

Key Points
Hb-conformation–dependent interaction with band 3 protein regulates glycolysis in RBCs.
In hypoxia, HbS disrupts this system, disabling RBC antioxidant defense.
Abstract
Energy metabolism in RBCs is characterized by O2-responsive variations in flux through the Embden Meyerhof pathway (EMP) or the hexose monophosphate pathway (HMP). Therefore, the generation of ATP, NADH, and 2,3-DPG (EMP) or NADPH (HMP) shift with RBC O2 content because of competition between deoxyhemoglobin and key EMP enzymes for binding to the cytoplasmic domain of the Band 3 membrane protein (cdB3). Enzyme inactivation by cdB3 sequestration in oxygenated RBCs favors HMP flux and NADPH generation (maximizing glutathione-based antioxidant systems). We tested the hypothesis that sickle hemoglobin disrupts cdB3-based regulatory protein complex assembly, creating vulnerability to oxidative stress. In RBCs from patients with sickle cell anemia, we demonstrate in the present study constrained HMP flux, NADPH, and glutathione recycling and reduced resilience to oxidative stress manifested by membrane protein oxidation and membrane fragility. Using a novel, inverted membrane-on-bead model, we illustrate abnormal (O2-dependent) association of sickle hemoglobin to RBC membrane that interferes with sequestration/inactivation of the EMP enzyme GAPDH. This finding was confirmed by immunofluorescent imaging during RBC O2 loading/unloading. Moreover, selective inhibition of inappropriately dispersed GAPDH rescues antioxidant capacity. Such disturbance of cdB3-based linkage between O2 gradients and RBC metabolism suggests a novel mechanism by which hypoxia may influence the sickle cell anemia phenotype.
Introduction
Sickle cell anemia (SCA) arises from a single amino acid substitution (Glu6Val) in the β-globin chain. Although the change to hemoglobin (Hb) is simple and uniform, SCA is characterized by broad differences in clinical manifestation. Phenotype variation in SCA is thought to arise from both environmental and genetic factors (eg, β-gene cluster haplotype, degree of HbF expression, or effects of other epistatic genes). The environmental factor that most clearly influences SCA phenotype is hypoxia, which drives sickle Hb (HbS) polymerization and the resulting well-characterized alterations in RBC physiology and the microcirculation. However, the influence of hypoxia on the SCA phenotype appears to be insufficiently explained by HbS polymerization alone.1 Moreover, we lack a clear mechanistic understanding of the significant oxidative stress complicating SCA, a key feature of phenotype variation, both at rest and in association with hypoxia.2 Nonpolymerized, solution-phase HbS may promote oxidative stress, even in RBCs under normal physiologic O2 gradients.3 Specifically, the low redox potential for heme in HbS4 and avid binding affinity of HbS for the cytoplasmic regulatory domain of the Band 3 membrane protein (cdB3)5,6 strongly affect RBC energetics and antioxidant systems7–9 and, notably, do so as a function of RBC O2 content. Therefore, both the genesis and the disposal of reactive oxygen species are abnormal in SCA, creating a baseline state of oxidative stress, which worsens in hypoxia.
In particular, consideration of metabolic control in RBCs suggests O2-dependent HbS-cdB3 interaction as a relatively unexplored means by which hypoxia might influence the SCA phenotype. Numerous RBC functions cycle with pO2 during circulation because of regulation by Hb-conformation–dependent control of the cdB3-based protein assembly, including: ion and amino acid transport,10 cytoskeleton-membrane interaction,11 processing/export of vasoactive effectors (eg, NO),12–14 and glycolysis.8 Accumulating evidence now affords detailed understanding of such cycling in glycolysis, in which the Embden Meyerhof pathway (EMP) flux is linked to O2 gradients via a reciprocal binding relationship between key EMP enzymes and deoxy-Hb for regulatory sites on cdB3.15,16 After RBC oxygenation, EMP enzymes bind to cdB3 and are inactivated; therefore, glycolysis (via the EMP) decelerates and metabolism is routed through the alternate hexose monophosphate pathway (HMP).16 With O2 unloading, deoxy-Hb displaces and activates EMP enzymes, limiting HMP substrate availability.8,17 This coupling between energy metabolism and Hb O2 saturation (HbSO2) conspires to limit antioxidant defense in hypoxia (as we have shown previously9), because the HMP is the sole means by which RBCs can recycle NADPH,8 a reducing equivalent essential for glutathione (GSH) regeneration, as well as for the ascorbate, catalase, and thioredoxin antioxidant systems.
We chose O2-responsive regulation of glycolysis in RBCs as a model system in which to study the influence of HbS on cdB3-based protein complex assembly. We hypothesized that increased affinity of HbS for cdB35,6 results in persistent masking of regulatory cdB3-binding sites, preventing pO2-responsive membrane recruitment and inactivation of EMP enzymes. In addition, denatured HbS (hemichrome) also binds strongly to cdB3, bridging Band 3 monomers into complex aggregates; this process may also interfere with inhibitory glycolytic complex assembly.6,7,18–20 Consequent loss of O2-dependent EMP control might then reduce HMP substrate availability, limiting NADPH and GSH recycling capacity and thereby creating vulnerability to oxidative stress, an important and highly variable manifestation of the SCA phenotype.2,21 Of the EMP enzymes under cdB3 control, we focused on GAPDH, which is notable for its location at the juncture of the HMP/EMP pathways. To address our hypothesis, we used 1H-nuclear magnetic resonance (1H-NMR) spectroscopy to precisely quantitate proportional HMP flux in intact RBCs and developed a novel, inverted membrane-on-bead model to study reciprocal binding between HbS and GAPDH on cdB3 under physiologic conditions. Herein, as others have shown, we report limited resilience to oxidative stress in RBCs from patients homozygous for HbS (SSRBCs) and newly illustrate O2-dependent coupling between impaired antioxidant capacity, specific disruption in energy metabolism, and reducing equivalent recycling. Moreover, we examined HbSO2-linked migration of GAPDH between cytosol and membrane with confocal immunofluorescence imaging and found that in SSRBCs, GAPDH remains cytosolic regardless of O2 content. In such cells, resilience to oxidative stress is restored by selective inhibition of inappropriately dispersed GAPDH. These findings are consistent with disruption of cdB3-based metabolic regulation by HbS and suggest a novel means by which hypoxia may influence the SCA phenotype (by promoting avid HbS-cdB3 interaction).
Methods
Reagents and antibodies
Unless otherwise stated, all reagents were purchased from Sigma-Aldrich. A custom peptide corresponding to the cdB3 NH2 terminus (MEELQDDYED)22 was obtained from Biosynthesis. Koningic acid (KA)23 was kindly provided by Keiji Hasumi (Tokyo Noko University, Tokyo, Japan). D2O, [2-13C] D-glucose, and trimethylsilyl propionic acid (TSP) were obtained from Cambridge Isotope. Primary antibodies to cdB3, GAPDH, pTyr, and the secondary anti–mouse FITC-linked antibody were obtained from Santa Cruz Biotechnology. Primary antibodies to glycophorin and Hb were obtained from Abcam. Secondary anti–rabbit IRDye680 and anti–mouse IRDye800CW were obtained from LI-COR; secondary Alexa Fluor 488–conjugated anti–mouse, Alexa Fluor 546–conjugated anti–rabbit, and Alexa Fluor 633–conjugated anti–chicken were obtained from Invitrogen.
Blood sampling, purification of RBC membranes and hemoglobin, and manipulation of O2
This project was approved by the Washington University institutional review board and written informed consent was obtained from participants in accordance with the Declaration of Helsinki. Blood was drawn from healthy volunteers or from patients homozygous for HbS; 40 children with SCA were recruited during routine clinic visits. The mean values at sampling were: age: 12.4 years (range, 5-22); hematocrit: 23.3% (range, 17.2%-34.5%); and reticulocytes: 14.1% (range, 2.0-26.9). None of the patients had active painful episodes, respiratory pathology, nor had been transfused within 3 months. RBCs were studied after washing and resuspension (PBS and 6mM glucose). Where indicated, RBC O2 content was manipulated in a rotating tonometer as follows: 37°C, flushed with 21% (or 0%) O2, 7% CO2, balance N2, to yield > 97% (or < 10%) HbSO2, as described previously.9,12 Deoxygenated RBCs, lysate fractions, and buffers/reagents were handled under N2 throughout (CO2/O2-controlled glove box; Coy Laboratories). RBC membranes were isolated after lysis and washing (5mM NaH2PO4, pH 8.0), unless otherwise noted. GAPDH-depleted membranes were prepared by washing in lysis buffer containing 150mM NaCl. Hb was purified by ion-exchange chromatography, as described previously.9
In vitro model of oxidative stress, osmotic fragility, and membrane free thiol density
The hypoxanthine/xanthine oxidase (HX/XO) system was used as a steady, gradable superoxide (O2−) source, as described previously.9 Briefly, washed RBCs (40% hematocrit, Krebs) were incubated with 1.5mM HX and XO (0 or 0.8 U/mL) for 2, 5, 10, 20, or 45 minutes, after which time O2− generation was quenched (1mM allopurinol). Where indicated, the EMP was blocked by RBC incubation with KA (15μM, 15 minutes, 37°C), a selective GAPDH inhibitor.23 To quantify osmotic fragility, RBCs were washed, suspended in NaCl (0-15g/L), pelleted, and the supernatant read at 540nm. A value of 100% lysis was assigned to the 0 g/L NaCl solution, against which others were indexed. In paired aliquots, RBCs were added to lysing/washing buffer (with 0.5mM EDTA and 10mM PMSF) and fractions were assayed for membrane protein, free (reduced) thiol density, and GSH and NADPH redox pairs (see next paragraph). As described previously,9 to determine the membrane free thiol concentration ([RSH]), extracted proteins (in guanidine, 8M) were incubated with 5,5′dithio bis(2-nitrobenzoic acid (10mM) and absorbance (412 nm) was compared with GSH standards. Free thiol density (μmol/mg) was expressed as moles of RSH per protein mass (ADV01 protein assay; Cytoskeleton).
GSH and NADPH redox couples and Ehc
RBC lysates were derivatized and the glutathione disulfide/glutathione redox couple (GSSG/GSH) components were quantified, as described previously.9 Calculation of the half-cell reduction potential (Ehc) permitted comparison of normal and SSRBCs with differing GSH content. The Nernst equation for the GSH Ehc (37°C, pH 7.4) has the form24:
The NADP+/NADPH couple in paired lysates from HX/XO experiments was assayed by the enzymatic cycling reaction, as described previously.9 Likewise, the Nernst equation for the NADPH Ehc (37°C, pH 7.4) has the form24:
1H-NMR Analysis of HMP and EMP flux in intact RBCs
Washed RBCs were depleted of endogenous glucose and lactate (incubation in 25mM HEPES, 120mM NaCl, 5.4mM KCl, 1.8mM CaCl2, 1mM NaH2PO4, pH 7.4, 60 minutes, 37°C)25; O2 content was manipulated in our rotating tonometer, and the suspension was spiked with [2-13C] D-glucose (11mM final concentration) with or without the following additional components: (1) methylene blue (MB; 6.7μM final concentration to stimulate the HMP25), (2) O-vanadate (250μM final concentration to increase cdB3 phosphorylation15), and (3) KA (15μM final concentration to block the EMP23). Note that O-vanadate and KA incubation preceded labeled glucose exposure. After further incubation to generate 13C-lactate isotopomers for analysis (8 hours at 37°C), RBCs were pelleted and 400-μL aliquots of supernatant were lyophilized and then equilibrated with TSP (1mM in 580 μL of D2O for 5 minutes) before delivery into 5-mm NMR tubes. 1H-NMR measurements were performed at 25°C on an Agilent/Varian Direct Drive-1 spectrometer operating at 11.75 Tesla as described previously25 with the following modifications: spectra were obtained within 2 hours of sample preparation, with water presaturation, a 6-second preacquisition delay, a 6-μsecond excitation pulse, and a 2-second acquisition period. Total lactate and glucose was established using the TSP concentration reference.
GAPDH and Hb binding to purified RBC membranes
Equal membrane volumes were simply washed, fully stripped, or enzymatically shaved. Stripping26 proceeded by washing serially in: (1) fructose 1,6-bisphosphate (1mM) in imidazole (10mM, pH 7.8); (2) stripping buffer (imidazole 120mM, 120mM NaOAc, 1mM DTT, 0.1mM EDTA, 2μM NAD+, and 0.08% BSA, pH 7.0); and (3) imidazole (10mM, pH 7.0). Shaving27 proceeded by washing membranes in NaOH (10mM), incubating with proteinase-K (3 mg/mL per 100-μL membrane aliquots for 15 hours at room temperature [RT]), stopping digestion with PMSF, and washing in NaOH (15mM). Fractions were reserved at each stage for heme measurements (below, this paragraph). To study GAPDH quenching by membrane, preparations were returned to start volume in GAPDH activity buffer26 (10mM NaOAc, 100μM NAD+, 100μM NaAsO4, 100μM EDTA, 1mM DTT, 10mM imidazole, and 60nM human RBC-derived GAPDH). Membranes were titrated to 1 mL of this solution, incubated (1 hour at 4°C), pelleted, and supernatant GAPDH activity was determined by adding the substrate glyceraldehyde-3-phosphate (G3P, 100μM, RT), and monitoring the absorbance (340 nm) increase during the second minute. True membrane heme (rather than ghost-associated Hb aggregates)18 was assayed by incubating membrane aliquots in formic acid (1:1 vol:vol, 5 minutes, RT); absorbance (398nm) was compared with purified HbAo standards. Membrane-bound [heme] was indexed to [membrane protein] measured in paired samples (BCA assay).
Immobilization of inverted RBC membrane on beads
Nonporous silica beads (3.14 μm; Bangs Laboratories) were dispersed into a suspension of RBC ghosts (± GAPDH depletion) for orientation-selective attachment, as described previously.28 Uniformity of bead coating between normal and SS batches was evaluated by: (1) measuring on-bead protein mass (BCA); (2) quantitating on-bead membrane volume by fluorometry (Vybrant DiI membrane-selective dye; Molecular Probes and a SynergyHT microplate reader; BioTek); and (3) determining on-bead B3 number (for fully stripped membranes) by quantitative immunofluorescence (SynergyHT). Inside-out membrane orientation was confirmed by immunofluorescent imaging of glycophorin, an exofacial marker, and cdB3 (AxioSkop; ZeissAG), a cytoplasmic marker. The binding relationship for GAPDH in solution was determined by GAPDH titration to beads, pelleting, membrane isolation, and immunoblotting/densitometry (OdysseyCLx; LI-COR). Specific GAPDH binding to cdB3 was confirmed by incubating beads ± antibody to cdB3, followed by ± GAPDH (75 pmoles/500 μL in 5mM NaH2PO4, pH 8.0, 2 minutes, RT), and then probing with antibody to GAPDH and imaging (AkioSkop, ZeissAG).
Quantitative comparison of GAPDH binding to normal and SSRBC membrane
Beads with inverted GAPDH-depleted membrane were counted in a hemocytometer, normalized (0-6 × 108), and incubated with GAPDH (75 pmol/500 μL in 5mM NaH2PO4, pH 8.0, 2 minutes, RT). GAPDH activity26 quenching was determined by measuring the enzyme activity remaining in supernatant after pelleting. In a complementary fashion, GAPDH binding to beads was measured after beads (3 × 108) were incubated with GAPDH (2.34-75 pmol/500 μL, 2 minutes, RT). After washing, proteins were released from beads by boiling in (reducing) loading buffer and then quantitated by immunoblotting/densitometry.
Competitive binding between GAPDH and Hb for cdB3 as a function of RBC O2 content
This experiment was performed under true intraerythrocytic molar ratios for GAPDH:cdB3:Hb (1:2.4:540).29,30 Beads coated with GAPDH-depleted membrane from oxygenated or deoxygenated normal RBCs were incubated with either oxygenated (HbO2 > 85%) or deoxygenated (HbO2 < 15%) HbAo or HbS and human 2,3-DPG (equimolar Hb:2,3-DPG, 100μM, 10 minutes, 37°C). After incubation, beads were washed and then incubated with GAPDH (75 pmol/500 μL, 2 minutes, RT) before pelleting and determination of GAPDH activity in supernatant (see previous paragraph).
Measurement of B3 pTyr
For measurement of tyrosine phosphorylation (pTyr), washed RBCs were suspended in phosphatase inhibitor buffer (25mM HEPES, 135mM NaCl, 10mM glucose, 100μM EGTA, and 250μM O-vanadate, pH 7.4). RBC O2 content was manipulated in our rotating tonometer (10 minutes) and incubation was continued (50 minutes, 37°C). Membrane proteins were extracted (25mM HEPES, 300mM NaCl, 1mM EGTA, 1mM PMSF, 1mM O-vanadate, 1% DDM, and 10% glycerol, pH 7.4, 45 minutes, 30°C) and collected for immunoblotting/densitometry (above).
Immunofluorescent imaging of intact RBCs
RBC O2 content was manipulated in our rotating tonometer and oxygenated RBCs were fixed (5 minutes, 0.5% acrolein, PBS). Deoxygenated RBCs were initially fixed in formaldehyde (1% final, 20 minutes)15 and then washed and resuspended in acrolein (as for oxy-RBCs). All cells were rinsed (0.1M glycine and PBS), permeabilized (0.1% Triton X-100 and rinsing buffer, 5 minutes), and rinsed further to ensure complete neutralization of unreacted aldehydes. RBCs were blocked (0.05mM glycine, 0.2% fish skin gelatin, 0.05% NaN3, PBS, 2 hours), probed with antibody to cdB3 or to GAPDH, and confocal images were obtained (FluoViewFV1000; Olympus).
Statistical comparisons
Key variables were tested for Gaussian distribution with the Kolmogrov-Smirnov test. Where appropriate, differences in means within groups were compared with either the paired t test or 1-way ANOVA. Differences in means between groups were compared with unpaired t test or 2-way RM-ANOVA. When nonparametric analyses were required, the Mann-Whitney, Friedman, or Kruskal-Wallis tests were used. Either the Bonferroni or Dunn post hoc tests were used for multiple comparisons. Nonlinear regression was used to determine the relationship between the percentage of GAPDH inhibition and membrane-bound heme. All data are reported as means ± SEM. Two-tailed P < .05 was considered significant (Prism Version 5 software; GraphPad).
Results
RBC resilience to oxidative stress
After oxidant loading, RBC membrane integrity was gauged functionally by monitoring RBC resilience to osmotic stress and quantitatively by monitoring free thiol (eg, RSH) loss. Osmotic fragility was analyzed at baseline and after exposure to O2− (generated by HX/XO). At baseline, SSRBCs demonstrated less osmotic fragility than normal RBCs, likely because of the higher surface area to volume ratio of (nonsickled) cells. Oxidant exposure did not alter resilience to osmotic stress in normal RBCs; however, SSRBC osmotic fragility increased significantly (eg, lysed at higher tonicity, as evidenced by a rightward shift in the SSRBC curve; the half-maximal effective concentration [EC50] increased from 3.17 ± 0.14 to 3.95 ± 0.30 g/L of NaCl; P < .05; Figure 1A). In addition, at baseline, the free membrane thiol fraction in SSRBCs was somewhat lower than normal, likely indicative of in vivo oxidative damage, as described previously by others.31 After oxidant loading, SSRBCs suffered greater membrane free thiol loss (from an already depleted pool; Figure 1B). This increase in membrane protein oxidation is consistent with the increase in osmotic fragility. These findings illustrate that SSRBCs have limited ability to preserve membrane integrity during oxidative stress.
Figure 1.

Comparison of osmotic fragility, fraction of reduced RBC membrane thiol, and reducing equivalent recycling in normal and SSRBCs after oxidant exposure. (A) NaCl-induced osmotic lysis at baseline and after oxidant exposure (HX/XO) for normal and SSRBCs; half-maximal effective concentration (EC50) increased for SSRBCs alone (n = 3-10). *P < .05, mean ± SEM, is plotted. (B) Free (reduced) membrane thiol for normal and SSRBCs at baseline and after oxidant exposure (HX/XO; n = 6-8). *P < .05 for % loss, normal versus sickle cell. (C) GSH and NADPH Ehc for normal and SSRBCs at baseline and during oxidant exposure (n = 4-6). *P < .05 for normal versus sickle cell (baseline); +P < .05 for normal versus sickle cell (HX/XO exposed comparison); #P < .05 for normal versus sickle cell (HX/XO exposed comparison). At baseline, note that relative to normal RBCs, SS membrane thiol redox poise favors oxidation and GSH and NADPH reducing power is diminished (ie, less negative Ehc). Antioxidant system failure in SSBRCs is evidenced by the rate and degree to which GSH and NADPH reducing power is lost under O2− loading. Moreover, after oxidant exposure, SSRBCs suffer membrane thiol depletion and lose resilience to osmotic fragility (ie, rightward shift).
Reducing equivalent recycling capacity
At baseline, total GSH content (GSH + GSSG) was lower in SSRBCs than in normal RBCs, as described previously by others21 (supplemental Figure 1C-D, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). Further, diminished GSH-based reducing power in SSRBCs was evidenced by a weaker (ie, less negative) reduction potential (Figure 1C). During oxidant loading, GSSG recycling capacity was estimated by monitoring the GSH half-cell (Ehc), which best accounts for the “redox ratio” (eg, GSSG/GSH) when total GSH is not uniform between groups.24 During oxidant loading, SSRBCs were less able to recycle GSH, evidenced by greater Ehc loss (Figure 1C); therefore, SSRBCs accumulated GSSG in a dramatic fashion that was not observed in normal RBCs (see supplemental Figure 1 for Ehc components). Because GSH regeneration from GSSG by GSH reductase requires NADPH, the GSH and NADPH redox couples are linked. At baseline, NADP+ levels were higher and NADPH levels lower in SSBRCs (supplemental Figure 2); demonstrating impaired pyridine nucleotide recycling at rest. As a result, baseline Ehc for NADPH was weaker (ie, less negative) in SSRBCs (Figure 1C). When Ehc was monitored during oxidant loading, further limitation in NADPH-recycling capacity in SSRBCs was revealed (Figure 1C; see supplemental Figure 2 for Ehc components). This finding suggests that HMP flux (the sole source of NADPH) is constrained in SSRBCs (please refer to Figure 7 for pathway schema).
Figure 7.

Simplified scheme of cdB3-based control of glucose metabolism in RBCs. (A) Energy metabolism in RBCs proceeds through either the EMP pathway (orange arrows) or the HMP pathway (blue arrows; also known as the pentose shunt). Both share G6P as initial the substrate. The HMP is the sole source of NADPH in RBCs and generates fructose-6-phosphate (F6P) or glyceraldehyde-3-phosphate (G3P), which rejoin the EMP before glyceraldehyde-3-phosphate dehydrogenase (G3PD/GAPDH), a key regulatory point. The EMP generates NADH (used by met-Hb reductase) and ATP (to drive ion pumps) and 2,3-DPG (to modulate Hb p50). Hydrogen peroxide (H2O2) and O2− are the principal endogenous reactive oxygen species (ROS) encountered by RBCs. Both ROS may be generated internally (not shown); however, only H2O2 can cross the membrane directly. O2− enters RBCs through Band 3 (anion exchange protein 1 or AE-1). H2O2 and O2− are ultimately reduced to water by catalase or GSH peroxidase (GPx). (B) O2 content modulates EMP and HMP balance via reciprocal binding for cdB3 between deoxy-Hb and key EMP enzymes (PFK, Aldo, GAPDH, PK, and LDH).16,17 In oxygenated RBCs (right half of stylized oxygen dissociation plot), sequestration to cdB3 inactivates these EMP enzymes, resulting in HMP dominance and maximal NADPH (and thus GSH) recycling capacity. In deoxygenated RBCs (left half of oxygen dissociation plot), deoxy-Hb binding to cdB3 disperses these EMP enzymes, creating G6P substrate competition and thereby constraining HMP flux, limiting NADPH and GSH recycling capacity, and weakening resilience to ROS such as O2−. (C) In SSRBCs, SSHb binds abnormally avidly to the RBC membrane at cdB3.6,7 We hypothesized that this biases normal EMP/HMP cycling (disfavoring HMP), rendering SSRBCs vulnerable to oxidant attack. In support of this, we found that by blocking the EMP with KA (at the point normally inhibited by cdB3-GAPDH binding) restored resilience to oxidative loads, presumably by lifting the G6PD substrate constraint.
Effect of glycolytic pathway dominance on NADPH generation, GSH recycling, and membrane thiol defense
We used 1H-NMR spectroscopy to determine positional 13C-enrichment in (HMP/EMP) pathway-specific lactate isotopomers generated by RBCs incubated with [2-13C]-glucose (± MB to stimulate the HMP).25 From these measurements and known stoichiometries,32 we calculated the maximum HMP flux and NADPH-generation capacity (Figure 2 and Table 1). Resting glucose consumption was greater for SSRBCs than for normal RBCs (and the lactate generated per glucose metabolized differed), likely reflecting pathway inefficiencies and a compensatory increase in substrate uptake. As expected, MB accelerated glucose consumption for both normal and SSRBCs (Figure 2B and supplemental Figure 7C). Specifically, in normal RBCs, HMP flux increased 15.2-fold during MB stimulation; deoxygenation blunted this increase (4.5-fold change, which is consistent with our previous results9). The response in SSRBCs was significantly less robust (3.2- and 1.8-fold increases in oxy- and deoxy-SSRBCs, respectively; Figure 2C and supplemental Figure 5). Moreover, maximum NADPH generation was also significantly limited in SSRBCs (4.54 ± 0.95 vs 1.13 ± 0.04 mol NADPH/mol glucose for normal and SSRBCs, respectively; Table 1 and supplemental Figure 5). These data are consistent with our direct measurements of impaired NADP+, GSSG, and oxidized membrane thiol recycling. To determine whether impaired SSRBC antioxidant capacity arose from lost EMP control, we used the selective GAPDH inhibitor KA23 to block inappropriately dispersed, active GAPDH in oxygenated SSRBCs (thus restoring HMP dominance). As expected, KA abrogated lactate generation (see pathway schema in Figure 7), precluding isotopomer analysis of HMP/EMP dominance (supplemental Figure 7D). In KA-treated SSRBCs under oxidant loading, however, GSH Ehc maintenance and membrane thiol recycling normalized fully (Figure 2D-E; see supplemental Figure 3 for Ehc components).
Figure 2.

Glucose metabolism in SSRBCs is dysregulated with EMP bias and resilience to oxidative loading is restored by EMP blockade. HMP flux as a function of O2 content was determined using 1H-NMR spectroscopy to monitor positional 13C-enrichment in lactate isotopomers generated by RBCs incubated with [2-13C]-glucose (± MB to stimulate the HMP). (A) Representative spectrum from normal, unstimulated RBCs is shown (supplemental Figure 5 illustrates spectra for all conditions studied). Per sample, 256 acquisitions were recorded, resulting in a lactate S/N exceeding 1000/1. During acquisition, continuous-wave 13C-decoupling of lactate C1 was performed using Bayesian algorithms.50 Lactate methyl isotopomer signals were deconvoluted and fit to: a doublet D (3-bond 1H-1H coupling only) arising from [12C]-lactate, a doublet of doublets Q3 (3-bond 1H-1H coupling and one bond 1H-13C coupling) arising from HMP-generated [3-13C]-lactate, and a doublet of doublets Q2 (3-bond 1H-1H coupling and 2 bond 1H-13C coupling) arising from EMP-generated [2-13C]-lactate. This approach yielded signal amplitude estimates with a 12% SD for the lowest magnitude (Q3) to ≤ 2% for the highest magnitude (D) peaks. Proportional HMP flux was calculated from peak areas attributable to the pathway-specific lactate isotopomers (eg, Q2→EMP, Q3→HMP, see insets and Table 1). (B) Glucose uptake rate plotted against lactate generation for all conditions studied; note grouping among normal and SSRBC populations, which demonstrate differing lactate/glucose relationships. Both glucose uptake and lactate generation accelerated with MB stimulation. (C) In normal RBCs, HMP flux increased significantly during MB stimulation and deoxygenation blunted this increase. SSRBCs demonstrated similar O2 content–dependent variation in HMP flux, but the HMP increase with MB stimulation was significantly less robust than in normal RBCs. HMP/EMP bias was rebalanced in SSRBCs by incubation with the selective GAPDH inhibitor KA (please refer to Figure 7 for pathway schema); RBCs were then presented with an oxidative challenge. (D) This manipulation stabilized GSH-reducing power in SSRBCs during oxidant loading (mean ± SEM; n = 3-4). *P < .05, reported as Ehc, see supplemental Figure 3 for Ehc components. (E) KA treatment also restored membrane thiol protection in SSRBCs under oxidant loading (n = 3-8; mean ± SEM). *P < .05.
Table 1.
Metabolic flux data from 1H-NMR spectroscopy after RBC incubation with [2-13C] D-glucose
| Positional 13C enrichment (lactate) | Normal |
SS |
||||||
|---|---|---|---|---|---|---|---|---|
| Oxygenated |
Deoxygenated |
Oxygenated |
Deoxygenated |
|||||
| − MB | + MB | − MB | + MB | − MB | + MB | − MB | + MB | |
| Raw data | ||||||||
| Carbon 2 | 0.29 ± 0.01 | 0.16 ± 0.02 | 0.31 ± 0.01 | 0.24 ± 0.02 | 0.31 ± 0.00 | 0.25 ± 0.01§ | 0.31 ± 0.01 | 0.27 ± 0.01 |
| Carbon 3 | 0.03 ± 0.00 | 0.10 ± 0.00 | 0.03 ± 0.00 | 0.06 ± 0.01 | 0.04 ± 0.01 | 0.08 ± 0.00§ | 0.01 ± 0.00§ | 0.05 ± 0.01 |
| Corrected* | ||||||||
| Carbon 2 | 0.29 ± 0.01 | 0.16 ± 0.02 | 0.31 ± 0.01 | 0.24 ± 0.02 | 0.31 ± 0.00 | 0.25 ± 0.01§ | 0.31 ± 0.01 | 0.27 ± 0.01 |
| Carbon 3 | 0.03 ± 0.00 | 0.09 ± 0.00 | 0.02 ± 0.00 | 0.06 ± 0.01 | 0.03 ± 0.01 | 0.07 ± 0.01§ | 0.00 ± 0.00§ | 0.05 ± 0.01 |
| Flux estimates | ||||||||
| Proportional [2-13C] D-glucose flux through HMP† | 0.05 ± 0.00 | 0.76 ± 0.16 | 0.04 ± 0.01 | 0.18 ± 0.06 | 0.06 ± 0.01 | 0.19 ± 0.01§ | 0.00 ± 0.00§ | 0.11 ± 0.02 |
| Maximum NADPH generation per glucose, mol/mol‡ | 0.31 ± 0.03 | 4.54 ± 0.95 | 0.23 ± 0.06 | 1.10 ± 0.38 | 0.36 ± 0.08 | 1.13 ± 0.04§ | 0.03 ± 0.02§ | 0.65 ± 0.10 |
Values are reported as mean ± SEM; for all groups, n = 3-5.
MB indicates methylene blue; HMP, hexose monophosphate shunt; G6P, glucose 6-phosphate; and NADPH, nicotinamide adenine dinucleotide phosphate.
With a natural abundance of [2-13C] lactate of 1.1% and an assumed random distribution of natural 13C among lactate carbons, the relative intensities of the signals are: [12C] lactate (D-peak): 97.8%; [2-13C] lactate (Q2): 1.1%; and [3-13C] lactate (Q3): 1.1%. Values for carbon 2 and 3 enrichment in the lactate methyl signal, corrected for natural 13C abundance, were therefore calculated as follows25: Q2corr = Q2 − ((D × 0.011)/0.978)).
The equation for estimating the proportion of [2-13C] D-glucose metabolized by the HMP relative to total (eg, HMP + EMP) was as follows25: Q3corr/Q2corr = 2(HMP fraction)/(1 + 2[HMP fraction]).
Moles of NADPH that may be generated per mole [2-13C] D-glucose metabolized were determined according to known stoichiometry for NADPH generation from glucose: due to pentose- and hexose-phosphate recycling within the HMP, each glucose molecule may be functionally recycled 3 times through the oxidative HMP branch. Because each molecule of G6P oxidized to ribose-P generates 2 NADPH, the maximum NADPH generation (and glutathione recycling) may reach 6 times the HMP flux estimate.32
Significant difference between normal and sickle cell (P < .05).
GAPDH binding to RBC membranes and influence of bound Hb
Membranes prepared from normal RBCs were washed, stripped (exposing cdB3), or shaved (removing cdB3), and then assessed for their ability to bind and inhibit free GAPDH in solution. Stripped membranes demonstrated significantly greater GAPDH inhibition compared with those simply washed, whereas shaved membranes displayed significantly less (Figure 3A), in expected relation to cdB3 availability for GAPDH binding. In addition, samples from each wash (containing progressively less membrane-bound heme) were likewise evaluated. Membranes inhibited GAPDH in inverse proportion to bound heme (Figure 3B), which is consistent with Hb masking the GAPDH-binding site on cdB3. The amount of membrane-bound heme was compared between normal and SSRBCs. Extensively washed SS membranes contained significantly more heme (and globin, see paragraph below and Figure 4A) compared with normal RBCs (Figure 3C), as described previously.18 Because cdB3 binds deoxy-Hb preferentially to oxy-Hb29 and the RBC samples were fully oxygenated (HbSO2 > 95%) before lysis, this observation may represent both abnormal release kinetics of deoxy-HbS from cdB35,6 and cdB3-associated hemichrome (although the cdB3-binding site differs for deoxy-Hb and hemichrome, both demonstrate competitive binding with GAPDH).7 To permit precise study of reciprocal binding between Hb and GAPDH for cdB3 under physiologic conditions, we developed the inverted membrane-on-bead model described in the next paragraph.
Figure 3.
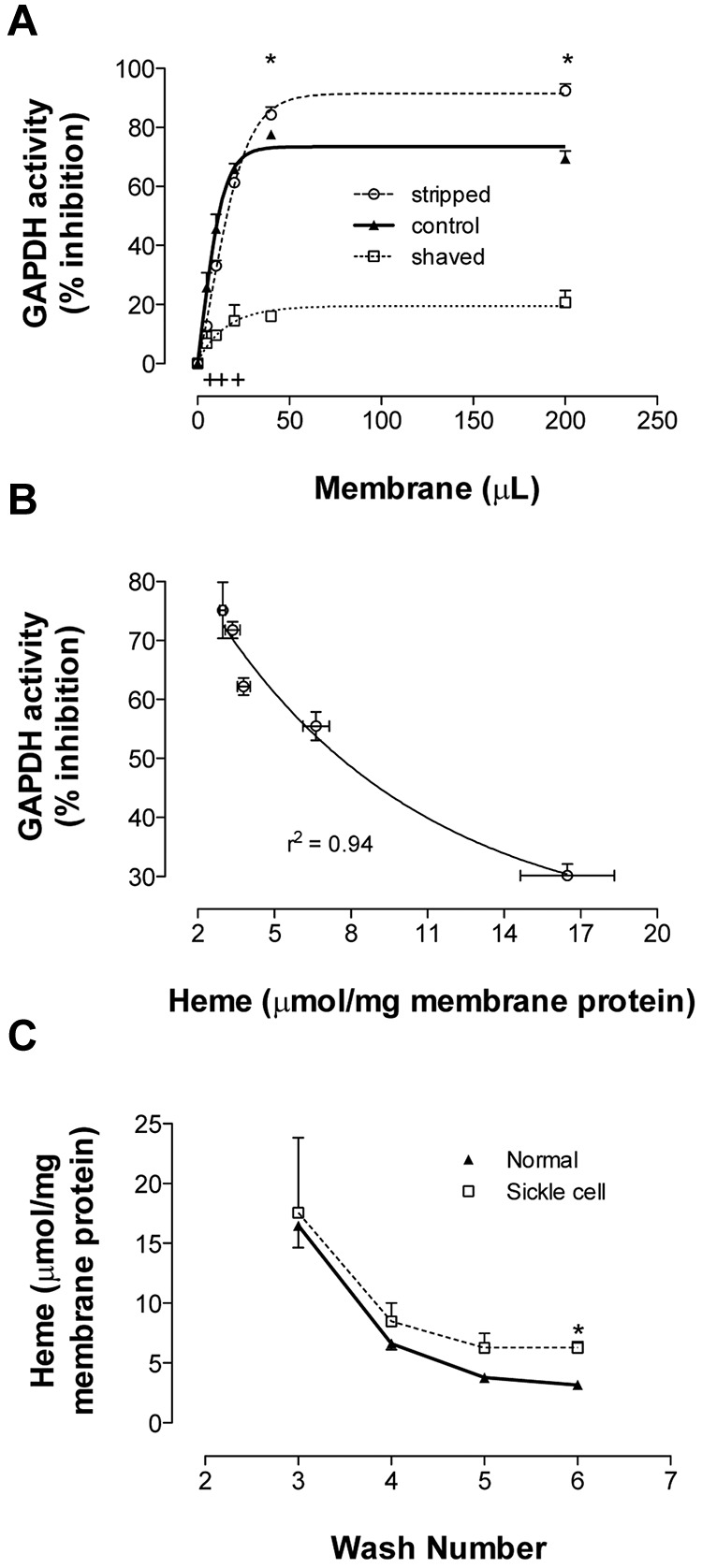
Binding of GAPDH and Hb to RBC membrane. Membranes from fully oxygenated normal RBCs were prepared by hypotonic lysis and alkaline washing. Membranes referred to as “control” were taken from the final (7th) wash. Equal volumes of these membranes were further processed to produce stripped (ie, removal of associated extrinsic membrane proteins) or shaved membranes (ie, digestion of exofacial and cytoplasmic protein domains). (A) To study GAPDH inhibition, the 3 preparations were incubated in enzyme activity buffer, pelleted, and the activity remaining in supernatant was determined (n = 3; mean ± SEM). +P < .05 for control versus shaved; *P < .05 for shaved versus stripped. (B) Samples from each wash (containing progressively less heme) were run in this GAPDH activity assay. The percentage GAPDH inhibition varied inversely with membrane-bound heme (n = 3; R2 = 0.94). (C) Membrane-bound heme was monitored during repeated alkaline washing of membranes purified from fully oxygenated normal or SSRBCs. SS membranes retained heme more than did normal membranes (n = 3-5; mean ± SEM). Note similar globin findings in Figure 4A. *P < .05.
Figure 4.

Model of inverted RBC membrane attached to silica beads. RBC membranes from normal and SSRBCs were prepared by hypotonic lysis and alkaline washing; GAPDH was depleted by further salt washing. (A) Differences were observed in membrane protein banding from normal and SSRBCs. Note that despite salt washing, Hb globin (Band 9) remained associated with SS membranes, which is consistent with membrane-associated heme in Figure 3C. Inverted GAPDH-depleted membranes were subsequently attached to silica beads. (B) Immunofluorescence imaging of glycophorin A (an exofacial label) and cdB3 (a cytoplasmic label) was used to determine membrane orientation (Zeiss Axioskop microscope, 40× objective; AxioCam with AxioVision acquisition software; imaging medium Prolong Gold). (C) Confocal immunofluorescence images of (non–GAPDH-depleted) inverted membrane coated beads demonstrate free access to cdB3, GAPDH, and Hb (FluoViewFV1000 Olympus confocal microscope, PlanApo 60×/1.40 oil objective, 2.4× zoom; Flouview acquisition software; imaging medium Prolong Gold). (D) The model was further characterized by comparing membrane protein, membrane volume, and B3 number (as a function of bead count) between preparations from normal and SSRBCs. Although the membrane protein load per bead was greater for SS preparations, the membrane volume and B3 count was invariant. (E) To evaluate GAPDH binding to the bead model, we determined the ratio between membrane B3 and membrane-bound GAPDH as a function of [GAPDH] in solution; as expected, this relationship is linear. (F) GAPDH binding specificity for B3 was qualitatively evaluated by confocal immunofluorescence, which illustrates variation in GAPDH binding ± preincubation with antibody to the cdB3 NH2 terminus (GAPDH-binding site; FluoViewFV1000 Olympus confocal microscope, PlanApo 60×/1.40 oil objective; Flouview acquisition software; imaging medium Prolong Gold).
Immobilization of inverted, GAPDH-depleted membranes on silica beads and model validation
Membranes from normal and SSRBCs underwent salt washing, which released both GAPDH and Hb from normal membranes; however, Hb was not fully released from SS membranes (Figure 4A band 9). This difference in the ratio of protein mass to membrane volume between normal and SS membrane limited our ability to standardize the membrane surface area for comparative cdB3-binding studies using a simple inside-out vesicle (IOV) model. Experiments were therefore performed on inverted membranes immobilized on silica beads; bead count was used to standardize membrane area and B3 number between groups (Figure 4D). Inverted membrane orientation was confirmed using antibodies to glycophorin A (an exofacial marker) or to cdB3 (a cytoplasmic marker; Figure 4B). Immunofluorescent confocal images of bead-attached (unwashed) membranes further illustrated the appropriate orientation of the proteins under study, demonstrating free access to cdB3, GAPDH, and Hb (Figure 4C). Titration of GAPDH against beads confirmed a linear binding relationship (Figure 4E), and specific GAPDH binding to cdB3 was confirmed by blocking with antibody to the cdB3 NH2 terminus (Figure 4F). Finally, inverted, bead-attached, GAPDH-depleted membranes were incubated with free GAPDH in solution, spun out, and residual supernatant enzyme activity was determined.26 Bead number and enzyme activity were inversely related; activity was confirmed to vary appropriately with added GAPDH or with an inhibitory, synthetic cdB3 peptide (supplemental Figure 4).
Quantitative comparison of GAPDH binding to bead-attached membranes from normal and SSRBCs
GAPDH-quenching capacity was determined for beads prepared from either normal or SSRBCs; beads coated with normal membrane (count: 0-6 × 108) quenched GAPDH more avidly than equivalent bead numbers coated with sickle cell membrane (Figure 5A), as would be anticipated from similar, less quantitative reports.33 The difference in GAPDH quenching by control and sickle cell membrane is concordant with the difference we observed in membrane-bound Hb (Figures 3C and 4A). In paired experiments, the bead number was held constant (3 × 108) while varying free [GAPDH] to near saturation; after incubation, beads were pelleted and washed, membrane-bound proteins were released, and GAPDH was quantitated by immunoblotting/densitometry. Sickle cell membrane–attached beads bound GAPDH less avidly than did normal membrane–attached beads (Figure 5B), consistent with our findings of diminished GAPDH quenching (Figure 5A) and increased membrane-bound Hb (Figures 3C and 4A).
Figure 5.

Avid O2-responsive HbS binding to RBC membrane defeats potential control of GAPDH activity. RBC membranes depleted of endogenous GAPDH were attached to silica beads. (A) To study membrane-based inhibition of free GAPDH, progressive numbers of membrane coated beads were incubated with a GAPDH activity buffer (containing a fixed amount of GAPDH). Beads were pelleted and residual enzyme activity in the supernatant was assayed (n = 3-9; mean ± SEM). *P < .05 for normal versus SS. (B) To quantitate GAPDH binding to membrane, progressive amounts of GAPDH (in activity buffer) were incubated with membrane-coated beads (bead number fixed at 3 × 108), beads were pelleted, and membrane-bound GAPDH was quantitated by immunoblot and densitometry. SSRBC membranes quenched and bound significantly less GAPDH than control RBC membranes (n = 4; mean ± SEM) *P < .05 for normal versus SS. In separate experiments, beads coated with membrane from oxygenated or deoxygenated normal RBCs were incubated with oxygenated (HbO2% > 85%) or deoxygenated (HbO2% < 15%) HbAo or HbS and then incubated with GAPDH. Enzyme activity was determined in the supernatant. (C) No difference was observed in GAPDH quenching after incubation of bead-attached membrane from oxygenated RBCs with either oxy-HbAo or HbS (n = 4; mean ± SEM). (D) However, beads coated with washed membrane from deoxygenated RBCs and incubated with deoxy-HbS quenched significantly less GAPDH than those incubated with deoxy-HbAo (n = 6; mean ± SEM). *P < .05 for deoxy-HbAo versus deoxy-HbS.
Competitive binding between GAPDH and Hb for cdB3: dependence on RBC O2 content and cdB3 pTyr density
cdB3 is known to undergo phosphorylation in a fashion that is linked to RBC O2 content34 and that diminishes GAPDH binding.15 We confirmed this and found that O2 content-responsive cdB3 phosphorylation in SSRBCs was significantly more robust than in normal RBCs (supplemental Figure 6A); however, increased cdB3pTyr in oxy-RBCs appeared insufficient to fully account for the substantial HMP constraint we observed in deoxy-RBCs by 1H-NMR (supplemental Figure 6B). We therefore sought to determine whether O2 content-based membrane modification differentially altered competition between deoxy-HbAo or deoxy-HbS with GAPDH for cdB3 binding. Membranes were purified from normal RBCs to avoid confounding by cdB3-associated hemichrome,18 which was not present in these preparations. No difference in GAPDH activity quenching was observed from HbAo- or HbS-exposed, membrane-attached beads made from oxygenated RBCs (Figure 5C). However, when membrane-attached beads made from deoxygenated RBCs were studied, those exposed to deoxy-HbS quenched significantly less GAPDH than those exposed to deoxy-HbAo (Figure 5D). Finally, we monitored O2-dependent GAPDH migration between membrane and cytosol in intact RBCs (Figure 6). The O2 content of freshly drawn human RBCs was manipulated (in our rotating tonometer) and then cdB3 and GAPDH location was determined by immunofluorescent confocal microscopy. After oxygenation of normal RBCs, GAPDH migrated from cytosol to membrane (as shown previously by others15). However, this pattern was not observed in SSRBCs, suggesting that the cdB3-binding site was not vacated by Hb on RBC oxygenation and consistent with our data above (Figures 2C-D, 3C, 4A, and 5, and supplemental Figure 5).
Figure 6.

O2-dependent cytosol/membrane migration of GAPDH is absent in SSRBCs. Confocal immunofluorescent imaging was used to observe GAPDH migration in intact RBCs during O2 loading/unloading. Washed RBCs were either fully oxygenated (HbO2% > 95%) or deoxygenated (HbO2% < 20%), fixed under appropriate gas tensions, probed for GAPDH (green) or cdB3 (yellow; see “Methods”), and confocal images were obtained (FluoViewFV1000 Olympus confocal microscope, PlanApo 60×/1.40 oil objective; Flouview acquisition software; imaging medium Prolong Gold). Membrane-based location of cdB3 is easily visualized in all images from both SS and normal RBCs. SSRBCs fail to demonstrate normal O2 content–dependent migration of GAPDH from cytosol to membrane, as demonstrated in normal RBCs on oxygenation.
Discussion
Our findings indicate that dysregulated glycolysis sensitizes SSRBCs to oxidative stress and that abnormal O2-dependent competition between HbS and GAPDH for cdB3 binding is a possible mechanism for this phenomenon (schema presented in Figure 7). Specifically, during steady O2− loading, SSRBCs demonstrated oxidative modification of membrane thiols and reduced membrane integrity with impaired GSH and NADPH recycling arising from constrained HMP flux and were rescued by blocking EMP glycolysis at GAPDH. Further investigation suggested that avid binding of HbS to cdB3 disturbs membrane-based control of HMP and EMP balance (disfavoring HMP flux). We used a novel, inverted membrane-on-bead model to test this possibility and found that purified sickle cell membranes demonstrated increased membrane-bound Hb, an inverse relationship between membrane-bound Hb and ability to both bind and quench GAPDH activity, and markedly attenuated GAPDH binding and quenching relative to membranes from normal RBCs. Furthermore, deoxy-HbS (more so than deoxy-HbAo) prevented GAPDH binding to normal RBC membrane and remarkably this difference was only apparent when studying washed membranes obtained from previously deoxygenated RBCs. This variation between Hb types in reciprocal binding with GAPDH appeared to be related to RBC O2 content–responsive cdB3pTyr density, which was strikingly different between normal and SSRBCs. Observations in our model were confirmed by imaging GAPDH migration in intact RBCs during O2 loading/unloading, in which oxygenated SSRBCs fail to exhibit normal GAPDH recruitment to membrane. Diminished GAPDH quenching after deoxy-HbS incubation with beads composed from normal Hb-free membranes indicates that dispersed GAPDH in oxygenated SSRBCs is unlikely to be attributable to hemichrome alone. Diminished GAPDH sequestration/inhibition would be expected to release EMP control, thereby creating an HMP substrate deficit, which undermines antioxidant systems in SSRBCs by limiting NADPH and GSH recycling capacity (as was observed).
These data are consistent with several lines of evidence indicating that HbS disturbs cdB3-based regulation of erythrocyte energy metabolism. HbS may prevent glycolytic complex assembly on cdB3 by either avid (solution-phase) O2-dependent binding5 or by binding of denatured HbS aggregates (hemichrome), which bridge cdB3 monomers.6,7,19,20 Either or both of these possibilities may explain the observation that HbS binds avidly5,6,18 and GAPDH binds poorly to SSRBC membranes.33 In addition, others have demonstrated abnormal constraint on maximal HMP flux in SSRBCs by NMR spectroscopy,35 or metabolomic analysis,36 and this is predicted by mathematical modeling of avid Hb-cdB3 binding in this system.17 Moreover, commonly appreciated signs of oxidative injury to SSRBCs are predicted (though not necessarily exclusive) consequences of restricted HMP flux such as limited GSH21 and NADPH37 recycling capacity and extensive membrane thiol oxidation.31
In the present study, we observed influence of O2 content on the HbS-cdB3 interaction and GAPDH regulation in both our membrane-on-bead model and in intact RBCs. Deoxygenation independently influenced both binding partners; specifically, differences between HbS and HbAo competition with GAPDH for cdB3 were apparent only when membranes isolated from deoxygenated RBCs were studied, suggesting that deoxygenation-dependent modification of cdB3 differentially influences affinity for (deoxy) HbS or Ao. On RBC deoxygenation, cdB3 is known to undergo phosphorylation (at multiple Tyr residues, possibly via p72Syk activation or PTP1B inhibition)34 and S-nitrosylation (at either/both Cys152 or Cys330).13 These modifications are well placed on cdB3 to inhibit either/both Hb and GAPDH binding,22,29 as has been observed previously.13,15,16,38,39 Both cdB3 Tyr-phosphorylation (as we observed) and Cys nitrosylation are reported to be dysregulated in SCA40,41; in ongoing work, we are exploring the role (and regulation) of these modifications with regard to O2-responsive metabolic control in SCA. Because it appears that the degree of cdB3 masking by HbS varies with O2 content and that cdB3 masking undermines antioxidant systems (shown herein and previously for normal RBCs under severe hypoxia9), we suggest that this may offer a plausible mechanism (beyond sickling alone) for the disproportionate prevalence of oxidative stress and hypoxia-associated phenotype variation in patients with SCA. Differing O2 loading/unloading histories of RBC subpopulations might therefore introduce phenotype variation within subjects over time or between groups with various respiratory pathologies. More detailed evaluation of the influence of HbS on other cdB3-based signaling systems7,8,13,14 may improve our understanding of hypoxia-dependent phenotype variation in SCA pathobiology.
Considering that the HbS-cdB3–based disturbance in RBC glycolysis (HMP constraint) is functionally similar to glucose 6-phosphate dehydrogenase (G6PD) deficiency, it is worth noting that coincident G6PD deficiency has been reported to influence SCA phenotype,42 although this is not a consistent observation.43 In light of our present findings, however, phenotype penetrance of G6PD deficiency in SCA is likely to be limited in the setting of HMP constraint arising from HbS-cdB3 interactions (eg, G6P substrate lack might render G6PD deficiency moot; see Figure 7). Therefore, by varying G6P availability, it may be more likely that EMP (rather than HMP) enzymopathies or variations in activity (with polymorphism) will influence the impact of dysfunctional cdB3 masking by HbS.
Several features of the models used herein merit discussion. We chose an O2−–generating system to model oxidative stress for several reasons. RBCs have robust systems to deal with the O2− generated by normal oxygenation/deoxygenation; this is notably increased in SCA,44 likely because of lower heme redox potential for HbS.4 Furthermore, O2− is a major oxidative product of activated endothelium and leukocytes and both are part of the microcirculatory disturbance in SCA.1 The HX/XO system we used to generate O2− is very well characterized, is commonly used in RBC studies, and the degree of O2− evolved is well within the physiologically relevant range produced by activated vascular NADPH oxidase.45 The resulting “oxidative footprint” on membrane was quantitated by gauging thiol status, principally because the pentose shunt and NADPH recycling are specifically coupled to thiol-based (GSH) antioxidant systems in RBCs; this antioxidant system also has demonstrated dysfunction in SCA.21 We focused specifically on GAPDH to examine the influence of HbS-cdB3 interactions on erythrocyte energetics and antioxidant systems. Among the EMP enzymes under cdB3 control (including phosphofructokinase, aldolase, pyruvate kinase, and lactate dehydrogenase; see Figure 7A-C), we chose GAPDH for its central location at the HMP/EMP juncture and its mapped and well-characterized cdB3-binding site.15,22 In addition, we were able to test “rescue” of SSRBCs by imitating cdB3 sequestration/inhibition with KA, a selective inhibitor of GAPDH activity.23
Overall, our data are consistent with an emerging model of RBC metabolic regulation in which Hb conformation–dependent interactions with cdB3 govern diverse metabolic streams and signaling pathways.8,15,17 Such key functions oscillate with O2 gradients encountered during arteriovenous transit. In addition to glycolysis,8,9 these include ion and amino acid transport,10 cytoskeleton-membrane interactions,11 and processing/export of vasoactive effectors.12–14 Coupling among regional O2 gradients, RBC metabolism, and vascular signaling reinforces the linkage between metabolic demand and the regulation of regional blood flow,12,14 which is disturbed in SCA.41 Although abnormally avid HbS-cdB3 binding may render RBCs vulnerable to oxidative injury,8,9 with direct consequences of altered rheology, hemolysis, and accelerated clearance, inappropriate cdB3 masking by HbS may further disrupt O2 delivery through maladaptive alterations in RBC signaling in the microcirculation, interfering with appropriate control of regional blood flow.12,14,41 Therefore, this paradigm extends the implications of our findings because abnormal HbS-cdB3 interactions are likely to influence the regulation of a diverse array of RBC functions.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (grants UL1 RR024992 to A.D. and 1S10 RR027184 to A.d.A.), the American Heart Association (grant GIA0950133G to A.D.), the Doris Duke Charitable Foundation CRF Program (to J.R.), the Children's Discovery Institute (grants PD-II-2006-01 to A.D. and PD-F-2009-203 to S.C.R.), and the Burroughs Wellcome Fund (TRANS Clinical Scientist Award to M.D. and S.G.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.C.R. analyzed the results and produced the figures; S.C.R., J.G.C.R., L.B.G., V.G., M.N.H., D.M., S.G., T.N., A.d.A., and M.D. performed the experiments; and S.C.R., M.R.D., A.d.A., and A.D. designed the research and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Dr Allan Doctor, Washington University School of Medicine, Campus Box 8208, 1 Children's Pl, St Louis, MO 63110; e-mail: doctor@wustl.edu.
References
- 1.Embury SH. The not-so-simple process of sickle cell vasoocclusion. Microcirculation. 2004;11(2):101–113. doi: 10.1080/10739680490278277. [DOI] [PubMed] [Google Scholar]
- 2.Chirico EN, Pialoux V. Role of oxidative stress in the pathogenesis of sickle cell disease. IUBMB Life. 2012;64(1):72–80. doi: 10.1002/iub.584. [DOI] [PubMed] [Google Scholar]
- 3.Riggs A, Wells M. The oxygen equilibrium of sickle-cell hemoglobin. Biochim Biophys Acta. 1961;50:243–248. doi: 10.1016/0006-3002(61)90322-5. [DOI] [PubMed] [Google Scholar]
- 4.Hebbel RP, Morgan WT, Eaton JW, Hedlund BE. Accelerated autoxidation and heme loss due to instability of sickle hemoglobin. Proc Natl Acad Sci U S A. 1988;85(1):237–241. doi: 10.1073/pnas.85.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shaklai N, Sharma VS, Ranney HM. Interaction of sickle cell hemoglobin with erythrocyte membranes. Proc Natl Acad Sci U S A. 1981;78(1):65–68. doi: 10.1073/pnas.78.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Platt OS, Falcone JF. Membrane protein interactions in sickle red blood cells: evidence of abnormal protein 3 function. Blood. 1995;86(5):1992–1998. [PubMed] [Google Scholar]
- 7.Low PS. Structure and function of the cytoplasmic domain of band 3: center of erythrocyte membrane–peripheral protein interactions. Biochim Biophys Acta. 1986;864(2):145–167. doi: 10.1016/0304-4157(86)90009-2. [DOI] [PubMed] [Google Scholar]
- 8.Messana I, Orlando M, Cassiano L, et al. Human erythrocyte metabolism is modulated by the O2-linked transition of hemoglobin. FEBS Lett. 1996;390(1):25–28. doi: 10.1016/0014-5793(96)00624-2. [DOI] [PubMed] [Google Scholar]
- 9.Rogers SC, Said A, Corcuera D, McLaughlin D, Kell P, Doctor A. Hypoxia limits antioxidant capacity in red blood cells by altering glycolytic pathway dominance. FASEB J. 2009;23(9):3159–3170. doi: 10.1096/fj.09-130666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibson JS, Cossins AR, Ellory JC. Oxygen-sensitive membrane transporters in vertebrate red cells. J Exp Biol. 2000;203(Pt 9):1395–1407. doi: 10.1242/jeb.203.9.1395. [DOI] [PubMed] [Google Scholar]
- 11.Chang SH, Low PS. Regulation of the glycophorin C-protein 4.1 membrane-to-skeleton bridge and evaluation of its contribution to erythrocyte membrane stability. J Biol Chem. 2001;276(25):22223–22230. doi: 10.1074/jbc.M100604200. [DOI] [PubMed] [Google Scholar]
- 12.Doctor A, Platt R, Sheram ML, et al. Hemoglobin conformation couples erythrocyte S-nitrosothiol content to O2 gradients. Proc Natl Acad Sci U S A. 2005;102(16):5709–5714. doi: 10.1073/pnas.0407490102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pawloski JR, Hess DT, Stamler JS. Export by red blood cells of nitric oxide bioactivity. Nature. 2001;409(6820):622–626. doi: 10.1038/35054560. [DOI] [PubMed] [Google Scholar]
- 14.Doctor A, Stamler JS. Nitric oxide transport in blood: a third gas in the respiratory cycle. Compr Physiol. 2011;1:611–638. doi: 10.1002/cphy.c090009. [DOI] [PubMed] [Google Scholar]
- 15.Campanella ME, Chu H, Low PS. Assembly and regulation of a glycolytic enzyme complex on the human erythrocyte membrane. Proc Natl Acad Sci U S A. 2005;102(7):2402–2407. doi: 10.1073/pnas.0409741102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewis IA, Campanella ME, Markley JL, Low PS. Role of band 3 in regulating metabolic flux of red blood cells. Proc Natl Acad Sci U S A. 2009;106(44):18515–18520. doi: 10.1073/pnas.0905999106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kinoshita A, Tsukada K, Soga T, et al. Roles of hemoglobin Allostery in hypoxia-induced metabolic alterations in erythrocytes: simulation and its verification by metabolome analysis. J Biol Chem. 2007;282(14):10731–10741. doi: 10.1074/jbc.M610717200. [DOI] [PubMed] [Google Scholar]
- 18.Kuross SA, Rank BH, Hebbel RP. Excess heme in sickle erythrocyte inside-out membranes: possible role in thiol oxidation. Blood. 1988;71(4):876–882. [PubMed] [Google Scholar]
- 19.Waugh SM, Willardson BM, Kannan R, Labotka RJ, Low PS. Heinz bodies induce clustering of band 3, glycophorin, and ankyrin in sickle cell erythrocytes. J Clin Invest. 1986;78(5):1155–1160. doi: 10.1172/JCI112696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kannan R, Labotka R, Low PS. Isolation and characterization of the hemachrome-stabilized membrane protein aggregates from sickle erythrocytes. Major site of autologous antibody binding. J Biol Chem. 1988;263(27):13766–13773. [PubMed] [Google Scholar]
- 21.Gizi A, Papassotiriou I, Apostolakou F, et al. Assessment of oxidative stress in patients with sickle cell disease: The glutathione system and the oxidant-antioxidant status. Blood Cells Mol Dis. 2011;46(3):220–225. doi: 10.1016/j.bcmd.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 22.Chu H, Low PS. Mapping of glycolytic enzyme-binding sites on human erythrocyte band 3. Biochem J. 2006;400(1):143–151. doi: 10.1042/BJ20060792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Endo A, Hasumi K, Sakai K, Kanbe T. Specific inhibition of glyceraldehyde-3-phosphate dehydrogenase by koningic acid (heptelidic acid). J Antibiot. 1985;38(7):920–925. doi: 10.7164/antibiotics.38.920. [DOI] [PubMed] [Google Scholar]
- 24.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30(11):1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 25.Delgado TC, Castro MM, Geraldes CF, Jones JG. Quantitation of erythrocyte pentose pathway flux with [2-13C]glucose and 1H NMR analysis of the lactate methyl signal. Magn Reson Med. 2004;51(6):1283–1286. doi: 10.1002/mrm.20096. [DOI] [PubMed] [Google Scholar]
- 26.Tsai IH, Murthy SN, Steck TL. Effect of red cell membrane binding on the catalytic activity of glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem. 1982;257(3):1438–1442. [PubMed] [Google Scholar]
- 27.Dupuy AD, Engelman DM. Protein area occupancy at the center of the red blood cell membrane. Proc Natl Acad Sci U S A. 2008;105(8):2848–2852. doi: 10.1073/pnas.0712379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaufmann S, Tanaka M. Cell adhesion onto highly curved surfaces: one-step immobilization of human erythrocyte membranes on silica beads. Chemphyschem. 2003;4(7):699–704. doi: 10.1002/cphc.200200537. [DOI] [PubMed] [Google Scholar]
- 29.Chu H, Breite A, Ciraolo P, Franco RS, Low PS. Characterization of the deoxyhemoglobin binding site on human erythrocyte band 3: implications for O2 regulation of erythrocyte properties. Blood. 2008;111(2):932–938. doi: 10.1182/blood-2007-07-100180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eaton JW, Brewer GJ. The relationship between red cell 2,3-diphosphoglycerate and levels of hemoglobin in the human. Proc Natl Acad Sci U S A. 1968;61(2):756–760. doi: 10.1073/pnas.61.2.756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rank BH, Carlsson J, Hebbel RP. Abnormal redox status of membrane-protein thiols in sickle erythrocytes. J Clin Invest. 1985;75(5):1531–1537. doi: 10.1172/JCI111857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katz J, Wood HG. The use of glucose-C14 for the evaluation of the pathways of glucose metabolism. J Biol Chem. 1960;235:2165–2177. [PubMed] [Google Scholar]
- 33.Vasseur C, Leclerc L, Hilly M, Bursaux E. Decreased G3PDH binding to erythrocyte membranes in sickle cell disease. Nouv Rev Fr Hematol. 1992;34(2):155–161. [PubMed] [Google Scholar]
- 34.Barbul A, Zipser Y, Nachles A, Korenstein R. Deoxygenation and elevation of intracellular magnesium induce tyrosine phosphorylation of band 3 in human erythrocytes. FEBS Lett. 1999;455(1-2):87–91. doi: 10.1016/s0014-5793(99)00822-4. [DOI] [PubMed] [Google Scholar]
- 35.Schrader MC, Simplaceanu V, Ho C. Measurement of fluxes through the pentose phosphate pathway in erythrocytes from individuals with sickle cell anemia by carbon-13 nuclear magnetic resonance spectroscopy. Biochim Biophys Acta. 1993;1182(2):179–188. doi: 10.1016/0925-4439(93)90139-r. [DOI] [PubMed] [Google Scholar]
- 36.Darghouth D, Koehl B, Madalinski G, et al. Pathophysiology of sickle cell disease is mirrored by the red blood cell metabolome. Blood. 2011;117(6):e57–e66. doi: 10.1182/blood-2010-07-299636. [DOI] [PubMed] [Google Scholar]
- 37.Zerez CR, Lachant NA, Lee SJ, Tanaka KR. Decreased erythrocyte nicotinamide adenine dinucleotide redox potential and abnormal pyridine nucleotide content in sickle cell disease. Blood. 1988;71(2):512–515. [PubMed] [Google Scholar]
- 38.Low PS, Allen DP, Zioncheck TF, et al. Tyrosine phosphorylation of band 3 inhibits peripheral protein binding. J Biol Chem. 1987;262(10):4592–4596. [PubMed] [Google Scholar]
- 39.Misiti F, Meucci E, Zuppi C, et al. O(2)-dependent stimulation of the pentose phosphate pathway by S-nitrosocysteine in human erythrocytes. Biochem Biophys Res Commun. 2002;294(4):829–834. doi: 10.1016/S0006-291X(02)00552-1. [DOI] [PubMed] [Google Scholar]
- 40.Siciliano A, Turrini F, Bertoldi M, et al. Deoxygenation affects tyrosine phosphoproteome of red cell membrane from patients with sickle cell disease. Blood Cells Mol Dis. 2010;44(4):233–242. doi: 10.1016/j.bcmd.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 41.Pawloski JR, Hess DT, Stamler JS. Impaired vasodilation by red blood cells in sickle cell disease. Proc Natl Acad Sci U S A. 2005;102(7):2531–2536. doi: 10.1073/pnas.0409876102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bernaudin F, Verlhac S, Chevret S, et al. G6PD deficiency, absence of alpha-thalassemia, and hemolytic rate at baseline are significant independent risk factors for abnormally high cerebral velocities in patients with sickle cell anemia. Blood. 2008;112(10):4314–4317. doi: 10.1182/blood-2008-03-143891. [DOI] [PubMed] [Google Scholar]
- 43.Steinberg MH, West MS, Gallagher D, Mentzer W. Effects of glucose-6-phosphate dehydrogenase deficiency upon sickle cell anemia. Blood. 1988;71(3):748–752. [PubMed] [Google Scholar]
- 44.Schacter LP. Generation of superoxide anion and hydrogen peroxide by erythrocytes from individuals with sickle trait or normal haemoglobin. Eur J Clin Invest. 1986;16(3):204–210. doi: 10.1111/j.1365-2362.1986.tb01330.x. [DOI] [PubMed] [Google Scholar]
- 45.Souza HP, Liu X, Samouilov A, Kuppusamy P, Laurindo FR, Zweier JL. Quantitation of superoxide generation and substrate utilization by vascular NAD(P)H oxidase. Am J Physiol Heart Circ Physiol. 2002;282(2):H466–474. doi: 10.1152/ajpheart.00482.2001. [DOI] [PubMed] [Google Scholar]
- 46.Bretthorst GL. Bayesian analysis. III. Applications to NMR signal detection, model selection, and parameter estimation. J Mag Res. 1990;88(3):571–595. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.