

Abstract
Genetic and genomic aberrations are the primary cause of cancer. Chromosome missegregation leads to aneuploidy and provides cancer cells with a mechanism to lose tumor suppressor loci and gain extra copies of oncogenes. Using cytogenetic and array-based comparative genomic hybridization data, we analyzed numerical chromosome aneuploidy in 43,205 human tumors and found that 68% of solid tumors are aneuploid. In solid tumors, almost all chromosomes are more frequently lost than gained with chromosomes 7, 12 and 20 being the only exceptions with more frequent gains. Strikingly, small chromosomes are lost more readily than large ones, but no such inverse size correlation is observed with chromosome gains. Due to increasing levels of proteotoxic stress, chromosome gains have been shown to slow cell proliferation in a manner proportional to the number of extra gene copies gained. However, we find that the extra chromosome in trisomic tumors does not preferentially have a low gene copy number, suggesting that a proteotoxicity-mediated proliferation barrier is not sustained during tumor progression. Paradoxically, despite a bias towards chromosome loss, gains of chromosomes are a poor prognostic marker in ovarian adenocarcinomas. In addition, we find that solid and non-solid cancers have markedly distinct whole-chromosome aneuploidy signatures, which may underlie their fundamentally different etiologies. Finally, preferential chromosome loss is observed in both early and late stages of astrocytoma. Our results open up new avenues of enquiry into the role and nature of whole-chromosome aneuploidy in human tumors and will redirect modeling and genetic targeting efforts in patients.
Keywords: Genomic instability, chromosome instability, aneuploidy, cancer
Introduction
Genetic, epigenetic and genomic alterations are the primary cause of cancer. Germ-line mutations in a number of genes have been shown to predispose to tumorigenesis. Several well-established examples include Li-Fraumeni syndrome (mutations in the TP53 gene), Cowden syndrome (PTEN), Von Hippel-Lindau syndrome (VHL), hereditary forms of increased breast and ovarian cancer susceptibility (BRCA1, BRCA2) and ataxia-telangiectasia (ATM). More frequently, though, cancer is caused by somatic mutations or chromosomal aberrations involving the tumor suppressor genes TP53 (encoding p53), CDKN2A (p16) and RB1 (pRB) or the oncogenes CCND1 (cyclin D1), CDK4, MYC (c-Myc) and the RAS genes1. All these mutations affect, directly or indirectly, several critical checkpoints in the cell that normally protect against genomic damage and unabated cell proliferation, such as the DNA damage checkpoint, the G1/S checkpoint and the mitotic checkpoint.
The mitotic checkpoint ensures faithful segregation of the chromosomes to daughter cells during cell division. The failure to do so is referred to as whole-chromosome instability (W-CIN) and results in the formation of aneuploid cells. W-CIN provides cancer cells with a mechanism to lose tumor suppressor loci and gain extra copies of oncogenes. Accordingly, aneuploidy is a hallmark of most cancers and is associated with increased malignancy, tumor recurrence and drug-resistance2–4. Recent studies have shown that W-CIN and aneuploidy also lead to additional forms of genomic aberrations and instability, thus accelerating further tumor progression5–8.
To date, only one cancer predisposition syndrome has been directly associated with mutations in mitotic checkpoint genes. Autosomal recessive mosaic variegated aneuploidy (MVA) is characterized by developmental abnormalities and increased cancer susceptibility, often leading to childhood cancers9. The patients exhibit chromosomal mosaisism: cells with various aneuploidies, primarily monosomic and trisomic. MVA is caused by biallelic missense and truncating mutations in the BUB1B gene, which encodes the mitotic checkpoint kinase BubR19. Additionally, a monoallelic deletion encompassing BUB1, encoding another key mitotic checkpoint component, Bub1, has been associated with elevated aneuploidy and the development of microsatellite-stable colon carcinoma10. These checkpoint gene mutations are thought to predispose to tumor development through the acquisition of aneuploidy as a result of premature sister-chromatid separation during mitosis.
Despite the paucity of both germ-line and somatically acquired mutations in mitotic checkpoint genes11, W-CIN and aneuploidy are hallmarks of most cancers and are strongly associated with poor prognosis2. This paradox has recently been explained by the fact that all of the most common mutations in human cancers affect the activities of the Rb (Retinoblastoma) and p53 pathways12. This, in turn, causes aberrant expression of E2F and p53 target genes and, with that, an acute stress on multiple mechanisms that are essential for orchestrating faithful chromosome segregation during mitosis12–15. One example is the E2F target gene MAD2L1, which encodes the mitotic checkpoint regulator Mad2. Overexpression of Mad2 occurs as a result of defects in the Rb and p53 pathway16,17 and causes chromosome missegregation and tumorigenesis in vivo18.
Whole-chromosome instability provides cancer cells with a mechanism that allows them to acquire additional copies of oncogenes or lose tumor suppressor loci13–15. As such, it constitutes a powerful driver for tumor progression, anti-cancer drug-resistance and tumor relapse after initially successful cancer therapies2,4–8,19. The development of strategies that specifically target aneuploid cells will therefore likely be effective for the treatment of a wide range of human cancers. However, the significant heterogeneity of aneuploid cancer cells constitutes a major hurdle for the development of aneuploidy-specific regimens. We set out to determine whether, and if so which, specific chromosomes are preferentially lost or gained by human tumor cells to enhance our understanding of the nature of aneuploidy in human cancer. We find that human solid tumor cells preferentially lose small chromosomes, whereas gains of chromosomes are a poor prognostic marker for ovarian adenocarcinoma.
Materials and Methods
Karyotype analysis
Karyotypes of human tumors from previous studies were collected from the Mitelman Database of Chromosome Aberrations in Cancer20. A total of 19,003 karyotypes from human solid tumors and 23,165 karyotypes from human non-solid tumors were analyzed and the percentages of these that had lost, gained or simultaneously lost and gained whole-chromosomes were calculated. Similarly, the gain and loss rates were determined for individual chromosomes. The mean values of gain and loss rates were calculated as follows: (blue bars in Figures 1b, 3b, d, 5b and Supporting Information Figure 4; see also Supporting Information Tables 1, 4, 5 and 6). Chromosome sizes were obtained from the Human Genome Browser, University of California Santa Cruz21. Numbers of genes on each chromosome (Figures 1c, d and Supporting Information Table 1 and Figures 1 and 2) were obtained from the Nature articles that reported the sequence analyses of each of the chromosomes (1999–2006) (PubMed ID numbers: 16710414 [chromosome 1] 15815621 [2, 4], 16641997 [3], 15372022 [5], 14574404 [6], 12853948 [7], 16421571 [8], 15164053 [9], 15164054 [10], 16554811 [11], 16541075 [12], 15057823 [13], 12508121 [14], 16572171 [15], 15616553 [16], 16625196 [17], 16177791 [18], 15057824 [19], 11780052 [20], 10830953 [21], 10591208 [22], 15772651 [X], 12815422 [Y]). Whole-chromosome aneuploidy rates and the separate rates for loss, gain and concomitant loss and gain were also calculated separately for selected types of cancer or organ sites at which the tumors developed.
Figure 1.

Human solid tumors preferentially lose small chromosomes. (a) Area-proportional Venn diagram indicating the numbers and fractions of a total of 19,003 human solid tumors that have lost, gained, concomitantly lost and gained or have neither lost nor gained whole-chromosomes. (b) Rates at which human solid tumors gain or lose individual whole-chromosomes. The blue bars indicate the means of the gain and loss rates. Chromosome 7 is gained significantly more frequently than any of the other chromosomes (p<0.01; two-sided Grubbs’ test). (c) Scatter plot of chromosome size in megabases (Mb) in relationship to the rate at which the chromosome is lost in human solid tumors. (d) Scatter plot of the number of genes on the chromosome in relationship to the rate at which the chromosome is lost in human solid tumors.
Figure 3.

Different types of human solid tumors have distinct whole-chromosome aneuploidy signatures. (a) Area-proportional Venn diagram as in Figure 1a. However, these data are derived from The Cancer Genome Atlas (TCGA) array-based comparative genomic hybridization (aCGH) data of 570 human ovarian serous cystadenocarcinomas. (b) Rates at which ovarian serous cystadenocarcinomas gain or lose individual whole-chromosomes. Blue bars indicate the means of the gain and loss rates. Chromosome 20 is gained significantly more frequently than any of the other chromosomes (p<0.01; two-sided Grubbs’ test), whereas chromosome 22 is significantly more frequently lost than any of the other chromosomes (p<0.05; two-sided Grubbs’ test). (c) Area-proportional Venn diagram of 520 human colorectal adenocarcinomas using aCHG-derived data from TCGA. The areas between (a) and (c) are not proportional. (d) Individual whole-chromosome gain and loss rates as in (b) but for colorectal adenocarcinomas. Chromosomes 13 and 18 are more frequently gained and lost, respectively, than the other chromosomes (p<0.01 and p<0.05, respectively; two-sided Grubbs’ tests).
Figure 5.

Human solid and non-solid tumors have markedly different whole-chromosome aneuploidy signatures. (a) Area-proportional Venn diagram as in Figure 1a but for non-solid tumors. (b) Whole-chromosome gain and loss rates for individual chromosomes in non-solid cancers. The blue bars indicate the mean of the gain and loss rates. Chromosome 21 is gained significantly more frequently than any of the other chromosomes (p<0.01; two-sided Grubbs’ test). (c) Comparison of the means of the gain and loss rates of each chromosome for solid (blue) and non-solid tumors (orange). These frequencies represent the respective blue bars in Figures 1b and 5b. Bars above the x-axis indicate a bias towards gain, those below the axis a bias towards loss. The average on the far right corresponds to the average of the plotted gain and negative loss rates of all chromosomes. See also main text for details.
Figure 2.

Whole-chromosome aneuploidy rates in human solid tumors vary among tumor types and tumor sites. (a) Whole-chromosome aneuploidy rates for selected tumor types. (b) Whole-chromosome aneuploidy rates for tumors that developed in various different organs.
Array-based comparative genomic hybridization data analysis
Array-based comparative genomic hybridization (aCGH) data, including segmented copy number (Affymetrix SNP6) and patient survival data, were obtained from The Cancer Genome Atlas (TCGA) from the National Cancer Institute22. For each individual tumor sample, whole-chromosome aneuploidy rates were extracted from these data by scoring individual chromosomes as gained or lost if at least 90% of the chromosome was gained or lost by aCGH (absolute log2 copy-number threshold >= 0.2), respectively. Data from the X and Y chromosomes were not available. Therefore, all analyses that involved aCGH data were limited to the 22 autosomes.
Statistical analyses
A two-tailed Chi-square test was used to assess whether chromosomes are more frequently lost than gained in various tumor types. To determine whether some chromosomes were significantly more frequently gained or lost than other chromosomes, a two-sided Grubbs’ test for outliers was used. GraphPad Prism software (GraphPad Software, Inc.) was used to approximate the p value for outliers (p<0.05 or p<0.01, if significant).
For linear regression analysis (Figure 1 and Supporting Information Figures 1 and 2), the best-fit regression line was determined by minimizing the sum of the squares of the differences between the actual sizes of the chromosomes or numbers of genes on the chromosomes and the linear regression line. The goodness-of-fit of the regression lines was expressed by R2. The p values express the probability that the slope of the linear regression line is zero.
For survival curves, we performed log-rank (Mantle-Cox) tests to determine whether differences were statistically significant. All events were considered significant if the p values were smaller than 0.05. Where exact p values are omitted, they are summarized as follows: ns, not significant (i.e., p>0.05); *, 0.01<p<0.05; **, 0.001<p<0.01; ***, p<0.001.
Database information
Data used in this article are publicly available from the National Cancer Institute’s Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer (http://cgap.nci.nih.gov/Chromosomes/Mitelman)20 and The Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov/)22. None of the data that we generated required databank submission.
Results
Human solid tumor cells preferentially lose chromosomes
We analyzed numerical chromosome aberrations in 19,003 human solid tumors using their karyotypes as previously reported in the literature20. This revealed that approximately two thirds of all human solid tumors, 67.9%, exhibit numerical chromosome aneuploidy (Figure 1a and Supporting Information Table 1). Importantly, chromosome loss occurs significantly more frequently than chromosome gain (p<0.0001; two-sided Chi-square test) and almost a fourth of all solid tumors have simultaneously lost and gained one or multiple chromosomes (Figure 1a). The latter is consistent with the observation that tumor cells often have complex karyotypes which, in addition to abnormal chromosome numbers, present with structural abnormalities such as deletions, amplifications and translocations20,22–24.
We also determined the individual rate at which each chromosome is lost or gained (Figure 1b and Supporting Information Table 1). This revealed that individual chromosomes differ in their likelihood to be lost or gained. When we determined the mean of the gain and loss rates for each chromosome (blue bars in Figure 1b), it became apparent that 18 out of the 24 chromosomes are more frequently lost than gained. Three chromosomes are lost and gained at approximately the same rates (2, 5 and 8) and only three others (7, 12 and 20) show a clear bias towards gain. At an incidence of about one in seven solid tumors (14.3%), or one in five solid aneuploid tumors (21.1%), chromosome 7 is significantly more frequently gained than any of the other chromosomes (p<0.01; two-sided Grubbs’ test). Interestingly, chromosome 7 is also lost at a lower frequency than any of the other chromosomes, although that difference is not statistically significant (p>0.05; two-sided Grubbs’ test; Figure 1b and Supporting Information Table 1). Nonetheless, this indicates that there is selective pressure for cancer cells to retain both copies of chromosome 7 and perhaps acquire extra copies of it.
Solid tumor cells preferentially lose – but not gain – small chromosomes
Next, we asked whether there is a relationship between the likelihood that a chromosome is lost or gained and its size. To investigate that, we independently plotted chromosome size against the rate of loss and gain. Linear regression analysis indicates that small chromosomes are lost significantly more readily than larger ones (p=0.0208; R2=0.2199; Figure 1c).
Recent studies have shown that trisomic yeast and mouse cells – i.e., cells that have a 2n+1 chromosome complement – proliferate more slowly than diploid cells and that that effect is greater as the size of the extra chromosome increases25,26. However, we do not find a significant inverse linear relationship between chromosome size and the likelihood that it is gained in human solid tumors (p=0.8238; R2=0.0023; Supporting Information Figure 1a). The observed phenomenon in yeast and mouse cells has been attributed to a proteotoxic effect that is caused by increased protein production25–27. Because chromosome size is not necessarily proportional to the number of genes on the chromosome – and therefore to the expected degree of a proteotoxic effect, – we also compared the number of genes on each chromosome to the respective likelihoods that the chromosomes are lost or gained. Again, we find that there is no correlation between the number of genes on a chromosome and the likelihood that it is gained (p=0.8010; R2=0.0030; Supporting Information Figure 1b). On the other hand, for chromosome loss, there is a significant inverse linear correlation between these parameters (p=0.0158; R2=0.2370; Figure 1d). Finally, we specifically analyzed trisomic (2n+1) tumors and compared the size of the extra chromosome and the number of genes it harbors to the frequency at which the trisomy occurs. We did not find significant correlations between these parameters (p=0.4052, R2=0.0317 and p=0.9723, R2=0.0001, respectively; Supporting Information Figure 1c and d). These data indicate that any proteotoxic effect of gains of large, gene-dense chromosomes is not strongly selected against during human tumor evolution (see also Discussion). Together, our analyses indicate that human cancer cells not only preferentially lose chromosomes, but also that they lose small chromosomes more readily than large ones.
Chromosome loss and gain rates vary between different tumor types and tumor sites
We next assessed whether the aneuploidy rate differs among various human tumor types. Most solid cancer types show an aneuploidy rate around the average of 67.9% (Figure 2a and Supporting Information Table 2). Among a number of selected tumor types, mantle cell lymphomas have the lowest rate of about 55%, whereas more than 86% of astrocytomas, glioblastomas and gliomas are aneuploid. The latter may be due to an inherent increased drive towards aneuploidy in these tumors or to the fact that brain tumor samples only become available at more advanced stages of disease.
To determine whether whole-chromosome aneuploidy also varies between tumors that develop at different sites, we analyzed the cytogenetic data for different organs. This revealed a wide range of aneuploidy rates, varying from slightly less than 40% in salivary gland and cervical tumors to more than 80% in renal and brain cancers (Figure 2b and Supporting Information Table 3). In addition, 18 out of the 20 tissues that we examined showed a bias towards chromosome loss. Only colon and liver tumors preferentially gain chromosomes (Supporting Information Table 3).
Taken together, the vast majority of human solid tumors preferentially lose chromosomes and aneuploidy rates markedly differ among tumor types and tumor sites. An analysis of losses and gains of individual chromosomes for each type of cancer – in particular gains of chromosome 7 in carcinomas – is likely to reveal an additional stratification (see also below).
Validation of whole-chromosome aneuploidy data by array-based comparative genomic hybridization
We next set out to validate our above observations using data that were generated by a second, independent method: array-based comparative genomic hybridization (aCGH). For this, we selected a tumor type that preferentially loses chromosomes, ovarian cancers, and a type that preferentially gains chromosomes, colon cancers (Supporting Information Table 3).
aCGH data from 570 ovarian serous cystadenocarcinomas were obtained from The Cancer Genome Atlas (TCGA)22. We find that the overall whole-chromosome aneuploidy rate in these tumors is 85% (Figure 3a). This is higher than the 60% of ovarian cancers that we found to be aneuploid by cytogenetic analysis (Figure 2b). The most likely explanation for this is that serous cystadenocarcinomas represent the most malignant form of ovarian tumors and high-grade tumors are known to be significantly more aneuploid than low-grade ones2. Consistent with our previous observations, our aCGH data analysis shows that ovarian serous cystadenocarcinomas lose chromosomes significantly more readily than they gain chromosomes (p<0.0001; two-sided Chi-square test; Figure 3a). The tendency to lose, rather than gain, chromosomes is in fact more manifest than in our analysis of all solid tumors, as only 7.5% of the ovarian tumors had exclusively gained chromosomes without having lost chromosomes, while 55% of the tumors had lost chromosomes without having gained chromosomes (Figure 3a). For all solid tumors these fractions were 16% and 28%, respectively (Figure 1a).
We also analyzed whole-chromosome gain and loss rates for individual chromosomes using this aCGH-derived data set. The overall trend of gain and loss rates was similar between the ovarian serous cystadenocarcinomas and our previous analysis of all solid tumors (Figures 1b and 3b and Supporting Information Table 4). The means of the gain and loss rates (blue bars in Figure 3b) indicate that 11 chromosomes had a clear preference to be lost. Chromosome 22 was lost significantly more frequently than any of the other chromosomes (p<0.05; two-sided Grubbs’ test). Six chromosomes showed a clear bias towards gain. Interestingly, both analyses showed the most explicit bias towards gain for chromosomes 7, 12 and 20. However, the ovarian adenocarcinomas gained chromosome 20 significantly more frequently than any of the other chromosomes (p<0.01; two-sided Grubbs’ test; Figure 3b). In addition, while chromosomes 1 and 3 are generally preferentially lost, they were not lost in any of the ovarian tumor samples.
For validation of our whole-chromosome aneuploidy data in colon cancers, we used TCGA aCGH data from 520 colorectal adenocarcinomas. This confirmed our previous observation as a significantly larger fraction of this type of cancer had gained (75%) than lost chromosomes (61%; p<0.0001; two-sided Chi-square test; Figure 3c and Supporting Information Table 5). More than half of the tumors had in fact concomitantly lost and gained whole-chromosomes.
An analysis of gain and loss rates of individual chromosomes in the colorectal tumor samples also showed that chromosomes 7, 12 and 20 contributed substantially to the chromosome gains (Figure 3d and Supporting Information Table 5). However, chromosome 13 was gained at the highest frequency (p<0.01; two-sided Grubbs’ test), in about half of all the tumors, while this chromosome is in general, including in ovarian adenocarcinomas, preferentially lost (Figures 1b, 3b and d). Five chromosomes were lost at high rates. Of these, chromosome 18 was lost at the highest frequency (p<0.05; two-sided Grubbs’ test).
Finally, the ovarian and colorectal aCGH data also confirmed our previous observation that solid tumor cells lose small chromosomes, or chromosomes with fewer genes, significantly more readily than larger chromosomes or chromosomes that encode more genes (p=0.0415, R2=0.1917 and p=0.0248, R2=0.2274, respectively; Supporting Information Figure 2a and b). Additionally, neither correlation exists for the gain of chromosomes (p=0.7834, R2=0.0039 and p=0.5664, R2=0.0167; Supporting Information Figure 2c and d).
The TCGA aCGH data that we used lacked information about copy number variations of the X and Y chromosomes. However, these chromosomes rank second and third in terms of most frequently lost chromosomes in solid tumors (Figure 1b). Therefore, our inability to include information from these chromosomes likely caused an overall underestimation of the observed effects. Despite that, using data from two independent sources, tumor karyotypes and TCGA aCGH data, we can conclude that human solid tumors preferentially lose small chromosomes, but that chromosomes 7, 12 and 20 are preferentially gained.
Clinical implications of whole-chromosome gains and losses
TCGA also documents patient survival data. We used that information to examine possible clinically relevant correlations between numerical chromosome aneuploidy and patient prognosis. Using the TCGA aCGH data from ovarian cystadenocarcinomas, we analyzed patient overall survival rates for different subgroups of tumors. Consistent with previous studies2, patients with whole-chromosome aneuploid cancers had a poorer overall survival than patients whose tumors had not gained or lost whole chromosomes (p=0.0030, log-rank test; median survival (m.s.) of 42.0 and 64.0 months, respectively; Figure 4). Additionally, patients whose tumors had lost chromosomes had essentially the same prognosis as patients from the overall aneuploidy group (p=0.8710, log-rank test; m.s. of 43.3 and 42.0 months, respectively). However, patients whose tumors had gained chromosomes had a significantly shorter overall survival than patients from the overall aneuploidy group (p=0.0446, log-rank test; m.s. of 35.4 and 42.0 months, respectively) or patients whose tumors were aneuploid, but had not gained any chromosomes (p=0.0339, log-rank test; m.s. of 43.3 months).
Figure 4.
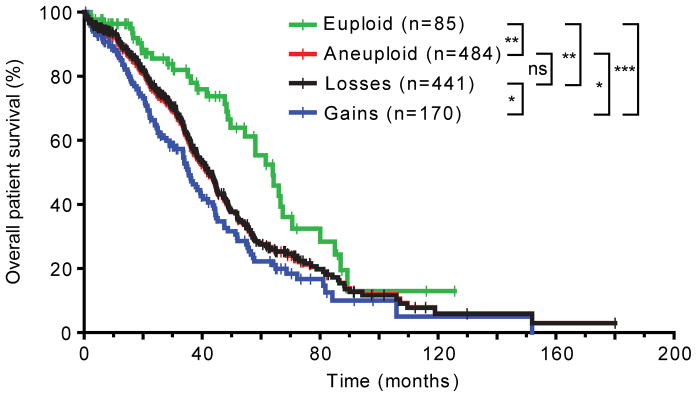
Whole-chromosome gains are more malignant than whole-chromosome losses in human ovarian cystadenocarcinoma. The overall survival of 569 ovarian serous cystadenocarcinoma patients is plotted. All p values are calculated using the log-rank test and summarized as follows: ns, not significant (i.e., p>0.05); *, 0.01<p<0.05; **, 0.001<p<0.01; ***, p<0.001. See also main text. Censored patients are indicated by vertical bars on the curves. The terms “euploid” and “aneuploid” exclusively refer to numerical chromosome abnormalities in this Figure.
We then subdivided the patients from the overall aneuploidy group into cohorts with exclusive chromosome loss (“losses only”), exclusive chromosome gain (“gains only”) and simultaneous chromosome loss and gain (“gains and losses”). Patients from the “gains and losses” group had the poorest prognosis (m.s. of 35.4 months), closely followed by the “gains only” cohort (35.5 months), and the “aneuploid” (42.0 months), “losses only” (44.9 months) and “euploid” groups (64.0 months), respectively (Supporting Information Figure 3). All differences between each of the groups were statistically significant, except between the “aneuploid” and “losses only” groups and between the “gains only” and the other whole-chromosome aneuploidy groups. In part, the latter is likely to be due to the relatively small size of the “gains only” group (n=43) in comparison to the other groups (127≤n≤484). Due to the lack of TCGA survival data for a sufficient number of colorectal cancer patients, we also could not study similar correlations for colorectal tumors. Nonetheless, our analysis indicates that in ovarian serous cystadenocarcinomas, chromosome gains confer a poorer prognosis than chromosome losses.
Distinct chromosome gain and loss rates between solid and non-solid cancers may underlie their fundamentally different etiologies
We also analyzed cytogenetic data of 23,165 non-solid cancers. With 59.9%, non-solid tumors have a lower whole-chromosome aneuploidy rate than the 67.9% in solid tumors (Figure 5a and Supporting Information Table 6). These tumors had also more frequently lost than gained chromosomes (p<0.0001; two-sided Chi-square test). Yet, the fractions of tumors that had only lost chromosomes or only gained chromosomes differed by only 2.7%, which is more than 4-fold lower than the 12.0% difference that we observed in solid tumors (Figures 1b, 5b and c ).
An analysis of the gain and loss rates of individual chromosomes showed that only 9 chromosomes had a clear bias towards loss, compared to 18 in solid tumors (Figures 1a, 5a, b and c). Eight chromosomes had a clear preference towards gain in these cancers, compared to only 3 in solid tumors. Overall, the gain and loss rates seemed much more balanced in non-solid tumors, in contrast to the clear bias towards loss in solid malignancies. This was confirmed by the average of all gain rates and the negative loss rates for each of the tumor types: −1.73% (i.e., a bias towards loss) for solid tumors and 0.07% (i.e., a negligible bias towards gain) for non-solid tumors (“average” in Figure 5c). Most strikingly, as compared to solid tumors, for half of all chromosomes the preference for loss or gain was reversed to gain and loss, respectively, in non-solid tumors (Figure 5c). The most outstanding chromosome in this regard is chromosome 7, which in solid tumors is gained at the highest frequency and lost at the lowest frequency, whereas it is lost at the highest rate in non-solid cancers.
Together, these observations reveal the existence of dramatically different biases of individual chromosome gains and losses in solid and non-solid cancers.
A comparison between early- and late-stage whole-chromosome aneuploidies in astrocytoma provides insights into tumor evolution
Finally, we wondered how whole-chromosome aneuploidy signatures may evolve during tumor progression. For astrocytomas, early- and late-stage were clearly defined as stage I–II (n=90) and stage III–IV (n=516) tumors, respectively. Using these data, we performed analyses similar to the ones above. This revealed a number of differences (Supporting Information Figure 4), of which the most prominent ones are summarized in Figure 6. For this tumor type, the preference to lose – rather than gain – chromosomes exists both at early and late stages of disease. Late-stage tumors are more frequently aneuploid and the fraction of tumors that concurrently sustains gains and losses increases from less than one in six to nearly half of the tumors. For almost all individual chromosomes both the gain and loss rates increase during tumor progression. Chromosome 8 is the most notable exception to this, as a preference towards gain changes to a preference towards loss from early to advanced stages of disease. Remarkably, loss of the X and Y chromosomes (and to some extend chromosome 22) occurs almost exclusively at early stages during tumor development, because their loss rates are only marginally increased at advanced stages. In contrast, chromosome 7 gain and chromosome 10 loss are enriched in stage III–IV cancers. Much like in glioblastomas23, they each occur in approximately 40% of the astrocytomas. Concurrence of these events increases from only 1% in the early stage tumors to more than a quarter in late stage astrocytomas (Figure 6a). Lastly, the number of whole-chromosome aberrations per tumor increases during cancer progression (Figure 6b). Together, these observations provide novel insights into the tumor evolution of astrocytomas.
Figure 6.
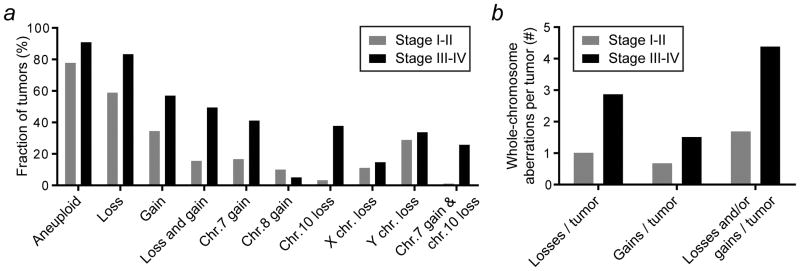
A comparison between early- and late-stage whole-chromosome aneuploidies in human astrocytoma provides insights into tumor evolution. (a) Summary of the most striking differences in whole-chromosome aneuploidy signatures between stage I–II and stage III–IV astrocytomas (see also main text and Supporting Information Figure 4). (b) The number of whole-chromosome aberrations per tumor increases during tumor progression.
Discussion
Various studies have reported deletions and amplifications of smaller or larger genomic regions in human cancers1,20,22–24. However, whole-chromosome instability, the missegregation of whole chromosomes during mitosis, has not been quantitatively examined in a large tumor data set prior to this study. Using cytogenetic data of 19,003 human solid tumors we found that solid cancer cells preferentially lose whole chromosomes. A bias towards loss – rather than gain or random missegregation – of chromosomes during mitosis and meiosis has also been observed in mouse models of whole-chromosome instability, fertility and development of the central nervous system28–30. These independent observations in different biological contexts suggest that the simultaneous gain of many loci is detrimental for cells, while a concomitant hemizygosity for a large number of genes can more readily be sustained. However, in these cases, the identity of the gained or lost chromosome, and hence the genes involved, is likely to be important (see also below).
To our knowledge, the relationship between chromosome size and its rate of loss in human solid cancers has not previously been studied. However, two groups cultured human lymphocytes to study chromosome loss rates in relation to age and gender and found that small chromosomes are lost more frequently than large chromosomes in adult, but not in newborn-derived cells31,32. Another in vitro study reported a significantly increased rate of chromosome gain, but not loss, of small chromosomes relative to large ones in patients with a non-solid form of cancer, chronic myeloid leukemia33. In human gametes, the most common aneuploidies involve chromosomes 16, 21 and 22 in oocytes and chromosomes 21, 22, X and Y in sperm cells34,35. We find that loss of chromosomes 22, X and Y also occur at the highest frequencies in solid tumors – including at early stages during astrocytomagenesis. This raises the possibility that loss of these chromosomes in tumors is simply due to a high spontaneous loss rate (perhaps due to size or other factors) independent of any oncogenic advantage per se. Alternatively, there may be a growth advantage that results from a preferential loss of these chromosomes in both normal and precancerous cells.
Tumor cells have been shown to exhibit endoreduplication, i.e., genome replication in the absence of cell division, thus leading to triploidy or tetraploidy36. Our data suggest that this phenomenon only plays a minor or temporary role, because we do not observe that the vast majority of chromosomes are gained. Instead, we find that cancer cells significantly more often lose chromosomes. Alternatively, if endoreduplication does play an important role, our results indicate that that is accompanied or followed by a massive loss of most chromosomes. Irrespective of the mechanism (e.g., more frequent gain of chromosome 7 or less frequent loss of that chromosome following endoreduplication), the observed endpoint (e.g., frequent gain of chromosome 7) is the most significant from a clinical perspective (see also our clinical data in Figure 4). Consistent with previous studies2, we find that euploidy supports better overall patient survival. Additionally, our analyses suggest that, during tumorigenesis, chromosome gains may generally be selected against. Only about 1 in 6.2 solid tumors exclusively sustain chromosome gains without losses, compared to 1 in 3.6 tumors with exclusive losses, and only three chromosomes are preferentially gained. Interestingly, trisomy 21, observed in Down syndrome patients, in fact protects, rather than predisposes to solid cancers37–39. Additionally, trisomies for larger chromosomes cause embryonic or perinatal lethality, such as in Patau (trisomy 13) and Edwards (trisomy 18) syndrome40, and trisomies for yet other autosomes are not observed in humans, suggesting that they may suppress, rather than enhance tumor development. Indeed, the gain of chromosomes has been shown to slow cell proliferation, which is postulated to be caused by a proteotoxic effect as a larger number of genes are concomitantly expressed in the cell25–27. Our data do not support this latter component of the hypothesis as we find that in trisomic tumors larger chromosomes or chromosomes with higher gene numbers are not underrepresented relative to their smaller or less gene-dense counterparts. Also, we observed that chromosome gains are significantly more frequent in more advanced astrocytomas and are a poorer prognostic marker than chromosome losses in a large cohort of ovarian serous cystadenocarcinoma patients. The enhanced degree of malignancy of tumors that have gained chromosomes raises the possibility that at advanced stages of disease, increases in oncogene copy numbers are more important drivers than the loss of tumor suppressor loci.
We find that chromosomes 7, 12 and 20 are the only three chromosomes that have a significant bias towards gain in solid tumors. Chromosome 7 is lost at the lowest frequency and is gained significantly more often than any of the other chromosomes. Gains of chromosome 7 have been reported in a variety of cancers, including those in skin41, lung42, breast43, colon44, prostate45, kidney46 and bone47. It has also been suggested to occur in early stages of brain cancer48 and colon tumorigenesis. In the latter, it is thought to precede APC loss and mutation of KRAS or TP5349. Chromosome 7 harbors a number of oncogenes, including EGFR, BRAF and SHH. EGFR overexpression occurs in a variety of cancers and the EGFR gene is located close to the centromere (at 7p11.2), thus making it less probable that it is lost by translocations or deletions of the chromosome arm on which it is located. BRAF-activating mutations and aberrant activation of the hedgehog pathway are common in various types of carcinoma. Similarly, chromosomes 12 and 20 harbor the KRAS, CDK4, MDM2, BCL2L1 (encoding Bcl-x), E2F1 and CDC25B genes, all of which have been shown to possess oncogenic properties. Thus, the acquisition of extra copies of chromosome 7, 12 or 20 may provide tumor cells with a mechanism to elevate the protein levels of these genes.
While solid tumors have a clear tendency to lose chromosomes, this bias is much less obvious in non-solid tumors. In solid cancers, 18 chromosomes have a clear bias towards loss and only 3 towards gain, whereas for non-solid malignancies these numbers are 10 and 8, respectively. For 12 out of the 24 chromosomes, the bias towards loss or gain is reversed. For chromosomes 2 and 5, this is only a minor difference, but for the other 10 chromosomes (4, 6, 7, 10, 11, 14 and 18–21) the reversal is dramatic. In this regard, chromosome 7 stands out again; not only is it lost at the lowest frequency and gained at the highest frequency in solid tumors, it is also lost at the highest frequency in non-solid cancers. This suggests that the gain of oncogenes located on chromosome 7 plays a far more important role in solid cancers than in non-solid cancers. Why such dramatic differences in chromosome gain and loss rates are so different between solid and non-solid tumors is a question of obvious interest.
We find that chromosome 21 is preferentially lost in solid tumors (Figure 1b), while it is the most frequently gained chromosome in non-solid cancers (Figure 5b). Interestingly, in Down syndrome patients, trisomy 21 protects against solid cancers, but predisposes to hematological malignancies37–39,50. This indicates that the gain or loss of specific whole-chromosomes can significantly accelerate tumor progression of specific subtypes of cancers. For instance, gain of chromosome 13, which we find occurs in about half of all colorectal adenocarcinomas (Figure 3d), may be critical during colorectal tumorigenesis.
Finally, understanding why small chromosomes are preferentially lost and why gains are an extremely poor prognostic marker in solid tumors may lead to new insights into the nature of human tumor evolution. We speculate that during early tumorigenesis, the loss of smaller chromosomes results in a lower probability of losing vital housekeeping functions, while at the same time specific tumor suppressor loci are lost on these chromosomes. Chromosome gains, which are generally disfavored for as yet unknown reasons, may be preferentially acquired following whole-chromosome loss or the acquisition of structural chromosomal aberrations, at which time increases in oncogene copy numbers may confer enhanced malignancy. Our observations comparing aneuploidy signatures of early- and late-stage astrocytoma are consistent with this possibility. In any event, our analysis expands our knowledge of the nature of whole-chromosome aneuploidy in human tumors, provides some insights into tumor evolution and may ultimately be used for prognostic or therapeutic purposes.
Supplementary Material
Novelty and impact.
Many studies cite the prevalence of aneuploidy in human cancers. However, a systematic study of whole-chromosome aneuploidy is lacking. We find that nearly all chromosomes, particularly small ones, are preferentially lost. Only three chromosomes show a significant preference towards chromosome gain. Despite the strong bias towards chromosome loss, chromosome gains constitute a poorer prognostic marker in ovarian carcinoma patients. Our results facilitate more accurate genetic cancer modeling and the development of more effective treatment strategies.
Acknowledgments
We thank Daniel Marks, Juan-Manuel Schvartzman and Rozario I. Thomas for critical reading of the manuscript. This work was supported by a Young Investigator Award from Alex’s Lemonade Stand Foundation (P.H.G.D.), the National Institutes of Health (U24CA143840; N.S.) and the Breast Cancer Research Foundation (R.B).
Abbreviations
- aCGH
array-based comparative genomic hybridization
- Mb
megabases
- m.s
median survival
- MVA
mosaic variegated aneuploidy
- TCGA
The Cancer Genome Atlas
- W-CIN
whole-chromosome instability
References
- 1.Albertson DG, Collins C, McCormick F, Gray JW. Chromosome aberrations in solid tumors. Nat Genet. 2003;34:369–76. doi: 10.1038/ng1215. [DOI] [PubMed] [Google Scholar]
- 2.Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006;38:1043–8. doi: 10.1038/ng1861. [DOI] [PubMed] [Google Scholar]
- 3.McClelland SE, Burrell RA, Swanton C. Chromosomal instability: a composite phenotype that influences sensitivity to chemotherapy. Cell Cycle. 2009;8:3262–6. doi: 10.4161/cc.8.20.9690. [DOI] [PubMed] [Google Scholar]
- 4.Sotillo R, Schvartzman JM, Socci ND, Benezra R. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature. 2010;464:436–40. doi: 10.1038/nature08803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guerrero AA, Gamero MC, Trachana V, Futterer A, Pacios-Bras C, Diaz-Concha NP, Cigudosa JC, Martinez AC, van Wely KH. Centromere-localized breaks indicate the generation of DNA damage by the mitotic spindle. Proc Natl Acad Sci U S A. 2010;107:4159–64. doi: 10.1073/pnas.0912143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janssen A, van der Burg M, Szuhai K, Kops GJ, Medema RH. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science. 2011;333:1895–8. doi: 10.1126/science.1210214. [DOI] [PubMed] [Google Scholar]
- 7.Sheltzer JM, Blank HM, Pfau SJ, Tange Y, George BM, Humpton TJ, Brito IL, Hiraoka Y, Niwa O, Amon A. Aneuploidy drives genomic instability in yeast. Science. 2011;333:1026–30. doi: 10.1126/science.1206412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53–8. doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanks S, Coleman K, Reid S, Plaja A, Firth H, Fitzpatrick D, Kidd A, Mehes K, Nash R, Robin N, Shannon N, Tolmie J, et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet. 2004;36:1159–61. doi: 10.1038/ng1449. [DOI] [PubMed] [Google Scholar]
- 10.de Voer RM, Hoogerbrugge N, Kuiper RP. Spindle-assembly checkpoint and gastrointestinal cancer. N Engl J Med. 2011;364:1279–80. doi: 10.1056/NEJMc1101053. [DOI] [PubMed] [Google Scholar]
- 11.Perez de Castro I, de Carcer G, Malumbres M. A census of mitotic cancer genes: new insights into tumor cell biology and cancer therapy. Carcinogenesis. 2007;28:899–912. doi: 10.1093/carcin/bgm019. [DOI] [PubMed] [Google Scholar]
- 12.Malumbres M. Oncogene-induced mitotic stress: p53 and pRb get mad too. Cancer Cell. 2011;19:691–2. doi: 10.1016/j.ccr.2011.05.023. [DOI] [PubMed] [Google Scholar]
- 13.Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. 2009;10:478–87. doi: 10.1038/nrm2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schvartzman JM, Sotillo R, Benezra R. Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat Rev Cancer. 2010;10:102–15. doi: 10.1038/nrc2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gordon DJ, Resio B, Pellman D. Causes and consequences of aneuploidy in cancer. Nat Rev Genet. 2012;13:189–203. doi: 10.1038/nrg3123. [DOI] [PubMed] [Google Scholar]
- 16.Hernando E, Nahle Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, Michel L, Mittal V, Gerald W, Benezra R, Lowe SW, Cordon-Cardo C. Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature. 2004;430:797–802. doi: 10.1038/nature02820. [DOI] [PubMed] [Google Scholar]
- 17.Schvartzman JM, Duijf PH, Sotillo R, Coker C, Benezra R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell. 2011;19:701–14. doi: 10.1016/j.ccr.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sotillo R, Hernando E, Diaz-Rodriguez E, Teruya-Feldstein J, Cordon-Cardo C, Lowe SW, Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 20.Mitelman F, Johansson B, Mertens F. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. 2012 http://cgap.nci.nih.gov/Chromosomes/Mitelman.
- 21.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.TCGARN Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.TCGARN Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, Mc Henry KT, Pinchback RM, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322:703–9. doi: 10.1126/science.1160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916–24. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- 27.Tang YC, Williams BR, Siegel JJ, Amon A. Identification of aneuploidy-selective antiproliferation compounds. Cell. 2011;144:499–512. doi: 10.1016/j.cell.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 29.Jin F, Hamada M, Malureanu L, Jeganathan KB, Zhou W, Morbeck DE, van Deursen JM. Cdc20 is critical for meiosis I and fertility of female mice. PLoS Genet. 2010;6:e1001147. doi: 10.1371/journal.pgen.1001147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaushal D, Contos JJ, Treuner K, Yang AH, Kingsbury MA, Rehen SK, McConnell MJ, Okabe M, Barlow C, Chun J. Alteration of gene expression by chromosome loss in the postnatal mouse brain. J Neurosci. 2003;23:5599–606. doi: 10.1523/JNEUROSCI.23-13-05599.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Richard F, Aurias A, Couturier J, Dutrillaux AM, Flury-Herard A, Gerbault-Seureau M, Hoffschir F, Lamoliatte E, Lefrancois D, Lombard M, et al. Aneuploidy in human lymphocytes: an extensive study of eight individuals of various ages. Mutat Res. 1993;295:71–80. doi: 10.1016/0921-8734(93)90003-l. [DOI] [PubMed] [Google Scholar]
- 32.Neurath P, DeRemer K, Bell B, Jarvik, Kato T. Chromosome loss compared with chromosome size, age and sex of subjects. Nature. 1970;225:281–2. doi: 10.1038/225281a0. [DOI] [PubMed] [Google Scholar]
- 33.Pegoraro L, Rovera G, Masera P, Gavosto F. Chromosome Size and Aneuploidy. Lancet. 1967;2:618. [Google Scholar]
- 34.Martin RH, Ko E, Rademaker A. Distribution of aneuploidy in human gametes: comparison between human sperm and oocytes. Am J Med Genet. 1991;39:321–31. doi: 10.1002/ajmg.1320390315. [DOI] [PubMed] [Google Scholar]
- 35.Martin RH. Meiotic errors in human oogenesis and spermatogenesis. Reprod Biomed Online. 2008;16:523–31. doi: 10.1016/s1472-6483(10)60459-2. [DOI] [PubMed] [Google Scholar]
- 36.King RW. When 2+2=5: the origins and fates of aneuploid and tetraploid cells. Biochim Biophys Acta. 2008;1786:4–14. doi: 10.1016/j.bbcan.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet. 2000;355:165–9. doi: 10.1016/S0140-6736(99)05264-2. [DOI] [PubMed] [Google Scholar]
- 38.Satge D, Sasco AJ, Carlsen NL, Stiller CA, Rubie H, Hero B, de Bernardi B, de Kraker J, Coze C, Kogner P, Langmark F, Hakvoort-Cammel FG, et al. A lack of neuroblastoma in Down syndrome: a study from 11 European countries. Cancer Res. 1998;58:448–52. [PubMed] [Google Scholar]
- 39.Yang Q, Rasmussen SA, Friedman JM. Mortality associated with Down’s syndrome in the USA from 1983 to 1997: a population-based study. Lancet. 2002;359:1019–25. doi: 10.1016/s0140-6736(02)08092-3. [DOI] [PubMed] [Google Scholar]
- 40.Lakovschek IC, Streubel B, Ulm B. Natural outcome of trisomy 13, trisomy 18, and triploidy after prenatal diagnosis. Am J Med Genet A. 2011;155A:2626–33. doi: 10.1002/ajmg.a.34284. [DOI] [PubMed] [Google Scholar]
- 41.Carless MA, Griffiths LR. Cytogenetics of melanoma and nonmelanoma skin cancer. Adv Exp Med Biol. 2008;624:227–40. doi: 10.1007/978-0-387-77574-6_18. [DOI] [PubMed] [Google Scholar]
- 42.Testa JR, Siegfried JM, Liu Z, Hunt JD, Feder MM, Litwin S, Zhou JY, Taguchi T, Keller SM. Cytogenetic analysis of 63 non-small cell lung carcinomas: recurrent chromosome alterations amid frequent and widespread genomic upheaval. Genes Chromosomes Cancer. 1994;11:178–94. doi: 10.1002/gcc.2870110307. [DOI] [PubMed] [Google Scholar]
- 43.Rohen C, Meyer-Bolte K, Bonk U, Ebel T, Staats B, Leuschner E, Gohla G, Caselitz J, Bartnitzke S, Bullerdiek J. Trisomy 8 and 18 as frequent clonal and single-cell aberrations in 185 primary breast carcinomas. Cancer Genet Cytogenet. 1995;80:33–9. doi: 10.1016/0165-4608(94)00164-7. [DOI] [PubMed] [Google Scholar]
- 44.Becher R, Gibas Z, Sandberg AA. Involvement of chromosomes 7 and 12 in large bowel cancer: trisomy 7 and 12q. Cancer Genet Cytogenet. 1983;9:329–32. doi: 10.1016/0165-4608(83)90080-8. [DOI] [PubMed] [Google Scholar]
- 45.Bandyk MG, Zhao L, Troncoso P, Pisters LL, Palmer JL, von Eschenbach AC, Chung LW, Liang JC. Trisomy 7: a potential cytogenetic marker of human prostate cancer progression. Genes Chromosomes Cancer. 1994;9:19–27. doi: 10.1002/gcc.2870090105. [DOI] [PubMed] [Google Scholar]
- 46.Hagenkord JM, Gatalica Z, Jonasch E, Monzon FA. Clinical genomics of renal epithelial tumors. Cancer Genet. 2011;204:285–97. doi: 10.1016/j.cancergen.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 47.Heyning FH, Jansen PM, Hogendoorn PC, Szuhai K. Array-based comparative genomic hybridisation analysis reveals recurrent chromosomal alterations in primary diffuse large B cell lymphoma of bone. J Clin Pathol. 2010;63:1095–100. doi: 10.1136/jcp.2010.078915. [DOI] [PubMed] [Google Scholar]
- 48.Sareen D, McMillan E, Ebert AD, Shelley BC, Johnson JA, Meisner LF, Svendsen CN. Chromosome 7 and 19 trisomy in cultured human neural progenitor cells. PLoS One. 2009;4:e7630. doi: 10.1371/journal.pone.0007630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ly P, Eskiocak U, Kim SB, Roig AI, Hight SK, Lulla DR, Zou YS, Batten K, Wright WE, Shay JW. Characterization of aneuploid populations with trisomy 7 and 20 derived from diploid human colonic epithelial cells. Neoplasia. 2011;13:348–57. doi: 10.1593/neo.101580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Malinge S, Izraeli S, Crispino JD. Insights into the outcomes, and mechanisms of leukemogenesis in Down syndrome. Blood. 2009;113:2619–28. doi: 10.1182/blood-2008-11-163501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.