo-6-carboxylate (7b)
(procedure A) from 0.53 mL of diisopropylamine (3.78 mmol), 2.0 mL of n-BuLi (3.24 mmol), 0.40 mL of propiophenone (3.01mmol) and 0.49 g of 5 (3.01 mmol) (hexane/EtOAc gradient 30:1 → 10:1): 0.24 g (29 %). 1H NMR (500 MHz, CDCl3) δ = 7.92 (d, J = 7.6 Hz, 2H), 7.56 (t, J = 7.4 Hz, 1H), 7.46 (t, J = 7.8 Hz, 2H), 6.34 (ddd, J = 8.0, 6.8, 1.2 Hz, 1H), 6.24 (t, J = 7.3 Hz, 1H), 3.73 (s, 3H), 3.26 (dt, J = 15.3, 7.1 Hz, 1H), 2.75-2.71 (m, 1H), 2.71-2.67 (m, 1H), 2.61 (ddd, J = 8.3, 5.7, 1.7 Hz, 1H), 2.01-1.99 (m, 1H), 1.67-1.61 (m, 2H), 1.35-1.27 (m, 1H), 1.17 (d J = 7.0 Hz, 3H), 1.14-1.06 (m, 1H). 13C NMR (500 MHz, CDCl3) δ = 204.0, 175.1, 137.0, 133.75, 133.0, 132.8, 128.6, 128.3, 51.7, 49.4, 46.1, 43.2, 33.4, 31.3, 25.3, 20.5, 15.8. HRMS (ESI/TOF) calcd for C19H23NaO3+ (MH+) 299.1642, found 299.1649.
Dimethyl spiro[endo-5-phenacylbicyclo[2.2.1]hept-2-ene-exo-5-endo-6-dicarboxylate-7,1′-cyclopropane] (7c)
(procedure B) from 1.4 mL of LDA (2.52 mmol), 0.25 mL of acetophenone (2.14 mmol) and 0.50 g of 4 (2.13 mmol) (hexane/EtOAc gradient 40:1 → 10:1): 0.23 g (30 %) colorless crystals, m.p. 81-83°C. 1H NMR (500 MHz, CDCl3) δ = 7.89 (d, J = 7.1 Hz, 2H), 7.53 (t, J = 7.4 Hz, 1H), 7.42 (t, J = 7.7 Hz, 2H), 6.47 (ddd, J = 5.8, 3.1, 0.9 Hz, 1H), 6.44 (ddd, J = 5.8, 2.8, 0.9 Hz, 1H), 4.38 (d, J = 3.5 Hz, 1H), 3.70 (s, 3H), 3.63 (d, J = 17.8 Hz, 1H), 3.50 (d, J = 17.8 Hz, 1H), 3.44 (s, 3H), 2.65 (m, 1H), 2.58 (m, 1H), 0.65 (ddd, J = 9.4, 5.8, 5.8 Hz, 1H), 0.49 (ddd, J = 9.6, 5.7, 5.7 Hz, 1H), 0.44-0.35 (m, 2H). 13C NMR (500 MHz, CDCl3) δ = 198.6, 175.4, 173.6, 138.6, 136.7, 135.1, 132.9, 128.5, 127.9, 57.4, 57.4, 53.2, 52.3, 51.5, 48.6, 44.3, 43.3, 10.2, 4.3. HRMS (ESI/TOF) calcd for C21H23O5+ (MH+) 355.1540, found 355.1561. See SI for xray data.
Dimethyl spiro[exo-5-phenacylbicyclo[2.2.1]hept-2-ene-endo-5-endo-6-dicarboxylate-7,1′-cyclopropane] (7c-exo)
0.15 g (20 %) colorless crystals, m.p. 89-92°C. 1H NMR (500 MHz, CDCl3) δ = 7.92 (d, J = 7.2 Hz, 2H), 7.57 (t, J = 7.4 Hz, 1H), 7.47 (t, J = 7.7 Hz, 2H), 6.43 (m, 2H), 3.98 (d, J = 15.5 Hz, 1H), 3.62 (s, 3H), 3.58 (d, J = 15.5 Hz, 1H), 3.43 (s, 3H), 3.17 (d, J = 3.2 Hz, 1H), 2.70 (m, 1H), 2.45 (m, 1H), 0.81-0.77 (m, 1H), 0.76-0.71 (m, 1H), 0.43 (m, 2H). 13C NMR (500 MHz, CDCl3) δ = 197.7, 173.0, 172.9, 137.3, 136.4, 135.9, 133.2, 128.7, 128.1, 59.6, 56.2, 54.8, 52.0, 51.6, 51.6, 46.0, 43.0, 10.8, 3.6. HRMS (ESI/TOF) calcd for C21H22NaO5+ (MNa+) 377.1359, found 377.1368. See SI for xray data.
Dimethyl (1RS, 4SR, 5RS, 6SR)-endo-5-((1′RS)-1′-benzoylethyl)bicyclo[2.2.1]hept-2-ene-exo-5-endo-6-dicarboxylate (7d)
(procedure A) from 0.23 mL of diisopropylamine (1.64 mmol), 1.0 mL of n-BuLi (1.60 mmol), 0.15 mL of propiophenone (1.13 mmol) and 0.23 g of 2 (1.10 mmol) (hexane/EtOAc gradient 30:1 → 20:1): 0.125 g (34 %), colorless crystals, m.p. 97-101°C. 1H NMR (500 MHz, CDCl3) δ = 7.90 (d, J = 7.2 Hz, 2H), 7.57 (t, J = 7.4 Hz, 1H), 7.47 (t, J = 7.6 Hz, 2H), 6.33 (dd, J = 5.6, 2.9 Hz, 1H), 5.81 (dd, J = 5.6, 3.2 Hz, 1H), 3.95 (q, J = 7.2 Hz, 1H), 3.84 (s, 3H), 3.72 (s, 3H), 3.35 (d, J = 3.3 Hz, 1H) overlaps with 3.34 (m, 1H), 3.02 (m, 1H), 1.52 (ddd, J = 9.2, 1.8, 1.8 Hz, 1H), 1.46 (d, J = 9.2 Hz, 1H), 1.08 (d, J = 7.1 Hz, 3H). 1H NMR (500 MHz, C6D6) δ = 7.98 (m, 2H), 7.07 (m, 1H), 7.00 (m, 2H), 6.35 (dd, J = 5.6, 3.0 Hz, 1H), 5.86 (dd, J = 5.6, 3.2 Hz, 1H), 4.24 (q, J = 7.1 Hz, 1H), 3.59 (s, 3H), 3.57 (m, 1H), 3.46 (d, J = 3.3 Hz, 1H), 3.33 (s, 3H), 2.69 (m, 1H), 1.41-1.33 (m, 2H) overlaps with 1.34 (d, J = 7.1 Hz, 3H). 13C NMR (500 MHz, CDCl3) δ = 202.7, 175.6, 174.0, 137.6, 136.4, 134.2, 133.1, 128.8, 128.4, 62.0, 52.1, 51.8, 51.3, 50.2, 49.5, 47.1, 43.6, 16.3. HRMS (ESI/TOF) calcd for C20H23O5+ (MH+) 343.1540, found 343.1546. See SI for xray data.
Dimethyl exo-5-(1′-benzoylethyl)bicyclo[2.2.1]hept-2-ene-endo-5-endo-6-dicarboxylate (7d-exo)
0.092 g (25 %). 1H NMR (500 MHz, CDCl3) δ = 7.91 (d, J = 7.2 Hz, 2H), 7.59 (t, J = 7.4 Hz, 1H), 7.1249 (t, J = 7.7 Hz, 2H), 6.34 (dd, J = 5.5, 3.0 Hz, 1H), 6.16 (dd, J = 5.5, 3.2 Hz, 1H), 3.90 (q, J = 7.0 Hz, 1H), 3.68 (s, 3H), 3.60 (s, 3H), 3.05 (d, J = 3.1 Hz, 1H), 3.00 (m, 1H), 2.76 (m, 1H), 1.72 (d, J = 9.1 Hz, 1H), 1.49 (t, J = 7.0 Hz, 3H) overlaps with 1.48 (ddd, J = 9.1, 1.7, 1.7 Hz, 1H). 13C NMR (500 MHz, CDCl3) δ = 201.8, 173.6, 171.7, 138.0, 136.6, 136.3, 133.3, 128.8, 128.1, 64.3, 54.5, 51.7, 51.4, 49.8, 48.9, 46.4, 46.0, 15.4. HRMS (ESI/TOF) calcd for C20H23O5+ (MH+) 343.1540, found 343.1555.
Dimethyl (1RS, 4RS, 5RS, 6SR)-spiro[(1″RS)-endo-5-(1″-benzoylethyl)bicyclo[2.2.1]hept-2-ene-exo-5-endo-6-dicarboxylate-7,1′-cyclopropane] (7e)
(procedure A) from 0.19 mL of diisopropylamine (1.36 mmol), 0.96 mL of n-BuLi (1.54 mmol), 0.14 mL of propiophenone (1.05 mmol) and 0.24 g of 4 (1.02 mmol) (hexane/EtOAc gradient 30:1 → 10:1): 0.23 g (61 %). 1H NMR (500 MHz, CDCl3) δ = 7.88 (d, J = 7.2 Hz, 2H), 7.57 (t, J = 7.4 Hz, 1H), 7.47 (t, J = 7.6 Hz, 2H), 6.41 (ddd, J = 5.8, 2.9, 0.8 Hz, 1H), 5.84 (ddd, J = 5.8, 3.2, 0.8 Hz, 1H), 4.02 (q, J = 7.2 Hz, 1H), 3.82 (s, 3H), 3.77 (d, J = 3.4 Hz, 1H), 3.74 (s, 3H), 2.98 (m, 1H), 2.39 (m, 1H), 1.03 (d, J = 7.2 Hz, 3H), 0.50 (m, 1H), 0.44-0.38 (m, 3H). 13C NMR (500 MHz, CDCl3) δ = 202.5, 174.5, 173.9, 137.4, 137.0, 134.4, 132.9, 128.7, 128.3, 63.8, 54.0, 52.6, 52.2, 51.8, 51.8, 45.1, 42.7, 15.9, 9.3, 5.4. HRMS (ESI/TOF) calcd for C22H25O5+ (MH+) 369.1697, found 369.1693.
Dimethyl (1RS, 4SR, 5RS, 6SR)-endo-5-((1′RS)-1′-benzoyl-2′-phenylethyl)bicyclo[2.2.1]hept-2-ene-exo-5-endo-6-dicarboxylate (7f)
(procedure B) from 0.80 mL of LDA (1.44 mmol), 0.20 g of 3-phenylpropiophenone (β-phenylpropiophenone) (0.95 mmol) and 0.20 g of 2 (0.96 mmol) (hexane/EtOAc gradient 30:1 → 20:1): 47.7 mg (12 %). 1H NMR (500 MHz, CDCl3) δ = 7.37 (m, 3H), 7.22 (m, 2H), 7.09 (m, 4H), 7.01 (m, 1H), 6.25 (dd, J = 5.7, 2.9 Hz, 1H), 5.46 (dd, J = 5.7, 3.2 Hz, 1H), 4.06 (dd, J = 12.0, 2.3 Hz, 1H), 3.91 (s, 3H), 3.81 (s, 3H), 3.45 (d, J = 3.3 Hz, 1H), 3.04 (m, 2H), 2.93 (dd, J = 13.5, 12.0 Hz, 1H), 2.74 (dd, J = 13.5, 2.3 Hz, 1H), 1.47 (ddd, J = 9.1, 1.8, 1.8 Hz, 1H), 1.35 (d, J = 9.1 Hz, 1H). 1H NMR (500 MHz, C6D6) δ = 7.58 (m, 2H), 7.40 (m, 2H), 6.98 (m, 2H), 6.90-6.80 (m, 4H), 6.30 (dd, J = 5.6, 3.0 Hz, 1H), 5.64 (dd, J = 5.6, 3.1 Hz, 1H), 4.43 (dd, J = 11.9, 2.3 Hz, 1H), 3.67 (s, 3H), 3.57 (d, J = 3.1 Hz, 1H), 3.48 (dd, J = 13.5, 11.9 Hz, 1H), 3.39 (s, 3H), 3.25 (m, 1H), 3.12 (dd, J = 13.5, 2.3 Hz, 1H), 2.70 (m, 1H), 1.31 (ddd, J = 9.1, 1.8, 1.8 Hz, 1H), 1.24 (d, J = 9.1 Hz, 1H). 13C NMR (500 MHz, CDCl3) δ = 203.8, 175.3, 173.9, 139.3, 138.8, 137.3, 134.4, 132.4, 129.2, 128.3, 128.2, 128.1, 126.8, 63.1, 52.3, 52.0, 51.7, 50.9, 50.0, 49.7, 47.1, 37.6. See SI for xray data.
Dimethyl (1RS, 4SR, 5RS, 6SR)-endo-5-((1′SR)-1′-benzoyl-2′-phenylethyl)bicyclo[2.2.1]hept-2-ene-exo-5-endo-6-dicarboxylate (7f’)
119 mg (30 %). 1H NMR (500 MHz, CDCl3) δ = 7.33 (m, 2H), 7.25 (m, 1H), 7.06 (m, 2H), 7.00-6.93 (m, 5H), 6.69 (dd, J = 5.6, 3.0 Hz, 1H), 6.46 (dd, J = 5.6, 2.9 Hz, 1H), 4.19 (d, J = 3.4 Hz, 1H), 3.98 (s, 3H), 3.68 (dd, J = 11.1, 3.6 Hz, 1H), 3.52 (m, 1H), 3.39 (dd, J = 13.6, 3.6 Hz, 1H), 3.21 (dd, J = 13.6, 11.1 Hz, 1H), 3.06 (m, 1H), 2.60 (s, 3H), 1.49 (ddd, J = 9.1, 1.8, 1.8 Hz, 1H), 1.43 (d, J = 9.1 Hz, 1H). 1H NMR (500 MHz, C6D6) δ = 7.54 (m, 2H), 6.94 (m, 2H), 6.85-6.75 (m, 6H) overlaps with 6.74 (dd, J = 5.6, 3.0 Hz, 1H), 6.31 (dd, J = 5.6, 2.9 Hz, 1H), 4.42 (d, J = 3.4 Hz, 1H), 3.95 (dd, J = 11.0, 3.9 Hz, 1H), 3.76 (s, 3H), 3.48 (dd, J = 13.6, 11.0 Hz, 1H) overlaps with 3.46 (m, 1H), 3.37 (dd, J = 13.6, 3.9 Hz, 1H), 2.81 (m, 1H), 2.46 (s, 3H), 1.40 (d, J = 8.9 Hz, 1H), 1.32 (ddd, J = 8.9, 1.8, 1.8 Hz, 1H). 13C NMR (500 MHz, CDCl3) δ = 201.7, 176.0, 173.6, 140.4, 139.0, 136.8, 132.4, 132.0, 129.3, 128.6, 128.1, 127.6, 126.3, 62.7, 52.7, 52.7, 52.3, 50.8, 50.2, 46.9, 46.5, 39.1. See SI for xray data.
Dimethyl exo-5-(1′-benzoyl-2′-phenylethyl)bicyclo[2.2.1]hept-2-ene-endo-5-exo-6-dicarboxylate (7f-exo)
71.5 mg (18 %). 1H NMR (500 MHz, CDCl3) δ = 7.51 (m, 2H), 7.34 (m, 1H), 7.18 (m, 2H), 7.11-7.02 (m, 5H), 6.32 (dd, J = 5.6, 3.0 Hz, 1H), 6.06 (dd, J = 5.6, 3.0 Hz, 1H), 4.22 (dd, J = 9.2, 4.3 Hz, 1H), 3.80 (s, 3H), 3.65 (dd, J = 14.3, 4.3 Hz, 1H), 3.47 (m, 1H), 3.37 (d, J = 2.0 Hz, 1H), 3.05 (dd, J = 14.3, 9.2 Hz, 1H), 2.97 (m, 1H), 2.74 (s, 3H), 2.36 (d, J = 9.3 Hz, 1H), 1.58 (dddd, J = 9.3, 1.8, 1.8, 1.8 Hz, 1H) overlaps with HOD. 1H NMR (500 MHz, C6D6) δ = 7.68 (m, 2H), 7.01 (m, 2H), 6.91-6.82 (m, 6H), 6.05 (m, 2H), 4.51 (dd, J = Hz, 1H), 3.68-3.62 (m, 2H), 3.59 (s, 3H), 3.45 (m, 1H), 3.31 (dd, J = 14.0, 9.6 Hz, 1H), 2.83 (m, 1H), 2.61 (s, 3H), 2.56 (d, J = 9.2 Hz, 1H), 1.51 (dddd, J = 9.2, 1.8, 1.8, 1.8 Hz, 1H). 13C NMR (500 MHz, CDCl3) δ = 201.3, 175.2, 174.7, 139.7, 139.5, 136.4, 136.0, 132.7, 129.2, 128.9, 128.4, 127.8, 126.3, 59.9, 52.1, 51.9, 51.1, 49.0, 48.9, 47.8, 45.9, 38.0. HRMS (ESI/TOF) calcd for C26H27O5+ (MH+) 419.1853, found 419.1849. See SI for xray data.
Dimethyl (1RS, 4RS, 5RS, 6SR)-spiro[(1″RS)-endo-5-(1″-benzoyl-2″-phenylethyl)bicyclo[2.2.1]hept-2-ene-exo-5-endo-6-dicarboxylate-7,1′-cyclopropane] (7g)
(procedure B) from 1.55 mL of LDA (2.79 mmol), 0.45 g of 3-phenylpropiophenone (2.14 mmol) and 0.50 g of 4 (2.13 mmol) (hexane/EtOAc gradient 30:1 → 5:1): 0.17 g (18 %). 1H NMR (500 MHz, CDCl3) δ = 7.38 (m, 3H), 7.25 (m, 2H), 7.10 (m, 4H), 7.02 (m, 1H), 6.33 (dd, J = 5.8, 3.0 Hz, 1H), 5.46 (ddd, J = 5.8, 3.2, 0.9 Hz, 1H), 4.17 (dd, J = 12.0, 2.4 Hz, 1H), 3.89 (s, 3H), 3.81 (s, 3H), 3.80 (d, J = 3.5 Hz, 1H), 2.85 (dd, J = 13.5, 12.0 Hz, 1H), 2.71 (dd, J = 13.5, 2.4 Hz, 1H), 2.60 (m, 1H), 2.40 (m, 1H), 0.49-0.33 (m, 4H). 1H NMR (500 MHz, C6D6) δ = 7.62 (m, 2H), 7.45 (m, 2H), 7.01 (m, 3H), 6.86 (m, 3H), 6.42 (ddd, J = 5.8, 3.0, 0.8 Hz, 1H), 5.76 (dd, J = 5.8, 3.1, 0.9 Hz, 1H), 4.58 (dd, J = 12.0, 2.2 Hz, 1H), 4.02 (d, J = 3.5 Hz, 1H), 3.66 (s, 3H), 3.44 (dd, J = 13.5, 12.0 Hz, 1H) overlaps with 3.42 (s, 3H), 3.13 (dd, J = 13.5, 2.2 Hz, 1H), 2.82 (m, 1H), 2.04 (m, 1H), 0.52 (m, 1H), 0.23-0.16 (m, 3H). 13C NMR (500 MHz, CDCl3) δ = 203.3, 174.1, 173.8, 139.4, 139.2, 137.0, 134.7, 132.3, 129.2, 128.3, 128.2, 127.9, 126.2, 64.5, 53.5, 52.8, 52.0, 51. 9, 51.5, 51.1, 45.6, 36.9, 9.5, 5.4. HRMS (ESI/TOF) calcd for C28H28NaO5+ (MNa+) 467.1829, found 467.1812. See SI for xray data.
Dimethyl (1RS, 4RS, 5RS, 6SR)-spiro[endo-5-((1″SR)-1″-benzoyl-2″-phenylethyl)bicyclo[2.2.1]hept-2-ene-exo-5-endo-6-dicarboxylate-7,1′-cyclopropane] (7g’)
0.22 g (23 %). 1H NMR (500 MHz, CDCl3) δ = 7.35 (m, 2H), 7.25 (m, 1H), 7.08 (m, 2H), 6.99-6.93 (m, 5H), 6.78 (dd, J = 5.8, 3.0 Hz, 1H), 6.58 (ddd, J = 5.8, 3.0, 0.9 Hz, 1H), 4.33 (d, J = 3.5 Hz, 1H), 3.96 (s, 3H), 3.75 (dd, J = 11.2, 3.7 Hz, 1H), 3.33 (dd, J = 13.5, 3.7 Hz, 1H), 3.20 (dd, J = 13.5, 11.2 Hz, 1H), 3.07 (m, 1H), 2.59 (s, 3H), 2.46 (m, 1H), 0.53-0.41 (m, 4H). 13C NMR (500 MHz, CDCl3) δ = 201.7, 175.4, 173.6, 140.6, 139.0, 137.2, 132.6, 132.4, 129.6, 128.8, 128.3, 127.8, 126.5, 64.3, 57.0, 52.6 overlaps with 52.6, 52.5, 51.3, 51.0, 43.0, 39.5, 9.6, 4.9. See SI for xray data.
Dimethyl (1RS, 4RS, 5RS, 6SR)-spiro[(1″RS)-endo-5-(1″-benzoylbutyl)bicyclo[2.2.1]hept-2-ene-exo-5-endo-6-dicarboxylate-7,1′-cyclopropane] (7h)
(procedure B) from 0.57 mL of LDA (1.03 mmol), 0.14 mL of valerophenone (0.85 mmol) and 0.20 g of 4 (0.85 mmol) (hexane/EtOAc gradient 50:1 → 10:1): 118 mg (35 %). 1H NMR (500 MHz, CDCl3) δ = 7.88 (d, J = 7.1 Hz, 2H), 7.58 (t, J = 7.4 Hz, 1H), 7.48 (t, J = 7.6 Hz, 2H), 6.34 (ddd, J = 5.9, 3.0, 0.7 Hz, 1H), 5.65 (ddd, J = 5.7, 3.2, 0.9 Hz, 1H), 3.89 (dd, J = 11.2, 2.0 Hz, 1H), 3.81 (s, 3H), 3.77 (d, J = 3.4 Hz, 1H), 3.72 (s, 3H), 2.62 (m, 1H), 2.35 (m, 1H), 1.64-1.58 (m, 1H) overlaps with HOD, 1.37-1.30 (m, 1H), 1.11 (m, 1H), 1.01 (m, 1H), 0.78 (t, J = 7.3 Hz, 3H), 0.43-0.31 (m, 4H). 1H NMR (500 MHz, C6D6) δ = 8.03 (m, 2H), 7.09 (m, 1H), 7.03 (m, 2H), 6.43 (ddd, J = 5.9, 3.0, 0.8 Hz, 1H), 5.89 (dd, J = 5.8, 3.2, 0.8 Hz, 1H), 4.25 (dd, J = 11.0, 1.8 Hz, 1H), 4.03 (d, J = 3.6 Hz, 1H), 3.60 (s, 3H), 3.38 (s, 3H), 2.82 (m, 1H), 2.13 (dddd, J = 13.8, 11.0, 11.0, 4.7 Hz, 1H), 2.03 (m, 1H), 1.69 (dddd, J = 13.8, 12.0, 5.1, 1.7 Hz, 1H), 1.39-1.29 (m, 1H), 1.22-1.13 (m, 1H), 0.79 (t, J = 7.3 Hz, 3H), 0.48-0.39 (m, 4H).
Photoinactive exo-compounds 7i – 7q
Dimethyl exo-5-phenacylbicyclo[2.2.1]hept-2-ene-bis-endo-5,6-dicarboxylate (7i)
(procedure A) from 0.13 mL of diisopropylamine (0.93 mmol), 0.50 mL of n-BuLi (0.80 mmol), 0.08 mL of acetophenone (0.68 mmol) and 0.14 g of 2 (0.67 mmol) (hexane/EtOAc gradient 30:1 → 10:1): 57 mg (26 %) colorless crystals, m.p. 142.5-144°C. 1H NMR (500 MHz, CDCl3) δ = 7.92 (d, J = 7.1 Hz, 2H), 7.56 (t, J = 7.4 Hz, 1H), 7.45 (t, J = 7.7 Hz, 2H), 6.35 (dd, J = 5.7, 3.1 Hz, 1H), 6.02 (dd, J = 5.6, 2.9 Hz, 1H), 3.98 (d, J = 18.1 Hz, 1H), 3.75 (d, J = 18.1 Hz, 1H), 3.62 (s, 3H), 3.43 (s, 3H) overlaps with 3.43 (m, 1H), 3.00 (m, 1H), 2.94 (m, 1H), 2.26 (d, J = 9.2 Hz, 1H), 1.56 (ddd, J = 9.2, 1.7, 1.7 Hz, 1H) overlaps with HOD. 1H NMR (500 MHz, C6D6) δ = 7.86 (d, J = 7.1 Hz, 2H), 7.09 (t, J = 7.3 Hz, 1H), 7.02 (t, J = 7.5 Hz, 2H), 6.09 (dd, J = 5.6, 3.1 Hz, 1H), 5.94 (dd, J = 5.6, 2.9 Hz, 1H), 4.08 (d, J = 18.0 Hz, 1H), 3.81 (d, J = 1.8 Hz, 1H), 3.68 (d, J = 18.0 Hz, 1H), 3.32 (s, 3H), 3.17 (s, 3H), 2.82 (m, 1H) overlaps with 2.80 (m, 1H), 2.36 (d, J = 9.0 Hz, 1H), 1.44 (dddd, J = 9.0, 1.7, 1.7, 1.7 Hz, 1H). 13C NMR (500 MHz, CDCl3) δ = 198.5, 175.5, 174.7, 138.7, 136.4, 134.8, 133.2, 128.6, 128.0, 53.9, 52.8, 52.3, 51.8, 48.6, 47.0, 46.4, 45.8. HRMS (ESI/TOF) calcd for C19H20NaO5+ (MNa+) 351.1203, found 351.1196.
Dimethyl exo-5-phenacylbicyclo[2.2.1]hept-2-ene-bis-endo-5,6-dicarboxylate (7i’)
33 mg (15 %). 1H NMR (500 MHz, CDCl3) δ = 7.87 (d, J = 7.2 Hz, 2H), 7.53 (t, J = 7.4 Hz, 1H), 7.43 (t, J = 7.6 Hz, 2H), 6.34 (m, 2H), 4.08 (d, J = 3.4 Hz, 1H), 3.70 (s, 3H), 3.55 (s, 2H), 3.45 (s, 3H), 3.17 (m, 1H), 3.10 (m, 1H), 1.79 (d, J = 8.9 Hz, 1H), 1.44 (ddd, J = 8.9, 1.7, 1.7 Hz, 1H). 1H NMR (500 MHz, C6D6) δ = 7.89 (d, J = 7.0 Hz, 2H), 7.08 (t, J = 7.3 Hz, 1H), 7.02 (t, J = 7.5 Hz, 2H), 6.28 (dd, J = 5.6, 2.9 Hz, 1H), 6.10 (dd, J = 5.7, 3.2 Hz, 1H), 4.41 (d, J = 3.2 Hz, 1H), 3.76 (d, J = 18.0 Hz, 1H), 3.50 (dd, J = 18.0 Hz, 1H), 3.38 (s, 3H), 3.16 (s, 3H), 2.97 (m, 2H), 1.89 (d, J = 8.8 Hz, 1H), 1.28 (ddd, J = 8.8, 1.7, 1.7 Hz, 1H). 13C NMR (500 MHz, CDCl3) δ = 198.9, 176.1, 173.9, 138.1, 136.5, 134.8, 133.0, 128.5, 127.9, 55.5, 52.9, 52.6, 51.5, 48.4, 47.5, 46.8, 43.9. HRMS (ESI/TOF) calcd for C19H20NaO5+ (MNa+) 351.1203, found 351.1209.
Methyl spiro[exo-6-phenacylbicyclo[2.2.1]hept-2-ene-endo-5-carboxylate-7,1′-cyclopropane] (7j)
21.9 mg (13 %). 1H NMR (500 MHz, CDCl3) δ = 8.00 (d, J = 7.1 Hz, 2H), 7.59 (t, J = 7.4 Hz, 1H), 7.49 (t, J = 7.6 Hz, 2H), 6.43 (ddd, J = 5.9, 3.2, 0.7 Hz, 1H), 6.15 (ddd, J = 5.8, 2.7, 0.7 Hz, 1H), 3.66 (s, 3H), 3.42 (dd, J = 16.2, 6.4 Hz, 1H), 3.30 (dd, J = 16.2, 8.7 Hz, 1H), 2.86 (dd, J = 4.8, 3.6 Hz, 1H), 2.56 (m, 1H), 2.47 (ddd, J = 8.7, 6.4, 5.0 Hz, 1H), 2.21 (m, 1H), 0.72 (ddd, J = 9.6, 5.3, 5.3 Hz, 1H), 0.65 (ddd, J = 9.4, 5.5, 5.5 Hz, 1H), 0.44 (ddd, J = 9.5, 5.2, 5.2 Hz, 1H), 0.38 (ddd, J = 9.5, 5.4, 5.4 Hz, 1H). 1H NMR (500 MHz, C6D6) δ = 7.94 (m, 2H), 7.13 (m, 1H), 7.08 (m, 2H), 6.28 (ddd, J = 5.8, 3.2, 0.7 Hz, 1H), 6.19 (ddd, J = 5.8, 2.8, 0.7 Hz, 1H), 3.37 (s, 3H), 3.17-3.12 (m, 1H), 2.99-2.93 (m, 1H), 2.73-2.69 (m, 2H), 2.38 (m, 1H), 2.06 (m, 1H), 0.41-0.36 (m, 1H), 0.28-0.16 (m, 3H). 13C NMR (500 MHz, CDCl3) δ = 199.49, 174.41, 138.51, 136.85, 133.51, 133.03, 128.61, 128.14, 51.58, 51.48, 51.26, 51.14, 43.16, 42.64, 41.68, 9.27, 3.34. HRMS (ESI/TOF) calcd for C19H21O3+ (MH+) 297.1485, found 297.1492.
Methyl exo-6-(1′-benzoylethyl)bicyclo[2.2.1]hept-2-ene-endo-5-carboxylate (7k)
(procedure A) from 0.33 mL of diisopropylamine (2.35 mmol), 1.70 mL of n-BuLi (2.72 mmol), 0.24 mL of propiophenone (1.80 mmol) and 0.27 g of 5 (1.80 mmol) (hexane/EtOAc gradient 30:1→10:1): 0.27 g (53 %). 1H NMR (500 MHz, CDCl3) δ = 7.97 (d, J = 7.1 Hz, 2H), 7.58 (t, J = 7.4 Hz, 1H), 7.49 (t, J = 7.6 Hz, 2H), 6.27 (dd, J = 5.6, 3.2 Hz, 1H), 6.00 (dd, J = 5.6, 2.8 Hz, 1H), 3.65 (s, 3H), 3.39 (dddd, J = 10.8, 7.0, 7.0, 7.0 Hz, 1H), 3.19 (m, 1H), 2.66 (dd, J = 4.6, 3.7 Hz, 1H), 2.46 (m, 1H), 2.33 (ddd, J = 10.8, 4.8, 1.6 Hz, 1H), 1.54 (d, J = 8.7 Hz, 1H), 1.44 (dddd, J = 8.7, 1.7, 1.7, 1.7 Hz, 1H), 1.27 (d, J = 7.0 Hz, 3H). 1H NMR (500 MHz, C6D6) δ = 7.89 (d, J = 7.0 Hz, 2H), 7.15 (m, 1H), 7.09 (t, J = 7.3 Hz, 2H), 6.12 (dd, J = 5.6, 3.2 Hz, 1H), 6.03 (dd, J = 5.6, 2.8 Hz, 1H), 3.34 (s, 3H), 3.10 (m, 1H), 3.04 (dddd, J = 10.9, 7.0, 7.0, 7.0 Hz, 1H), 2.68 (ddd, J = 10.9, 4.7, 1.6 Hz, 1H), 2.44 (m, 2H), 1.25 (dddd, J = 8.7, 1.7, 1.7, 1.7 Hz, 1H), 1.20 (d, J = 7.0 Hz, 3H) overlaps with 1.20 (m, 1H). 13C NMR (500 MHz, CDCl3) δ = 204.1, 174.6, 138.8, 137.1, 133.5, 133.1, 128.8, 128.2, 51.7, 49.1, 46.6, 46.5, 46.5, 46.1, 45.7, 17.7. HRMS (ESI/TOF) calcd for C18H21O3+ (MH+) 285.1485, found 285.1492.
Dimethyl exo-5-(1′-indanon-2′-yl)bicyclo[2.2.1]hept-2-ene-bis-endo-5,6-dicarboxylate (7l)
(procedure A) from 0.26 mL of diisopropylamine (1.86 mmol), 1.35 mL of n-BuLi (2.16 mmol), 0.20 mL of 1-indanone (1.51 mmol) and 0.30 g of 2 (1.44 mmol) (hexane/EtOAc gradient 30:1 → 5:1): 0.14 g (28 %). 1H NMR (500 MHz, CDCl3) δ = 7.76 (d, J = 7.7 Hz, 1H), 7.57 (ddd, J = 7.5, 7.5, 1.1 Hz, 1H), 7.45 (d, J = 7.7 Hz, 1H), 7.36 (t, J = 7.5 Hz, 1H), 6.50 (dd, J = 5.5, 3.0 Hz, 1H), 6.06 (dd, J = 5.5, 3.2 Hz, 1H), 4.22 (m, 1H), 3.70 (s, 3H), 3.51 (dd, J = 17.7, 4.1 Hz, 1H), 3.43 (dd, J = 17.7, 7.9 Hz, 1H), 3.16 (s, 3H), 3.13 (m, 1H), 3.00 (d, J = 3.1 Hz, 1H), 2.91 (dd, J = 7.9, 4.1 Hz, 1H), 1.79 (d, J = 9.4 Hz, 1H), 1.63 (ddd, J = 9.4, 1.7, 1.7 Hz, 1H). 1H NMR (500 MHz, C6D6) δ = 7.83 (d, J = 7.6 Hz, 1H), 7.09 (ddd, J = 7.4, 7.4, 1.2 Hz, 1H), 7.03 (d, J = 7.6 Hz, 1H), 6.94 (t, J = 7.4 Hz, 1H), 6.62 (dd, J = 5.5, 3.0 Hz, 1H), 6.10 (d, J = 5.5, 3.1 Hz, 1H), 4.53 (m, 1H), 3.57 (dd, J = 17.5, 4.1 Hz, 1H), 3.46 (s, 3H), 2.99 (dd, J = 17.5, 8.1 Hz, 1H), 2.87 (m, 1H), 2.81 (s, 3H), 2.59 (d, J = 3.1 Hz, 1H), 2.44 (dd, J = 8.1, 4.1 Hz, 1H), 1.45 (ddd, J = 9.2, 1.8, 1.8 Hz, 1H), 1.39 (d, J = 9.2 Hz, 1H). 13C NMR (500 MHz, CDCl3) δ = 204.7, 173.4, 172.1, 152.4, 138.2, 137.3, 134.5, 134.3, 127.4, 126.4, 123.7, 63.3, 55.9, 54.3, 51.8, 51.3, 48.5, 46.2, 46.2, 31.7. HRMS (ESI/TOF) calcd for C20H21O5+ (MH+) 341.1384, found 341.1390.
Dimethyl exo-5-(1′-indanon-2′-yl)bicyclo[2.2.1]hept-2-ene-bis-endo-5,6-dicarboxylate (7l’)
trace amount. 1H NMR (500 MHz, CDCl3) δ = 7.77 (d, J = 7.7 Hz, 1H), 7.55 (ddd, J = 7.5, 7.5, 1.2 Hz, 1H), 7.41 (d, J = 7.7 Hz, 1H), 7.36 (t, J = 7.6 Hz, 1H), 6.54 (dd, J = 5.5, 3.1 Hz, 1H), 6.09 (dd, J = 5.5, 3.1 Hz, 1H), 4.27 (d, J = 3.4 Hz, 1H), 3.64 (s, 3H), 3.33 (dd, J = 17.5, 8.2 Hz, 1H), 3.17 (m, 1H), 3.12 (s, 3H), 3.09 (m, 1H), 2.96 (dd, J = 17.5, 4.1 Hz, 1H), 2.85 (dd, J = 8.2, 4.1 Hz, 1H), 1.77 (d, J = 9.3 Hz, 1H), 1.55 (ddd, J = 9.3, 1.7, 17 Hz, 1H). 13C NMR (500 MHz, CDCl3) δ = 205.1, 173.3, 172.7, 151.5, 137.7, 137.3, 134.4, 133.7, 127.5, 126.2, 123.8, 63.2, 52.7, 51.8, 51.6, 51.0, 50.9, 47.8, 46.7, 31.3. HRMS (ESI/TOF) calcd for C20H21O5+ (MH+) 341.1384, found 341.1390.
Dimethyl (1RS, 4SR, 5SR, 6RS)-exo-5-((2′SR)-1′-indanon-2′-yl)bicyclo[2.2.1]hept-2-ene-endo-5-exo-6-dicarboxylate (7m)
58.8 mg (12 %). 1H NMR (500 MHz, CDCl3) δ = 7.74 (d, J = 7.8 Hz, 1H), 7.53 (ddd, J = 7.5, 7.5, 1.1 Hz, 1H), 7.35 (m, 2H), 6.27 (dd, J = 5.6, 3.1 Hz, 1H), 6.02 (dd, J = 5.6, 3.1 Hz, 1H), 4.22 (m, 1H), 3.77 (m, 3H), 3.25-3.19 (m, 2H), 3.07-2.97 (m, 5H), 2.84 (dd, J = 17.5, 2.7 Hz, 1H), 2.53 (d, J = 9.5 Hz, 1H), 1.67 (dddd, J = 9.5, 1.7, 1.7, 1.7 Hz, 1H). 13C NMR (500 MHz, CDCl3) δ = 175.1, 173.3, 151.8, 139.1, 137.4, 136.8, 134.4, 127.5, 126.4, 123.5, 52.0, 51.7, 51.3, 49.1, 49.0, 48.2, 45.7, 45.0. HRMS (ESI/TOF) calcd for C20H21O5+ (MH+) 341.1384, found 341.1386.
Dimethyl (1SR, 4RS, 5RS, 6RS)-exo-5-((2′SR, 3′RS)-3′-methyl-1′-indanon-2′-yl)bicyclo[2.2.1]hept-2-ene-bis-endo-5,6-dicarboxylate (7n)
(procedure B) from 0.60 mL of LDA (1.08 mmol), 0.10 mL of 3-methyl-1-indanone (0.74 mmol) and 0.15 g of 2 (0.72 mmol) (hexane/EtOAc gradient 30:1 → 20:1): 66.3 mg (26 %). 1H NMR (500 MHz, CDCl3) δ = 7.73 (d, J = 7.7 Hz, 1H), 7.57 (ddd, J = 7.5, 7.5, 1.3 Hz, 1H), 7.41 (d, J = 7.7 Hz, 1H), 7.36 (t, J = 7.5 Hz, 1H), 6.53 (dd, J = 5.5, 3.2 Hz, 1H), 6.10 (dd, J = 5.5, 3.0 Hz, 1H), 4.21 (d, J = 3.4 Hz, 1H), 3.63 (s, 3H), 3.30 (dddd, J = 7.0, 7.0, 7.0, 3.0 Hz, 1H), 3.23 (m, 1H), 3.16 (m, 1H), 3.04 (s, 3H), 2.41 (d, J = 3.0 Hz, 1H), 1.79 (d, J = 9.3 Hz, 1H), 1.57 (ddd, J = 9.3, 1.7, 1.7 Hz, 1H) overlaps with HOD, 1.41 (d, J = 7.0 Hz, 3H). 13C NMR (500 MHz, CDCl3) δ = 205.2, 173.3, 172.7, 157.0, 137.1, 136.8, 134.6, 134.2, 127.5, 125.1, 123.5, 63.8, 61.0, 51.8, 51.5, 50.8, 50.3, 47.8, 46.4, 38.4, 23.1. HRMS (ESI/TOF) calcd for C21H23O5+ (MH+) 355.1540, found 355.1548.
Dimethyl exo-5-(3′-methyl-1′-indanon-2′-yl)bicyclo[2.2.1]hept-2-ene-endo-5,exo-6-dicarboxylate (7n’)
51 g (20 %). 1H NMR (500 MHz, CDCl3) δ = 7.69 (d, J = 7.6 Hz, 1H), 7.53 (m, 1H), 7.39-7.33 (m, 2H), 6.22 (m, 1H), 6.01 (m, 1H), 3.82 (m, 4H), 3.20 (m, 1H) overlaps with 3.14 (m, 1H), 2.95 (m, 1H), 2.88 (m, 1H), 2.74 (m, 3H), 2.59 (m, 1H), 1.64 (dddd, J = 9.4, 1.8, 1.8, 1.8 Hz, 1H), 1.15 (m, 3H). 13C NMR (500 MHz, CDCl3) δ = 174.9, 173.2, 158.2, 138.9, 137.6, 136.0, 134.5, 127.6, 125.7, 125.6, 123.2, 63.9, 58.1, 52.1, 51.0, 49.1, 48.7, 46.3, 45.4, 38.7, 24.5. HRMS (ESI/TOF) calcd for C21H23O5+ (MH+) 355.1540, found 355.1542.
Dimethyl (1SR, 4RS, 5RS, 6RS)-exo-5-((2′SR, 3′SR)-3′-phenyl-1′-indanon-2′-yl)bicyclo[2.2.1]hept-2-ene-endo-bis-5,6-dicarboxylate (7o)
(procedure A) from 0.10 mL of diisopropylamine (0.71 mmol), 0.50 mL of t-BuLi (1.7 M, 0.85 mmol), 0.10 mL of 3-phenyl-1-indanone (0.48 mmol) and 0.10 g of 2 (0.48 mmol) at -78°C → 20°C gradient (hexane/EtOAc gradient 30:1 → 4:1): 96 mg (48 %), colorless crystals, m.p. 168.5-170.0°C. 1H NMR (500 MHz, CDCl3) δ = 7.80 (d, J = 7.7 Hz, 1H), 7.50 (ddd, J = 7.5, 7.5, 1.3 Hz, 1H), 7.38 (t, J = 7.5 Hz, 1H), 7.32-7.28 (m, 2H), 7.23 (m, 1H), 7.11 (m, 3H), 6.48 (dd, J = 5.5, 3.1 Hz, 1H), 6.08 (dd, J = 5.5, 3.1 Hz, 1H), 4.41 (d, J = 3.8 Hz, 1H), 4.18 (d, J = 3.4 Hz, 1H), 3.66 (s, 3H), 3.16 (m, 1H), 3.14 (s, 3H), 3.09 (m, 1H), 2.98 (d, J = 3.8 Hz, 1H), 1.48 (d, J = 9.4 Hz, 1H), 1.31 (ddd, J = 9.4, 1.8, 1.8 Hz, 1H). 1H NMR (500 MHz, C6D6) δ = 7.86 (d, J = 7.6 Hz, 1H), 7.01-6.95 (m, 4H), 6.91 (t, J = 7.4 Hz, 1H), 6.84 (m, 2H), 6.80 (d, J = 7.6 Hz, 1H), 6.70 (dd, J = 5.5, 3.1 Hz, 1H), 6.14 (dd, J = 5.5, 3.0 Hz, 1H), 4.55 (d, J = 3.9 Hz, 1H), 4.37 (d, J = 3.4 Hz, 1H), 3.40 (s, 3H), 3.27 (m, 1H), 3.01 (s, 3H), 2.94 (d, J = 3.9 Hz, 1H), 2.87 (m, 1H), 1.16 (d, J = 9.3 Hz, 1H), 1.06 (ddd, J = 9.3, 1.7, 1.7 Hz, 1H). 13C NMR (500 MHz, CDCl3) δ = 205.0, 173.3, 172.8, 155.6, 143.8, 137.1, 136.7, 135.0, 134.2, 129.1, 127.9, 127.8, 127.1, 126.4, 123.3, 63.9, 62.2, 51.9, 51.8, 51.0, 50.4, 50.2, 47.6, 46.5. HRMS (ESI/TOF) calcd for C26H25O5+ (MH+) 417.1697, found 417.1679.
Dimethyl (1RS, 4SR, 5SR, 6RS)-exo-5-((2′SR, 3′SR)-3′-phenyl-1′-indanon-2′-yl)bicyclo[2.2.1]hept-2-ene-endo-5-exo-6-dicarboxylate (7o’)
56 mg (28 %), colorless crystals, m.p. 151.0-154.5°C. 1H NMR (500 MHz, CDCl3) δ = 7.73 (d, J = 7.6 Hz, 1H), 7.43 (t, J = 7.4 Hz, 1H), 7.32 (t, J = 7.4 Hz, 1H), 7.23 (m, 3H), 7.15 (m, 1H), 7.06 (m, 2H), 6.24 (dd, J = 5.5, 3.1 Hz, 1H), 6.00 (m, 1H), 4.32 (m, 1H), 3.95 (m, 1H), 3.57 (m, 1H), 3.22 (m, 1H), 3.03 (m, 4H), 2.92 (m, 1H), 2.84 (m, 2H), 2.56 (m, 1H), 1.61 (m, 1H). HRMS (ESI/TOF) calcd for C26H25O5+ (MH+) 417.1697, found 417.1684.
Methyl (1RS, 4SR, 5RS, 6SR)-exo-6-((2′RS)-1′-indanon-2′-yl)bicyclo[2.2.1]hept-2-ene-endo-5-carboxylate (7p)
(procedure A) from 0.40 mL of diisopropylamine (2.85 mmol), 2.0 mL of n-BuLi (3.2 mmol), 0.18 mL of 1-indanone (1.36 mmol) and 0.20 g of 5 (1.33 mmol) (hexane/EtOAc gradient 30:1 → 2:1): 52.5 mg (14 %), colorless crystals, m.p. 99.5-102.0°C. 1H NMR (500 MHz, CDCl3) δ = 7.77 (d, J = 7.6 Hz, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.62 (m, 1H), 7.42 (t, J = 7.3 Hz, 1H), 6.24 (dd, J = 5.6, 3.3 Hz, 1H), 6.02 (d, J = 5.7, 2.8 Hz, 1H), 3.61 (s, 3H), 3.40 (ddd, J = 9.9, 7.5, 2.8 Hz, 1H), 3.23 (m, 1H), 3.09 (m, 1H), 2.88 (dd, J = 10.0, 7.5 Hz, 1H), 2.71 (dd, J = 4.6, 3.6 Hz, 1H), 2.67 (dd, J = 18.6, 2.9 Hz, 1H), 2.03 (ddd, J = 9.5, 4.7, 1.6 Hz, 1H), 1.67 (d, J = 8.9 Hz, 1H), 1.55 (dddd, J = 8.9, 1.7, 1.7, 1.7 Hz, 1H). 13C NMR (500 MHz, CDCl3) δ = 205.5, 174.5, 157.1, 138.3, 137.4, 134.4, 134.3, 127.9, 127.1, 123.9, 51.8, 50.0, 49.3, 46.2, 46.0, 45.8, 43.3, 43.0. HRMS (ESI/TOF) calcd for C18H22NO3+ (MNH4+) 300.1594, found 300.1583.
Dimethyl exo-5-(1′-benzoyl-1′-phenylmethyl)bicyclo[2.2.1]hept-2-ene-endo-5-exo-6-dicarboxylate (7q)
(procedure B) from 1.04 mL of LDA (1.87 mmol), 0.28 g of 2-phenylacetophenone (deoxybenzoin) (1.43 mmol) and 0.30 g of 2 (1.44 mmol) (hexane/EtOAc gradient 30:1 → 5:1): 0.13 g (23 %). 1H NMR (500 MHz, CDCl3) δ = 7.84 (m, 2H), 7.43 (m, 1H), 7.34-7.27 (m, 7H) overlaps with CDCl3, 6.32 (dd, J = 5.6, 3.1 Hz, 1H), 5.88 (dd, J = 5.6, 3.0 Hz, 1H), 5.16 (s, 1H), 3.73 (s, 3H), 3.54 (d, J = 2.0 Hz, 1H), 3.09 (s, 3H), 2.95 (m, 1H), 2.76 (m, 1H), 2.48 (d, J = 9.2 Hz, 1H), 1.64 (dddd, J = 9.2, 1.8, 1.8, 1.8 Hz, 1H). 13C NMR (500 MHz, CDCl3) δ = 196.5, 175.7, 174.4, 139.8, 135.9, 135.7, 135.1, 133.0, 130.9, 128.9, 128.5, 128.3, 127.9, 59.2, 57.7, 51.6, 51.6, 50.3, 49.4, 47.5, 46.3. HRMS (ESI/TOF) calcd for C25H25O5+ (MH+) 405.1697, found 405.1687.
Dimethyl exo-5-(1′-benzoyl-1′-phenylmethyl)bicyclo[2.2.1]hept-2-ene-bis-endo-5,6-dicarboxylate (7q’)
(procedure B, same reaction as for 7q) 81.5 mg (14 %). 1H NMR (500 MHz, CDCl3) δ = 7.78 (m, 2H), 7.38 (m, 1H), 7.33-7.21 (m, 7H) overlaps with CDCl3, 6.57 (m, 2H), 4.86 (s, 1H), 4.42 (d, J = 3.6 Hz, 1H), 3.79 (s, 3H), 3.13 (m, 1H), 3.05 (s, 3H), 2.94 (m, 1H), 1.41 (m, 2H). 13C NMR (500 MHz, CDCl3) δ = 197.5, 175.5, 174.0, 139.4, 135.8, 135.7, 134.2, 132.6, 131.3, 128.8, 128.3, 128.0, 127.6, 62.9, 58.5, 52.2, 52.1, 51.3, 49.7, 47.3, 47.1. HRMS (ESI/TOF) calcd for C25H25O5+ (MH+) 405.1697, found 405.1690.
Irradiation experiments. General procedure
Approximately 1-5 mM solution of the endo-Michael adduct in benzene was irradiated in Pyrex™ or borosilicate glass reaction vessels in a Rayonet reactor equipped with RPR-3500 UV lamps (broadband 300-400 nm UV source with peak emission at 350 nm) for 1-24 hours. The crude reaction mixture was purified on a silica gel column using a mixture of hexane and EtOAc as an eluent.
Dimethyl (1RS, 5SR, 6RS, 8SR)-spiro[6-formyl-3-phenylbicyclo[3.2.1]oct-2-ene-5,8-dicarboxylate-7,1′-cyclopropane] (9c)
from 140 mg of 7c (0.40 mmol) for 20 h (hexane/EtOAc gradient 30:1 → 5:1): 74 mg (53 %). 1H NMR (500 MHz, CDCl3) δ = 9.49 (d, J = 4.9 Hz, 1H), 7.40 (m, 2H), 7.33 (m, 2H), 7.28 (m, 1H) overlaps with CDCl3, 6.41 (d, J = 6.6 Hz, 1H), 3.86 (d, J = 4.0 Hz, 1H), 3.82 (s, 3H), 3.64 (s, 3H), 3.36 (d, J = 18.8 Hz, 1H), 2.87 (d, J = 18.8 Hz, 1H), 2.72 (d, J = 4.9 Hz, 1H), 2.48 (dd, J = 6.6, 4.0 Hz, 1H), 0.82 (m, 2H), 0.71-0.66 (m, 1H), 0.49-0.44 (m, 1H). 13C NMR (500 MHz, CDCl3) δ = 201.9, 174.1, 170.2, 139.0, 134.5, 128.5, 127.8, 126.7, 125.1, 62.3, 54.0, 52.7, 52.4, 51.9, 46.4, 31.2, 30.7, 14.5, 5.9. HRMS (ESI/TOF) calcd for C21H22NaO5+ (MNa+) 377.1359, found 377.1354.
Dimethyl (1RS, 5SR, 6RS, 8SR)-spiro[6-(1,3-dioxolan-2-yl)-3-phenylbicyclo[3.2.1]oct-2-ene-5,8-dicarboxylate-7,1′-cyclopropane] (12c)
from 70 mg of 9c (0.20 mmol), one drop of HCl (4.0 M solution in 1,4-dioxane) in 5 mL of THF/ethylene glycol mixture (1:1) (hexane/EtOAc gradient 30:1 → 5:1): 77 mg (98 %). 1H NMR (500 MHz, CDCl3) δ = 7.45 (m, 2H), 7.32 (m, 2H), 7.25 (m, 1H), 6.33 (d, J = 6.7 Hz, 1H), 4.76 (d, J = 8.5 Hz, 1H), 3.81 (s, 3H), 3.80 (d, J = 4.2 Hz, 1H), 3.76-3.70 (m, 4H), 3.60 (s, 3H), 3.32 (d, J = 18.8 Hz, 1H), 3.01 (d, J = 18.8 Hz, 1H), 2.64 (d, J = 8.5 Hz, 1H), 2.31 (dd, J = 6.7, 4.2 Hz, 1H), 1.20 (m, 1H), 0.73 (m, 1H), 0.45 (m, 2H). 13C NMR (500 MHz, CDCl3) δ = 176.0, 170.9, 139.9, 133.8, 128.3, 127.3, 126.9, 125.1, 104.8, 64.7, 64.5, 56.3, 53.3, 52.9, 52.4, 51.7, 46.4, 30.0, 29.9, 15.5, 6.0. HRMS (ESI/TOF) calcd for C23H27O6+ (MH+) 399.1802, found 399.1817.
Methyl (1RS, 3SR, 4RS, 5RS, 6SR, 9RS, 10SR)-11-oxa-3-phenyltetracyclo[4.3.1.13,5.04,9]undecane-10-carboxylate (10a)
from 0.18 g of 7a (0.63 mmol) irradiated for 20 h; (hexane/EtOAc gradient 30:1 → 5:1): 79 mg (44 %). 1H NMR (500 MHz, CDCl3) δ = 7.40-7.36 (m, 3H), 2H in aromatic region obscured by CDCl3, 4.67 (dd, J = 8.0, 5.1 Hz, 1H), 3.78 (s, 3H), 3.33 (d, J = 4.5 Hz, 1H), 3.12 (dd, J = 6.4, 5.1 Hz, 1H), 3.05-3.01 (m, 1H), 2.52 (q, J = 9.4, 4.7 Hz, 1H), 2.22-2.18 (m, 1H), 2.16-2.02 (m, 1H), 1.91-1.83 (m, 1H), 1.65-1.58 (m, 1H), 1.56-1.50 (m, 1H), 1.14-1.06 (m, 1H). 13C NMR (500 MHz, CDCl3) δ = 176.6, 142.4, 128.2, 126.9, 124.0, 96.0, 51.7, 47.4, 44.1, 44.1, 36.2, 32.6, 17.2, 17.0. HRMS (ESI/TOF) calcd for C18H20NaO3+ (MNa+) 307.1310, found 307.1317. See SI for xray data.
Methyl (1SR, 2SR, 3SR, 4RS, 5RS, 6SR, 9RS, 10SR)-1-methyl-11-oxa-3-phenyltetracyclo[4.3.1.13,5.04,9] undecane-10-carboxylate (10b)
from 55 mg of 7b (0.18 mmol) irradiated for 20 h; (hexane/EtOAc gradient 30:1 → 5:1): 36 mg (65 %).1H NMR (500 MHz, CDCl3) δ = 7.38 (t, J = 7.6 Hz, 2H), 7.30 (d, J = 8.5 Hz, 2H), 7.27 (t, J = 7.2 Hz, 1H), 4.66 (dd, J = 7.9, 5.1 Hz, 1H), 3.78 (s, 3H), 3.42 (d, J = 4.7 Hz, 1H), 3.07 (ddd, J = 7.8, 4.9, 1.3 Hz, 1H), 2.85 (t, J = 5.1 Hz, 1H), 2.50 (q, J = 9.4, 4.7 Hz, 1H), 2.27-2.18 (m, 2H), 1.91-1.82 (m, 1H), 1.67-1.60 (m, 1H), 1.54-1.47 (m, 1H), 1.13-1.05 (m, 1H), 0.92, (d, J = 6.9 Hz, 3H). 13C NMR (500 MHz, CDCl3) δ = 176.9, 143.9, 128.0, 126.6, 126.0, 124.0, 96.1, 51.7, 48.1, 47.8, 42.8, 37.6, 35.1, 32.5, 14.7, 17.1, 6.4. HRMS (ESI/TOF) calcd for C19H22NaO3+ (MNa+) 321.1467, found 321.1450.
Methyl endo-3-(1,3-dioxalan-2-yl)-8-phenylbicyclo[4.3.0]non-7-ene-exo-2-carboxylate (13a)
A solution of 0.18 g of 7a (0.63 mmol), 0.22 mL HCl solution (4M in dioxane, 0.89 mmol) and 0.08 g ethylene glycol (1.26mmol) was prepared in dichloromethane (130 mL) in a Pyrex™ reaction vessel. This solution was irradiated for 29 hours. The crude reaction mixture was purified on a silica gel column using a mixture of hexane and EtOAc as an eluent. The reaction mixture was then concentrated in vacuo to a volume of 20 mL, washed with saturated sodium bicarbonate solution (2×15mL), and the aqueous layer extracted with dichoromethane (2×20mL). The combined organic layers were dried over anhydrous sodium sulfate, and the solvent removed in vacuo to give a crude yellow oil. The crude mixture was purified on a silica gel column using hexane and ethyl acetate as the eluent (hexane/EtOAc gradient 30:1 → 5:1), 63 mg (30%). 1H NMR (500 MHz, CDCl3) δ = 7.44 (d, J = 7.9 Hz, 2H), 7.34 (t, J = 7.4 Hz, 2H), 7.25 (t, J = 7.4 Hz, 1H), 6.01 (t, J = 2.2 Hz, 1H), 4.71 (d, J = 3.9 Hz, 1H), 3.94-3.77 (m, 4H), 3.72 (s, 1H), 3.11 (s, 1H), 2.80 (dddd, J = 15.4, 6.2, 3.7, 2.6 Hz, 1H), 2.59 (dt, J = 12.8, 6.5 Hz, 1H), 2.53 (d, J = 15.4 Hz, 1H), 2.32 (t, J = 11.2 Hz, 1H), 2.05-2.96 (m, 2H), 1.77-1.67 (m, 2H), 1.35-1.24 (m, 2H). 13C NMR (500 MHz, CDCl3) δ = 176.9, 142.0, 136.6, 129.6, 128.3, 127.1, 125.5, 106.1, 65.0, 64.9, 51.5, 45.2, 45.0, 41.2, 41.1, 38.9, 26.1, 22.0. HRMS (ESI/TOF) calcd for C20H24LiO4+ (MLi+) 335.1835, found 335.1829.
Methyl 6-(1,3-dioxalan-2-yl)-3-phenylbicyclo[3.3.1]non-2-ene-syn-9-carboxylate (12a)
From the reaction mixture above - isolated as 6-epimeric mixture, 38 mg (18%). HRMS (ESI/TOF) calcd for C20H24NaO4+ (MNa+) 351.1572, found 351.1558.
Methyl endo-3-(1,3-dioxalan-2-yl)-endo-10-methyl-8-phenylbicyclo[4.3.0]non-7-ene-exo-2-carboxylate (13b)
A solution of 0.21 g of 10b (0.70 mmol), 0.25 mL HCl solution (4M in dioxane, 0.99 mmol) and 0.09 g ethylene glycol (1.40mmol) was prepared in chloroform (10 mL), then heated to 70°C while stirring for 4 h. The reaction mixture was then concentrated in vacuo to remove chloroform, diluted with dichoromethane (10 mL), washed with saturated sodium bicarbonate solution (2×15mL), and the aqueous layer extracted with dichoromethane (2×10mL). The combined organic layers were dried over anhydrous sodium sulfate, and the solvent removed in vacuo to give a crude yellow oil. The crude mixture was purified on a silica gel column using hexane and ethyl acetate as the eluent (hexane/EtOAc gradient 30:1 → 5:1), 0.17 g (71%). 1H NMR (500 MHz, CDCl3) δ = 7.35-7.31 (m, 4H), 7.25 (t, J = 7.2 Hz, 1H), 5.87 (t, J = 2.1 Hz, 1H), 4.79 (d, J = 3.7 Hz, 1H), 4.01-3.85 (m, 3H), 3.71 (s, 3H), 3.31 (tt, J = 7.7, 1.7 Hz, 1H), 2.95-2.82 (m, 2H), 2.68 (t, J = 11.5 Hz, 1H), 2.08-2.00 (m, 2H), 1.78-1.71 (m, 2H), 1.56-1.48 (m, 2H), 1.08 (d, J = 7.4 Hz, 3H).
Methyl endo-5-hydroxy-2-methyl-3-phenyltricyclo[4.3.1.04,9]dec-2-ene-exo-10-carboxylate (14)
A solution of 0.11 g of 10b (0.37 mmol), 0.13 mL HCl solution (4M in dioxane, 0.51 mmol) was prepared in chloroform (10 mL), stirred at ambient temperature for 4 h. The reaction mixture was then concentrated in vacuo to remove chloroform, diluted with dichoromethane (10 mL), washed with saturated sodium bicarbonate solution (2×15mL), and the aqueous layer extracted with dichoromethane (2×10mL). The combined organic layers were dried over anhydrous sodium sulfate, and the solvent removed in vacuo to give a crude yellow oil. The crude mixture was purified on a silica gel column using hexane and ethyl acetate as the eluent (hexane/EtOAc gradient 30:1 → 5:1), 80 mg (73%). The structure is characterized by xray (see supporting information). 1H NMR (500 MHz, CDCl3) δ = 7.45 (d, J = 8.4 Hz, 2H), 7.37 (t, J = 7.7 Hz, 2H), 7.24 (t, J = 7.4 Hz, 1H), 4.03 (dt, J = 8.5, 2.2 Hz, 1H), 3.75 (s, 3H), 3.15 (dd, J = 8.7, 4.3 Hz, 1H), 2.88 (d, J = 4.5 Hz, 1H), 2.76-2.74 (m, 1H), 2.54-2.50 (m, 1H), 2.32-2.28 (m, 1H), 2.03 (s, 3H), 1.80-1.59 (m, 4H) . 13C NMR (500 MHz, CDCl3) δ = 175.9, 145.5, 137.7, 137.3, 128.2, 128.1, 126.5, 69.3, 51.8, 51.4, 48.2, 42.3, 38.3, 38.1, 21.9, 18.1, 14.6. HRMS (ESI/TOF) calcd for C19H22LiO3+ (MLi+) 305.1728, found 305.1729. See SI for xray data.
Dimethyl (1SR, 2RS, 3RS, 5SR, 6SR, 7RS, 9SR, 10SR)-3-hydroxy-3-phenyltetracyclo[3.3.1.17,9.02,6]decane-9,10-dicaboxylate (15)
from 120 mg of 7d (0.35 mmol) for 3 h (hexane/EtOAc gradient 30:1 → 2:1): 90 mg (75 %), colorless crystals, m.p. 101.5-103.5°C. 1H NMR (500 MHz, CDCl3) δ = 7.47 (d, J = 7.3 Hz, 2H), 7.34 (t, J = 7.5 Hz, 2H), 7.24 (t, J = 7.3 Hz, 1H), 3.82 (s, 3H), 3.68 (s, 3H), 3.02 (s, 1H), 2.92 (ddd, J = 2.7, 2.7, 2.7 Hz, 1H), 2.87 (m, 1H), 2.75 (ddd, J = 3.0, 3.0, 3.0 Hz, 1H), 2.56 (m, 1H) overlaps with 2.56 (s, OH), 2.46 (m, 1H), 2.41 (d, J = 14.9 Hz, 1H), 2.24 (dd, J = 14.9, 4.6 Hz, 1H), 1.78 (dd, J = 10.4, 3.0 Hz, 1H), 1.61 (dd, J = 10.4, 2.7 Hz, 1H). 1H NMR (500 MHz, C6D6) δ = 7.34 (m, 2H), 7.19 (m, 2H), 7.10 (m, 1H), 3.46 (s, 3H), 3.34 (s, 3H), 2.91 (m, 2H), 2.65 (m, 1H), 2.55 (d, J = 14.5 Hz, 1H) overlaps with 2.56 (ddd, J = 3.0, 3.0, 3.0 Hz, 1H) overlaps with 2.52 (m, 1H), 2.07 (dd, J = 14.5, 4.7 Hz, 1H) overlaps with 2.05 (m, 1H), 1.42 (dd, J = 10.3, 3.0 Hz, 1H), 1.13 (dd, J = 10.3, 2.7 Hz, 1H). 13C NMR (500 MHz, CDCl3) δ = 173.9, 172.4, 148.9, 128.3, 126.8, 124.6, 78.7, 67.2, 63.8, 54.0, 52.0, 51.9, 51. 8, 51.2, 46.0, 42.6, 41.0, 40.1. HRMS (ESI/TOF) calcd for C20H23O5+ (MH+) 343.1540, found 343.1544. See SI for xray data.
Dimethyl (1SR, 3RS, 4RS, 5SR, 6SR, 7SR, 8SR, 9SR)-6-hydroxy-5-methyl-6-phenyltetracyclo [3.2.1.13,8.04,7]nonane-8,9-dicaboxylate (16)
18 mg (15 %). 1H NMR (500 MHz, CDCl3) δ = 7.49 (m, 2H), 7.34 (m, 2H), 7.26 (m, 1H) overlaps with CDCl3, 4.61 (s, OH), 3.86 (s, 3H), 3.59 (s, 3H), 3.10-3.07 (m, 3H), 2.66 (m, 1H), 2.41 (ddd, J = 3.1, 3.1, 3.1 Hz, 1H), 1.78 (dd, J = 10.4, 2.6 Hz, 1H), 1.69 (dd, J = 10.4, 2.7 Hz, 1H) overlaps with 1.68 (d, J = 7.3 Hz, 1H), 0.89 (s, 3H).
Dimethyl (1RS, 2RS, 3RS, 5SR, 6RS, 7RS, 9SR, 10SR)-spiro[3-hydroxy-3-phenyltetracyclo[3.3.1.17,9.02,6] decane-9,10-dicaboxylate-8,1′-cyclopropane] (17)
from 65 mg of 7e (0.18 mmol) for 12 h (hexane/EtOAc gradient 30:1 → 1:1): 50 mg (77 %). 1H NMR (500 MHz, CDCl3) δ = 7.49 (m, 2H), 7.36 (m, 2H), 7.25 (m, 1H), 3.82 (s, 3H), 3.68 (s, 3H), 3.58 (m, 1H), 2.95 (m, 1H), 2.88 (m, 1H), 2.62 (m, 1H), 2.52 (s, OH), 2.40 (d, J = 14.8 Hz, 1H), 2.36 (ddd, J = 2.7, 2.7, 1.0 Hz, 1H), 2.25 (dd, J = 14.8, 4.7 Hz, 1H), 2.17 (dd, J = 3.3, 3.3 Hz, 1H), 0.87-0.82 (m, 1H), 0.80-0.76 (m, 1H), 0.55-0.48 (m, 2H). 1H NMR (500 MHz, C6D6) δ = 7.43 (m, 2H), 7.20 (m, 2H), 7.11 (m, 1H), 3.63 (m, 1H), 3.48 (s, 3H), 3.38 (s, 3H), 2.77 (m, 1H), 2.73 (m, 1H) overlaps with 2.71 (m, 1H), 2.58 (d, J = 14.7 Hz, 1H), 2.39 (m, 1H), 2.19 (br. s, OH), 2.13 (dd, J = 14.7, 4.7 Hz, 1H), 2.03 (dd, J = 3.2. 3.2 Hz, 1H), 0.62 (ddd, J = 9.7, 5.0, 5.0 Hz, 1H), 0.39 (ddd, J = 9.7, 5.0, 5.0 Hz, 1H), 0.18 (m, 1H), 0.12 (m, 1H). 1H NMR (500 MHz, CD3OD) δ = 7.45 (m, 2H), 7.32 (m, 2H), 7.20 (m, 1H), 3.78 (s, 3H), 3.66 (s, 3H), 3.55 (m, 1H), 2.88 (m, 1H), 2.82 (m, 1H), 2.43 (m, 1H), 2.37 (d, J = 14.6 Hz, 1H), 2.34 (ddd, J = 2.7, 2.7, 1.0 Hz, 1H), 2.15 (m, 1H) overlaps with 2.13 (dd, J = 14.6, 4.9 Hz, 1H), 0.88-0.79 (m, 2H), 0.57-0.48 (m, 2H). 13C NMR (500 MHz, CDCl3) δ = 173.9, 172.6, 149.0, 128.3, 126.7, 124.6, 78.3, 65.7, 62.7, 55.3, 52.2, 52.0, 52.0, 51.8, 51.1, 46.5, 40.8, 38.9, 9.8 (two overlapping peaks). 13C NMR (500 MHz, CD3OD) δ = 176.3, 174.6, 143.5, 129.0, 129.0, 128.4, 77.8, 64. 6, 64.5, 61.8, 58.1, 57.6, 52.6, 52.1, 51.8, 46.2, 39.3, 13. 7, 8.4, 8.3. HRMS (ESI/TOF) calcd for C22H24NaO5+ (MNa+) 391.1516, found 391.1527.
Dimethyl (1RS, 3RS, 4RS, 5SR, 6SR, 7RS, 8SR, 9SR)-spiro[6-hydroxy-5-methyl-6-phenyltetracyclo [3.2.1.13,8.04,7]nonane-8,9-dicaboxylate-2,1′-cyclopropyl] (18)
13 mg (20 %). 1H NMR (500 MHz, CDCl3) δ = 7.53 (m, 2H), 7.35 (m, 2H), 7.27 (m, 1H) overlaps with CDCl3, 4.62 (s, OH), 3.87 (s, 3H), 3.68 (m, 1H), 3.59 (s, 3H), 3.33 (dd, J = 3.1, 1.9 Hz, 1H), 2.51 (ddd, J = 2.3, 2.3, 2.3 Hz, 1H), 2.48 (m, 1H), 2.07 (dd, J = 3.1, 3.1 Hz, 1H), 0.98-0.94 (m, 1H), 0.92 (s, 3H), 0.82-0.75 (m, 2H), 0.69-0.64 (m, 1H), 0.59-0.54 (m, 1H). 13C NMR (500 MHz, CDCl3) δ = 175.8, 172.6, 141.9, 128.0, 127.8, 127.2, 77.0 overlaps with CDCl3, 63.7, 62.5, 60.7, 57.3, 56.7, 52.5, 51.5, 50.0, 44.2, 38.2, 12.1, 7.8, 7.6. HRMS (ESI/TOF) calcd for C22H24NaO5+ (MNa+) 391.1516, found 391.1526.
Dimethyl (1SR, 2RS, 3SR, 4SR, 5RS, 6SR, 7RS, 9RS, 10SR)-3-hydroxy-3,4-diphenyltetracyclo[3.3.1.17,9.02,6] decane-9,10-dicaboxylate (19)
from 0.23 g of 7f (0.55 mmol) for 6 h (hexane/EtOAc gradient 30:1 → 10:1): 0.15 g (65 %). 1H NMR (500 MHz, CDCl3) δ = 7.67 (m, 2H), 7.40 (m, 2H), 7.26 (m, 1H) overlaps with CDCl3, 7.23-7.18 (m, 4H), 7.13 (m, 1H), 3.39 (d, J = 3.3 Hz, 1H), 3.93 (s, OH), 3.90 (ddd, J = 3.0, 3.0, 1.5 Hz, 1H), 3.81 (s, 3H), 3.33 (s, 3H), 3.01 (m, 2H), 2.98 (s, 1H), 2.80 (ddd, J = 3.1, 3.1, 3.1 Hz, 1H), 2.64 (m, 1H), 1.72 (dd, J = 10.4, 2.8 Hz, 1H), 1.59 (dd, J = 10.5, 2.8 Hz, 1H). 13C NMR (500 MHz, CDCl3) δ = 174.1, 172.7, 149.5, 137.3, 128.3, 128.2, 127.8, 126.5, 125.9, 124.7, 76.9 overlaps with CDCl3, 70.9, 62.7, 55.2, 53.9, 53.6, 51.9, 51.8, 49.8, 46.1, 42.1, 41.5. HRMS (ESI/TOF) calcd for C26H26NaO5+ (MNa+) 441.1672, found 441.1668.
Dimethyl (1SR, 3RS, 4RS, 5SR, 6SR, 7SR, 8SR, 9SR)-5-benzyl-6-hydroxy-6-phenyltetracyclo[3.2.1.13,8.04,7] nonane-8,9-dicaboxylate (20)
58mg, (25%). 1H NMR (500 MHz, CDCl3) δ = 7.39 (m, 2H), 7.11-7.05 (m, 3H), 6.88-6.83 (m, 3H), 6.67 (m, 2H), 4.31 (s, OH), 3.83 (s, 3H), 3.58 (s, 3H), 3.15 (m, 2H), 3.03 (d, J = 16.2 Hz, 1H), 2.91 (m, 1H), 2.84 (m, 2H), 2.69 (d, J = 16.2 Hz, 1H), 1.83 (dd, J = 10.3, 2.7 Hz, 1H), 1.69 (dd, J = 10.3, 1.6 Hz, 1H). 13C NMR (500 MHz, CDCl3) δ = 175.6, 172.1, 141.8, 138.0, 129.9, 127.7, 127.3, 127.1, 127.0, 124.7, 75.1, 69. 5, 63.6, 59.7, 58.1, 52.5, 51.7, 50.9, 48.34, 41.5, 38.5, 32.6.
Dimethyl (1RS, 2SR, 3RS, 4SR, 5RS, 6SR, 7RS)-6-benzoyl-5-phenyltricyclo[2.2.2.13,7]nonane-1,2-dicaboxylate (21)
from 400 mg of 7f’ (0.95 mmol) for 72 h (hexane/EtOAc gradient 30:1 → 1:1): 80mg (20%). 1H NMR (500 MHz, CDCl3) δ = 7.98 (m, 2H), 7.53 (m, 1H), 7.47 (m, 2H), 7.30 (m, 2H), 7.25 (m, 2H) overlaps with CDCl3, 7.14 (m, 1H), 5.28 (d, J = 2.8 Hz, 1H), 3.59 (s, 3H), 3.48 (m, 1H), 3.08 (m, 1H), 3.03 (m, 1H), 2.90 (dddd, J = 7.8, 5.1, 3.4, 1.6 Hz, 1H), 2.84 (s, 3H), 2.75 (s, 1H), 1.87-1.80 (m, 2H), 1.74-1.69 (m, 2H). 13C NMR (500 MHz, CDCl3) δ = 205.5, 175.2, 171.0, 142.9, 138.8, 132.0, 128.6, 128.2, 128.0, 127.4, 126.1, 124.7, 57.1, 52.0, 50.9, 50.8, 50.1, 46.7, 45.6, 43.7, 40.2, 39.7, 33.6. HRMS (ESI/TOF) calcd for C26H26NaO5+ (MNa+) 441.1672, found 441.1659.
Dimethyl (1RS, 2RS, 3SR, 4SR, 5RS, 6SR, 7RS, 9SR, 10SR)-spiro[3-hydroxy-3,4-diphenyltetracyclo [3.3.1.17,9.02,6]decane-9,10-dicaboxylate-8,1′-cyclopropane] (22)
from 48 mg of 7g (0.11 mmol) for 5 h (hexane/EtOAc gradient 30:1 → 1:1): 17 mg (35%), colorless crystals, m.p. 173.5-176.5°C. 1H NMR (500 MHz, CDCl3) δ = 7.68 (m, 2H), 7.41 (m, 2H), 7.27 (m, 1H) overlaps with CDCl3, 7.21 (m, 4H), 7.13 (m, 1H), 3.97 (m, 2H), 3.86 (s, OH), 3.80 (s, 3H), 3.55 (s, 1H), 3.34 (s, 3H), 3.10 (m, 1H), 3.05 (m, 1H), 2.45 (ddd, J = 2.8, 2.8, 1.0 Hz, 1H), 2.21 (dd, J = 3.3, 3.3 Hz, 1H), 0.79-0.72 (m, 2H), 0.58-0.49 (m, 2H). 13C NMR (500 MHz, CDCl3) δ = 174.1, 172.8, 149.6, 137.3, 128.3, 128.2, 127.8, 126.5, 125.9, 124.8, 69.5, 61.6, 54.8, 54.6, 53.6, 52.2, 51.9, 51.9, 50.7, 47.9, 38.5, 9.8, 9.7. HRMS (ESI/TOF) calcd for C28H28NaO5+ (MNa+) 467.1829, found 467.1813. See SI for xray data.
Dimethyl (1RS, 2SR, 3RS, 4SR, 5SR, 6SR, 7RS)-spiro[6-benzoyl-5-phenyltricyclo[2.2.2.13,7]nonane-1,2-dicaboxylate-9,1′-cyclopropyl] (24)
from 0.22 mg of 7g’ (0.49 mmol) for 24-48 h (hexane/EtOAc gradient 20:1 → 1:1): 7mg (30%). 1H NMR (500 MHz, CDCl3) δ = 7.98 (m, 2H), 7.53 (m, 1H), 7.47 (m, 2H), 7.31 (m, 2H), 7.25 (m, 2H), 7.14 (m, 1H), 5.29 (d, J = 2.7 Hz, 1H), 3.60 (s, 3H), 3.52 (m, 1H), 3.46 (s, 1H), 3.02 (m, 1), 2.83 (s, 3H), 2.40 (m, 2H), 2.20 (ddd, J = 11.5, 7.8, 1.5 Hz, 1H), 1.81 (d, J = 11.5 Hz, 1H), 1.05-1.00 (m, 1H), 0.93-0.88 (m, 1H), 0.68-0.60 (m, 2H). 13C NMR (500 MHz, CDCl3) δ = 205.5, 175.1, 171.1, 142.8, 138.7, 132.0, 128.6, 128.2, 128.0, 127.4, 126.1, 56.2, 52.0, 51.2 (two overlapping peaks), 50.8 (two overlapping peaks), 46.5 (two overlapping peaks), 40.4, 38.8, 34.4, 10.3, 9.7.
Dimethyl (1RS, 2RS, 3RS, 4SR, 5RS, 6SR, 7RS, 9SR, 10SR)-spiro[4-ethyl-3-hydroxy-3-phenyltetracyclo [3.3.1.17,9.02,6]decane-9,10-dicaboxylate-8,1′-cyclopropane] (26)
from 118 mg of 7h (0.30 mmol) for 20 h (hexane/EtOAc gradient 30:1 → 10:1): 41 mg (35 %). 1H NMR (500 MHz, CDCl3) δ = 7.46 (d, J = 7.9 Hz, 2H), 7.33 (t, J = 7.7 Hz, 2H), 7.23 (t, J = 7.3 Hz, 1H), 3.82 (s, 3H), 3.69 (s, 3H), 3.60 (s, 1H), 3.14 (m, 1H), 2.97 (m, 1H), 2.62 (s, OH), 2.46 (m, 1H), 2.38 (m, 1H), 2.28 (dd, J = 11.1, 4.4 Hz, 1H), 2.24 (dd, J = 3.3, 3.3 Hz, 1H), 0.88-0.76 (m, 3H), 0.66 (t, J = 7.1 Hz, 3H), 0.61-0.53 (m, 2H), 0.51-0.46 (m, 1H). 13C NMR (500 MHz, CDCl3) δ = 174.0, 172.6, 145.4, 127.7, 126.7, 126.1, 81.8, 66.6, 62.9, 55.9, 54.5, 52.2, 52.0, 51.8 (two overlapping peaks), 51.2, 46.7, 38.7, 25.2, 13.0, 9.8 (two overlapping peaks). HRMS (ESI/TOF) calcd for C24H28NaO5+ (MNa+) 419.1829, found 419.1818. See SI for xray data.
Compounds 25 and 27 were only partially purified, as we were able to obtain only enriched fractions described with 1H NMR; additionally isomers 26 and 27 were characterized by xray crystallography.
Dimethyl (1RS, 2RS, 3RS, 4RS, 5RS, 6SR, 7RS, 9SR, 10SR)-spiro[4-ethyl-3-hydroxy-3-phenyltetracyclo [3.3.1.17,9.02,6]decane-9,10-dicaboxylate-8,1′-cyclopropane] (25)
1H NMR (500 MHz, CDCl3) δ = 7.49 (m, 2H), 7.34 (m, 2H), 7.23 (m, 1H), 3.78 (s, 3H), 3.71 (s, 3H), 3.48 (m, 1H), 3.01 (ddd, J = 3.1, 3.1, 1.4 Hz, 1H), 2.98 (ddd, J = 2.7, 1.3, 1.3 Hz, 1H), 2.92 (m, 1H), 2.55 (br. s, OH), 2.47 (ddd, J = 2.9, 2.9, 1.0 Hz, 1H), 2.14 (ddd, J = 9.6, 6.3, 3.4 Hz, 1H) overlaps with 2.12 (dd, J = 3.3, 3.3 Hz, 1H), 1.55-1.42 (m, 1H), 1.00-0.85 (m, 2H), 0.81 (t, J = 7.4 Hz, 3H) overlaps with 0.81-0.73 (m, 2H), 0.58-0.46 (m, 1H). HRMS (ESI/TOF) calcd for C24H28NaO5+ (MNa+) 419.1829, found 419.1815.
Dimethyl (1RS, 3RS, 4RS, 5SR, 6SR, 7RS, 8SR, 9SR)-spiro[6-hydroxy-6-phenyl-5-propyltetracyclo [3.2.1.13,8.04,7]nonane-8,9-dicaboxylate-2,1′-cyclopropyl] (27)
1H NMR (500 MHz, CDCl3) δ = 7.58 (m, 2H), 7.34 (m, 2H), 7.27 (m, 1H) overlaps with CDCl3, 4.85 (s, OH), 3.87 (s, 3H), 3.67 (m, 1H), 3.58 (s, 3H), 3.30 (dd, J = 3.2, 1.8 Hz, 1H), 2.63 (m, 1H), 2.49 (ddd, J = 2.3, 2.3, 2.3 Hz, 1H), 2.03 (dd, J = 3.7, 2.6 Hz, 1H), 1.55-1.42 (m, 2H), 1.07 (ddd, J = 15.7, 13.2, 4.2 Hz, 1H), 1.00-0.85 (m, 1H), 0.81-0.73 (m, 1H), 0.67-0.63 (m, 1H), 0.58-0.46 (m, 2H) overlaps with 0.53 (t, J = 7.2 Hz, 3H). See SI for xray data.
Dimethyl 7-oxabicyclo[2.2.1]hepta-2,5-diene-2,3-dicarboxylate (28):15
(procedure A) from 0.5 mL of dimethyl acetylenedicarboxylate (4.07 mmol) (hexane/EtOH gradient 40:1→10:1 and 4:1): 0.79 g (92 % in 24 h), 0.64 g (75 % in 48 h). 1H NMR (500 MHz, CDCl3) δ = 7.23 (dd, J = 1.1, 1.1 Hz, 2H), 5.69 (dd, J = 1.1, 1.1 Hz, 2H), 3.83 (s, 6H).
syn-Methyl 11,12-dioxatetracyclo[6.2.1.13,6.02,7.]dodeca-4.9-diene-2,7-dicarboxylate (29) and anti-Methyl 11,12-dioxatetracyclo[6.2.1.13,6.02,7.]dodeca-4.9-diene-2,7-dicarboxylate (30):17
Dimethyl acetylenedicarboxylate (0.5 mL, 4.07 mmol) was dissolved in 5 ml furan (large excess), which was also used as a solvent. The resulting reaction mixture was stirred for 24 h. The solvent was removed to take NMR. The solvent was removed in vacuo and the crude reaction mixture was purified on a flash silica gel column using a mixture of hexane and EtOH as an eluent (1:30 = EtOAc : Hexane, then 1:10). The two dimers were isolated in an endo:exo ratio of 3:1, 0.12 g (11%). Syn isomer (29): 1H NMR (500 MHz, CDCl3) δ = 6.63 (s, 4H), 5.09 (s, 4H), 3.65 (s, 6H). Anti isomer (30): 1H NMR (500 MHz, CDCl3) δ = 6.69 (s, 2H), 6.52 (s, 2H), 5.20 (s, 2H), 4.55 (s, 2H), 3.75 (s, 6H). 13C NMR of the mixture (500 MHz, CDCl3) δ = 172.3, 170.3, 143.3, 138.9, 138.7, 135.6, 85.1, 83.9, 83.5, 79.2, 70.2, 70.0, 52.4, 52.1. The two compounds have an unusual 1:1 crystalline packing – see xray data below.
Methyl 6-phenylpyran-2-one-4-carboxylate (31):18
Diisopropylamine (0.13 mL, 0.93mmol) was dissolved in THF at -30°C, then n-BuLi (0.5 mL, 0.80mmol) was added and the resulting mixture was stirred for 30 min at -30°C. After that, acetophenone was added (0.08 g, 0.66mmol) and the mixture was kept at the same temperature (-30°C) for additional 40 min. After the anion was generated, 28 was added (0.14 g, 0.66mmol) and the reaction mixture was allowed to warm to ambient temperature and stirred for an additional 30 min. After the reaction was complete, the mixture was quenched with saturated aqueous solution of NH4Cl (10 mL), dichloromethane was added (15 mL). The organic layer was washed with water (1×10 mL) and brine (1×10 mL). The organic layer was dried over anhydrous NaSO4. The solvent was removed in vacuo and the crude reaction mixture was purified on a flash silica gel column using a mixture of hexane and EtOH as an eluent (1:30 = EtOAc : Hexane, then 1:10) 70 mg (45%). 1H NMR (500 MHz, CDCl3) δ = 7.92-7.89 (m, 2H), 7.53-7.49 (m, 3H), 7.16 (d, J = 1.4 Hz, 1H), 6.93 (d, J = 1.4 Hz, 1H), 4.00 (s, 3H). 13C NMR (500 MHz, CDCl3) δ = 164.0, 161.8, 161.3, 144.3, 131.3, 131.0, 129.1, 125.8, 115.7, 99.3, 53.3.
Supplementary Material
Scheme 7.

Scheme 8.

Scheme 15.

Scheme 16.
Acknowledgements
Support of this research by the NIH (GM093930) and NSF (CHE-1057800) is gratefully acknowledged.
Footnotes
Dedicated to Howard E. Zimmerman
Supporting Information. Additional experimental data, NMR spectra, xray data, computational details (150 pages). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a Rawal VH, Dufour C. J. Am. Chem. Soc. 1994;116:261–2614. [Google Scholar]; b Dvorak CA, Dufour C, Iwasa S, Rawal VH. J. Org. Chem. 1998;63:5302–5303. [Google Scholar]
- 2.Sauers RR, Kelly KW, Sickles BR. J. Org. Chem. 1972;37:537–543. [Google Scholar]
- 3.Mudryk B, Cohen T. J. Am. Chem. Soc. 1991;113:1866–1867. [Google Scholar]
- 4.Valiulin RA, Arisco TM, Kutateladze AG. Org. Lett. 2010;12(15):3398–3401. doi: 10.1021/ol101297b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones G, Schwartz SB, Marton MT. J. Chem. Soc., Chem. Comm. 1973;11:374–375. [Google Scholar]; b Jones G, Acquadro MA, Carmody MA. J. Chem. Soc., Chem. Comm. 1975;6:206–207. [Google Scholar]
- 6.Perez-Ruiz R, Miranda MA, Alle R, Meerholz K, Griesbeck A. Photochem. Photobiol. Sci. 2006;5:51–55. doi: 10.1039/b513875b. [DOI] [PubMed] [Google Scholar]
- 7.a Valiulin RA, Kutateladze AG. Org. Lett. 2009;11(17):3886–3889. doi: 10.1021/ol901456m. [DOI] [PubMed] [Google Scholar]; b Valiulin RA, Kutateladze AG. Tetrahedron Lett. 2010;51(29):3803–3806. doi: 10.1016/j.tetlet.2010.05.078. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Valiulin RA, Arisco TM, Kutateladze AG. J. Org. Chem. 2011;76(5):1319–1332. doi: 10.1021/jo102221q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diels-Alder adducts 1-6 are described; Trost BM, Balkovec JM, Angle SR. Tetrahedron Lett. 1986;27:1445–1448. 1.Kaydos JA, Smith DL. J. Org. Chem. 1983;48:1096–1099. 2.Prinzbach H, Auge W, Basbudak M. Chemische Berichte. 1973;106:1822–1835. 3.Carman RM, Derbyshire RPC, Hansford KA, Kadirvelraj R, Robinson WT. Aust. J. Chem. 2001;54:117–126.Wilcox CF, Jr., Craig RR. J. Am. Chem. Soc. 1961;83:3866–3871. 4.Boucher J-L, Stella L. Tetrahedron. 1988;44:3607–3615. 5.Kaper H, Antonietti M, Goettmann F. Tetrahedron Lett. 2008;49:4546–4549. 6.
- 9.Gilmore K, Alabugin IV. Chem. Rev. 2011;111:6513–6556. doi: 10.1021/cr200164y. [DOI] [PubMed] [Google Scholar]
- 10.For conformational analysis of spin-orbit coupling in Paternò-Büchi diradicals see Kutateladze AG. J. Am. Chem. Soc. 2001;123(38):9279–9282. doi: 10.1021/ja016092p.Griesbeck AG, Fiege M. In: Molecular and Supramolecular Photochemistry. Ramamurthy V, Schanze KS, editors. Vol. 6. Marcel Dekker; New York: 2000. pp. 33–100.
- 11.a Freeman PK, Kinnel RB, Ziebarth TD. Tetrahedron Lett. 1970;15:1059. [Google Scholar]; b Voget BR, Suter SR, Hoover JRE. Tetrahedron Lett. 1968;13:1609. [Google Scholar]; c Sauers RR, Kelly KW. J.Org. Chem. 1970;35:3286. [Google Scholar]
- 12.Gleiter R, Gaa B, Sigwart C, Lange H, Borzyk O, Rominger F, Irngartinger H, Oeser T. Eur. J. Org. Chem. 1998:171–176. [Google Scholar]
- 13.a Sauers RR, Kelly KW. J. Org. Chem. 1970;35:498–501. [Google Scholar]; b Sauers RR, Whittle JA. J. Org. Chem. 1969;34:3579–3582. [Google Scholar]; c Sauers RR, Schinski W, Mason MM. Tetrahedron Lett. 1969:79–82. [Google Scholar]; d Sauers RR, Kelly KW, Sickles BR. J. Org. Chem. 1972;37:537–543. [Google Scholar]; e Sauers RR. J. Org. Chem. 1974;39:1850–1853. [Google Scholar]
- 14.a Yang NC, Yang D-DH. Photochemical Reactions of Ketones in Solution. J. Am. Chem. Soc. 1958;80:2913–2914. [Google Scholar]; b Brumfield MA, Agosta WC. J. Am. Chem. Soc. 1988;110:6790–6794. [Google Scholar]; c Prathapan S, Robinson KE, Agosta WC. J. Am. Chem. Soc. 1992;114:1838–1843. [Google Scholar]; d Rao CJ, Agosta WC. J. Org. Chem. 1994;59:2125–2131. [Google Scholar]; e Roth WJ, El Raie MN. Tetrahedron Lett. 1970:2445–2446. [Google Scholar]; f Kraus GA, Chen L. Tetrahedron Lett. 1991;32:7151–7154. [Google Scholar]; g Weigel W, Schiller S, Henning H-G. Tetrahedron. 1997;53:7855–7866. [Google Scholar]
- 15.Shinohara H, Sonoda M, Atobe S, Masuno H, Ogawa A. Tetrahedron Lett. 2011;52:6238–6241. [Google Scholar]
- 16.Slee JD, LeGoff E. J. Org. Chem. 1970;35:3897–3901. [Google Scholar]
- 17.Masters K-S, Nieger M, Brase S. Synlett. 2011;3:399–401. [Google Scholar]
- 18.Hendrickson JB, Hall C, Rees R, Templeton JF. J. Org. Chem. 1965;30:3312–3316. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.