

Abstract
Increased intracellular levels of α-synuclein are implicated in Parkinson's disease and related disorders and may be caused by alterations in the ubiquitin–proteasome system (UPS) or the autophagy–lysosomal pathway (ALP). A critical question remains how α-synuclein is degraded by neurons in vivo. To address this, our study uses α-synuclein transgenic mice, expressing human α-synuclein or α-synuclein-eGFP under the (h)PDGF-β promoter, in combination with in vivo pharmacologic and multiphoton imaging strategies to systematically test degradation pathways in the living mouse brain. We demonstrate that the UPS is the main degradation pathway for α-synuclein under normal conditions in vivo while with increased α-synuclein burden the ALP is recruited. Moreover, we report alterations of the UPS in α-synuclein transgenic mice and age dependence to the role of the UPS in α-synuclein degradation. In addition, we provide evidence that the UPS and ALP might be functionally connected such that impairment of one can upregulate the other. These results provide a novel link between the UPS, the ALP, and α-synuclein pathology and may have important implications for future therapeutics targeting degradation pathways.
Introduction
Progressive accumulation of α-synuclein has been implicated in many neurodegenerative diseases, including Parkinson's disease (PD) and dementia with Lewy bodies. The cascade by which this natively unfolded protein aggregates and leads to neuronal degeneration is not yet fully understood (Dauer and Przedborski, 2003). One clear predisposing factor, however, is an increase in intracellular levels of α-synuclein due to increased expression or reduced degradation. Enhanced expression of α-synuclein through gene multiplication (Singleton et al., 2003; Chartier-Harlin et al., 2004; Ibáñez et al., 2004) or polymorphisms in its promoter (Maraganore et al., 2006) can lead to autosomal-dominant PD or increased susceptibility to sporadic PD, respectively. In addition, genetic or viral-mediated overexpression can induce features of parkinsonism in a variety of animal models (Chesselet, 2008). In addition to abnormal expression of α-synuclein, alterations in protein degradation pathways have been implicated in PD by both genetic and pathological studies in PD patients as well as experimental studies in disease models. While earlier investigations have mainly focused on dysfunction of the ubiquitin–proteasome system (UPS) (Olanow and McNaught, 2006; Cook and Petrucelli, 2009), more recent reports also provide evidence for malfunction of the autophagy–lysosomal pathway (ALP) (Martinez-Vicente and Cuervo, 2007; Wong and Cuervo, 2010). The UPS and ALP are the major degradation pathways that neurons depend on to maintain protein homeostasis. While the UPS degrades most short-lived, soluble proteins (Goldberg, 2003), the ALP is the bulk degradation process by which longer-lived macromolecules and dysfunctional organelles are cleared (Klionsky, 2007). Within the ALP, delivery of targets to the lysosome occurs in three distinct ways that distinguish the respective subtype: macroautophagy, chaperone-mediated autophagy (CMA), and microautophagy. Relevant to PD, either genetic depletion of 26S proteasomes or macroautophagy function in knock-out mouse models leads to profound neurodegeneration and the formation of inclusion bodies (Hara et al., 2006; Komatsu et al., 2006; Bedford et al., 2008). Current literature, based mainly on different cell culture models, provides evidence for both the UPS (Bennett et al., 1999; Tofaris et al., 2001; Webb et al., 2003) and the ALP (Webb et al., 2003; Cuervo et al., 2004; Vogiatzi et al., 2008) as the mechanism by which α-synuclein is degraded in neurons. These cell culture studies, in addition to the few in vivo studies that have been reported (Mak et al., 2010), have unfortunately produced conflicting results.
In this context, the main objective of our study is to use novel in vivo pharmacologic and imaging strategies to systematically test the role of the UPS and ALP in α-synuclein degradation in the living mouse brain. We find that the UPS is the main degradation pathway for α-synuclein under normal conditions in vivo while under conditions of increased α-synuclein expression the ALP is recruited to degrade α-synuclein. In addition, we report age dependence to the role of the UPS and that both the UPS and macroautophagy are functionally coupled, such that selective impairment of one can upregulate the other in vivo. These results suggest specific roles for the UPS and ALP in α-synuclein degradation and provide insight into how neurons respond to pathogenic states in which protein levels are increased or either degradation pathway is dysfunctional.
Materials and Methods
Animals.
Human wild-type α-synuclein transgenic mice (Masliah et al., 2000), human wild-type α-synuclein-eGFP transgenic mice [“αSyn-GFP” (Rockenstein et al., 2005)], and nontransgenic littermates (background strain BDF-1; Charles River Laboratories) were used. Animals were held in a light/dark cycle, temperature- and humidity-controlled animal vivarium with ad libitum food and water. All experiments were approved by the Subcommittee on Research Animal Care at Massachusetts General Hospital. For all experiments, animals were littermates or age-matched controls. Both females and male animals were studied after no differences were noted.
Cranial window surgery and topical application of proteasome and autophagy inhibitors.
The general techniques for cranial window preparation and in vivo multiphoton imaging have been published previously (Skoch et al., 2005; Holtmaat et al., 2009). In this study, we have used a modified approach, developed by our group, to optimize the application of blood–brain barrier-impermeant agents to cortical areas (Unni et al., 2011). This protocol allows reliable drug application and incubation over time in combination with in vivo multiphoton imaging or tissue recovery for subsequent biochemical assays. For imaging experiments, the drug was incubated for 90–120 min, the temporary wax or silicon chamber was discarded, and a 8 mm coverslip was placed directly over the treated cortex. Next, the animal was immediately taken to the imaging set-up to acquire time point 0 (T0) images (for details, see below, In vivo multiphoton imaging). The animal was then returned to its home cage, allowed to recover from anesthesia, and housed in the animal facility until 24 h later when the animal was reimaged to acquire time point 24 h (T24) images. For biochemical assays, the animal was allowed to recover from anesthesia and returned to its home cage, and the drug was incubated topically for 24 h as described previously (Unni et al., 2011) until tissue recovery. Clasto-lactacystin-β-lactone (CLBL) (20 μm), Z-Leu-Leu-Leu-CHO (MG132) (100 μm; both Cayman Chemical), and Bafilomycin A1 (BafA1) (2.5 μm; LC Laboratories) were reconstituted with DMSO (Sigma-Aldrich) and prepared in sterile PBS. Hydroxychloroquine sulfate (HCQ) (100 μm; Sigma-Aldrich) was dissolved in sterile PBS.
Stereotaxic viral injections.
For gene delivery, intracortical injections of adenoassociated virus (AAV) were used as described previously (Spires et al., 2005; Spires-Jones et al., 2011). The gene for the ubiquitin–proteasome reporter GFPu (construct: pAAV-CBA-GFPu) was introduced into neocortical neurons by an AAV8-based system. Using a Hamilton syringe (Hamilton Company), 4 μl of virus (titer, 1.62 × 1011 genome copies/ml) were infused 1.2 mm deep into the somatosensory cortex of each hemisphere at a rate of 0.2 μl/min. After a 4–6 week incubation period to allow adequate expression of GFPu, a cranial window was installed and in vivo multiphoton imaging was performed as described below (see In vivo multiphoton imaging). Twelve animals (six for each mouse line) were injected and subjected to analysis.
In vivo multiphoton imaging.
In vivo multiphoton imaging of GFP-tagged α-synuclein (αSyn-GFP) and virally expressed GFPu was performed as described previously (Unni et al., 2010). After drug incubation and coverslipping, the anesthetized animal was placed on an in vivo multiphoton imaging setup (Olympus Fluoview 1000MPE with prechirp excitation optics and an acousto-optical modulator on an Olympus BX61WI upright microscope). A mode-locked titanium sapphire laser (MaiTai; Spectra-Physics) was set to 860 nm excitation and a z-stack was acquired (step size, 1–2 μm). Image size (x–y dimension) was 512 × 512 pixels. αSyn-GFP-expressing neurons were sampled up to ∼200–300 μm below the cortical surface and subject to analysis using NIH ImageJ software. The somatic αSyn-GFP signal was measured by creating a region of interest (ROI) outlining the cell body for each αSyn-GFP-expressing neuron and calculating the mean fluorescence intensity within this ROI. For each animal, images were obtained immediately after drug application (T0) and after a 24 h incubation period (T24) using the same ROI for each individual αSyn-GFP-positive neuron, therefore comparing the fluorescent signal of the exact same neuronal cell bodies at both time points. αSyn-GFP expression at synaptic terminals was analyzed using the analyze particles routine (size, 0.2–5 μm2; default automatic threshold) for complete stacks. Fluorescent signal of presynaptic terminals was plotted along with the signal in corresponding cell bodies of the same z-plane to provide that the same cortical volume was compared at T0 and T24.
Tissue preparation and immunoblotting.
After 24 h, animals were deeply anesthetized by intraperitoneal injection with ketamine (10 mg/ml; Phoenix Pharmaceuticals)/xylazine (1 mg/ml; Lloyd Laboratories). Following decapitation, the brain was quickly removed and mounted onto a vibrating microtome stage (Leica VT1000S) ventral surface down in ice-cold PBS with additional oxygen supply. Three serial horizontal sections of the treated cortex, each 100 μm thick, were prepared and transferred to the appropriate buffer solution for homogenization. For all experiments, only tissue from the top 300 μm of treated cortex was used. For immunoblotting, the recovered tissue was homogenized in ice-cold lysis buffer [50 mm Tris-HCl, pH 7.5, 5 m guanidinium, protease inhibitor mixture (Roche Complete)] and agitated overnight, followed by centrifugation (13,000 × g, 10 min). The supernatant was collected and dialyzed against urea (6 m; 1 h) using membrane filters (MF-Millipore; Millipore; 0.025 μm). Total protein concentration was determined using a BCA assay. Total cell lysates (12 μg of protein) were solubilized in LDS (lithium dodecyl sulfate) buffer under reducing conditions. Proteins were separated by gel electrophoresis, using a 4–12 or 12% Bis-Tris gel and MOPS [3-(N-morpholino)propanesulfonic acid] or MES [2-(N-morpholino)ethanesulfonic acid] buffer (Invitrogen) and transferred to a PVDF membrane (PerkinElmer). Following blocking with blocking buffer (LI-COR Biosciences), membranes were incubated overnight with the respective primary antibody. Immunoreactivity was determined for the following: α-synuclein using a pan-α-synuclein (1:1000; Syn-1; BD Biosciences) and a human α-synuclein specific antibody (1:1000; clone 4B12; Covance), ubiquitin (1:1000; Dako), LC3 (1:500; Novus Biologicals), p62 (1:1000; Progen Biotechnik), GAPDH (1:10,000; Millipore), and α-tubulin (1:100,000; Sigma-Aldrich). Near infrared fluorescent-labeled secondary antibodies (1:5000; IR800CW, IR680LT; LI-COR Biosciences) were used and quantification was done using the Odyssey infrared imaging system (LI-COR Biosciences) and NIH ImageJ.
Proteasome activity assay.
For determining proteasomal activity, the recovered brain tissue was immediately homogenized manually and by sonication in ice-cold Tris-HCl (50 mm), pH 7.5, containing EDTA (1 mm). Homogenates were frozen in a dry-ice bath and stored at −80°C until all samples for each mouse line were collected and could be run on the same assay to avoid artifacts from repeated freeze-thaw cycles. Samples were thawed and kept on ice at all times. After centrifugation (13,000 × g) at 4°C for 10 min, total protein concentration was determined using a BCA protein assay. Thirty micrograms of total protein were incubated with Suc-Leu-Leu-Val-Tyr-AMC as a substrate to measure the chymotrypsin-like activity of the 20S and/or the 26S proteasome in a fluorescence-based assay in the presence or absence of SDS [assay buffer for 20S activity: 250 mm HEPES, pH 7.5, 5 mm EDTA, and 0.01% SDS (all reagents Millipore); assay buffer for 26S activity as described previously (Kisselev and Goldberg, 2005): 50 mm Tris-HCl, pH 7.5, 40 mm KCl, 5 mm MgCl2, 0.5 mm ATP, 1 mm DTT, and 0.05 mg/ml BSA (all reagents Thermo Fisher Scientific or Sigma-Aldrich)]. A standard curve for 7-amino-4-methylcoumarin (AMC) was generated to evaluate substrate turnover in the samples, and specificity of the proteasomal assay was ascertained by the ability of additional lactacystin (25 μm) to inhibit fluorescence change and by a parallel assay in a serial dilution of purified 20S proteasomes (all reagents Millipore). After 2 h incubation at 37°C, fluorescence was read at 380/460 nm, and proteolytic activity was expressed as the amount of free AMC after cleavage.
RNA extraction and quality evaluation.
Total RNA was extracted using Tri reagent (Sigma-Aldrich) according to specifications. To assess RNA quality, 1 μl of the RNA was analyzed using an Agilent Bioanalyzer (Agilent) with a nanoRNA Chip (Buesa et al., 2004). Only the brains that showed RNA integrity numbers of 7.5 and higher were considered for subsequent quantitative PCR (qPCR).
Primer design and real-time qPCR.
To discriminate between human and mouse α-synuclein, short synthetic (20- to 22-mer) PCR primers were designed to amplify small (100–350 bp) amplicons in mRNA regions with the lowest homology between the two species, using Primer3 software (http://frodo.wi.mit.edu/primer3/). First-strand cDNA synthesis was performed on the extracted total RNA extracted with an Invitrogen SuperScript first-strand synthesis kit (Invitrogen). Quantitative real-time PCR was performed in a 96-well plate using an iCycler (Bio-Rad), and IQ SYBR Green PCR Master Mix (Bio-Rad) with the following protocol: 2 min hot start at 95°C; denaturation, 95°C, 30 s; annealing, 60°C, 30 s; elongation, 72°C, for 45 cycles; followed by 80 0.5°C increases in temperature (starting at 55°C) to collect melting curve data. For each primer set, we used a curve with known concentrations of cDNA that was used to calculate the efficiency of the primers, and to quantitate products. All qPCR results were calculated with the ΔΔCt method (Fink et al., 1998; Livak and Schmittgen, 2001) and normalized to nanograms of total RNA. Mouse α-synuclein primers were designed to transcribe both Mus musculus transcript variants [i.e., NM_001042451 (tv1) and NM_009221 (tv2)]: forward primer, CAT CTT TAG CCA TGG ATG TG (position tv1 289–308; tv2 201–220); reverse primer, CCC ATC TGG TCC TTC TTG AC (position tv1 582–301; tv2 494–513); annealing temperature, 60°C; product size, 313 bp; efficiency, 99%. Human α-synuclein primers were designed to transcribe within the region of lowest homology with mouse within the translated region (present on the transgene): forward primer, GCA GGG AGC ATT GCA GCA GC; reverse primer, GGC TTC AGG TTC GTA GTC TTG; annealing temperature, 60°C; product size, 168 bp; efficiency, 100%. The specificity of the primer was ensured by in silico analysis by matching the primers against the Homo sapiens and Mus musculus nucleotide databases with BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi), followed by testing the human and mouse specific primers using qPCR with wild-type mouse cDNA and human cDNA. The PCR products were then analyzed with agarose gel electrophoresis (2%).
Statistics.
Data are presented as mean ± SEM. Statistical analysis was done using GraphPad Prism5 (GraphPad Software). Groups were compared using two-tailed, unpaired or paired Student's test or ANOVA with Dunnett's or Bonferroni's post hoc correction. Statistical significance is indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001.
Results
Published studies provide evidence for both the UPS and ALP being involved in the degradation of α-synuclein. These findings, however, are conflicting and mostly based on different cell culture systems. Here, we systematically investigated the role of the UPS and ALP in α-synuclein degradation in vivo, in the living brain, using transgenic mice overexpressing human α-synuclein or α-synuclein-GFP, and nontransgenic littermates.
To explore the role of the UPS in vivo, we used a modified cranial window approach to topically apply the irreversible, selective, and cell-permeable proteasome inhibitor CLBL to the exposed cortex. To confirm that CLBL can penetrate the brain and block proteasome function, we assayed immunoblot levels of ubiquitin, chymotrypsin-like proteasome activity, and the UPS reporter GFPu. Following CLBL incubation, serial cortical sections were prepared and subjected to immunoblot analysis for monomeric ubiquitin and polyubiquitinated high-molecular-weight (HMW) protein species. As shown in Figure 1, CLBL led to a robust accumulation of polyubiquitinated HMW conjugates, mainly in the first 600 μm from the surface, compared with vehicle treatment [integrated density of HMW ubiquitinated protein smear (in folds of vehicle control): non-tg: 2.89 ± 0.12, p < 0.0001; (h)αSyn tg: 2.26 ± 0.08, p < 0.0001; αSyn-GFP tg: 3.82 ± 0.07, p < 0.0001; Fig. 1A–C]. Of note, vehicle-treated mice overexpressing α-synuclein showed significantly higher levels of HMW ubiquitinated protein species than nontransgenic controls or mice overexpressing the fusion protein α-synuclein-GFP (p < 0.0001; p < 0.0001; Fig. 1B). To further confirm that proteasome activity was being inhibited by CLBL, we used a fluorescence-based assay to measure chymotrypsin-like proteasome activity and found that the catalytic activity of both the 20S and 26S proteasome was significantly impaired, particularly in the first 300 μm of subsurface (Fig. 1C).
Figure 1.

Topically applied UPS inhibitor CLBL leads to accumulation of polyubiquitinated proteins and reduced 20S and 26S proteasome activity. A, Representative immunoblot showing levels of polyubiquitinated HMW species and monomeric ubiquitin in homogenates of recovered cortical tissue of human α-synuclein transgenic mice treated with proteasome inhibitor CLBL or vehicle. B, Densitometric analysis of HMW polyubiquitinated protein smear (100–250 kDa) in all mouse lines after treatment with CLBL or vehicle: non-tg (vehicle): 7863 ± 1151, n = 12 (vehicle); 22,729 ± 2435, n = 12 (CLBL); t test, p < 0.0001; (h)αSyn tg: 26,277 ± 1706, n = 8 (vehicle); 59,418 ± 4204, n = 10 (CLBL); t test, p < 0.0001; αSyn-GFP tg: 8848 ± 538, n = 11 (vehicle); 33,792 ± 2271, n = 11 (CLBL); t test, p < 0.0001. C, Levels of polyubiquitinated HMW species and monomeric ubiquitin by immunoblot analysis (left axis) and chymotrypsin-like catalytic activity of the 20S/26S proteasome measured by turnover of a specific fluorogenic substrate (Suc-Leu-Leu-Val-Try-AMC) (right axis) in homogenates of serial cortical sections of nontransgenic mice treated with CLBL (horizontal sections, each 100 μm thick). D, Chymotrypsin-like catalytic activity of the 26S proteasome and both the 20S and 26S proteasome in nontransgenic, human α-synuclein transgenic, and human α-synuclein-GFP transgenic mice treated with vehicle as measured by cleavage of Suc-Leu-Leu-Val-Try-AMC in homogenates of treated cortex in the presence (20S/26S) and absence (26S) of SDS. Baseline proteasome activity is significantly reduced in α-synuclein transgenic mice compared with nontransgenic controls. [AMC] (in μm): non-tg (20S and 26S): 1.48 ± 0.11, n = 5; (h)αSyn tg (20S and 26S): 0.71 ± 0.04, n = 9; αSyn-GFP tg (20S and 26S): 1.26 ± 0.16, n = 5; ANOVA, p < 0.0001; Dunnett's posttest, p < 0.001 [non-tg vs (h)αSyn tg], p < 0.01 [(h)αSyn tg vs αSyn-GFP tg], p > 0.05 (non-tg vs αSyn-GFP tg); non-tg (26S): 1.72 ± 0.13, n = 3; (h)αSyn tg (26S): 0.94 ± 0.09, n = 4; αSyn-GFP tg: 1.04 ± 0.07, n = 4; ANOVA, p = 0.0011; Dunnett's posttest, p < 0.001 [non-tg vs (h)αSyn tg], p > 0.05 [(h)αSyn tg vs αSyn-GFP tg], p < 0.01 (non-tg vs αSyn-GFP tg). E, Chymotrypsin-like catalytic activity of the 26S proteasome and both the 20S and 26S proteasome in nontransgenic, human α-synuclein transgenic and human α-synuclein-GFP transgenic mice treated with CLBL as measured by cleavage of Suc-Leu-Leu-Val-Try-AMC in homogenates of treated cortex in the presence (20S/26S) and absence (26S) of SDS. Non-tg (20S and 26S): 1.48 ± 0.11, n = 5 (vehicle); 0.42 ± 0.14, n = 7 (CLBL); t test, p = 0.0003; (h)αSyn tg: (20S and 26S): 0.71 ± 0.04, n = 9 (vehicle); 0.30 ± 0.07, n = 7 (CLBL); t test, p = 0.0002; αSyn-GFP tg (20S and 26S): 1.26 ± 0.16, n = 5 (vehicle); 0.54 ± 0.09, n = 6 (CLBL); t test, p = 0.0028; non-tg (26S): 1.72 ± 0.13, n = 3 (vehicle); 0.37 ± 0.001, n = 3 (CLBL); t test, p = 0.0004; (h)αSyn tg (26S): 0.94 ± 0.09, n = 4 (vehicle); 0.69 ± 0.04, n = 4 (CLBL); t test, p = 0.04; αSyn-GFP tg (26S): 1.04 ± 0.07, n = 4 (vehicle); 0.75 ± 0.05, n = 4 (CLBL); t test, p = 0.01. Tissue of the first 300 μm of subsurface was used unless noted otherwise. *p < 0.05, **p < 0.01, ***p < 0.001. Error bars indicate SEM.
Interestingly, we found that baseline proteasome activity, as measured in vehicle-treated controls, was significantly impaired in animals overexpressing either human α-synuclein or α-synuclein-GFP [non-tg: (20S and 26S), 1.48 ± 0.11; (26S), 1.72 ± 0.13; (h)αSyn tg: (20S and 26S), 0.71 ± 0.04, p < 0.001; (26S), 0.94 ± 0.09, p < 0.01; αSyn-GFP tg (20S and 26S), 1.26 ± 0.16, p > 0.05; (26S), 1.04 ± 0.07, p < 0.01; Fig. 1D]. This reduction of proteasome activity at baseline, together with increased levels of HMW polyubiquitinated protein species (Fig. 1B), suggests that α-synuclein overexpression impairs proteasome function in the mouse models examined (Fig. 1B,D). With topical CLBL application, nontransgenic as well as human α-synuclein and α-synuclein-GFP transgenic mice showed a significant reduction of both 20S and 26S proteasome activity [in folds of vehicle-treated controls: non-tg: (20S and 26S), 0.28 ± 0.13, p = 0.0003; (26S), 0.21 ± 0.07, p = 0.0004; (h)αSyn tg: (20S and 26S), 0.42 ± 0.12, p = 0.0002; (26S), 0.73 ± 0.11, p = 0.04; αSyn-GFP tg (20S and 26S), 0.43 ± 0.14, p = 0.003; (26S), 0.72 ± 0.09, p = 0.02; Fig. 1E]. Finally, we took advantage of the UPS reporter GFPu to assess proteasome inhibition in the living mouse brain. GFPu contains a degron sequence (CL1) that targets it for ubiquitin-dependent unfolding and degradation by the proteasome (Bence et al., 2001). Using a combination of AAV-mediated [serotype 2/8, displaying neuronal specific expression (McFarland et al., 2009a,b)] delivery of GFPu to the neocortex and in vivo multiphoton imaging, we studied UPS inhibition in the brain over time. At baseline, no or background signal (reflecting autofluorescence at the injection site) was present (Fig. 2A–C), making baseline comparisons of proteasomal function between individual animals difficult. Topical application of CLBL, however, induced robust accumulation of GFPu after 24 h as demonstrated by the development of GFP-positive neurons containing GFP-labeled inclusions and an overall increase in GFP signal. In vehicle-treated animals, no accumulation of GFPu, consistent with homeostatic proteasome function, was observed. The increase of GFPu after CLBL treatment was similarly seen in human α-synuclein transgenic mice and nontransgenic littermates (Fig. 2A–C). Together, this set of data indicates that CLBL can successfully inhibit the UPS in the region of application.
Figure 2.
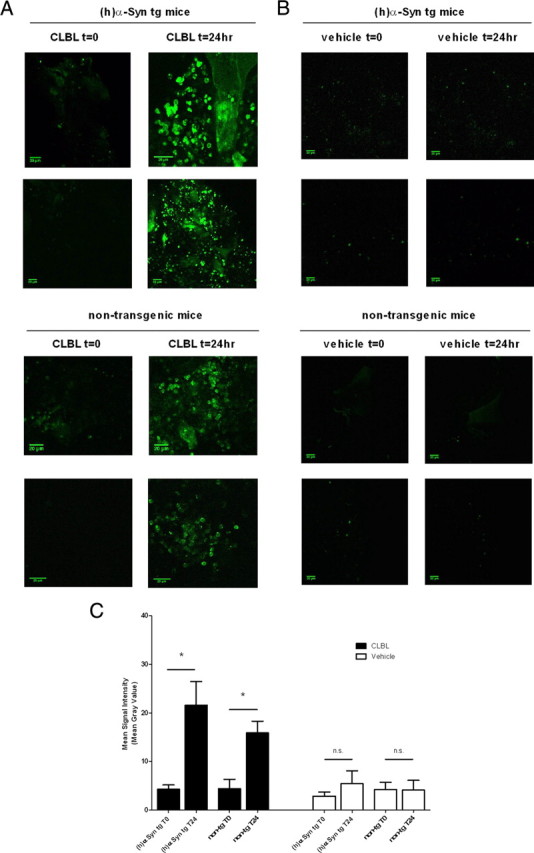
Topically applied UPS inhibitor CLBL leads to enhanced fluorescence of the UPS reporter GFPu in vivo. AAV8-mediated delivery of the UPS reporter GFPu to neocortex in combination with in vivo multiphoton imaging demonstrates CLBL-mediated proteasome inhibition in the living mouse brain. Baseline GFP signal immediately after cranial window installation and CLBL (A) or vehicle (B) application. GFPu accumulates in a granular pattern in neurons after topical incubation with CLBL for 24 h, demonstrating proteasome inhibition. GFPu does not accumulate in vehicle-treated animals after 24 h, consistent with homeostatic proteasome function. Signal at T0 in vehicle-treated animals is due to autofluorescence at the injection site. At total of 12 animals were injected and subjected to analysis (C): (h)αSyn tg (CLBL): 4.30 ± 0.8 (T0), 21.56 ± 4.9 (T24), n = 3; t test, p = 0.03; non-tg (CLBL): 4.403 ± 1.9 (T0), 15.91 ± 2.4 (T24); n = 3; t test, p = 0.02; (h)αSyn tg (vehicle): 2.88 ± 0.8 (T0), 5.44 ± 2.7 (T24); n = 3; t test, p > 0.05; non-tg (vehicle): 4.23 ± 1.5 (T0), 4.14 ± 2.0 (T24); n = 3; t test, p > 0.05. *p < 0.05. Error bars indicate SEM.
To evaluate the effects of CLBL-mediated UPS inhibition on α-synuclein in vivo, CLBL or MG132, as an alternative UPS inhibitor, were topically applied to the cortex of nontransgenic control mice, human α-synuclein transgenic, and α-synuclein-GFP transgenic mice using the cranial window-based approach described above. We found that, compared with vehicle treatment in littermates, immunoblot levels of endogenous α-synuclein in nontransgenic mice were moderately but significantly increased to 1.35 ± 0.09-fold following CLBL treatment (p < 0.05; Fig. 3A,B). In agreement with this finding in nontransgenic littermates, UPS inhibition with CLBL in human α-synuclein transgenic and αSyn-GFP transgenic mice led to a strong increase of cortical α-synuclein levels. Levels of overexpressed human α-synuclein increased to 2.05 ± 0.14-fold of vehicle-treated littermates with CLBL (p < 0.001) and 2.73 ± 0.12-fold with MG132 (p < 0.001), while levels of αSyn-GFP and endogenous α-synuclein increased to 2.87 ± 0.11- and 4.56 ± 0.17-fold of control, respectively (p < 0.001; p < 0.001; Fig. 3A–D). Topical treatment with a third UPS inhibitor epoxomicin unfortunately did not cause significant proteasomal inhibition as assayed by the development of HMW ubiquitinated species on immunoblot, nor did it cause an increase in cortical α-synuclein levels (data not shown), likely due to its decreased ability to penetrate the brain. Next, we determined the depth dependence of the increase in human α-synuclein measured after topical treatment with CLBL. As expected from our analysis of proteasomal inhibition with depth described above (Fig. 1C), CLBL treatment produced a detectable increase in human α-synuclein in the first 300 μm of subsurface, the exact region where proteasome inhibition is the strongest in our paradigm (Fig. 3E).
Figure 3.

UPS inhibition using the specific proteasome inhibitor CLBL results in significantly elevated α-synuclein levels in vivo independent of the preexisting α-synuclein in burden. A, C, Immunoblot levels of α-synuclein in homogenates of recovered cortical tissue of nontransgenic, human α-synuclein transgenic, and α-synuclein-GFP transgenic mice after treatment with proteasome inhibitor CLBL (20 μm; 24 h), MG132 (100 μm; 24 h), or vehicle. Non-tg: 0.43 ± 0.03, n = 17 (vehicle), 0.58 ± 0.04, n = 14 (CLBL); ANOVA, p < 0.0001; Dunnett's posttest, p < 0.05; (h)αSyn tg: 0.42 ± 0.05, n = 15 (vehicle), 0.85 ± 0.11, n = 14 (CLBL), 1.14 ± 0.09, n = 2 (MG132); ANOVA, p = 0.0001; Dunnett's posttest, p < 0.001; αSyn-GFP tg: αSyn-GFP: 0.25 ± 0.04, n = 11 (vehicle), 0.72 ± 0.07, n = 11 (CLBL); ANOVA, p < 0.0001; Dunnett's posttest, p < 0.001; endogenous α-synuclein: 0.25 ± 0.05, n = 11 (vehicle), 1.15 ± 0.18, n = 11 (CLBL); ANOVA, p = 0.0001; Dunnett's posttest, p < 0.001. B, D, Representative immunoblots of α-synuclein levels. E, CLBL treatment in human α-synuclein transgenic mice produces a detectable increase in human α-synuclein in the first 300 μm of subsurface, the exact region where proteasome inhibition is the strongest [1.52-fold the level of vehicle-treated littermates within the first 300 μm in this set of experiments; n = 3 (vehicle); n = 3 (CLBL)]. F, Comparison of immunoblot levels of α-synuclein in different age groups in human α-synuclein transgenic mice after proteasome inhibition with CLBL shows a linear increase with age. Aged 3–6 months: 0.30 ± 0.05, n = 5 (vehicle), 0.48 ± 0.03, n = 4 (CLBL); aged 6–9 months: 0.39 ± 0.09, n = 5 (vehicle), 0.67 ± 0.06 n = 4 (CLBL); aged >9 months: 0.56 ± 0.10, n = 5 (vehicle), 1.22 ± 0.14, n = 6 (CLBL); CLBL: ANOVA, p = 0.0017; posttest for linear trend: p = 0.0008; vehicle: ANOVA, p = 0.13; posttest for linear trend, p > 0.05. G, H, Immunoblot levels of the autophagosome marker LC3 in different age groups in human α-synuclein transgenic mice treated with CLBL show no change. Aged 3–6 months: 0.80 ± 0.11, n = 3; aged 6–9 months: 0.72 ± 0.06, n = 3; aged >9 months: 0.76 ± 0.08, n = 3; ANOVA, p > 0.05; posttest for linear trend, p > 0.05. *p < 0.05, **p < 0.01, ***p < 0.001 compared with vehicle-treated littermates. Error bars indicate SEM.
Because the functional capacity of protein degradation pathways declines with aging (Cuervo and Dice, 2000; Keller et al., 2004), we examined the consequences of proteasome inhibition in human α-synuclein transgenic mice as they age. Interestingly, we found that significantly more α-synuclein accumulated after proteasome inhibition in old mice (>9 months) compared with younger animals (3–6 months, p < 0.01; 6–9 months, p < 0.05; Fig. 3F). This linear trend (posttest for linear trend, p=0.0008; Fig. 3F) was not significant in vehicle-treated animals (p > 0.05), suggesting that proteasome function is increasingly relevant for α-synuclein degradation in aged animals. Of interest, levels of the autophagosome marker LC3 (see below) did not change with age in these animals (Fig. 3G,H).
Because the ALP (macroautophagy and CMA) has also been implicated in the degradation of α-synuclein, we next investigated the effect of ALP inhibition on α-synuclein levels in vivo. BafA1 is a selective inhibitor of the vacuolar-type H+-ATPase leading to reduced acidification of lysosomes, decreasing the fusion with autophagosomes and consequently reducing protein clearance via autophagy (Yoshimori et al., 1991; Yamamoto et al., 1998; Kawai et al., 2007; Jahreiss et al., 2008). ALP inhibition with BafA1 can be monitored by immunoblot analysis of LC3, a marker of autophagosomes (Kabeya et al., 2000), and p62/SQSTM1, a selective substrate of macroautophagy (Bjørkøy et al., 2005; Komatsu et al., 2007). Following guidelines for assays monitoring autophagy (Klionsky et al., 2008), we measured the levels of LC3II (ratio with α-tubulin) and p62 in nontransgenic, human α-synuclein transgenic, and α-synuclein-GFP transgenic mice treated with BafA1. In all three mouse lines, levels of LC3II and p62 increased significantly compared with vehicle-treated controls, indicating an abundance of autophagosomes due to reduced downstream fusion with lysosomes and hence reduced autophagic flux, consistent with a significant impairment of macroautophagy [non-tg: p = 0.0004 (LC3II), p = 0.01 (p62); (h)αSyn tg: p < 0.0001 (LC3II), p < 0.0001 (p62); αSyn-GFP tg: p < 0.05 (LC3II), p < 0.01 (p62); Fig. 4A,B].
Figure 4.

Topical application of ALP inhibitor Bafilomycin A1 leads to elevated levels of LC3II and p62 and increased α-synuclein levels in vivo depending on the preexisting synuclein burden. A, B, Immunoblot levels of autophagosome marker LC3II and macroautophagy substrate p62 in homogenates of cortex of nontransgenic, human α-synuclein transgenic, and α-synuclein-GFP transgenic mice treated with autophagy inhibitor Bafilomycin A1 (2.5 μm; 24 h) or vehicle. Non-tg: LC3II: 0.17 ± 0.03, n = 8 (vehicle), 0.46 ± 0.04, n = 4 (BafA1); t test, p = 0.0004; p62: 0.83 ± 0.16, n = 8 (vehicle), 1.54 ± 0.06, n = 4 (BafA1); t test, p = 0.01; (h)αSyn tg: LC3II: 0.15 ± 0.05, n = 8 (vehicle), 1.09 ± 0.19, n = 4 (BafA1); t test, p < 0.0001; p62: 0.67 ± 0.05, n = 9 (vehicle), 1.35 ± 0.11, n = 4 (BafA1); t test, p < 0.0001; αSyn-GFP tg: LC3II: 0.29 ± 0.03, n = 3 (vehicle), 0.48 ± 0.06, n = 3 (BafA1); t test, p = 0.04; p62: 0.38 ± 0.06, n = 3 (vehicle), 1.08 ± 0.07, n = 3 (BafA1); t test, p = 0.0017. C–F, Immunoblot levels of α-synuclein in homogenates of recovered cortical tissue of nontransgenic, human α-synuclein transgenic, and α-synuclein-GFP transgenic mice after treatment with ALP inhibitor BafA1 (2.5 μm; 24 h), HCQ (100 μm; 24 h), or vehicle. Non-tg: 0.43 ± 0.03, n = 17 (vehicle), 0.33 ± 0.02, n = 10 (BafA1); ANOVA, p < 0.0001; Dunnett's posttest, p > 0.05, not significant; (h)αSyn tg: 0.42 ± 0.05, n = 15 (vehicle), 0.90 ± 0.08, n = 7 (BafA1), 1.44 ± 0.01, n = 2 (HCQ); ANOVA, p = 0.0001; Dunnett's posttest, p < 0.01; αSyn-GFP tg: αSyn-GFP: 0.25 ± 0.04, n = 11 (vehicle), 0.69 ± 0.04, n = 3 (BafA1); ANOVA, p < 0.0001; Dunnett's posttest, p < 0.01; endogenous α-synuclein: 0.25 ± 0.05, n = 11 (vehicle), 0.21 ± 0.02, n = 3 (BafA1); ANOVA, p < 0.0001; Dunnett's posttest, p > 0.05, not significant. *p < 0.05, **p < 0.01, ***p < 0.001 compared with vehicle-treated littermates. Error bars indicate SEM.
To assess the effect of ALP inhibition on α-synuclein degradation, we topically treated nontransgenic, human α-synuclein transgenic, and α-synuclein-GFP transgenic mice with BafA1, HCQ, or vehicle. Surprisingly, in nontransgenic animals, we did not observe any change in α-synuclein levels following BafA1-mediated ALP inhibition (p > 0.05, not significant; Fig. 4C,D). By contrast, in human α-synuclein transgenic mice, α-synuclein levels increased strongly to 2.15 ± 0.10-fold of vehicle controls (p < 0.01; Fig. 4C,D). This increase was confirmed using the lysosomotropic drug HCQ as an alternative agent with inhibitory effects on lysosomal degradation downstream of BafA1 (3.43 ± 0.10-fold of vehicle controls; p < 0.0001; Fig. 4E,F). Immunoblot analysis in recovered cortical tissue of α-synuclein-GFP transgenic mice demonstrated an increase in αSyn-GFP to 2.76 ± 0.12-fold of vehicle-treated controls (p < 0.001), whereas untagged endogenous α-synuclein levels from the same tissue showed no significant change (p > 0.05, not significant; Fig. 4C,D), indicating that levels of excess αSyn-GFP are affected by ALP inhibition while levels of endogenous α-synuclein remain stable.
α-Synuclein-GFP transgenic mice in combination with in vivo multiphoton imaging provide a valuable tool to study intracellular and synaptic α-synuclein levels in the living mouse brain by measuring GFP signal intensity in these compartments. Following craniotomy, CLBL was topically applied to the brain surface and incubated for 90–120 min. Images were obtained at the time of CLBL application (T0) and again 24 h later (T24) covering the exact same cortical volume (Fig. 5A–E). Multiple z-stacks, ∼200–300 μm deep, were taken, each encompassing ∼50–100 αSyn-GFP-labeled cell bodies and innumerable synaptic terminals, mostly in cortical layer I–III. When the signal intensity of αSyn-GFP-expressing cell bodies was analyzed, we found that CLBL-mediated proteasome inhibition increased αSyn-GFP signal to 1.53 ± 0.11-fold at T24 compared with T0 (p < 0.0001; Fig. 5A). By contrast, the mean signal intensity in vehicle-treated littermates showed a small but significant decrease to 0.76 ± 0.14 at T24 (p < 0.0001; Fig. 5A). Importantly, no cell loss was observed in either condition (Fig. 5B,C,E). Similar to the cell body, analysis of αSyn-GFP signal at synaptic terminals showed an increase with UPS inhibition at T24 indicating increased amounts of the fusion protein at the synaptic compartment (p < 0.05 compared with vehicle-treated littermates; Fig. 5D). Interestingly, the increase in αSyn-GFP signal detected by in vivo imaging (Fig. 5A) is somewhat less than that which was detected after immunoblotting for the same protein (Fig. 3A). This may reflect the fact that in vivo imaging is only sensitive to αSyn-GFP species where the GFP moiety is appropriately folded, and not sensitive to species where GFP is not yet appropriately folded or even misfolded. This is in contrast to immunoblotting, which is done against α-synuclein, and therefore will detect all species regardless of the intramolecular state of GFP.
Figure 5.
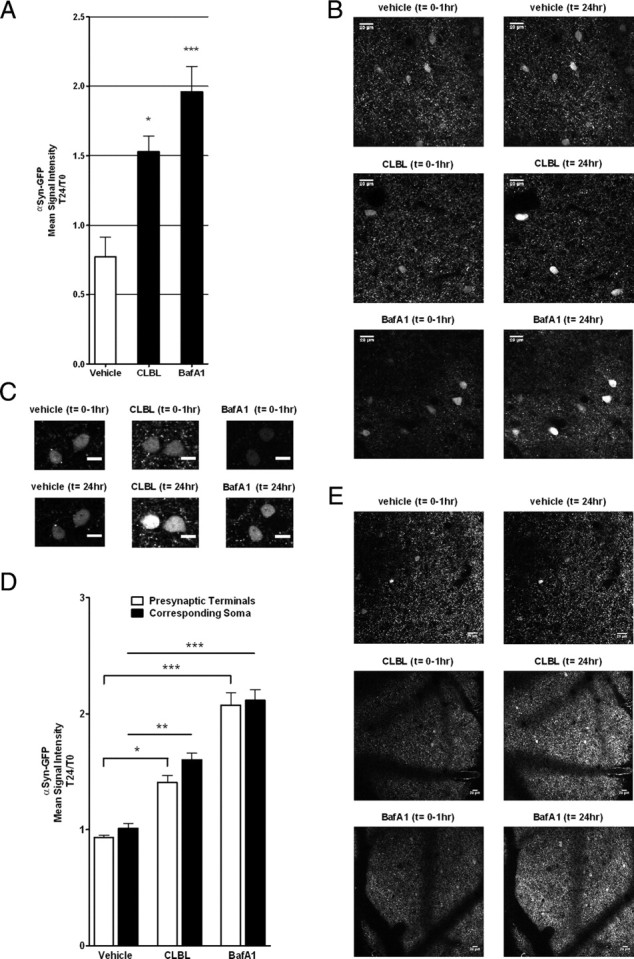
In vivo multiphoton imaging in α-synuclein-GFP transgenic mice reveals increased αSyn-GFP signal in individual neurons and presynaptic terminals. A, Mean αSyn-GFP signal intensity of cell bodies 24 h after topical application of UPS inhibitor CLBL, ALP inhibitor BafA1, or vehicle. αSyn-GFP signal intensity was measured in corresponding neurons immediately after cranial window installation (T0) and after 24 h (T24). Soma: vehicle: 0.77 ± 0.14 (T24/T0), n = 4 animals, n > 150 neurons; CLBL: 1.53 ± 0.11 (T24/T0), n = 4 animals, n > 300 neurons, ANOVA, p = 0.001; Dunnett's posttest, p < 0.05; BafA1: 1.96 ± 0.18 (T24/T0), n = 4 animals, n > 150 neurons; ANOVA, p = 0.001; Dunnett's posttest, p < 0.001. Representative low- (B) and high-power (C) in vivo multiphoton images of individual αSyn-GFP-positive neurons in neocortical neurons (layer II/III) immediately after CLBL, BafA1, or vehicle application and 24 h later. Scale bars: B, 20 μm; C, 10 μm. *p < 0.05, **p < 0.01, ***p < 0.001. D, Mean αSyn-GFP signal intensity of presynaptic terminals and corresponding cell bodies of the same z-section 24 h after topical application of UPS inhibitor CLBL, ALP inhibitor BafA1, or vehicle. αSyn-GFP signal intensity was measured in corresponding neurons and presynaptic terminals immediately after cranial window installation (T0) and after 24 h (T24). Presynaptic terminals: vehicle: 0.93 ± 0.02 (T24/T0), n = 4 animals, n > 200,000 presynaptic terminals; CLBL: 1.40 ± 0.06 (T24/T0), n = 4 animals, n > 400,000 presynaptic terminals; BafA1: 2.08 ± 0.11 (T24/T0), n = 4 animals, n > 600,000 presynaptic terminals; corresponding soma: vehicle: 1.0 ± 0.04 (T24/T0), n = 4 animals, n > 30 neurons; CLBL: 1.6 ± 0.06 (T24/T0), n = 4 animals, n > 40 neurons; BafA1: 2.12 ± 0.09 (T24/T0), n = 4 animals, n > 90 neurons. E, Representative low-power images showing the distribution of αSyn-GFP signal in presynaptic terminals immediately after drug incubation and cranial window installation and 24 h later. Scale bar, 20 μm. Blood vessels appear as negative contrast. *p < 0.05, **p < 0.01, ***p < 0.001. Error bars indicate SEM.
To confirm the effects of ALP inhibition on a cellular level in the intact, living mouse brain, we, again, took advantage of our ability to use in vivo multiphoton imaging in α-synuclein-GFP transgenic mice. The signal intensity of αSyn-GFP-positive cell bodies after BafA1 treatment increased to 1.96 ± 0.18 at T24 compared with T0 (p = 0.002; Fig. 5A), confirming the accumulation of αSyn-GFP observed by immunoblotting. Similarly, signal intensity in the synaptic compartment increased by 2.08 ± 0.11-fold, suggesting that ALP inhibition increases α-synuclein levels at presynaptic terminals (p < 0.001; Fig. 5D).
Our data suggest that application of specific UPS or ALP inhibitors can increase α-synuclein levels in cortical tissue treated in vivo. Presumably, this is because α-synuclein is being degraded by these pathways, but it is also formally possible that increases in α-synuclein detected after protein degradation is inhibited could be due to indirect changes on α-synuclein expression. For example, if the level of a transcription factor that is rate-limiting for α-synuclein transcription accumulated because of UPS or ALP inhibition, this could indirectly lead to increases in α-synuclein mRNA levels and therefore increased protein expression. To test for this, we measured levels of both mouse and human α-synuclein mRNA independently in the same cortical samples from human α-synuclein transgenic mice using specifically designed primers and qPCR. Primers designed to selectively detect human or mouse αSyn were tested by performing qPCR on both human cDNA and wild-type mouse cDNA. Human specific primers transcribing human cDNA produced a band of the expected size (168 bp) (Fig. 6A), but showed no cross-reactivity with mouse cDNA (Fig. 6A). Similarly, mouse-specific primers transcribing mouse cDNA produced a band of the expected size (313 bp) and showed no cross-reactivity with human cDNA (Fig. 6A). Human-specific primers and mouse-specific primers produced only one band of the expected size when transgenic mouse cDNA was used in the qPCR (Fig. 6A). This approached showed that treatment with CLBL or BafA1 caused no detectible changes in human α-synuclein mRNA levels, and therefore increased expression cannot explain the increases in protein levels we observe (Fig. 6B). The measurement of endogenous mouse α-synuclein mRNA levels showed a nonsignificant trend to decreased levels after both CLBL and BafA1 treatment (Fig. 6B), suggesting there may be compensatory downregulation of mRNA levels under control of the endogenous promoter when protein levels increase, but also arguing against any indirect increase in transcription explaining these results.
Figure 6.

UPS or ALP inhibition does not indirectly increase transcription of either mouse or human α-synuclein mRNA. Human- and mouse-specific primers were designed to independently measure each species of α-synuclein in the same sample. When each set of primers was tested on either a WT mouse or human brain sample, only the expected product was observed by gel electrophoresis (A, left panel). When the same primers were used on brain samples from human α-synuclein transgenic mice, again only the expected product was detected with each primer set (A, right panel). Analysis of cortical tissue from human α-synuclein transgenic mice treated either with vehicle, CLBL, or BafA1 demonstrated that human α-synuclein mRNA levels were unchanged by either treatment (B, left panel). The analysis of mouse α-synuclein mRNA levels in the same samples demonstrated a nonsignificant trend toward decreased levels in both CLBL and BafA1 treatment conditions (B, right panel). Error bars indicate SEM.
Because our data suggest a role for the UPS and ALP in the degradation of α-synuclein in vivo, we next investigated the potential cross talk between the two degradation pathways by measuring levels of LC3II and p62 in animals that had been topically treated with CLBL. In nontransgenic mice, no significant difference in LC3II or p62 levels was observed in comparison with vehicle-treated littermates (p > 0.05, not significant; Fig. 7A–C). In human α-synuclein transgenic mice, however, CLBL-mediated UPS inhibition led to a strong increase in LC3II to 5.06 ± 0.09-fold of control (p < 0.001), while levels of p62 were decreased to 0.23 ± 0.07-fold of vehicle control (p < 0.0001; Fig. 7A–C), indicating an increase in autophagosomes and autophagic flux. To further confirm this finding, we measured levels of LC3II in mice treated simultaneously with CLBL and BafA1. We found that the LC3II/α-tubulin ratio was increased to 7.40 ± 0.08-fold of control (p < 0.001; Fig. 7A,B). This large increase in the presence of an ALP inhibitor like BafA1, in combination with the decrease of p62 observed with CLBL alone, suggests enhanced macroautophagy (Mizushima and Yoshimori, 2007; Klionsky et al., 2008; Mizushima et al., 2010). In nontransgenic mice, LC3II levels increased to 2.05 ± 0.17-fold of vehicle control (p = 0.0077; Fig. 7A,B), indicating an accumulation of autophagosomes consistent with a block in autophagic clearance by BafA1 (Mizushima and Yoshimori, 2007; Klionsky et al., 2008; Mizushima et al., 2010). To determine whether this coupling between protein degradation pathways can occur in the other direction, we measured proteasome activity following BafA1 treatment. In nontransgenic mice, no change in the 26S chymotrypsin-like catalytic activity was present (p > 0.05, not significant; Fig. 7D), whereas in human α-synuclein transgenic mice BafA1 treatment led to an increase in 26S proteasome function (p < 0.001; Fig. 7D). In both mouse lines, simultaneous treatment with BafA1 and CLBL resulted in significantly reduced 26S proteasome activity, showing that the upregulation of proteasome activity was reversed by this specific proteasome inhibitor (p < 0.001). Together, this set of experiments supports a model whereby proteasome activity is increased upon autophagy inhibition under conditions of increased α-synuclein expression, although future studies are needed to decipher the exact mechanism involved. In the opposite direction, macroautophagy is enhanced following proteasome inhibition, again only in the presence of increased α-synuclein burden.
Figure 7.

Cross talk between autophagy and the UPS is induced in the presence of increased α-synuclein levels in vivo. Immunoblot levels of autophagosome marker LC3II and macroautophagy substrate p62 in homogenates of cortex of nontransgenic and human α-synuclein transgenic mice treated with UPS inhibitor CLBL (20 μm; 24 h) alone or in combination with ALP inhibitor BafA1 (2.5 μm; 24 h) (A–C) or vehicle. Non-tg: LC3II: 0.17 ± 0.03, n = 8 (vehicle), 0.29 ± 0.04, n = 4 (CLBL); ANOVA, p = 0.0004; Dunnett's posttest, p > 0.05, not significant; 0.35 ± 0.04, n = 4 (CLBL+BafA1); ANOVA, p = 0.0004; Dunnett's posttest, p < 0.05; p62: 0.83 ± 0.16, n = 8 (vehicle), 0.99 ± 0.05, n = 4 (CLBL); t test, p > 0.5, not significant; (h)αSyn tg: LC3II: 0.15 ± 0.05, n = 8 (vehicle), 0.76 ± 0.05, n = 13 (CLBL); ANOVA, p < 0.0001, Dunnett's posttest, p < 0.001; 1.11 ± 0.09, n = 4 (CLBL+BafA1); ANOVA, p < 0.0001, Dunnett's posttest, p < 0.001; p62: 0.67 ± 0.05, n = 9 (vehicle), 0.16 ± 0.02, n = 9 (CLBL); t test, p < 0.0001. D, Chymotrypsin-like catalytic activity of the 26S proteasome in homogenates of recovered cortical tissue of nontransgenic and human α-synuclein transgenic mice treated with vehicle, autophagy inhibitor BafA1, or a combination of UPS inhibitor CLBL and BafA1. Data are presented as folds of vehicle-treated littermates: non-tg: 1.06 ± 0.05, n = 3 (BafA1); ANOVA, p < 0.0001; Dunnett's posttest, p > 0.05, not significant; 0.47 ± 0.03, n = 3 (CLBL+BafA1); ANOVA, p < 0.0001; Dunnett's posttest, p < 0.001; (h)αSyn tg: 1.78 ± 0.12, n = 4 (BafA1); ANOVA, p < 0.0001; Dunnett's posttest, p < 0.001; 0.71 ± 0.07, n = 5 (CLBL+BafA1); ANOVA, p < 0.0001; Dunnett's posttest, p < 0.01. C+B, CLBL plus Bafilomycin A1. *p < 0.05, ***p < 0.001 compared with vehicle-treated littermates. Error bars indicate SEM.
Finally, simultaneous application of both CLBL and BafA1, causing a block of proteasome and autophagy function, resulted in increases levels of α-synuclein in nontransgenic and human α-synuclein transgenic mice (Fig. 8). This increase was not additive compared with CLBL or BafA1 alone, suggesting that a block of the compensatory upregulation of either pathway cannot further increase levels of α-synuclein at 24 h (p > 0.05, not significant). Only in nontransgenic mice, an additive effect of concomitant inhibition compared with BafA1-mediated ALP inhibition alone was evident, again supporting the importance of the UPS under normal conditions (p < 0.001).
Figure 8.

Combined UPS and autophagy inhibition with CLBL and BafA1 leads to increased levels of α-synuclein independent of the preexisting α-synuclein burden. Immunoblot levels of α-synuclein in homogenates of recovered cortical tissue of nontransgenic and human α-synuclein transgenic mice after simultaneous treatment with UPS inhibitor CLBL and ALP inhibitor BafA1. Non-tg: 0.43 ± 0.03, n = 17 (vehicle), 0.65 ± 0.06, n = 10 (CLBL+BafA1); ANOVA, p < 0.0001; Dunnett's posttest, p < 0.01; (h)αSyn tg: vehicle: 0.42 ± 0.05, n = 15; CLBL+BafA1: 1.04 ± 0.09, n = 4; ANOVA, p = 0.0001; Dunnett's posttest, p < 0.01. **p < 0.01, compared with vehicle-treated littermates. Error bars indicate SEM.
Discussion
Our study provides evidence for distinct roles of the UPS and ALP in the degradation of α-synuclein in vivo. Using a combination of different approaches, including topical treatment with specific pharmacological inhibitors and multiphoton imaging in different transgenic mouse models, we demonstrate that the mechanism used to degrade α-synuclein depends on the burden of this protein in the cell. Furthermore, we show that there is an age dependence to the relevance of the UPS in α-synuclein degradation and that the two clearance pathways appear to be functionally coupled under conditions of increased α-synuclein expression.
Topical treatment of the cortex with the specific proteasome inhibitor CLBL in nontransgenic animals as well as human α-synuclein transgenic and α-synuclein-GFP transgenic mice (Masliah et al., 2000; Rockenstein et al., 2005) leads to significantly elevated α-synuclein levels and thus provides direct evidence that the UPS degrades both endogenous murine and overexpressed human α-synuclein in vivo. These mouse lines were selected because they display abnormal accumulation of α-synuclein and develop α-synuclein inclusion-like structures in the brain, enabling us to explore degradation pathways in a scenario with pathologically elevated α-synuclein expression. In addition, we show that changes in intracellular α-synuclein levels can be explored in individual neurons and presynaptic terminals in vivo by combining pharmacological manipulation of degradation pathways with multiphoton imaging in α-synuclein-GFP transgenic mice. Using this technique, we find that αSyn-GFP signal increases robustly in neuronal cell bodies and presynaptic terminals following UPS inhibition. Intriguingly, αSyn-GFP-positive cellular processes and presynaptic terminals became increasingly prominent with UPS inhibition. These findings suggest that α-synuclein can accumulate in the synaptic compartment, which may contribute to the formation of synaptic aggregates described previously (Kramer and Schulz-Schaeffer, 2007; Scott et al., 2010).
Our results demonstrate that degradation by the UPS is independent of the preexisting α-synuclein burden and is likely to be active both under normal conditions and in the disease state, where intracellular α-synuclein levels are raised. These data are consistent with previous in vitro studies that indicate a role for the proteasome in α-synuclein degradation (Bennett et al., 1999; McLean et al., 2001; Webb et al., 2003; Shin et al., 2005). However, other studies have produced conflicting results (Ancolio et al., 2000; Rideout et al., 2001; Cuervo et al., 2004; Vogiatzi et al., 2008). Our work, in the living mouse brain, provides direct evidence that the UPS is important for α-synuclein degradation using two powerful and complementary strategies, protein biochemistry and in vivo fluorescence imaging. Our findings also extend two previously published in vivo reports addressing this issue using genetic depletion of proteasome function during development (Bedford et al., 2008) or chronic toxin exposure (Fornai et al., 2005).
Interestingly, our findings in vehicle-treated α-synuclein transgenic mice also confirm previous reports that showed that stably increased levels of α-synuclein can lead to impaired proteasome function, potentially paving the way for subsequent pathological changes (Stefanis et al., 2001; Tanaka et al., 2001; Petrucelli et al., 2002; Snyder et al., 2003; Lindersson et al., 2004; Q. Chen et al., 2005; L. Chen et al., 2006; Jiang et al., 2007; Zhang et al., 2008; Emmanouilidou et al., 2010). The reciprocal interaction between α-synuclein and proteasome function suggests a self-perpetuating process in which permanently raised α-synuclein levels impair the UPS, which can in turn lead to further α-synuclein accumulation. The detailed molecular mechanism by which α-synuclein is degraded by the proteasome is still unanswered. Evidence exists in cell culture that degradation can occur via a proteasome-dependent but ubiquitin-independent pathway (Tofaris et al., 2001; Liu et al., 2003; Machiya et al., 2010). Our own data suggest that this may also be the case in vivo since specific polyubiquitin-tagged α-synuclein species were not observed under control conditions or when the UPS was inhibited. Even though it has been notoriously difficult to detect ubiquitinated α-synuclein species after substantial proteasome inhibition (Bennett et al., 1999; McLean et al., 2001; Stefanis et al., 2001; Tofaris et al., 2001; Machiya et al., 2010), our findings do not rule out ubiquitination of α-synuclein during degradation, but further studies will be required to elucidate the exact mechanism involved.
Of interest, we observed that the increase of α-synuclein with UPS inhibition in human α-synuclein transgenic mice occurred in an age-dependent manner, potentially reflecting impaired proteasome function due to elevated α-synuclein levels in aged mice. Many studies have shown a decline in degradation capacity in normal aging and in protein conformational disorders associated with aging. A functional decay with age has been shown for both autophagy (Cuervo and Dice, 2000; Martinez-Vicente et al., 2005) and the UPS (Cuervo and Dice, 2000; Keller et al., 2004), although in the latter the decrease does not seem to be universal (Cook et al., 2009). We, however, did not find any detectible relationship between autophagy function as measured by LC3II levels and age in our animals, suggesting a different explanation for the age-dependent increase in importance of UPS function we observed. Age-dependent changes in alternative protein clearance pathways like CMA (Cuervo and Dice, 2000) or secretion via exosomes (Emmanouilidou et al., 2010; Alvarez-Erviti et al., 2011) could underlie this observation and remain an open question for further studies.
The role of autophagy in α-synuclein degradation is uncertain. Recent cell culture studies have shown the importance of macroautophagy in the degradation of α-synuclein (Webb et al., 2003; Lee et al., 2004; Vogiatzi et al., 2008), but no role for macroautophagy (as opposed to CMA) was seen in other studies (Cuervo et al., 2004). To our knowledge, systematic in vivo approaches to answer this question have been limited to one report (Mak et al., 2010) that argued for a role for lysosome-based degradation of α-synuclein, mediated by CMA, in a genetic and a toxin-induced mouse model. In our study, we found no significant change in levels of endogenous α-synuclein in nontransgenic mice following ALP inhibition. This indicates that, at least in our model system, autophagy is not the primary mechanism for degradation of α-synuclein in nontransgenic mice under normal conditions. By contrast, in human α-synuclein transgenic mice, ALP inhibition led to a robust accumulation of α-synuclein. Similarly, in α-synuclein-GFP transgenic mice, αSyn-GFP signal intensity increased strongly in neuronal cell bodies and presynaptic terminals after ALP inhibition. These results suggest that, in an in vivo scenario with increased α-synuclein burden in neurons, autophagy is recruited to degrade α-synuclein. Our findings are in agreement with data from the study by Mak et al. (2010) that showed that α-synuclein is increasingly associated with lysosomes when α-synuclein expression is enhanced. BafA1 blocks lysosomal degradation by two mechanisms, initially by inhibiting acidification (Yoshimori et al., 1991; Yamamoto et al., 1998) and indirectly by blocking the fusion of autophagosomes with lysosomes (Yamamoto et al., 1998; Kawai et al., 2007; Jahreiss et al., 2008). In agreement with the latter, we observed a strong increase in LC3II, a marker for autophagosomes, accompanied by a marked increase in p62, a substrate of macroautophagy. We therefore conclude that impaired macroautophagy is primarily responsible for the effects on α-synuclein levels we observe, although CMA could also be a possible contributor to synuclein pathology as has been shown previously (Cuervo et al., 2004; Vogiatzi et al., 2008; Mak et al., 2010). Our study does not specifically address the role of CMA because, unfortunately, pharmacological agents that selectively target CMA are not currently available.
We systematically investigated potential cross talk between the UPS and autophagy. Interestingly, we found evidence for cross talk between the two pathways, but only under conditions of increased α-synuclein burden. The compensatory upregulation of proteasome activity after autophagy inhibition stands in contrast to our findings in nontransgenic mice as well as to previous studies in cell culture models (Korolchuk et al., 2009; Qiao and Zhang, 2009), suggesting that the level of α-synuclein in the cell modulates cross talk between degradation pathways. The finding that UPS inhibition can upregulate autophagic flux is consistent with previous studies in cell culture (Rideout et al., 2004; Iwata et al., 2005; Ding et al., 2007) and in Drosophila melanogaster (Pandey et al., 2007), in which autophagy was induced in response to impaired UPS activity. It should be noted, however, that our findings are observational and future studies are needed to decipher the exact mechanism involved.
Together, our study demonstrates distinct roles for the UPS and autophagy in the degradation of α-synuclein in vivo and emphasizes collaborative interactions when α-synuclein levels are raised. Specifically, we demonstrate that the UPS degrades α-synuclein under conditions with endogenous and increased α-synuclein burden and that, in contrast, autophagy is recruited to degrade α-synuclein only when intracellular α-synuclein levels are elevated. These findings elucidate a link between the proteasome, autophagy, and α-synuclein pathology by providing the first in vivo evidence for a relationship between protein burden and the pathways recruited to maintain homeostasis. Our study may help to clarify contradictory findings from previous in vitro studies and importantly extends our knowledge to the in vivo level. In the context of synucleinopathies, our results have implications for future therapeutics targeting specific degradation pathways to counteract α-synuclein pathology (Ravikumar et al., 2004; Sarkar et al., 2007; Spencer et al., 2009; Crews et al., 2010; Lee et al., 2010; Malagelada et al., 2010).
Footnotes
This work was supported by National Institutes of Health Grants NS038372 (B.T.H.), NS063963 (P.J.M.), T32 AG000222, T32 NS048005, K08 NS069625 (V.K.U.), and AG18840, AG022074, AG10435 (E.R., E.M.); The Michael J. Fox Foundation (I.C.-C.); the German National Academic Foundation (D.E.-F.); the Hamburg Foundation for International Research and Studies (D.E.-F.); and the Parkinson's Disease Foundation (D.E.-F.). We thank Dr. Bakhos A. Tannous and the Vector Production Core (National Institutes of Health Grant P30NS045776) at Massachusetts General Hospital.
The authors declare no competing financial interests.
References
- Alvarez-Erviti L, Seow Y, Schapira AH, Gardiner C, Sargent IL, Wood MJ, Cooper JM. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis. 2011;42:360–367. doi: 10.1016/j.nbd.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancolio K, Alves da Costa C, Uéda K, Checler F. Alpha-synuclein and the Parkinson's disease-related mutant Ala53Thr-alpha-synuclein do not undergo proteasomal degradation in HEK293 and neuronal cells. Neurosci Lett. 2000;285:79–82. doi: 10.1016/s0304-3940(00)01049-1. [DOI] [PubMed] [Google Scholar]
- Bedford L, Hay D, Devoy A, Paine S, Powe DG, Seth R, Gray T, Topham I, Fone K, Rezvani N, Mee M, Soane T, Layfield R, Sheppard PW, Ebendal T, Usoskin D, Lowe J, Mayer RJ. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J Neurosci. 2008;28:8189–8198. doi: 10.1523/JNEUROSCI.2218-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradian MM. Degradation of α-synuclein by proteasome. J Biol Chem. 1999;274:33855–33858. doi: 10.1074/jbc.274.48.33855. [DOI] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buesa C, Maes T, Subirada F, Barrachina M, Ferrer I. DNA chip technology in brain banks: confronting a degrading world. J Neuropathol Exp Neurol. 2004;63:1003–1014. doi: 10.1093/jnen/63.10.1003. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Chen L, Thiruchelvam MJ, Madura K, Richfield EK. Proteasome dysfunction in aged human alpha-synuclein transgenic mice. Neurobiol Dis. 2006;23:120–126. doi: 10.1016/j.nbd.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Chen Q, Thorpe J, Keller JN. Alpha-synuclein alters proteasome function, protein synthesis, and stationary phase viability. J Biol Chem. 2005;280:30009–30017. doi: 10.1074/jbc.M501308200. [DOI] [PubMed] [Google Scholar]
- Chesselet MF. In vivo alpha-synuclein overexpression in rodents: a useful model of Parkinson's disease? Exp Neurol. 2008;209:22–27. doi: 10.1016/j.expneurol.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C, Petrucelli L. A critical evaluation of the ubiquitin-proteasome system in Parkinson's disease. Biochim Biophys Acta. 2009;1792:664–675. doi: 10.1016/j.bbadis.2009.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C, Gass J, Dunmore J, Tong J, Taylor J, Eriksen J, McGowan E, Lewis J, Johnston J, Petrucelli L. Aging is not associated with proteasome impairment in UPS reporter mice. PLoS One. 2009;4:e5888. doi: 10.1371/journal.pone.0005888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews L, Spencer B, Desplats P, Patrick C, Paulino A, Rockenstein E, Hansen L, Adame A, Galasko D, Masliah E. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One. 2010;5:e9313. doi: 10.1371/journal.pone.0009313. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Cuervo AM, Dice JF. Age-related decline in chaperone-mediated autophagy. J Biol Chem. 2000;275:31505–31513. doi: 10.1074/jbc.M002102200. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–524. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmanouilidou E, Melachroinou K, Roumeliotis T, Garbis SD, Ntzouni M, Margaritis LH, Stefanis L, Vekrellis K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink L, Seeger W, Ermert L, Hänze J, Stahl U, Grimminger F, Kummer W, Bohle RM. Real-time quantitative RT-PCR after laser-assisted cell picking. Nat Med. 1998;4:1329–1333. doi: 10.1038/3327. [DOI] [PubMed] [Google Scholar]
- Fornai F, Schlüter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, Pellegrini A, Nicoletti F, Ruggieri S, Paparelli A, Südhof TC. Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc Natl Acad Sci U S A. 2005;102:3413–3418. doi: 10.1073/pnas.0409713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Holtmaat A, Bonhoeffer T, Chow DK, Chuckowree J, De Paola V, Hofer SB, Hübener M, Keck T, Knott G, Lee WC, Mostany R, Mrsic-Flogel TD, Nedivi E, Portera-Cailliau C, Svoboda K, Trachtenberg JT, Wilbrecht L. Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc. 2009;4:1128–1144. doi: 10.1038/nprot.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibáñez P, Bonnet AM, Débarges B, Lohmann E, Tison F, Pollak P, Agid Y, Dürr A, Brice A. Causal relation between alpha-synuclein gene duplication and familial Parkinson's disease. Lancet. 2004;364:1169–1171. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–40292. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- Jahreiss L, Menzies FM, Rubinsztein DC. The itinerary of autophagosomes: from peripheral formation to kiss-and-run fusion with lysosomes. Traffic. 2008;9:574–587. doi: 10.1111/j.1600-0854.2008.00701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Wu YC, Nakamura M, Liang Y, Tanaka Y, Holmes S, Dawson VL, Dawson TM, Ross CA, Smith WW. Parkinson's disease genetic mutations increase cell susceptibility to stress: mutant alpha-synuclein enhances H2O2- and Sin-1-induced cell death. Neurobiol Aging. 2007;28:1709–1717. doi: 10.1016/j.neurobiolaging.2006.07.017. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai A, Uchiyama H, Takano S, Nakamura N, Ohkuma S. Autophagosome-lysosome fusion depends on the pH in acidic compartments in CHO cells. Autophagy. 2007;3:154–157. doi: 10.4161/auto.3634. [DOI] [PubMed] [Google Scholar]
- Keller JN, Dimayuga E, Chen Q, Thorpe J, Gee J, Ding Q. Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain. Int J Biochem Cell Biol. 2004;36:2376–2391. doi: 10.1016/j.biocel.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Kisselev AF, Goldberg AL. Monitoring activity and inhibition of 26S proteasomes with fluorogenic peptide substrates. Methods Enzymol. 2005;398:364–378. doi: 10.1016/S0076-6879(05)98030-0. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–937. doi: 10.1038/nrm2245. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, Hamazaki J, Nishito Y, Iemura S, Natsume T, Yanagawa T, Uwayama J, Warabi E, Yoshida H, Ishii T, Kobayashi A, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33:517–527. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer ML, Schulz-Schaeffer WJ. Presynaptic α-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci. 2007;27:1405–1410. doi: 10.1523/JNEUROSCI.4564-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Lee MJ, Park S, Oh DC, Elsasser S, Chen PC, Gartner C, Dimova N, Hanna J, Gygi SP, Wilson SM, King RW, Finley D. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Khoshaghideh F, Patel S, Lee SJ. Clearance of α-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci. 2004;24:1888–1896. doi: 10.1523/JNEUROSCI.3809-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindersson E, Beedholm R, Højrup P, Moos T, Gai W, Hendil KB, Jensen PH. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J Biol Chem. 2004;279:12924–12934. doi: 10.1074/jbc.M306390200. [DOI] [PubMed] [Google Scholar]
- Liu CW, Corboy MJ, DeMartino GN, Thomas PJ. Endoproteolytic activity of the proteasome. Science. 2003;299:408–411. doi: 10.1126/science.1079293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔC(T) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Machiya Y, Hara S, Arawaka S, Fukushima S, Sato H, Sakamoto M, Koyama S, Kato T. Phosphorylated alpha-synuclein at Ser-129 is targeted to the proteasome pathway in a ubiquitin-independent manner. J Biol Chem. 2010;285:40732–40744. doi: 10.1074/jbc.M110.141952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak SK, McCormack AL, Manning-Bog AB, Cuervo AM, Di Monte DA. Lysosomal degradation of alpha-synuclein in vivo. J Biol Chem. 2010;285:13621–13629. doi: 10.1074/jbc.M109.074617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson's disease. J Neurosci. 2010;30:1166–1175. doi: 10.1523/JNEUROSCI.3944-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maraganore DM, de Andrade M, Elbaz A, Farrer MJ, Ioannidis JP, Krüger R, Rocca WA, Schneider NK, Lesnick TG, Lincoln SJ, Hulihan MM, Aasly JO, Ashizawa T, Chartier-Harlin MC, Checkoway H, Ferrarese C, Hadjigeorgiou G, Hattori N, Kawakami H, Lambert JC, et al. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA. 2006;296:661–670. doi: 10.1001/jama.296.6.661. [DOI] [PubMed] [Google Scholar]
- Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007;6:352–361. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- Martinez-Vicente M, Sovak G, Cuervo AM. Protein degradation and aging. Exp Gerontol. 2005;40:622–633. doi: 10.1016/j.exger.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- McFarland NR, Lee JS, Hyman BT, McLean PJ. Comparison of transduction efficiency of recombinant AAV serotypes 1, 2, 5, and 8 in the rat nigrostriatal system. J Neurochem. 2009a;109:838–845. doi: 10.1111/j.1471-4159.2009.06010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland NR, Fan Z, Xu K, Schwarzschild MA, Feany MB, Hyman BT, McLean PJ. Alpha-synuclein S129 phosphorylation mutants do not alter nigrostriatal toxicity in a rat model of Parkinson disease. J Neuropathol Exp Neurol. 2009b;68:515–524. doi: 10.1097/NEN.0b013e3181a24b53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean PJ, Kawamata H, Hyman BT. Alpha-synuclein-enhanced green fluorescent protein fusion proteins form proteasome sensitive inclusions in primary neurons. Neuroscience. 2001;104:901–912. doi: 10.1016/s0306-4522(01)00113-0. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow CW, McNaught KS. Ubiquitin-proteasome system and Parkinson's disease. Mov Disord. 2006;21:1806–1823. doi: 10.1002/mds.21013. [DOI] [PubMed] [Google Scholar]
- Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, O'Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR. Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36:1007–1019. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- Qiao L, Zhang J. Inhibition of lysosomal functions reduces proteasomal activity. Neurosci Lett. 2009;456:15–19. doi: 10.1016/j.neulet.2009.03.085. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- Rideout HJ, Larsen KE, Sulzer D, Stefanis L. Proteasomal inhibition leads to formation of ubiquitin/alpha-synuclein-immunoreactive inclusions in PC12 cells. J Neurochem. 2001;78:899–908. doi: 10.1046/j.1471-4159.2001.00474.x. [DOI] [PubMed] [Google Scholar]
- Rideout HJ, Lang-Rollin I, Stefanis L. Involvement of macroautophagy in the dissolution of neuronal inclusions. Int J Biochem Cell Biol. 2004;36:2551–2562. doi: 10.1016/j.biocel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Schwach G, Ingolic E, Adame A, Crews L, Mante M, Pfragner R, Schreiner E, Windisch M, Masliah E. Lysosomal pathology associated with alpha-synuclein accumulation in transgenic models using an eGFP fusion protein. J Neurosci Res. 2005;80:247–259. doi: 10.1002/jnr.20446. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–5652. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S. A pathologic cascade leading to synaptic dysfunction in α-synuclein-induced neurodegeneration. J Neurosci. 2010;30:8083–8095. doi: 10.1523/JNEUROSCI.1091-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin Y, Klucken J, Patterson C, Hyman BT, McLean PJ. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J Biol Chem. 2005;280:23727–23734. doi: 10.1074/jbc.M503326200. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, et al. alpha-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Skoch J, Hickey GA, Kajdasz ST, Hyman BT, Bacskai BJ. In vivo imaging of amyloid-beta deposits in mouse brain with multiphoton microscopy. Methods Mol Biol. 2005;299:349–363. doi: 10.1385/1-59259-874-9:349. [DOI] [PubMed] [Google Scholar]
- Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B. Aggregated and monomeric alpha-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. J Biol Chem. 2003;278:11753–11759. doi: 10.1074/jbc.M208641200. [DOI] [PubMed] [Google Scholar]
- Spencer B, Potkar R, Trejo M, Rockenstein E, Patrick C, Gindi R, Adame A, Wyss-Coray T, Masliah E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-synuclein models of Parkinson's and Lewy body diseases. J Neurosci. 2009;29:13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spires-Jones TL, de Calignon A, Meyer-Luehmann M, Bacskai BJ, Hyman BT. Monitoring protein aggregation and toxicity in Alzheimer's disease mouse models using in vivo imaging. Methods. 2011;53:201–207. doi: 10.1016/j.ymeth.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type α-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001;21:9549–9560. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Engelender S, Igarashi S, Rao RK, Wanner T, Tanzi RE, Sawa A, Dawson VL, Dawson TM, Ross CA. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet. 2001;10:919–926. doi: 10.1093/hmg/10.9.919. [DOI] [PubMed] [Google Scholar]
- Tofaris GK, Layfield R, Spillantini MG. alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 2001;509:22–26. doi: 10.1016/s0014-5793(01)03115-5. [DOI] [PubMed] [Google Scholar]
- Unni VK, Weissman TA, Rockenstein E, Masliah E, McLean PJ, Hyman BT. In vivo imaging of alpha-synuclein in mouse cortex demonstrates stable expression and differential subcellular compartment mobility. PLoS One. 2010;5:e10589. doi: 10.1371/journal.pone.0010589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unni VK, Ebrahimi-Fakhari D, Vanderburg CR, McLean PJ, Hyman BT. Studying protein degradation pathways in vivo using a cranial window-based approach. Methods. 2011;53:194–200. doi: 10.1016/j.ymeth.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem. 2008;283:23542–23556. doi: 10.1074/jbc.M801992200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13:805–811. doi: 10.1038/nn.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H+-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem. 1991;266:17707–17712. [PubMed] [Google Scholar]
- Zhang NY, Tang Z, Liu CW. alpha-Synuclein protofibrils inhibit 26S proteasome-mediated protein degradation: understanding the cytotoxicity of protein protofibrils in neurodegenerative disease pathogenesis. J Biol Chem. 2008;283:20288–20298. doi: 10.1074/jbc.M710560200. [DOI] [PubMed] [Google Scholar]