

Abstract
Objective
To determine the efficacy of long-term anti-miR-33 therapy on the progression of atherosclerosis in high fat, high cholesterol-fed Ldlr−/− mice.
Methods and Results
Ldlr−/− mice received saline, or control or anti-miR-33 oligonucleotides once a week for 14 weeks. The treatment was effective, as measured by reduced levels of hepatic miR-33 and increased hepatic expression of miR-33 targets. Analysis of plasma samples revealed an initial elevation in HDL-cholesterol after two weeks of treatment that was not sustained by the end of the experiment. Additionally, we found a significant increase in circulating triglycerides in anti-miR-33-treated mice, compared to controls. Finally, examination of atheromata revealed no significant changes in the size or composition of lesions between the three groups.
Conclusion
Prolonged silencing of miR-33 fails to maintain elevated plasma HDL-cholesterol and does not prevent the progression of atherosclerosis in Ldlr−/− mice.
Keywords: miR-33, atherosclerosis, HDL, VLDL, ABCA1
Recently, we and others reported on miR-33 (also known as miR-33a or miR-33a-5p), an intragenic miRNA encoded within SREBP-2 that modulates the expression of several transmembrane transporters, that include ABCA1, ABCG1, ABCB11 and ATP8B11–6. These studies suggested that miR-33 plays important roles in fine-tuning both high-density lipoprotein (HDL) and biliary metabolism. Indeed, we and others showed that short-term silencing of hepatic miR-33 with antisense oligonucleotides increases both HDL-cholesterol (HDL-c)1–5 and hepatic bile secretion6. Additionally, treatment of mice with anti-miR-33 oligonucleotides led to increased reverse cholesterol transport (RCT) in vivo6, 7. The RCT pathway mobilizes extrahepatic cholesterol into HDL back to the liver, where both cholesterol and its metabolite bile acids are secreted into bile for final excretion through the feces. Hence, RCT is regarded as atheroprotective and strategies that promote/accelerate the flow of cholesterol through this pathway, e.g. raising HDL levels and/or promoting hepatobiliary removal of sterols, are predicted to be effective in reducing the risk of cardiovascular disease8.
Collectively, the initial reports on miR-33 suggested that patients with hypercholesterolemia might benefit from a therapy that incorporated silencing of miR-33 (e.g. anti-miR-33 oligonucleotides). In agreement with this idea, Rayner at al. reported that anti-miR-33 oligonucleotides were able to increase HDL-c and accelerate the regression of atherosclerotic lesions in hyperlipidemic Ldlr−/− mice switched to standard chow diet7. However, no reports are yet available regarding the impact of miR-33 silencing on the progression of atherosclerosis. Hence, our purpose was to determine the efficacy of long-term anti-miR-33 therapy on the development of arterial disease in high fat, high cholesterol-fed Ldlr−/− mice.
METHODS
Male, 10 week-old Ldlr−/− mice (Jackson Laboratories) were maintained on a 12h/12h light/dark cycle with unlimited access to food and water, and injected i.p. with 200 μL saline (n=5), or 7 mg/Kg/week control (5′-TCCTAGAAAGAGTAGA; n=13) or anti-miR-33 (5′-TGCAACTACAATGCA; n=11) LNA oligonucleotides (a kind gift from Miragen Therapeutics, Inc.) once a week. After two weeks on chow, mice were switched to and maintained on a western diet (WD) containing 21% fat and 1.25% cholesterol (Research Diets D12108) for 12 weeks. Mice were fasted for 6 h before sacrifice. Detailed materials and methods appear in supplemental materials.
RESULTS
Anti-miR-33 treatment did not affect body weight (Fig. 1A), although a trend was observed for accelerated weight gain in the anti-miR-33 group after 10 weeks of treatment. Analysis of plasma samples showed that both total cholesterol and HDL-c increased in the first 2 weeks of treatment with anti-miR-33 oligonucleotides, compared to control treatments (Fig. 1B, D). These data agree with previous reports1–3, 5, 7, 9 and provide evidence that the anti-miR-33 treatment was efficacious. As expected, at the end of the WD feeding circulating cholesterol levels increased in all groups (Fig. 1C); however, no changes in HDL-c were noted between groups (Fig. 1E). Furthermore, we noted that mice injected with anti-miR-33 oligonucleotides showed increased plasma triglycerides (TG), both on chow and after WD (Fig. 1F, G). FPLC analysis of plasma samples confirmed the changes in HDL-c on chow, but not on WD, in the anti-miR-33 group (Fig. I). Concomitant changes in the levels of APOA1 and APOB48/100 were also noted in these plasma samples (Fig. I), further verifying the changes in HDL-c and triglycerides. Collectively, these data suggest that the increase in HDL-c following miR-33 silencing is transient. This is also the first study to report changes in circulating triglycerides after treatment with anti-miR-33 oligonucleotides. These latter results were unexpected since a previous study showed a decline in plasma VLDL-tg in monkeys receiving anti-miR-33 treatment9.
Figure 1. Long-term anti-miR-33 therapy alters plasma and hepatic lipid contents.

A. Ldlr−/− mice were injected weekly (arrows) and fed a WD for 12 weeks. Body weight did not change with treatments. B–G. Plasma lipid contents. H, I. Hepatic RNA and protein contents at time of sacrifice. J–M. Hepatic lipid contents. Data shown as mean ± s.e.m., *P ≤ 0.05
One possibility to explain the absence of changes in HDL-c at week 12 in the anti-miR-33 group is that the bioavailability and/or potency of the oligonucleotides decline over time or in combination with the WD. Additionally, hepatic miR-33 levels are known to decrease in mice fed a WD2,3, which might also compromise the success of the antisense treatment. However, RNA and protein analysis of the livers showed that miR-33 was indeed effectively silenced in the anti-miR-33 group, since levels of miR-33 were decreased ~75% while those of miR-33 targets (ABCA1, CPT1α, ABCB11) were significantly increased in these mice, compared to controls (Fig. 1H, I and II-A). RNA and protein levels of other genes declined or remained unchanged in the same livers (Fig. 1H, I and II-A), suggesting that the changes in several miR-33 targets were specific. It is important to stress that the changes in hepatic ABCA1 noted in the livers of anti-miR-33 mice after 12 weeks on WD were not paralleled by increased levels of HDL-c in plasma, as noted above. The reasons behind these latter paradoxical results remain to be elucidated. Further analysis revealed no significant changes in liver weight, and hepatic cholesterol or triglyceride contents (Fig. 1J–L). However, hepatic free fatty acid levels were significantly reduced in the anti-miR-33 group (Fig. 1M). This latter result could be explained by the significant increase in CPT1α and/or decrease in FAS expression in the same livers (Fig. 1H, I). Additionally, we measured the hepatic expression of miRNAs that are known to regulate sterol and triglyceride metabolism10. Data in Fig. II-B show that miR-378, miR-27a, and miR-122 (but not miR-758, miR-370, miR-335, and miR-125a-5p) were modestly elevated in mice receiving anti-miR-33 treatment. The functional consequences of deregulated expression of these latter miRNAs on triglyceride and sterol homeostasis in mice receiving anti-miR-33 oligonucleotides are unknown. Nevertheless, taken together the data suggest that the anti-miR-33 oligonucleotides were effective in silencing hepatic miR-33 at the end of the 12 weeks of WD feeding.
Finally, analysis of atherosclerotic lesions both in the aortic root and in en face preparations of the whole aorta revealed no significant differences between groups (Fig. 2A–D). Likewise, assessment of macrophage and collagen contents in aortic root sections of selected mice in each group with similar lesion sizes revealed no discernible differences between treatments (Fig. 2B).
Figure 2. Long-term anti-miR-33 therapy does not attenuate the progression of atherosclerosis.
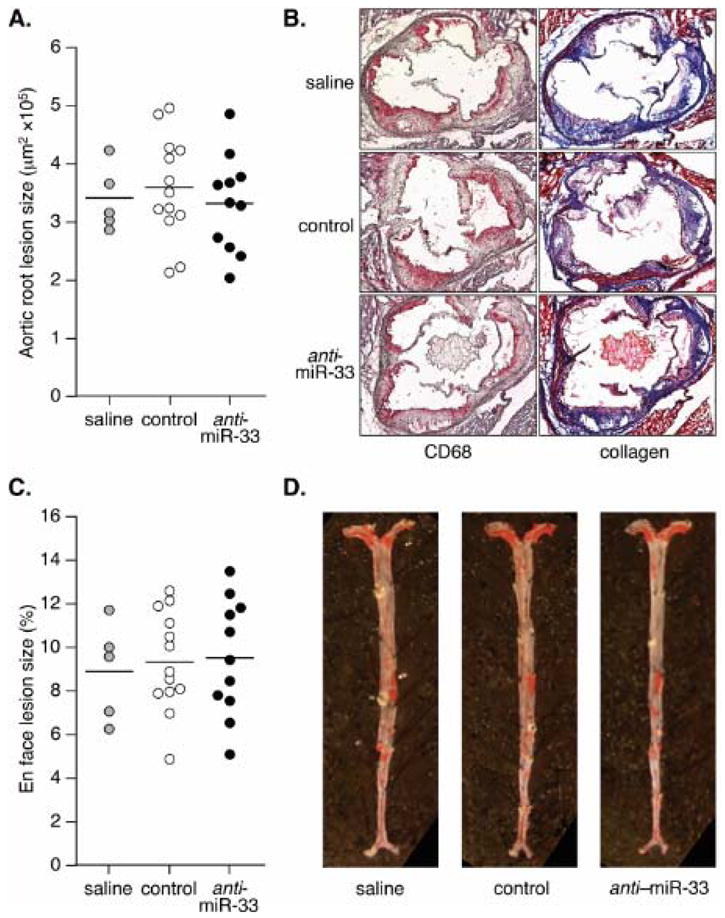
A. Lesion size in the aortic root. B. Representative sections stained to reveal macrophage and collagen content. C. Lesion size in en face whole aortas. D. Representative aortas stained with oil red O. Each dot denotes a single animal; horizontal lines indicate average lesion size.
DISCUSSION
This is the first report on the impact of long-term anti-miR-33 treatment on the progression of atherosclerosis in Ldlr−/− mice. Previous regression studies7 employed mice that received anti-miRs for only 4 weeks, and no data is yet available regarding the continuing efficacy of the oligonucleotides in this regression model. Our data show conclusively that anti-miR-33 oligonucleotides, while effective in suppressing hepatic miR-33 expression and increasing the expression of hepatic miR-33 targets, did not raise HDL-c after mice were switched to the WD and did not provide atheroprotection.
Primates, but not rodents, express a second miR-33 gene (miR-33b) from an intron of SREBP-1. Notably, SREBP-1 and -2 are differentially regulated by hormones, dietary challenges or statin treatment. In any case, the expression of miR-33a and miR-33b is expected to follow that of their corresponding hosting gene. Whether the 2 nt mismatch (outside of the seed sequence) between miR-33a and miR-33b results in differential targeting remains to be established, although studies in human HepG2 cells11 and in non-human primates9 suggest that both miRNAs target the same set of genes with similar specificity. Nevertheless, anti-miR-33 oligonucleotides are expected to block the action of both miR-33a and miR-33b9. A limitation of our study is that the lack of rodent miR-33b prevents the direct translation of our results in Ldlr−/− mice to humans. Indeed, in marked contrast to our study, a longitudinal study in non-human primates did show a sustained elevation of HDL-c, as well as a strong decrease in triglycerides. Whether the differences in cholesterol and triglyceride metabolism between mice and primates in response to anti-miR-33 treatment are due to miR-33b remains to be established. The discrepancies between rodent and primate models thus raise an important concern regarding the direct translation of data from rodent models to human physiology and metabolic disorders. Further studies using humanized mice, in which a miR-33b transgene is inserted within an intron of Srebf-1, will help us address this important question.
The mechanism behind the loss of elevated HDL-c in the anti-miR-33 group remains to be elucidated. It is likely that the potential atheroprotective effect of anti-miR-33-based therapies rely, at least in part, in their ability to promote and sustain such an increase in HDL-c. The fact that hepatic ABCA1 expression is still induced after 14 weekly anti-miR-33 injections suggests the existence of homeostatic compensatory mechanisms controlling plasma HDL. Interestingly, Brewer and colleagues reported that hepatic overexpression of ABCA1 results in enhanced atherosclerosis in Ldlr−/− mice12, while van Eck et al. showed that macrophage ABCA1 overexpression reduces atheromata in Ldlr−/− mice13. It is conceivable that the lack of atheroprotection in our model is the result of both pro- and anti-atherogenic effects that follow long-term silencing of miR-33 both in the liver and in macrophages in the lesion. Remarkably, Rayner et al. showed that the expression of ABCA1 was induced in macrophages recovered from atheromata in 2′F/MOE anti-miR-33-treated mice7. Whether 2′F/MOE and LNA oligonucleotides have different pharmacokinetic properties that result in distinctive bioavailability in plaques remains to be established. While this paper was under review, Horie et al. reported that WD-fed miR-33/ApoE double knock-out mice had decreased atheromata, compared to ApoE−/− mice14. However, no significant changes in atherosclerotic lesion size were observed in ApoE−/− mice transplanted with miR-33/ApoE double knock-out bone marrow14, suggesting that loss of miR-33 in bone marrow-derived cells is not atheroprotective per se. Interestingly, the double knock-out mice also showed a tendency towards increased circulating triglycerides14, although the changes did not reach statistical significance. Differences in the progression of atheromata following whole-body miR-33 deficiency or treatment with anti-miR-33 oligonucleotides are intriguing, and likely reflect the impact of miR-33 expression in several cell types (e.g. hepatocytes, macrophages, endothelial and smooth muscle cells). It is conceivable that anti-miR therapy could be effective in some, but not all, cell types where miR-33 is normally expressed. Tissue/cell-specific knock-out mice for miR-33 will help us understand the relative contribution of hepatocyte and plaque (macrophage, endothelial and smooth muscle cell) miR-33 to atherogenesis, and the anti-atherogenic potential of anti-miR-33 oligonucleotides. It will also be important to establish whether different chemistries alter the bioavailability and potency of anti-miR-33 oligonucleotides in either liver or lesions.
Supplementary Material
Acknowledgments
We thank Dr. Maureen J. Donlin (Saint Louis University) for statistical analysis assistance.
SOURCES OF FUNDING
This work was supported in part by NIH Grants HL107794 (to Á.B.), HL095154 and HL030568 (to A.J.L.).
Footnotes
DISCLOSURE
T.J.M. and Á.B. are pursuing a patent related to anti-miR-33 silencing.
References
- 1.Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, Näär AM. MicroRNA-33 and the srebp host genes cooperate to control cholesterol homeostasis. Science. 2010;238:1566–1569. doi: 10.1126/science.1189123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rayner KJ, Suárez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernández-Hernando C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;238:1570–1573. doi: 10.1126/science.1189862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marquart TJ, Allen RM, Ory DS, Baldán A. MiR-33 links SREBP-2 induction to repression of sterol transporters. Proc Natl Acad Sci USA. 2010;107:12228–12232. doi: 10.1073/pnas.1005191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gerin I, Clerbaux LA, Haumont O, Lanthier N, Das AK, Burant CF, Leclercq IA, MacDougald OA, Bommer GT. Expression of miR-33 from an SREBP2 intron inhibits cholesterol export and fatty acid oxidation. J Biol Chem. 2010;285:33652–33661. doi: 10.1074/jbc.M110.152090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horie T, Ono K, Horiguchi M, Nishi H, Nakamura T, Nagao K, Kinoshita M, Kuwabara Y, Marusawa H, Iwanaga Y, Hasegawa K, Yokode M, Kimura T, Kita T. MicroRNA-33 encoded by an intron of sterol regulatory element-binding protein 2 (SREBP2) regulates HDL in vivo. Proc Natl Acad Sci USA. 2010;107:17321–17326. doi: 10.1073/pnas.1008499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allen RM, Marquart TJ, Albert CJ, Suchy FJ, Wang DQ, Ananthanarayanan M, Ford DA, Baldán A. MiR-33 controls the expression of biliary transporters, and mediates statin- and diet-induced hepatotoxicity. EMBO Mol Med. 2012;4:882–895. doi: 10.1002/emmm.201201228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suárez Y, Fernández-Hernando C, Fisher EA, Moore KJ. Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. 2011;121:2921–2931. doi: 10.1172/JCI57275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rader DJ, Tall AR. The not-so-simple HDL story: Is it time to revise the HDL cholesterol hypothesis? Nature Med. 2012;18:1344–1346. doi: 10.1038/nm.2937. [DOI] [PubMed] [Google Scholar]
- 9.Rayner KJ, Esau CC, Hussain FN, et al. Inhibition of miR-33a/b in non-human primates raises plasma HDL and lowers VLDL-triglycerides. Nature. 2011;478:404–407. doi: 10.1038/nature10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sacco J, Adeli K. Micrornas: Emerging roles in lipid and lipoprotein metabolism. Curr Opin Lipidol. 2012;23:220–225. doi: 10.1097/MOL.0b013e3283534c9f. [DOI] [PubMed] [Google Scholar]
- 11.Davalos A, Goedeke L, Smibert P, et al. MiR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc Natl Acad Sci USA. 2011;108:9232–9237. doi: 10.1073/pnas.1102281108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joyce CW, Wagner EM, Basso F, et al. ABCA1 overexpression in the liver of LDLR-ko mice leads to accumulation of pro-atherogenic lipoproteins and enhanced atherosclerosis. J Biol Chem. 2006;281:33053–33065. doi: 10.1074/jbc.M604526200. [DOI] [PubMed] [Google Scholar]
- 13.Van Eck M, Singaraja RR, Ye D, Hildebrand RB, James ER, Hayden MR, Van Berkel TJ. Macrophage ATP-Binding Cassette transporter A1 overexpression inhibits atherosclerotic lesion progression in low-density lipoprotein receptor knockout mice. Arterioscler Thromb Vasc Biol. 2006;26:929–934. doi: 10.1161/01.ATV.0000208364.22732.16. [DOI] [PubMed] [Google Scholar]
- 14.Horie T, Baba O, Kuwabara Y, et al. MicroRNA-33 deficiency reduces the progression of atherosclerotic plaque in ApoE−/− mice. J Am Heart Assoc. 2012;1:e003376. doi: 10.1161/JAHA.112.003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.