

Abstract
The kinetics and thermodynamics of formation of Cu(II)-superoxo (Cu-O2) complexes by the reaction of Cu(I) complexes with dioxygen (O2) and the reduction of Cu(II)-superoxo complexes to dinuclear Cu-peroxo complexes are discussed. In the former case, electron transfer from a Cu(I) complex to O2 occurs concomitantly with binding of O2•− to the corresponding Cu(II) species. This is defined as an inner-sphere Cu(II) ion-coupled electron transfer process. Electron transfer from another Cu(I) complex to preformed Cu(II)-superoxo complexes also occurs concomitantly with binding of the the Cu(II)-peroxo species with the Cu(II) species to produce the dinuclear Cu-peroxo (Cu2-O2) complexes. The kinetics and thermodynamics of outer-sphere electron-transfer reduction of Cu2-O2 complexes are also been discussed in light of the Marcus theory of outer-sphere electron transfer.
Keywords: copper-dioxygen complexes, metal ion-coupled electron transfer, Marcus theory of electron transfer, outer-sphere electron transfer, inner-sphere electron transfer
1. Introduction
Reactions of copper(I) complexes with molecular oxygen (O2) play fundamental roles in many chemical and biological processes [1–8]. Copper dependent proteins perform a diverse array of oxidative and oxygenative reactions. In particular, dopamine-β-monooxygenase (DβM) and peptidylglycine-α-hydroxylating monooxygenase (PHM) [9–11] enzymes possess a so-called noncoupled binuclear active site [12], which comprises two Cu centers separated by ~11 Å. Dioxygen binding and substrate hydroxylation occur at one of the copper sites designated as CuM. In an important PHM X-ray structure, a dioxygen-derived species presumed to be an end-on bound cupric superoxide species (i.e., CuII-O-O•−) resides adjacent to an inhibitory substrate analogue [10]. Many researchers regard the cupric superoxo species as the reactive intermediate responsible for initiating oxidation via hydrogen-atom abstraction. Thus, extensive efforts have so far been devoted to elucidate structures, electronic characteristics, and substrate reactivities of Cu-O2 complexes [2–6,13–15]. This review focuses on kinetics and thermodynamics of formation of mononuclear copper-superoxo Cu-O2 complexes as well as dinuclear copper-peroxo Cu2-O2 complexes. Whether the formation of Cu-oxygen complexes occurs via (1) stepwise outer-sphere electron transfer from Cu(I) complexes to O2, followed by fast binding of O2•− to the resulting Cu(II) complex fragment or (2) binding of O2 to Cu(I) complexes, followed by inner-sphere electron transfer from Cu(I) complexes to O2 is discussed in light of the Marcus theory of electron transfer [16,17]. The favorable thermodynamics and kinetics of formation and redox reactions of Cu-O2 and Cu2-O2 complexes are rationalized as the primary goals of this review.
2. Kinetics and thermodynamics of formation of Cu-O2 complexes
2.1. Mechanism of electron transfer from Cu(I) complexes to O2
Outer-sphere electron transfer from Cu(I) complexes with LH (tris(2-pyridyl-methyl) amine = TMPA) ligands shown in Fig. 1 to O2 in acetonitrile (MeCN) at 298 K is highly endergonic, i.e., the free energy of electron transfer (ΔGet) is largely positive (ΔGet ≫ 0), judging from the less negative one-electron oxidation potentials of [(LH)CuI(MeCN)]+ (E(CuII/CuI) vs. SCE = −0.03 V for LH, −0.09 V for LtBu, −0.12 V for LMeO) [18] as compared with the one-electron reduction potential of O2 (E(O2/O2•−) vs. SCE = −0.86 V) [19]. Despite these extremely unfavorable energetics of electron transfer from [(LH)CuI]+ to O2, [(LH)CuI]+ (λmax = 338 nm, εmax = 5600 M−1 cm−1) reacts rapidly with O2 in propionitrile (EtCN) forming the 1:1 adduct [(LH)CuII(O2•−)]+, (λmax = 410 nm, εmax = 4700 M−1 cm−1) [20,21]. At low temperature, [(LR)Cu(RCN)]+ reacts in a reversible fashion with O2 to form [(LR)CuII(O2•−)]+, which further reacts reversibly with [(LR)Cu(RCN)]+ ion to form the dinuclear 2:1 Cu2O2 adduct, as discussed later.
Fig. 1.
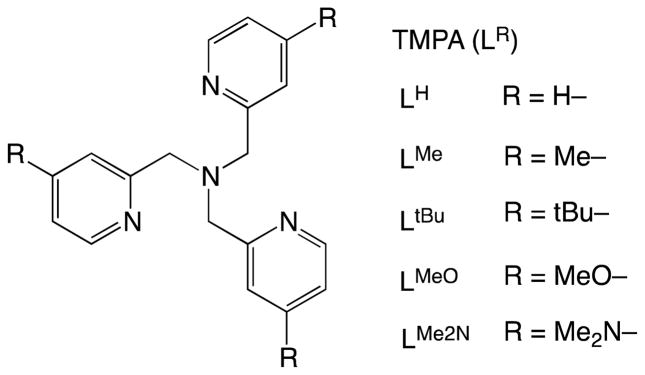
TMPA ligands (LR) employed for Cu complexes.
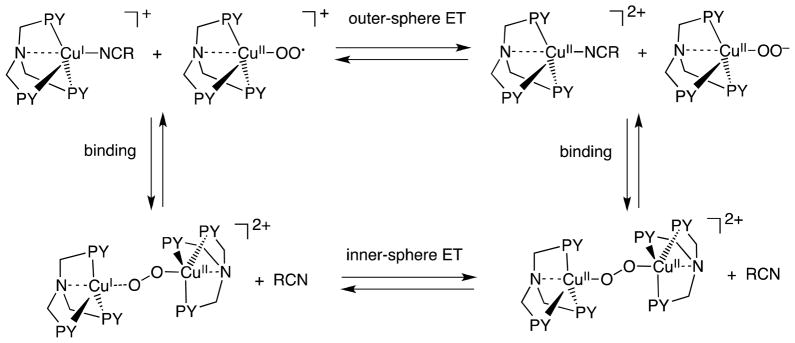
There are two possible reaction pathways for the formation of the Cu-superoxo complex: (1) outer-sphere electron transfer from [(LR)CuI(MeCN)]+ to O2, followed by binding of the resulting O2•− to the Cu(II) center to produce [(LR)CuII(O2•−)]+ and (2) binding of O2 to Cu(I) complexes, followed by inner-sphere electron transfer from Cu(I) complexes to O2 as shown in Scheme 1.
Scheme 1.

The rate constant (ket) of outer-sphere electron transfer is given by Eqn. (1),
| (1) |
where Z is the frequency factor that is taken as 1 × 1011 M−1 s−1 at 298 K [16,17]. The free energy change (ΔG≠) of electron transfer is given by the Marcus equation [Eqn. (2)] [16,17],
| (2) |
where λ is the reorganization energy of electron transfer and ΔG is the free energy change of electron transfer, which is given by Eqn. (3),
| (3) |
where e is the elementary charge, and E(D•+/D) and E(A/A•−) are the one-electron oxidation potential and the one-electron reduction potential of an electron donor (D) and acceptor (A), respectively.
The reorganization energy of electron transfer (λ) is given as an average of the reorganization energies of electron exchange between D and D•+ (λD) and that between A and A•− (λA) [Eqn. (4)] [16]. The λD value of [(LH)CuII]2+/[(LH)CuI]+ can be
| (4) |
determined from the λ value of electron transfer from decamethylferrocence (Fc*) to [(LH)CuII]2+ as follows. The second-order rate constant (ket) of electron transfer from Fc* to [(LH)CuII]2+ is 1.1 × 105 M−1 s−1 at 298 K [22]. The λvalue is 1.29 eV by using Eqns. (1) and (2) from the ΔG value (= e(E(D•+/D) – E(A/A•−) = −0.05 eV). The λD value of [(LH)CuII]2+/[(LH)CuI]+ is 1.58 eV by using Eqn. (4) using the λA value of Fc*/Fc*+ (1.01 eV) [23]. Since the λA value of O2/O2•− is 1.89 eV [23], the λ value of electron transfer from [(LH)CuI]+ to O2 is estimated to be 1.74 eV by using Eqn. (4). Then, the ket value of outer-sphere electron transfer from [(LH)CuI]+ to O2 is evaluated to be 5 × 10−23 M−1 s−1 at 298 K. The observed ket value for formation of [(LH)CuII(O2•−)]+ is 5.8 × 107 M−1 s−1 in EtCN at 298 K [18]. Because this ket value is 1030 times larger than the estimated value of outer-sphere electron transfer from [(LH)CuI]+ to O2, the latter cannot be the rate-determining step in formation of [(LH)CuII(O2•−)]+. Thus, binding of O2 to [(LH)CuI]+ may occur first, accompanied by dissociation of a solvent molecule (RCN); this is followed by electron transfer from [(LH)CuI]+ to O2 within the complex, Scheme 1. Because there was no intermediate observed in formation of [(LH)CuII(O2•−)]+ [18], binding of O2 to [(LH)CuI]+ may be too weak to be detected. Once electron transfer occurs in [(LH)CuI(O2)]+, binding of O2•− to [(LH)CuI]+ in [(LH)CuII(O2•−)]+ is much stronger than binding of O2 to [(LH)CuI]+ in [(LH)CuI(O2)]+. In such a case, electron transfer from [(LH)CuI]+ to O2 and the stronger binding to O2 should occur in a concerted manner. Such an electron transfer is classified as a “Metal ion-Coupled Electron Transfer” (MCET) [24–28]. The electron-transfer process is also defined as an inner-sphere pathway rather than an outer-sphere pathway, because there is significant interaction prior to electron transfer and the ligand is exchanged from MeCN to O2•− upon electron transfer [29,30].
The fast binding of O2•− to [(LH)CuII]2+ was confirmed by Smirnov and Roth using stopped-flow measurements of the reaction of O2•− with [(LH)CuII]2+ in 4:1 DMF:THF at 193 K [31]. Within the dead-time of the stopped-flow, the reaction of [(LH)CuII]2+ (5.5 × 10−4 M) and O2•− (1.25 × 10−4 M) gives [(LH)CuII(O2•−)]+ and [{(LH)CuII}2(O22−)]2+ [31]. The rate constant of outer-sphere electron transfer from O2•− to [(LH)CuII]2+ was also estimated by using Eqns. (1) and (2) to be 2.4 × 104 M−1 s−1, which is much smaller than the observed rate constant (> 1 × 108 M−1 s−1) [31]. Thus, electron transfer from O2•− to [(LH)CuII]2+, which is reverse of electron transfer from [(LH)CuI]+ to O2 in Scheme 1, also occurs via an inner-sphere pathway following binding O2•− to [(LH)CuII]2+ rather than occurring via an outer-sphere pathway.
Cyclic voltammetry measurements carried out on [(LR)CuI]+ revealed that the E(CuII/CuI) values vary such that as the R group becomes more electron-donating (i.e., R = OMe, tBu vs. H), more negative E(CuII/CuI) values are observed making it easier to oxidize Cu(I) to Cu(II); thermodynamically more stable Cu(II) complexes result [18]. The observed ligand effects upon E(CuII/CuI) correlate to the difference in ligand basicity. The pKa values for pyridine, 4-Me-pyridine (similar to 4-tBu-pyridine), 4-MeO-pyridine, and 4-Me2N-pyridine are 5.21, 6.03, 6.58, and 9.70, respectively [32]. In contrast to the change in E(CuII/CuI), the rate constants for formation of [(LR)CuII(O2•−)]+ fall into a small range between 1.2 × 104 and 3.1 × 104 M−1 s−1 at 183 K with ligand LR having different substituent groups (R = H, Me, tBu, MeO) [18]. This indicates that the O2 association rates are hardly affected by these variations in ligand electronic environment. The O2-binding processes are accompanied by significant activation enthalpies (7.0–7.6 kcal mol−1) and small positive activation entropies (0–5 cal K−1 mol−1) [18]. The insensitivity of the O2-binding rate constant toward variations of substituents in LR in R groups may be rationalized by the importance of the accompanying dissociation of a coordinated EtCN from the Cu(I)-complex during the O2 binding process. This is supported by the significant solvent effect on the O2 binding process. When the reaction of [(LH)CuI]+ with O2 was performed in weakly coordinating solvents such as acetone and tetrahydrofuran (THF), the stopped-flow kinetics revealed that a superoxocopper(II) species [(LH)CuII(O2•−)]+ with λmax = 420 nm formed within the stopped-flow mixing time (even at 183 K) [18]. The rate constant in THF at 183 K is estimated to be larger than 2 × 106 M−1 s−1, which is two-orders magnitude larger than the value in EtCN (1.2 × 104 M−1 s−1) [18].
The fast reaction of [(LH)CuI]+ with O2 in a weakly coordinating solvent such as THF was monitored using techniques originally developed by Gibson and co-workers [33,34], as well as diffusion controlled “flash-and-trap” experiments [35]. The photoinitiated ejection of carbon monoxide from heme-CO adducts in the presence of O2 has led to a wealth of important information for proteins such as hemoglobin, myoglobin, and cytochrome c oxidase [36–39], as well as synthetic iron-porphyrinate model compounds [35,40,41]. As is observed for many heme proteins or complexes where CO binding to the reduced metal ion (Fe(II)) protects against rapid O2-reaction [35,40,41], [(LH)CuI(CO)]+ in THF solvent is inert when appropriate mixtures of O2 and CO are introduced at low temperatures (188–218 K) [42]. Under these conditions, light excitation (λex = 355 nm, 8–10 ns pulse) into the [(LH)CuI(CO)]+ metal-to-ligand charge transfer (MLCT) band results in the immediate (within 10 ns) formation of the solvated species [(LH)CuI(thf)]+ (λmax = 333 nm; Fig. 2A) [43]. Subsequently, fast competing reactions occur (kfast, s−1 in Scheme 2), either regenerating [(LH)CuI(CO)]+ [42] or forming [(LH)CuII(O2•−)]+ (λmax = 425 nm, 600 nm in THF) [42]. The kfast is a combination of two rate constants [Eqn. (5)], and CO rebinding dominates over
Fig. 2.

Difference spectra {[(LH)CuI(X)]+ - [(LH)CuI(CO)]+} recorded after 355 nm excitation of [(LH)CuI(CO)]+ in tetrahydrofuran at 198 K under a 1 atm mixture of O2:CO = 7:3. The spectra were recorded at differing delay times: (a, top). 0 to 2 μs where [(LH)CuI(thf)]+ (λmax, 333 nm) converts to a mixture of [(LH)CuI(CO)]+ and [(LH)CuII(O2•−)]+ (λmax, 425 nm): squares (black spectrum), 0 μs; circles (red spectrum), 0.5 μs; diamonds (blue spectrum), 2.0 μs. The inset is an absorption transient monitored at 425 nm with a superimposed first-order fit (red), kobs = 3.0 × 106 s−1 and (b, bottom), 2 μs to 40 ms where [(LH)CuII(O2•−)]+ converts to [(LH)CuI(CO)]+: squares (black spectrum), 2 μs; circles (red spectrum) 5 ms; diamonds (blue spectrum), 20 ms. The inset is an absorption transient monitored at 425 nm with a superimposed first-order fit, kobs = 130 s−1. The spectrum shown in solid green is that calculated as {Abs[(LH)CuII(O2•−)]+ - Abs [(LH)CuI(CO)]+}. Figure adapted from reference 42.
Scheme 2.

| (5) |
O2-binding to [(LH)CuI(thf)]+, kCO > kO2 (Scheme 2) [42]. Values for kCO were determined in the absence of O2. The rate constant for the O2-reaction was thus determined from the slope of the plot, kfast-kCO[CO] vs. [O2].
Temperature-dependent kinetic studies on the O2-binding reaction of [(LH)CuI(thf)]+ afforded activation parameters ΔH≠ = 7.6 kJ mol−1 and ΔS≠ = −45 J mol−1 K−1 [42]. The negative ΔS≠ value suggests that the reaction proceeds via an inner-sphere electron transfer following O2-binding, which is controlled by the lability of the solvent molecule (Scheme 1). That O2-binding controls the mechanisms is also supported by the fact that the activation entropy for the same reaction of the same complex in RCN is close to zero (14 ± 18 J mol−1 K−1) [20], because with the more strongly coordinating RCN (compared to thf) is substituted by O2 in an interchange rather than in an associative mechanism. Consequently, in THF, without the interference of a coordinating solvent, the small ΔH≠ and negative ΔS≠ value indicate that O2-binding is basically unhindered. The temperature-dependent data allowed a determination of the room-temperature second-order rate constant, kO2 = 1.3 × 109 M−1 s−1, in fact greater than observed for O2-binding to any heme [42].
2.2. Binding Free Energy of O2•− to [(LH)CuII]2+
The free energy of formation of [(LH)CuII(O2•−)]+ (ΔG) is +0.02 eV at 298 K [18]. From the thermochemical cycle in Scheme 1, the binding free energy of O2•− to the Cu(II) center of [(LH)CuII]2+ (ΔGb) is estimated to be −0.81 eV (ΔGb = ΔG − ΔGet). Such strong binding of O2•− to a Lewis acidic Cu(II) center lowers the free energy of the entire process, making Cu(II)-superoxide formation possible. MCET from one-electron reductants to oxygen is related to the Lewis acidity of metal ions [45,46]. Charges and ion radii are important factors to determine the Lewis acidity of metal ions. The end-on coordination of O2•− to Sc3+ has been confirmed by observation of two different sets of six lines (14 and 21 G) due to the hyperfine splitting of two different 17O atoms (I = 5/2), because the electron spin is more localized at the terminal oxygen [47].
The actual binding energies (ΔE) for a variety of metal ions with superoxide ion (O2•−) have been derived from the gzz-values of the EPR spectra of O2•−–Mn+, providing a quantitative measure of Lewis acidity of the metal ions [45]. The gzz-values of O2•−–Mn+ complexes produced in the presence of a variety of closed shell metal ions become significantly smaller than the value for O2•− itself due to the binding of metal ion with O2•− as ligand [47]. The deviation of the gzz-value from the free spin value (ge = 2.0023) is caused by the spin-orbit interaction as given by Eqn. (5) [48,49], where λO is the spin-orbit coupling constant (0.014 eV) for oxygen [50] and ΔE is the energy splitting of πg levels due to the binding of Mn+ to O2•−.
| (5) |
The ΔE value obtained from the deviation of the gzz-value from the free spin value increases in order: monovalent cations (M+) < divalent cations (M2+) < trivalent cations (M3+) [45]. The ΔE value also increases with decreasing the ion radius when the oxidation state of the metal ion is the same. The same trend is observed for O2•− adsorbed on the surface of various metal oxides, which as well act as Lewis acids [52,53]. Scandium ion, which has the smallest ion radius among the trivalent metal cations, gives the largest ΔE value (1.00 eV) [45]. The ΔE values are directly correlated with the promoting effects of metal ions in electron-transfer reactions [45,46]. However, this method can only be applied to diamagnetic metal ions, since paramagnetic metal ion complexes with O2•− do not generally give EPR signals because of antiferromagnetic coupling or unfavorable conditions due to the presence of an S = 1 ground state, as in the case of [(TMG)3tren]CuII(O2•−)]+ ((TMG)3tren = tris(2-(N-tetramethylguanidyl)ethyl)-amine [54].
A much more general and convenient method to provide a quantitative measure of the Lewis acidity of metal ion salts is the fluorescence maxima of 10-methylacridone (AcrCO)-metal ion salt complexes [55]. The fluorescence energy (hνf) decreases with increasing the Lewis acidity of metal ion salts, and the hνf value provides quantitative measure of the Lewis acidity of metal ions including paramagnetic and redox active species. The linear correlation between the ΔE values and the hνf values was obtained as given by Eqn. (6) [55]. The stronger the acidity of the Lewis acid metal ion salts, the
| (6) |
larger is the ΔE value, the more red-shifted is the λmax value, and the smaller is the hνf value. The ΔE value of Cu(ClO4)2 is 0.96 eV from the hνf value, which is comparable to the ΔE values of Fe(ClO4)2 (0.96 eV) and Co(ClO4)2 (0.95 eV) [55]. The large ΔE value of Cu(ClO4)2 is consistent with the large binding energy of O2•− to the Cu(II) center of [(LH)CuII]2+ (ΔE = 0.77 eV).
3. Reduction of Cu-O2 complexes
3.1. Kinetics and thermodynamics of formation of Cu2-O2 complexes
The cupric-superoxo complex [(LR)CuII(O2•−)]+ is further reduced by [(LH)CuI]+ to form the dinuclear 2:1 Cu2O2 adduct, [{(LR)CuII}2(O22−)]2+ [18]. The X-ray structure (the first in Cu2-O2 chemistry) of [{(LR)CuII}2(O22−)]2+ revealed that [{(LR)CuII}2(O22−)]2+ has a bound dioxygen ligand coordinated as a μ-1,2-peroxodicopper(II) moiety, λmax = 525 nm (ε = 10,500 M−1 cm−1), and νO-O = 831 cm-1 (resonance Raman spectroscopy) [20, 21]. Fig. 3 shows the UV-vis behavior when, for example, [(LMeO)Cu(EtCN)]+ reacts with excess O2 at 179 K [18]. Upon mixing of solutions of O2 and [(LMeO)Cu(EtCN)]+, there is a fast formation of the superoxo species [(LMeO)CuII(O2•−)]+ with λmax at 413 and 598 nm; this quickly decays (~20 s) and is transformed into the dinuclear peroxo complex [{(LMeO)CuII}2(O22−)]2+ with 525 and 595 nm absorptions. The inset of Fig. 3 shows the absorbance versus time trace at 413 nm. The second-order rate constant of formation of [{(LMeO)CuII}2(O22−)]2+ was 1.9 × 104 M−1 s−1 in EtCN at 183 K [18]. Electron transfer from [(LMeO)CuI(EtCN)]+ to [(LMeO)CuII(O2•−)]+ may also occur via binding of the O2•− moiety of [(LMeO)CuII(O2•−)] to the CuI complex, followed by inner-sphere electron transfer from the CuI complex to the O2•− moiety to produce [{(LMeO)CuII}2(O22−)]2+, as opposed to outer-sphere electron transfer and binding (Scheme 3), because the activation entropy (ΔS≠) was large and negative (−11 cal mol−1 K−1) [18].
Fig. 3.

Absorption spectroscopic changes for the reaction of [(LMeO)CuI(EtCN)]+ with O2 in EtCN solvent at 179 K. Inset: Time profile for the absorbance at 413 nm due to [(LMeO)CuII(O2•−)]+. Adapted from reference 18.
Scheme 3.
If outer-sphere electron transfer from [(LMeO)CuI(EtCN)]+ to [(LMeO)CuII(O2•−)]+ is the rate-determining step, the activation entropy would be close to zero [17]. The one-electron reduction potential of [(LR)CuII(O2•−)]+ (R = H, tBu, MeO) has yet to be determined because of the instability of [(LR)CuII(O2•−)]+, precluding the determination of the binding energy of [(LR)CuII(O22−)] with [(LR)CuII]2+. The overall reactions to form the peroxodicopper(II) complexes ([{(LR)CuII}2(O22−)]2+) are accompanied by strongly negative reaction enthalpies and activation entropies, which indicates that these reactions proceed via an inner-sphere electron transfer following O2-binding, as shown in Scheme 3. This analysis was also discussed and applied to Cu(II)-superoxo complex formation, as described in Scheme 1. The thermodynamic stability for [{(LR)CuII}2(O22−)]2+ shows relatively small differences with ΔH° in the range −18 to −19 kcal mol−1 [18].
3.2. Hydride Reduction of Cu-O2 complexes
As described above, Cu(II)-superoxo complexes are mostly unstable and usually are converted to corresponding dinuclear Cu2-O2 peroxo complexes. However, Cu(II)-superoxo complexes can in some cases oxidize substrates prior to their reaction with corresponding Cu(I) species to form the Cu2-O2 peroxo complexes. Itoh and coworkers reported that mononuclear copper(II)-superoxo complexes obtained by the reactions of copper(I) complexes supported by ligands LX [1-(2-p-X-phenethyl)-5-(2-pyridin-2-ylethyl)-1,5-diazacyclooctane; X =MeO, H, NO2] undergo hydroxylation of their ligand phenethyl sidearm [56]. Such reactivity is another form of instability of Cu(II)-superoxo complexes with respect to investigation of the reactivity of Cu(II)-superoxo complexes on C–H bond activation with external substrates, although Cu(II)-superoxo complexes exhibit phenol O-H bond cleavage reactions [57,58]. When the CuI complex [LCuI]+ of a ligand L, previously employed by Masuda and co-workers [59], namely, [bis(pyrid-2-ylmethyl) {[6-(pivalamido)pyrid-2-yl]methyl}amine], was exposed to O2 in 2-methyltetrahydrofuran (MeTHF) solvent at −125 °C, however, a stable superoxo adduct [LCuII(O2•−)]+ (Fig. 4) was formed, exhibiting UV-vis absorptions [λmax = 410 nm (3700 M−1 cm−1), 585 nm (900 M−1 cm−1), 741 nm (1150 M−1 cm−1)] characteristic of a mononuclear end-bound CuII superoxo complex (Fig. 5a) [60]. Resonance Raman spectroscopic data confirmed the formulation of [LCuII(O2•−)]+ as an end-on superoxo-containing complex with O-O and Cu-O stretches of 1130 and 482 cm−1, respectively [60]. These values are higher than those for all cupric superoxo complexes previously described: ν(O-O) for the one structurally defined side-on bound cupric superoxo complex is 1043 cm−1 [61], while those for the end-on bound species range up to 1122 cm−1;. (Cu-O) varies from 422 to 474 cm−1 [54,58,60–62]. DFT calculations suggested that the superoxo moiety in [LCuII(O2•−)]+ forms an intramolecular H-bond with either the α or β oxygen, which would contribute to the relative stability of [LCuII(O2•−)]+ and can account for the higher O-O and Cu-O frequencies [60].
Fig. 4.
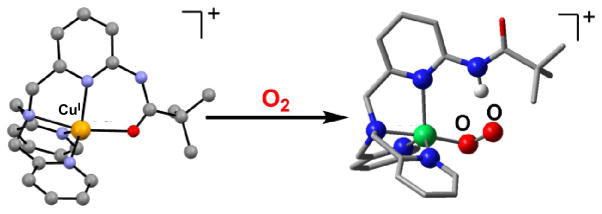
(left) Representation of the cationic portion of the X-ray structure of [LCuI]+(B(C6F5)4−), with N4O(amide) coordination (Cu-O = 2.190 Å). (right) Calculated structure of [LCuII(O2•−)]+, indicating the H-bonding interaction between the ligand and the anionic superoxo ligand. Adapted from reference 60.
Fig. 5.

(a) Spectroscopic changes of 0.4 mM [LCuII(O2•−)]+ in the presence of 8 mM BNAH at −125 °C in MeTHF. The first spectrum recorded (green) is that immediately following bubbling of O2 through a solution containing [LCuI]+ and BNAH. Inset: pseudo-first-order kinetics fit of the 741 nm data. (b) Plots of kobs as a function of BNAH, BNAD, or BzIMH concentration, used to determine the second-order rate constants. Adapted from reference 60.
The [LCuII(O2•−)]+ complex was EPR silent and quite stable, decaying only very slowly (half life > 4 h) with conversion to [{(LCuII)}2(O22−)]2+, a μ-1,2-peroxodicopper(II) complex [λmax = 541 nm (9900 M−1 cm−1)] that is otherwise observed when [LCuI]+ is oxygenated at −80 °C [59]. The stability of [LCuII(O2•−)]+ allowed to investigate the reactivity on C–H bond activation with external substrates (vide infra).
[LCuII(O2•−)]+ is unreactive toward a number of commonly employed C-H substrates, such as dihydroanthracene, xanthene, and 10-methyl-9,10-dihydroacridine, which are substrates possessing C-H bonds that are significantly weaker than those found for the DβM and PHM substrates (dopamine, 85 kcal mol−1; hippuric acid, 87 kcal mol−1) [12]. However, the addition of an excess of 1-benzyl-1,4-dihydro-nicotinamide (BNAH), an NADH analogue that is both a strong H atom (H•) and hydride (H−) donor [63,64], to solutions of [LCuII(O2•−)]+, leads to the decay of the latter, as observed by UV-vis spectroscopy (Fig. 5a) [60]. The decay dynamics of [LCuII(O2•−)]+ with large excess BNAH at −125 °C obeyed pseudo-first-order kinetics [60]. The decay was also first-order in [BNAH] and thus the second-order rate constant could be determined to be 0.19 M−1 s−1 (Fig. 5b). When BNAH was replaced by the 4,4′-dideuterated analogue, 1-benzyl-1,4-dihydro[4,4′-2H2]-nicotinamide (BNAD), a significant slowing of the reaction occurred with the second-order rate constant of 0.016 M−1 s−1; Fig. 5b) [60]. This gives a large kinetic deuterium isotope effect (KIE) of 12.1. This KIE value is comparable to the KIE of 10 reported for C–H bond cleavage of BNAH by a trans-dioxomanganese(V) porphyrin, which proceeds via proton-coupled electron transfer rather than simple electron transfer [65]. The oxidized product of BNAH was BNA+. The overall stoichiometry of the reaction observed is shown in Scheme 4, where the reduced product of [LCuII(O2•−)]+ was the μ-1,2-peroxo-dicopper(II) complex [{(LCuII)}2(O22−)]2+, which was identified by its characteristic UV-vis absorption bands (vide supra).
Scheme 4.
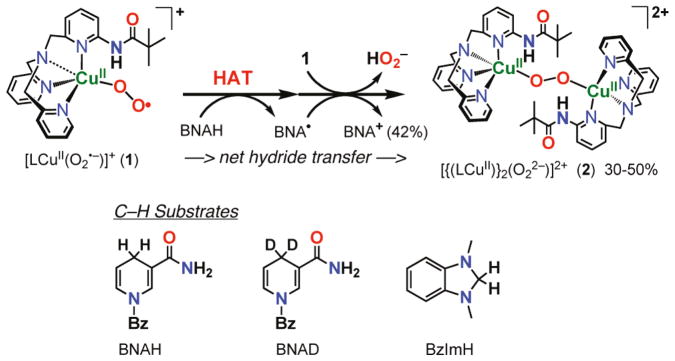
Like BNAH, 1,3-dimethyl-2,3-dihydrobenzimidazole (BzImH) possesses a weak C–H bond but has markedly different bond strengths in comparison to BNAH (homolytic, bond dissociation energy = 73.4 kcal mol−1 vs. 70.7 kcal mol−1 for BNAH; heterolytic, hydride affinity = 49.5 kcal mol−1 vs. 64.2 kcal mol−1 for BNAH) [66]. Kinetic studies revealed that the oxidation of BzImH occurs ~2.4 times slower than that of BNAH, with a second-order rate constant of 0.078 M−1 s−1 (Fig. 5b). The slower rate of C-H oxidation of BzImH (stronger H− donor) than of BNAH (stronger H• donor) thus suggests that at least for these substrates, the preferred mode of C-H activation by [LCuII(O2•−)]+ is via rate-limiting homolytic C-H bond cleavage, i.e., hydrogen atom transfer (HAT) or concerted proton-coupled electron transfer. Thus, C-H activation by [LCuII(O2•−)]+ occurs much faster than an alternative inner-sphere electron transfer pathway which would produce the dinuclear peroxo complex shown in Scheme 3.
4. Electron-transfer reduction of Cu2O2 complexes
As described above, the μ-1,2-peroxodicopper(II) complex [{(LCuII)}2(O22−)]2+ was not further reduced by BNAH. Similarly no electron transfer from a one-electron reductant Fc* to [{(LHCuII)}2(O22−)]2+ occurred in MeCN [22]. In contrast with these μ-1,2-peroxodicopper(II) complexes, a side-on bound μ-η2:η2-peroxodicopper(II) complex can be readily reduced by electron transfer from Fc* (vide infra) [67]. A binuclear copper(I) complex, [CuI2(N3)(MeCN)2](BArF)2 (N3 = (-(CH2)3-linked bis[2-(2-pyridyl)ethyl]-amine), BArF = tetrakis(pentafluorophenyl)borate), reacts with O2 to afford the μ-η2:η2-peroxodicopper(II) complex [CuII2(N3)(O22−)]2+ (Scheme 5 where L here is a solvent molecule) [68]. Electron transfer from Fc* to [CuII2(N3)(O22−)]2+ occurs to produce two equivalents of Fc*+ in acetone at 193 K [Eqn. (7)] as shown in Fig. 6, where the decay of absorbance at 490 nm due to [CuII2(N3)(O2)]2+ is accompanied by the appearance of absorbance at 780 nm due to Fc*+ (inset of Fig. 6) [67]. The one-step reaction in Fig. 6 suggests that initial electron transfer from Fc* to [CuII2(N3)(O2)]2+ is the rate-determining step followed by another rapid electron transfer from Fc* to [CuII2(N3)(O)2]+. The second-order rate constant (ket) of electron transfer from Fc* to [CuII2(N3)(O2)]2+ is 18 M−1 s−1 at 193 K [67].
Scheme 5.
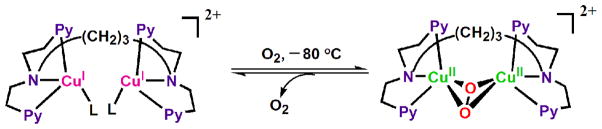
Fig. 6.

Formation and decay of the μ-η2:η2-peroxodicopper(II) complex [CuII2(N3)(O22−)]2+ (λmax = 490 nm) in the reaction of [CuI2(N3)]2+ (0.10 mM) with O2 in the presence of Fc* (0.80 mM) in acetone at 193 K. The inset shows the time profiles of the absorbance at 490 nm (black circles) and 780 nm (red circles) due to [CuII2(N3)(O22−)]2+ and Fc*+, respectively. Adapted from reference 67.
| (7) |
When Fc* was replaced by a weaker electron donor (Me2Fc), no electron transfer from Me2Fc to [CuII2(N3)(O22−)]2+ occurred at 193 K [67]. When Me2Fc is replaced by N,N,N′,N′-tetramethylphenylenediamine (TMPD), however, electron transfer from TMPD to [CuII2(N3)(O22−)]2+ occurred efficiently to completion as indicated by disappearance of the absorption band at 490 nm due to [CuII2(N3)(O22−)]2+, which was accompanied by appearance of the absorption band at 600 nm due to TMPD•+ [69,70]. Based on the one-electron oxidation potentials of TMPD (E(TMPD•+/TMPD) = 0.12 V vs. SCE) [70] and Me2Fc (E(Me2Fc+/Me2Fc) = 0.26 V vs. SCE) [71,72], the oneelectron reduction potential of [CuII2(N3)(O22−)]2+ is estimated to be 0.19 ± 0.07 V vs. SCE [67].
The μ-η2:η2-peroxodicopper(II) complex [CuII2(N3)(O22−)]2+ was also produced by electron transfer from Fc* and octamethylferrocene (Me8Fc) to [CuII2(N3)]4+ followed by the reaction of [CuI2(N3)]2+ with O2 [67]. The temperature dependence of rate constants of electron transfer from Fc* to [CuII2(N3)]4+ and [CuII2(N3)(O22−)]2+ was examined and the Eyring plots obtained are shown in Fig. 7 while the activation parameters obtained from these Eyring plots are listed in Table 1 [67].
Fig. 7.

Eyring plots of the rate constants (ket1 and ket2) of electron transfer from Fc* and Me8Fc to [CuII2(N3)]4+ and [CuII2(N3)(O22−)]2+ in acetone. Adapted from reference 67.
Table 1.
Activation parameters for electron transfer from Fc* and Me8Fc to [CuII2(N3)]4+ and [CuII2(N3)(O22−)]2+ in acetone.
| Activation parameter | [CuII2 (N3)]4+ | [CuII2(N3)(O22−)]2+ | ||
|---|---|---|---|---|
| Fc* | Me8Fc | Fc* | Me8Fc | |
| ΔH≠, kcal mol−1 | 8.7 ± | 8.5 ± 0.2 | 9.6 ± 0.3 | 10.3 ± 0.3 |
| ΔS≠, cal K−1 | 0 ± 2 | −3 ± 2 | −3 ± 2 | −1 ± 2 |
With regard to copper–dioxygen intermediates,μ-η2:η2-peroxodicopper(II) complexes are well known to be capable of reversible conversion to corresponding bis-μ-oxodicopper(III) complex isomers [73–76]. If electron transfer from Fc* and Me8Fc to [CuII2(N3)(O2)]2+occurred via the conversion to the putative isomeric bis-μ-oxo intermediate ([CuIII2(N3)(O)2]2+), the observed rate constant (kobs) would be given by Eqn. (8),
| (8) |
where Ko is the equilibrium constant for isomerization between [CuII2(N3)(O2)]2+ and [CuIII2(N3)(O)2]2+, that shown in Scheme 6 (via bis-μ-oxo complex). The Ko value is expected to be much smaller than 1 (Ko ≪ 1), because no bis-μ-oxo intermediate has been observed in this case. In such a case, the observed rate constant (kobs) would be much smaller than the rate constant of electron transfer to the putative bis-μ-oxo intermediate (kobs ≪ ket) and the observed activation entropy would not be the same as that of electron transfer, because it would be given as the sum of the activation entropy of electron transfer (ΔSet≠ ≈ 0) and the entropy of the formation of the bis-μ-oxo intermediate (ΔSo < 0), ΔSobs≠ = ΔSet≠ + ΔSo (< 0). Virtually the same ΔS≠ values (≈ 0) for electron transfer from Fc* and Me8Fc to [CuII2(N3)]4+ and [CuII2(N3)(O2)]2+ given in Table 1 clearly indicate that electron transfer from Fc* and Me8Fc to [CuII2(N3)(O2)]2+ occurs directly rather than via the interconversion from [CuII2(N3)(O2)]2+ to the corresponding bis-μ-oxo intermediate followed by rapid electron transfer from Fc* and Me8Fc (Scheme 6).
Scheme 6.

Because the one-electron reduction potential of [CuII2(N3)(O22−)]2+ was evaluated as 0.19 ± 0.07 V (vide supra), the ΔG values for electron transfer from Fc* and Me8Fc were evaluated to be −0.27 ± 0.07 and −0.23 ± 0.07 eV, respectively. Then, the λ value of electron transfer from Fc* and Me8Fc to [CuII2(N3)(O22−)]2+ was estimated from the ket and ΔGet values using Eqns. (1) and (2) to be 2.2 ± 0.1 eV, which is significantly larger than the value of electron transfer from ferrocene derivatives to [CuII2(N3)]4+ [67]. The larger λ value of [CuII2(N3)(O22−)]2+ as compared with [CuII2(N3)]4+ is consistent with direct electron transfer from ferrocene derivatives to [CuII2(N3)(O22−)]2+, which would require a large reorganization energy associated with the cleavage of the O–O bond.
As concerns the chemistry of another ligand-copper system we’ve studied, a bis-μ-oxodicopper(III) complex is produced by the reaction of dioxygen with a mononuclear copper complex, [CuI(BzPY1)]+ (BzPY1 = N,N-bis[2-(2-pyridyl)ethyl]benzylamine) [77] following its electron transfer reduction from Fc* to a copper(II) complex precursor [CuII(BzPY1)]2+ (Scheme 7) [67]. The spectroscopic monitoring of the formation of the bis-μ-oxodicopper(III) complex [{CuIII(BzPY1)}2(O2)]2+ (λmax = 390 nm) by the reaction of the CuI complex with O2 is shown in Fig. 8 [67]. Electron transfer from Fc* to [{CuIII(BzPY1)}2(O)]]2+ occurs rapidly upon mixing in acetone even at 193 K to produce two equiv of Fc*+ (Fig. 8). The same immediate reaction occurs with the weaker electron donors such as 1,2′-dimethylferrocene (Me2Fc) and even ferrocene (Fc) as well. [CuII(BzPY1)]2+ acts as an efficient catalyst for the four-electron reduction of O2 by Fc* in the presence of protons (Scheme 7) [67].
Scheme 7.

Fig. 8.

(a) Formation of the bis-μ-oxo complex [{CuIII(BzPY1)}2(O)2]2+ (λmax = 390 nm) in the reaction of [CuI(BzPY1)]+ (0.10 mM) with O2 in acetone at 193 K. (b) Formation of Fc*+ by addition of Fc* (0.70 mM) to the bis-μ-oxo complex generated. The reaction occurred upon mixing. Adapted from reference 67.
5. Concluding remarks
Reactions of Cu(I) complexes with O2 occur via concomitant electron transfer from Cu(I) complexes to O2 and binding of O2•− to Cu(II) complexes rather than stepwise electron transfer followed by binding processes. This is defined as inner-sphere Cu(II) ion-coupled electron transfer to produce the corresponding Cu(II)-superoxo complexes. The further reduction of these by Cu(I) compounds may also occur via Cu(II) ion-coupled electron transfer from Cu(I) to the Cu(II)-superoxo species to produce dinuclear Cu2-O2 complexes. Different types of Cu2-O2 complexes, μ-1,2-peroxodicopper(II), μ-η2:η2-peroxodicopper(II) and bis-μ-oxodicopper(III) complexes, are produced depending on the ligands utilized. The reactivity of the electron-transfer reduction of Cu2-O2 complexes is the greatest for bis-μ-oxodicopper(III) complexes, although the electron-transfer reduction of a μ-η2:η2-peroxodicopper(II) complex occurs directly without prior conversion to an isomeric bis-μ-oxodicopper(III) complex. In any case, inner-sphere Cu(II) ion-coupled electron transfer from Cu(I) complexes to O2 is much faster than the outer-sphere electron transfer pathway, enabling the catalytic reduction of O2 by outer-sphere one-electron reductants such as Fc*, which is a coordinatively saturated compound. It is expected that the range of copper complexes which are effective in the catalytic O2 reduction will be further expanded by a fuller understanding of Cu(O2) and Cu2(O2) compound reduction properties, along with elaborated ligand design, leading to varying O2 adduct Cu2O2 structures and chemistry.
Highlights.
Dioxygen binding to copper(I) complexes gives superoxo-copper(II), peroxodicopper(II) or bis-μ-oxodicopper(III) adducts.
Electron-transfer from copper(I) occurs concomitant with superoxide anion binding to copper(II) rather than by stepwise electron-transfer followed by ligation.
The kinetics and thermodynamics of electron-transfer reduction of various Cu2-O2 species is discussed in terms of Marcus theory.
Electron-transfer reduction of Cu2-O2 complexes is the fastest for bis-μ-oxodicopper(III) complexes and for at least one case, electron-transfer reduction of a μ-η2:η2-peroxodicopper(II) complex occurs directly and without prior conversion to an isomeric bis-μ-oxodicopper(III) species.
Acknowledgments
The authors gratefully acknowledge the contributions of their collaborators and coworkers mentioned in the references. The authors also acknowledge continuous support of their study by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan (S.F.), the KOSEF/MEST through WCU project (R31-2008-000-10010-0) in Korea (S.F. and K.D.K.) and the USA National Institutes of Health (GM 28962) (K.D.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Holm RH, Kennepohl P, Solomon EI. Chem Rev. 1996;96:2239. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 2.Mirica LM, Ottenwaelder X, Stack TDP. Chem Rev. 2004;104:1013. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- 3.Lewis EA, Tolman WB. Chem Rev. 2004;104:1047. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]
- 4.Solomon EI, Chen P, Metz M, Lee S-K, Palmer AE. Angew Chem, Int Ed. 2001;40:4570. doi: 10.1002/1521-3773(20011217)40:24<4570::aid-anie4570>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 5.Solomon EI, Szilagyi RK, George SD, Basumallick L. Chem Rev. 2004;105:419. doi: 10.1021/cr0206317. [DOI] [PubMed] [Google Scholar]
- 6.Itoh S. Curr Opin Chem Biol. 2006;10:115. doi: 10.1016/j.cbpa.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 7.Itoh S, Fukuzumi S. Acc Chem Res. 2007;40:592. doi: 10.1021/ar6000395. [DOI] [PubMed] [Google Scholar]
- 8.Kim E, Chufan EE, Kamaraj K, Karlin KD. Chem Rev. 2004;104:1077. doi: 10.1021/cr0206162. [DOI] [PubMed] [Google Scholar]
- 9.Lucas HR, Karlin KD. Met Ions Life Sci. 2009;6:295. doi: 10.1039/BK9781847559159-00295. [DOI] [PubMed] [Google Scholar]
- 10.Klinman JP. Chem Rev. 1996;96:2541. doi: 10.1021/cr950047g. [DOI] [PubMed] [Google Scholar]
- 11.Prigge ST, Eipper BA, Mains RE, Amzel LM. Science. 2004;304:864. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- 12.Klinman JP. J Biol Chem. 2006;281:3013. doi: 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]
- 13.Chen P, Solomon EI. Proc Natl Acad Sci USA. 2004;101:13105. doi: 10.1073/pnas.0402114101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quant HL, Karlin KD. J Biol Inorg Chem. 2004;9:669. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]
- 15.Himes RA, Karlin KD. Curr Opin Chem Biol. 2009;13:119. doi: 10.1016/j.cbpa.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marcus RA. Annu Rev Phys Chem. 1964;15:155. [Google Scholar]
- 17.Marcus RA. Angew Chem, Int Ed Engl. 1993;32:1111. [Google Scholar]
- 18.Zhang CX, Kaderli S, Costas M, Kim E-i, Neuhold Y-M, Karlin KD, Zuberbühler AD. Inorg Chem. 2003;42:1807. doi: 10.1021/ic0205684. [DOI] [PubMed] [Google Scholar]
- 19.Sawyer DT, Calderwood TS, Yamaguchi K, Angelis CT. Inorg Chem. 1983;22:2577. [Google Scholar]
- 20.Karlin KD, Kaderli S, Zuberbühler AD. Acc Chem Res. 1997;30:139. [Google Scholar]
- 21.Karlin KD, Wei N, Jung B, Kaderli S, Niklaus P, Zuberbühler AD. J Am Chem Soc. 1993;115:9506. [Google Scholar]
- 22.Fukuzumi S, Kotani H, Lucas HR, Doi K, Suenobu T, Peterson RL, Karlin KD. J Am Chem Soc. 2010;132:6874. doi: 10.1021/ja100538x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukuzumi S, Mochizuki S, Tanaka T. Inorg Chem. 1989;28:2459. [Google Scholar]
- 24.Fukuzumi S, Ohkubo K, Morimoto Y. Phys Chem Chem Phys. 2012 doi: 10.1039/C2CP40459A. in press. [DOI] [PubMed] [Google Scholar]
- 25.Fukuzumi S. Prog Inorg Chem. 2009;56:49. [Google Scholar]
- 26.Fukuzumi S, Bull S. Chem Soc Jpn. 1997;70:1. [Google Scholar]
- 27.Fukuzumi S, Ohtsu H, Ohkubo K, Itoh S, Imahori H. Coord Chem Rev. 2002;226:71. [Google Scholar]
- 28.Fukuzumi S, Ohkubo K. Coord Chem Rev. 2010;254:372. [Google Scholar]
- 29.Fukuzumi S, Wong CL, Kochi JK. J Am Chem Soc. 1980;102:2928. [Google Scholar]
- 30.Taube H. Science. 1984;226:1028. doi: 10.1126/science.6494920. [DOI] [PubMed] [Google Scholar]
- 31.Smirnov VV, Roth JP. J Am Chem Soc. 2006;128:3683. doi: 10.1021/ja056741n. [DOI] [PubMed] [Google Scholar]
- 32.Höfle G, Steglich W, Vorbrüggen H. Angew Chem, Int Ed Engl. 1978;17:569. [Google Scholar]
- 33.Gibson QH, Greenwood C. Biochem J. 1963;86:541. doi: 10.1042/bj0860541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greenwood C, Gibson QH. J Biol Chem. 1967;242:1782. [PubMed] [Google Scholar]
- 35.Momenteau M, Reed CA. Chem Rev. 1994;94:659. [Google Scholar]
- 36.Ferguson-Miller S, Babcock GT. Chem Rev. 1996;96:2889. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 37.Liebl U, Lipowski G, Négrerie M, Lambry J-C, Martin J-L, Vos MH. Nature. 1999;401:181. doi: 10.1038/43699. [DOI] [PubMed] [Google Scholar]
- 38.Okuno D, Iwase T, Shinzawa-Itoh K, Yoshikawa S, Kitagawa T. J Am Chem Soc. 2003;125:7209. doi: 10.1021/ja021302z. [DOI] [PubMed] [Google Scholar]
- 39.Stavrakis S, Koutsoupakis K, Pinakoulaki E, Urbani A, Saraste M, Varotsis C. J Am Chem Soc. 2002;124:3814. doi: 10.1021/ja0169825. [DOI] [PubMed] [Google Scholar]
- 40.David S, James BR, Dolphin D, Traylor TG, Lopez MA. J Am Chem Soc. 1994;116:6. [Google Scholar]
- 41.Collman JP, Brauman JI, Iverson BL, Sessler J, Morris RM, Gibson QH. J Am Chem Soc. 1983;105:3052. [Google Scholar]
- 42.Fry HC, Scaktruti DV, Karlin KD, Meyer GJ. J Am Chem Soc. 2003;125:11866. doi: 10.1021/ja034911v. [DOI] [PubMed] [Google Scholar]
- 43.Scaltrito DV, Fry HC, Showalter BM, Thompson DW, Liang H-C, Zhang CX, Kretzer RM, Kim E-i, Toscano JP, Karlin KD, Meyer GJ. Inorg Chem. 2001;40:4514. doi: 10.1021/ic010684r. [DOI] [PubMed] [Google Scholar]
- 44.Lucas HR, Meyer GJ, Karlin KD. J Am Chem Soc. 2010;132:12927. doi: 10.1021/ja104107q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fukuzumi S, Ohkubo K. Chem Eur J. 2000;6:4532. doi: 10.1002/1521-3765(20001215)6:24<4532::aid-chem4532>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 46.Ohkubo K, Menon SC, Orita A, Otera J, Fukuzumi S. J Org Chem. 2003;68:4720. doi: 10.1021/jo034258u. [DOI] [PubMed] [Google Scholar]
- 47.Fukuzumi S, Patz M, Suenobu T, Kuwahara Y, Itoh S. J Am Chem Soc. 1999;121:1605. [Google Scholar]
- 48.Bagchi RN, Bond AM, Scholz F, Stösser R. J Am Chem Soc. 1989;111:8270. [Google Scholar]
- 49.Känzig W, Cohen MH. Phys Rev Lett. 1959;3:509. [Google Scholar]
- 50.Zeller HR, Känzig W. Helv Phys Acta. 1967;40:845. [Google Scholar]
- 51.Kasai PH. J Chem Phys. 1965;43:3322. [Google Scholar]
- 52.Lunsford JH. Catal Rev. 1973;8:135. [Google Scholar]
- 53.Che M. Chem Rev. 1997;97:305. doi: 10.1021/cr950259d. [DOI] [PubMed] [Google Scholar]
- 54.Woertink JS, Tian L, Maiti D, Lucas HR, Himes RA, Karlin KD, Neese F, Würtele C, Holthausen MC, Bill E, Sundermeyer J, Schindler S, Solomon EI. Inorg Chem. 2010;49:9450–9459. doi: 10.1021/ic101138u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fukuzumi S, Ohkubo K. J Am Chem Soc. 2002;124:10270. doi: 10.1021/ja026613o. [DOI] [PubMed] [Google Scholar]
- 56.Kunishita A, Kubo M, Sugimoto H, Ogura T, Sato K, Takui T, Itoh S. J Am Chem Soc. 2009;131:2788. doi: 10.1021/ja809464e. [DOI] [PubMed] [Google Scholar]
- 57.Maiti D, Fry HC, Woertink JS, Vance MA, Solomon EI, Karlin KD. J Am Chem Soc. 2007;129:264. doi: 10.1021/ja067411l. [DOI] [PubMed] [Google Scholar]
- 58.Maiti D, Lee D-H, Gaoutchenova K, Würtele C, Holthausen MC, Narducci Sarjeant AA, Sundermeyer J, Schindler S, Karlin KD. Angew Chem, Int Ed. 2008;47:82. doi: 10.1002/anie.200704389. [DOI] [PubMed] [Google Scholar]
- 59.Yamaguchi S, Wada A, Funahashi Y, Nagatomo S, Kitagawa T, Jitsukawa K, Masuda H. Eur J Inorg Chem. 2003:4378. doi: 10.1021/ic035080x. [DOI] [PubMed] [Google Scholar]
- 60.Peterson RL, Himes RA, Kotani H, Suenobu T, Tian L, Siegler MA, Solomon EI, Fukuzumi S, Karlin KD. J Am Chem Soc. 2011;133:1702. doi: 10.1021/ja110466q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schatz M, Raab V, Foxon SP, Brehm G, Schneider S, Reiher M, Holthausen MC, Sundermeyer J, Schindler S. Angew Chem, Int Ed. 2004;43:4360. doi: 10.1002/anie.200454125. [DOI] [PubMed] [Google Scholar]
- 62.Sarangi R, Aboelella N, Fujisawa K, Tolman WB, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 2006;128:8286. doi: 10.1021/ja0615223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fukuzumi S, Koumitsu S, Hironaka K, Tanaka T. J Am Chem Soc. 1987;109:305. [Google Scholar]
- 64.Yuasa J, Fukuzumi S. J Am Chem Soc. 2006;128:14281. doi: 10.1021/ja0604562. [DOI] [PubMed] [Google Scholar]
- 65.Lee JY, Lee Y-M, Kotani H, Nam W, Fukuzumi S. Chem Commun. 2009:704. doi: 10.1039/b814928c. [DOI] [PubMed] [Google Scholar]
- 66.Zhu X-Q, Zhang M-T, Yu A, Wang C-H, Cheng J-P. J Am Chem Soc. 2008;130:2501. doi: 10.1021/ja075523m. [DOI] [PubMed] [Google Scholar]
- 67.Tahsini L, Kotani H, Lee Y-M, Cho J, Nam W, Karlin KD, Fukuzumi S. Chem–Eur J. 2012;18:1084. doi: 10.1002/chem.201103215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karlin KD, Tyeklár Z, Farooq A, Haka MS, Ghosh P, Cruse RW, Gultneh Y, Hayes JC, Toscano PJ, Zubieta J. Inorg Chem. 1992;31:1436. [Google Scholar]
- 69.Chaka G, Bakac A. Dalton Trans. 2009:318. doi: 10.1039/b811140e. [DOI] [PubMed] [Google Scholar]
- 70.Rosokha SV, Kochi JK. J Am Chem Soc. 2007;129:3683. doi: 10.1021/ja069149m. [DOI] [PubMed] [Google Scholar]
- 71.Lee Y-M, Kotani H, Suenobu T, Nam W, Fukuzumi S. J Am Chem Soc. 2008;130:434. doi: 10.1021/ja077994e. [DOI] [PubMed] [Google Scholar]
- 72.Fukuzumi S, Kotani H, Prokop KA, Goldberg DP. J Am Chem Soc. 2011;133:1859. doi: 10.1021/ja108395g. [DOI] [PubMed] [Google Scholar]
- 73.Halfen JA, Mahapatra S, Wilkinson EC, Kaderli S, Young VG, Jr, Que L, Jr, Zuberbühler AD, Tolman WB. Science. 1996;271:1397. doi: 10.1126/science.271.5254.1397. [DOI] [PubMed] [Google Scholar]
- 74.Tolman WB. Acc Chem Res. 1997;30:227. [Google Scholar]
- 75.Itoh S, Fukuzumi S. Bull Chem Soc Jpn. 2002;75:2081. [Google Scholar]
- 76.Rolff M, Schottenheim J, Decker H, Tuczek F. Chem Soc Rev. 2011;40 doi: 10.1039/c0cs00202j. [DOI] [PubMed] [Google Scholar]
- 77.Lucas HR, Li L, Sarjeant AAN, Vance MA, Solomon EI, Karlin KD. J Am Chem Soc. 2009;131:3230. doi: 10.1021/ja807081d. [DOI] [PMC free article] [PubMed] [Google Scholar]