

Abstract
3,4-Methylenedioxymethamphetamine (MDMA; Ecstasy) is a popular drug of abuse with well-documented acute effects on serotonergic, dopaminergic, and cholinergic transmitter systems, as well as evidence of long-term disruption of serotoninergic systems in the rat brain. Recently, it was demonstrated that MDMA evokes a delayed and sustained increase in glutamate release in the hippocampus. The purpose of the present study was to determine the role of inflammatory mediators in the MDMA-induced increase in glutamate release, as well as the contribution of inflammatory pathways in the persistent neurochemical toxicity associated with repeated MDMA treatment. Treatment with the non-selective cyclooxygenase (COX) inhibitor ketoprofen and the COX-2 selective inhibitor nimesulide attenuated the increase in extracellular glutamate in the hippocampus evoked by repeated MDMA exposure (10 mg/kg, i.p., every 2 h); no attenuation was observed in rats treated with the COX-1 selective inhibitor piroxicam. Reverse dialysis of a major product of COX activity, prostaglandin E2, also resulted in a significant increase in extracellular glutamate in the hippocampus. Repeated exposure to MDMA diminished the number of parvalbumin-positive GABA interneurons in the dentate gyrus of the hippocampus, an effect that was attenuated by ketoprofen treatment. However, COX inhibition with ketoprofen did not prevent the long-term depletion of 5-HT in the hippocampus evoked by MDMA treatment. These data are supportive of the view that cyclooxygenase activity contributes to the mechanism underlying both the increased release of glutamate and decreased number of GABA interneurons in the rat hippocampus produced by repeated MDMA exposure.
Keywords: MDMA, glutamate, cyclooxygenase, GABA
Introduction
3,4-Methylenedioxymethamphetamine (MDMA, Ecstasy) has generally been viewed as selectively neurotoxic for 5-HT axon terminals in view of the well documented reductions in brain 5-HT concentrations, 5-HT uptake sites, 5-HT transporter immunoreactivity and reductions in 5-HT immunoreactive fibers following repeated exposure to MDMA (c.f., Green et al., 2003; Gudelsky and Yamamoto, 2003). Moreover, MDMA-induced 5-HT neurotoxicity has been considered to resemble a distal axotomy affecting 5-HT axon terminals (Xie et al., 2006).
However, there are several reports that treatment of rats with MDMA produces neuronal degeneration in several brain regions, including the hippocampus (Kermanian et al., 2012; Riezzo et al., 2010; Schmued 2003; Wang et al., 2009; Warren et al., 2007). Investigators from these and other studies have concluded that MDMA promotes apoptotic cell death on the basis of increases in caspase-3, TUNEL staining and altered Bcl-2 family gene expression produced by MDMA (Meyer et al., 2004; Asi et al. 2012; Riezzo et al., 2010; Wang et al., 2009). Consistent with these data, Capela et al. (2007; 2012) have reported that MDMA promotes apoptotic cell death in cultured cortical and hippocampal neurons in vitro. Although these data support the view that MDMA neurotoxicity extends beyond 5-HT axon terminals to neuronal cell bodies themselves within the hippocampus, the identity of neurons potentially damaged by MDMA has not been identified.
A recent report from our lab documented the finding that MDMA produces a delayed and sustained increase in the extracellular concentration of glutamate in the hippocampus (Anneken and Gudelsky 2012). The MDMA-induced increase in hippocampal glutamate release was suppressed by fluoxetine and the 5-HT2 antagonist ketanserin, but was still evident in the presence of tetrodotoxin. Anneken and Gudelsky (2012) concluded that 5-HT, released by MDMA, activates 5-HT2A/C receptors, thereby promoting the release of glutamate, presumably from astrocytes, in the hippocampus.
Although the consequences of an increased release of hippocampal glutamate by MDMA are unknown, several studies suggest that hippocampal neurons are vulnerable to excitotoxic effects of elevated glutamate through the activation of glutamate receptors. For example, parvalbumin-positive GABA neurons in the hippocampus express Group 1 metabotropic glutamate receptors (Kerner et al., 1997), as well as GluR1 and GluR3 receptors lacking GluR2 immunoreactivity (Moga et al., 2003). In support of this vulnerability, parvalbumin-positive GABAergic neurons are selectively vulnerable to the toxic effects of kainic acid, an effect blocked by glutamate antagonists (Sanon et al., 2005).
Given the aforementioned reports that 1) MDMA produces neuronal damage in the hippocampus, 2) MDMA increases glutamate release in the hippocampus and 3) parvalbumin-positive GABA neurons in the hippocampus are sensitive to glutamate-mediated neurotoxicity, the present study was undertaken to evaluate the effects of repeated exposure to MDMA on parvalbumin-positive GABA neurons in the dorsal hippocampus. In view of the findings that MDMA induced glutamate release is dependent upon 5-HT2 receptor activation and that 5-HT2 receptor activation appears to increase the activity of cyclooxygenase (COX), we also sought to ascertain the role of COX in the effects of MDMA on hippocampal glutamate release and PV-positive GABAergic neurons.
Materials and Methods
Animals and Drug Treatments
Adult male Sprague-Dawley rats (250-350g) (Harlan Laboratories, Indianapolis, IN) were used in this study. Animals were given free access to food and water in a temperature and humidity controlled room. The animals were singly housed following cannula implantation until the day of the experiment. All procedures were performed in adherence to the National Institutes of Health guidelines and were approved by the institutional animal care and use committee.
MDMA was generously provided by the National Institute on Drug Abuse (Bethesda). Ketoprofen and piroxicam were obtained from Sigma-Aldrich (St. Louis, MO), while nimesulide and prostaglandin E2 were obtained from Tocris Bioscience (Ellisville, MO). MDMA was dissolved in 0.15M NaCl and injected i.p. at a dose of 10 mg/kg at two h intervals for a total of 2 or 3 injections. Ketoprofen was dissolved in a 30% Transcutol solution and administered in 3 injections at a dose of 5 mg/kg, s.c., 1 h prior to and 1h and 3h following the first MDMA injection. Both piroxicam and nimesulide were delivered in a 2% polyvinylpyrrolidone suspension. Piroxicam was administered as 3 injections at a dose of 3 mg/kg, i.p., in the same time course as ketoprofen. Nimesulide was administered as 3 injections at a dose of 7.5 mg/kg, i.p., also on the same time course. Prostaglandin E2 (PGE2) was dissolved in the dialysis buffer and was administered by reverse dialysis through the probe at a concentration of 30 μM for 1.5h and then at 100 μM for an additional 1.5h. Doses of COX inhibitors were based on those used in previous studies (Asanuma et al., 2003; Candelario-Jalil et al., 2004; Terao et al., 1998).
Microdialysis
Rats were implanted with a stainless steel guide cannula under ketamine/xylazine (70/6 mg/kg, i.p.) anesthesia 48-72 h prior to the insertion of the dialysis probe. On the evening prior to the experiment, a concentric style dialysis probe was inserted through the guide cannula into the dorsal hippocampus; the coordinates for the tip of the probe were: A/P, −3.6 mm, L, 2.0 mm, and D/V −4.0 mm. The active portion of the membrane for the probes was 2.0 mm. The probes were connected to an infusion pump set to deliver modified Dulbecco’s phosphate buffered saline containing 1.2 mM CaCl2 and 5 mM glucose at a flow rate of 1 μl/min overnight. On the morning of the experiment, the flow rate was increased to 2 μl/min and the probes were allowed to equilibrate for 1.5 h. Three collections were then taken at 30 min intervals to establish values; thereafter, samples were collected every hour for the duration of the experiment. Data were calculated as a percentage of the baseline value for glutamate which was obtained by averaging the three baseline samples.
HPLC glutamate analysis
Glutamate was derivitized according to the method described by Donzanti and Yamamoto (1988) and quantified by HPLC with electrochemical detection, as described previously (Anneken and Gudelsky 2012).
Analysis of Tissue Serotonin (5-HT)
All tissue samples were homogenized in 0.2N perchloric acid. Concentrations of 5-HT were determined via HPLC in 20μL aliquots of supernatants from tissue homogenates, as described previously (Shankaran et al., 2001).
Tissue preparation for GABA neuron counts
Rats were deeply anesthetized with ketamine hydrochloride (70mg/kg, ip) and xylazine (6mg/kg, ip) and were transcardially perfused with 0.1 M PBS (100 mL) prior to the perfusion with 4% paraformaldehyde (400 mL). Brains were removed and post-fixed for 2 hr in 4% paraformaldehyde before cryoprotection in a series of glycerol solutions; 10% glycerol with 2% dimethylsufide (DMS0) in 0.1 M PBS overnight, followed by 20% glycerol with 2% DMS0 in 0.1 M PBS overnight. The following day, brains were flash frozen in 2-methylbutane for 30 minutes. Coronal sectioning was at a thickness of 50 μm through the dorsal extent of the hippocampus (approximately −3.30 mm to −4.80 from bregma, (Paxinos, G. and Watson, C. 1998) as described previously (Muller et al., 2001). Each section was collected in four uninterrupted series with a 200 μm section interval and stored in 15% glycerol in 0.1 M PBS at −80°C until immunostaining.
Parvalbumin immunostaining
Free-floating brain sections were stained for parvalbumin. Sections were washed in 0.1 M PBS and then treated with 1% H2O2 for 20 min room at temperature (RT). After several washes, the sections were blocked for 1 h at RT with 3% normal goat serum (NGS; Invitrogen, Carlsbad, CA, USA) in 0.1 M PBS containing 0.5% Triton-X 100 and Avidin block (Vector Laboratories, Burlingame, CA, USA). Sections were then incubated for 36 h at 4°C with a mouse monoclonal parvalbumin antibody (1:3000; Swant, Bellinzona, Switzerland) in 0.1 M PBS containing 0.5% Triton-X 100, 1% NGS and Biotin block (Vector Laboratories). Sections were washed and incubated in goat anti-mouse secondary antibody (Millipore, Billerica, MA, USA) for 2 h at RT followed by incubation in avidin-biotin-horseradish peroxidase (Vectastain Elite ABC Kit; Vector Laboratories) for 2 h at RT. Sections were washed and developed in diaminobenzidine.
Quantitative Cell Counts
Stereological estimations of the total number of parvalbumin-immunoreactive (PV-ir) interneurons were assessed using a BX51 Olympus microscope equipped with a DVC camera interfaced with StereoInvestigator 8.21 software (MBF Bioscience, Williston, VT, USA). A modified optical fractionator technique (Gundersen et al., 1999; West et al., 1991) was used to quantify neurons throughout the right dorsal hippocampus. Pilot studies were used for sampling procedures to ensure that the coefficient of error (Gundersen et al., 1999) was less than 1% for all counts. All slides were coded and the code not broken until the end of quantitative analysis. Every fourth section was systematically sampled and all subfields (dentate gyrus [DG] and cornu ammonis 1 and 3 [CA1 & CA3]) were outlined using a 5X objective. The DG included the hilus and subgranular zone, but not the principle cell layer of the CA3. Counting was performed using a 60X oil objective (NA 0.16). Due to the limited number of PV-ir interneurons in the DG, exhaustive sampling was performed with grid dimensions of 100 μm × 100 μm and a sampling area of 100 μm × 100 μm. For both the CA1 and CA3 subfield, the grid dimensions were 200 μm × 300 μm with a sampling area of 300 μm × 300 μm. Guard zones above and below the dissector were not used due to the massive tissue shrinkage during processing, as previously described (Czeh et al., 2005; Lister et al., 2006).
Statistical Analysis
All microdialysis data were analyzed using two-way repeated measures ANOVA, and multiple pairwise comparisons were performed using post-hoc analysis with the Student-Newman-Keuls test. Tissue 5-HT data were analyzed using a two-way ANOVA, as were the stereological neuron counts. Treatment differences were considered statistically significant at p<0.05.
Results
Ketoprofen and nimesulide, but not piroxicam, attenuate the MDMA-induced increase in extracellular glutamate concentrations in the rat hippocampus
The involvement of cyclooxygenase (COX) activity in the MDMA-induced increase in extracellular glutamate in the rat hippocampus was investigated using the non-selective COX inhibitor ketoprofen. Rats received injections of vehicle or ketoprofen (5 mg/kg, s.c.) 1 h prior to and 1 h and 3 h following the first injection of MDMA (10 mg/kg, i.p.). As shown in Figure 1, ketoprofen significantly attenuated (p<0.05) the increase in the extracellular concentration of glutamate in the hippocampus evoked by 2 injections of MDMA. A two-way repeated measures ANOVA revealed a significant effect of treatment (F(3,286) = 3.27; p<0.05).
Figure 1. Effect of the COX 1/2 inhibitor ketoprofen. COX-1 inhibitor piroxicam, and COX-2 inhibitor nimesulide on the MDMA-induced efflux of glutamate in the rat hippocampus.

Rats received ketoprofen (5 mg/kg, s.c.), piroxicam (3 mg/kg, i.p.) (inset), nimesulide (7.5 mg/kg, i.p.) (inset) or vehicle 1 h prior to and 1 h and 3 h following the first injection of MDMA (10 mg/kg, i.p.) or vehicle. (n= 4-14 per group). Average basal glutamate for VEH-MDMA group was 2.54 ± 0.34 ng/20μL (uncorrected for recovery). Arrows indicate injections with MDMA or Veh. * Indicates values that differ significantly (p<0.05) from Veh-MDMA animals.
The relative contribution of each isoform of COX to the increased efflux of hippocampal glutamate elicited by MDMA was assessed using the relatively specific inhibitors piroxicam (COX-1) and nimesulide (COX-2) (Figure 1, inset). Piroxicam (3 mg/kg, i.p.), nimesulide (7.5 mg/kg, i.p.), or vehicle was administered 1 h prior to and 1 h and 3 h following the initiation of MDMA treatment (10 mg/kg, i.p.). A two-way repeated measures ANOVA revealed a significant treatment effect (F(3,169) = 12.00; p<0.001) as well as a significant effect of time (F(10,169) = 2.86; p<0.01) and a significant treatment × time interaction (F(30,169) = 2.44; p<0.001). Results of the ANOVA revealed no significant difference in the glutamate reponses of animals treated with piroxicam + MDMA and those receiving MDMA alone (p>0.05). In contrast, nimesulide significantly diminished (p<0.05) the MDMA-induced increase in the extracellular concentration of glutamate. Treatment with the COX inhibitors alone did not significantly alter glutamate concentrations (data not shown).
Reverse dialysis of prostaglandin E2 increases extracellular glutamate concentration in the rat hippocampus
The local effect of inflammatory mediators on hippocampal glutamate was assessed using reverse dialysis of prostaglandin E2 (PGE2), the most biologically active product of COX activity. PGE2 was delivered in the dialysis buffer at a concentration of 30 M for 1.5 h and was continued at 100 M for another 1.5 h. As shown in Figure 2, PGE2 produced a significant elevation (p<0.05) in the extracellular concentration of glutamate in the hippocampus when compared to control animals; this was evident at the end of the 30 μM perfusion and during the 100 M perfusion. Results of the ANOVA revealed a significant main effect of drug treatment (F(1,87) = 15.18; p<0.01).
Figure 2. Extracellular glutamate concentrations in the hippocampus following reverse dialysis of PGE2.
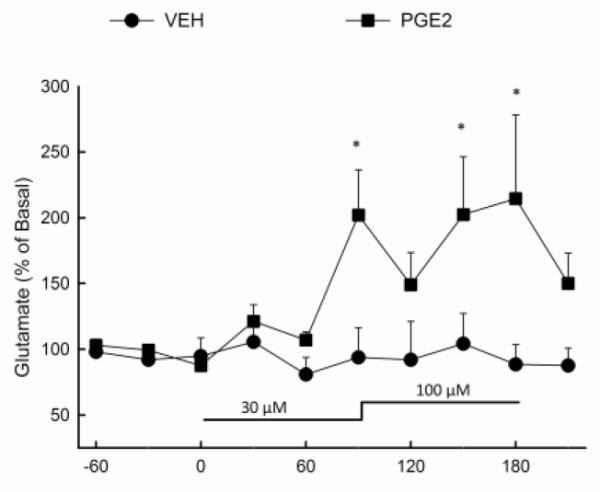
PGE2 (30μM) or vehicle was infused via the dialysis buffer into the hippocampus for a duration of 90 min after three 30 min baseline samples were taken. The concentration of PGE2 was then increased to 100μM for an additional 90 min. (n=5-8 per group) *Indicates values that differ significantly (p<0.05) from those animals that received vehicle.
Ketoprofen prevents the MDMA-induced reduction in parvalbumin-positive GABA neurons in the dentate gyrus
The effects of MDMA on a subpopulation of GABAergic neurons in the hippocampus was determined. Figure 3A,B illustrates the effect of MDMA on parvalbumin-immunoreactive GABA neurons in the dentate gyrus. Repeated treatment with MDMA (Panel B) resulted in a significant (p<0.05) reduction of approximately 36% in pavralbumin-positive GABA neurons in the dentate gyrus compared to control animals (Panel A). There was no significant effect of MDMA on these cells in the CA1 or CA3 regions (data not shown). Notably, the MDMA-induced reduction in parvalbumin-immunoreactive GABA neurons was not evident in rats treated with ketoprofen (Figure 3C). The number of parvalbumin-positive neurons in the dentate gyrus of rats treated with ketoprofen + MDMA was significantly (p<0.05) greater than that in rats treated with vehicle + MDMA.
Figure 3. Effect of ketoprofen on the MDMA-induced reduction of parvalbumin-reactive GABAergic neurons in the rat hippocampus.

Panel A is a representative photomicrograph depicting parvalbumin-immunoreactive cells in the dentate gyrus in control animals, as highlighted by the arrows, while panel B is representative of the dentate gyrus of animals given repeated doses of MDMA. Panel C depicts the quantitative assessment of PV-ir neurons in the dentate gyrus of vehicle- and MDMA-treated rats. Rats received ketoprofen (5 mg/kg, s.c.) or vehicle 1 h prior to and 1 h and 3 h following the first of 4 injections of MDMA (10 mg/kg, i.p.) or vehicle. (n = 4-8 per group) * indicates values that differ significantly (p<0.05) from those for animals that received VEH-VEH. # indicates values that differ significantly (p<0.05) from those for animals that received VEH-MDMA.
Ketoprofen does not prevent the MDMA-induced depletion of 5-HT in the rat hippocampus
In order to ascertain whether inhibition of COX activity also would afford protection against the long-term reductions in biochemical markers of 5-HT axon terminals produced by MDMA, rats were treated with the previously described regimen of ketoprofen or vehicle, and concomitantly administered MDMA (2 × 10 mg/kg, i.p.) or vehicle. Concentrations of 5-HT in the hippocampus were determined 7 days following MDMA treatment. The hippocampal concentration of 5-HT was depleted by approximately 25% (p<0.05) in rats treated with MDMA, and there was no significant difference in the depletion of hippocampal 5-HT produced by MDMA in vehicle- and ketoprofen-treated animals (p>0.05). The values (ng/mg tissue) for hippocampal 5-HT were: vehicle-vehicle, 0.33 ± 0.02; ketoprofen-vehicle, 0.34 ± 0.02; vehicle-MDMA, 0.21 ± 0.02; ketoprofen-MDMA, 0.21 ± 0.01. (n=6-12)
Discussion
The key findings of the present study include the demonstration that 1) the MDMA-induced increase in the extracellular concentration of glutamate in the hippocampus is attenuated in rats treated with ketoprofen and nimesulide, but not piroxicam, 2) infusion of PGE2 produces a significant increase in the extracellular concentration of glutamate, 3) MDMA reduces the number of parvalbumin-positive neurons in the dentate gyrus of the hippocampus, an effect attenuated by ketoprofen and 4) ketoprofen does not attenuate the depletion of 5-HT in the hippocampus produced by MDMA.
We have previously reported that MDMA produces a delayed and sustained increase in the extracellular concentration of glutamate in the hippocampus, but not in the striatum or prefrontal cortex (Anneken and Gudelsky 2012). Moreover, fluoxetine and ketanserin, a 5-HT2A/C antagonist, were found to suppress the glutamate response to MDMA, and it was suggested that MDMA increases glutamate release in the hippocampus subsequent to an increased release of 5-HT and increased activation of 5-HT2A/C receptors (Anneken and Gudelsky 2012). The present study extends the previous findings to further investigate additional mechanisms and consequences of the increased release of hippocampal glutamate produced by MDMA.
In the present study, the MDMA-evoked increase in extracellular glutamate was suppressed by ketoprofen and nimesulide, but not piroxicam, which is supportive of the involvement of COX-2, rather than COX-1, in the mechanism of MDMA-induced glutamate release. Although the doses of COX inhibitors used in this study were based on those used previously by other investigators (Asanuma et al., 2003; Terao et al., 1998), we cannot exclude the possibility that these dosage regimens did not selectively and/or effectively inhibit the respective COX isoforms. Nevertheless, the present results are consistent with previous findings that an increase in COX-2 activity in hippocampal tissue results in an increased extracellular concentration of glutamate and exacerbation of glutamate-associated excitotoxicity (Bezzi et al., 1998; Kelley et al., 1999; Sang et al., 2011). Moreover, reverse dialysis of a major product of COX activity, PGE2, also produced a significant increase in extracellular glutamate in the hippocampus. To our knowledge, this is the first in vivo demonstration of glutamate release evoked by PGE2 exposure. This finding is in accord with data from in vitro studies (Bezzi et al., 1998; Sanzgiri et al., 1999) wherein it was demonstrated that multiple prostaglandins, including PGE2, evoke calcium-dependent glutamate release from astrocytes in brain tissue culture.
There are multiple signaling pathways through which MDMA may increase COX activity and the subsequent production of prostaglandins. MDMA may directly or indirectly activate 5-HT2A/C receptors to increase the release of arachidonic acid, thereby increasing COX activity and prostanoid formation. Indeed, activation of 5-HT2 receptors on glia has been shown to result in arachidonic acid release, presumably through the activation of phospholipase A2 (Garcia and Kim, 1997; Mackowiak et al., 2002). Furthermore, 5-HT2 receptor stimulation has been shown to increase glutamate release from astrocytes in a calcium dependent manner (Bezzi et al., 1998; Meller et al., 2002). These findings together with our report that MDMA-induced glutamate release is dependent upon 5-HT2 receptor activation and is independent of neuronal activity (Anneken and Gudelsky, 2012) support the view that the MDMA-induced increase in extracellular glutamate is the result of: 1) increased 5-HT release and a resulting 5-HT2A/C receptor-dependent activation of COX, 2) an increased formation of prostanoids and 3) a prostanoid-induced release of glutamate from astrocytes.
Until recently, there has been little evidence that MDMA produces persistent deficits in neurotransmitter systems beyond 5-HT axon terminals. Perrine et al. (2010) has demonstrated that repeated treatment with MDMA reduces the hippocampal concentration of GABA. Armstrong and Noguchi (2004) reported that chronic MDMA administration reduced the binding of [3H]-flunitrazapam in the hippocampus and concluded that GABAergic terminals or interneurons may be damaged by MDMA. In the present study, MDMA treatment resulted in a reduction in parvalbumin-positive GABA neurons in the dentate gyrus. No effect of MDMA was evident in the CA1 or CA3 regions of the hippocampus. Thus, it appears that repeated treatment with MDMA results in a persistent reduction in markers of GABAergic neurons. Future studies are needed to examine the effects of MDMA on other subpopulations of GABAergic cells such as calbindin- and calretinin-positive neurons.
Previous studies have indicated that MDMA produces neuronal degeneration in the hippocampus under in vivo and in vitro conditions (Meyer et al., 2004; Riezzo et al., 2010; Wang et al., 2009). MDMA treatment increases caspase-3 and TUNEL staining and promotes apoptotic cell death in cultured hippocampal neurons (Capela et al., 2012). On the basis of the results of the present study, it is tempting to propose that MDMA-induced cell death occurs in GABAergic neurons within the hippocampus.
In the present study, ketoprofen suppressed not only the MDMA-induced increase in glutamate release but also the MDMA-induced reduction in parvalbumin-positive GABA neurons in the hippocampus. Thus, COX activity and neuroinflammatory products also appear to contribute to the mechanism of MDMA-induced deficits in GABA neurons. In view of the inhibitory effect of ketoprofen on MDMA-stimulated glutamate release and the sensitivity of parvalbumin-positive GABA neurons to glutamate-mediated damage (Sanon et al., 2005), it is suggested that MDMA-induced damage to GABA neurons involves glutamate-mediated excitotoxicity. Additional studies are necessary to establish a causal relationship between these two phenomena.
Although inhibition of COX activity with ketoprofen greatly attenuated the MDMA-induced release of glutamate, it did not prevent the long term depletion of 5-HT in the hippocampus following repeated MDMA treatment. Thus, it appears that neither neuroinflammatory mediators nor glutamate excitotoxicity is a contributing factor in MDMA-induced 5-HT depletion. This finding is in agreement with earlier work by Farfel and colleagues (1995) who reported MDMA-induced 5-HT depletion to be insensitive to NMDA receptor antagonists under hyperthermic conditions.
Repeated exposure of rats to MDMA has been to shown to produce deficits in reference memory (Able et al., 2006; Asi et al., 2011; Cunningham et al., 2009; Kay et al., 2011; Skelton et al., 2006), a function subserved by the hippocampus, and in particular the dentate gyrus (Morris et al., 2012; Okada and Okaichi 2009). Moreover, human abusers of MDMA exhibit deficits in verbal memory (Burgess et al., 2011; de Sola Llopis et al., 2008; Raj et al., 2010), and altered function of GABA interneurons in the hippocampus (Jacobsen et al., 2004). Further studies are warranted to investigate the hypothesis that MDMA-induced impairment of GABA interneurons in the hippocampus underlies the cognitive impairments associated with repeated exposure to MDMA.
In summary, the data are consistent with the hypothesis that MDMA produces damage to GABA neurons within the hippocampus through a mechanism involving enhanced formation of neuroinflammatory mediators and a subsequent increase in glutamate release.
Acknowledgements
This work was supported by awards from the National Institute on Drug Abuse DA 07427 (GG) and DA016886 (BY).
Footnotes
Conflict of Interest Statement:
The authors declare no conflicts of interest.
References
- Able JA, Gudelsky GA, Vorhees CV, Williams MT. 3,4-methylenedioxymethamphetamine in adult rats produces deficits in path integration and spatial reference memory. Biol Psychiatry. 2006;59:1219–1226. doi: 10.1016/j.biopsych.2005.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anneken JH, Gudelsky GA. MDMA produces a delayed and sustained increase in the extracellular concentration of glutamate in the rat hippocampus. Neuropharmacology. 2012 doi: 10.1016/j.neuropharm.2012.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong BD, Noguchi KK. The neurotoxic effects of 3,4-methylenedioxymethamphetamine (MDMA) and methamphetamine on serotonin, dopamine, and GABA-ergic terminals: An in-vitro autoradiographic study in rats. Neurotoxicology. 2004;25:905–914. doi: 10.1016/j.neuro.2004.06.003. [DOI] [PubMed] [Google Scholar]
- Asanuma M, Tsuji T, Miyazaki I, Miyoshi K, Ogawa N. Methamphetamine-induced neurotoxicity in mouse brain is attenuated by ketoprofen, a non-steroidal anti-inflammatory drug. Neurosci Lett. 2003;352:13–16. doi: 10.1016/j.neulet.2003.08.015. [DOI] [PubMed] [Google Scholar]
- Asi SS, Farhadi HM, Mousavizadeh K, Samadikuchaksaraei A, Soleimani M, Jameie SB, Joghataei MT, Samzadeh-Kermani A, Hashemi-Nasl H, Mehdizadeh M. Evaluation of Bcl-2 Family Gene Expression in Hippocampus of 3,4-methylenedioxymethamphetamine Treated Rats. Cell Journal. 2012 2012 Winter;13(4) [PMC free article] [PubMed] [Google Scholar]
- Asi SS, Farhadi HM, Naghdi N, Choopani S, Samzadeh-Kermani A, Mehdizadeh M. Non-acute effects of different doses of 3,4-methylenedioxymethamphetamine on spatial memory in the morris water maze in sprague-dawley male rats. Neural Regeneration Research. 2011;6:1715. [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A. Prostaglandins stimulate calcium-dependent glutamate release in astrocytes. Nature. 1998;391:281–285. doi: 10.1038/34651. [DOI] [PubMed] [Google Scholar]
- Burgess AP, Venables L, Jones H, Edwards R, Parrott AC. Event related potential (ERP) evidence for selective impairment of verbal recollection in abstinent recreational methylenedioxymethamphetamine (“ecstasy”)/polydrug users. Psychopharmacology (Berl) 2011;216:545–556. doi: 10.1007/s00213-011-2249-9. [DOI] [PubMed] [Google Scholar]
- Candelario-Jalil E, Gonzalez-Falcon A, Garcia-Cabrera M, Leon OS, Fiebich BL. Wide therapeutic time window for nimesulide neuroprotection in a model of transient focal cerebral ischemia in the rat. Brain Res. 2004;1007:98–108. doi: 10.1016/j.brainres.2004.01.078. [DOI] [PubMed] [Google Scholar]
- Capela JP, da Costa Araujo S, Costa VM, Ruscher K, Fernandes E, Bastos MD, Dirnagl U, Meisel A, Carvalho F. The neurotoxicity of hallucinogenic amphetamines in primary cultures of hippocampal neurons. Neurotoxicology. 2012 doi: 10.1016/j.neuro.2012.09.005. in press. [DOI] [PubMed] [Google Scholar]
- Capela JP, Fernandes E, Remiao F, Bastos ML, Meisel A, Carvalho F. Ecstasy induces apoptosis via 5-HT(2A)-receptor stimulation in cortical neurons. Neurotoxicology. 2007;28:868–875. doi: 10.1016/j.neuro.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Cunningham JI, Raudensky J, Tonkiss J, Yamamoto BK. MDMA pretreatment leads to mild chronic unpredictable stress-induced impairments in spatial learning. Behav Neurosci. 2009;123:1076–1084. doi: 10.1037/a0016716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czeh B, Simon M, van der Hart MG, Schmelting B, Hesselink MB, Fuchs E. Chronic stress decreases the number of parvalbumin-immunoreactive interneurons in the hippocampus: Prevention by treatment with a substance P receptor (NK1) antagonist. Neuropsychopharmacology. 2005;30:67–79. doi: 10.1038/sj.npp.1300581. [DOI] [PubMed] [Google Scholar]
- de Sola Llopis S, Miguelez-Pan M, Pena-Casanova J, Poudevida S, Farre M, Pacifici R, Bohm P, Abanades S, Verdejo Garcia A, Langohr K, Zuccaro P, de la Torre R. Cognitive performance in recreational ecstasy polydrug users: A two-year follow-up study. J Psychopharmacol. 2008;22:498–510. doi: 10.1177/0269881107081545. [DOI] [PubMed] [Google Scholar]
- Donzanti BA, Yamamoto BK. An improved and rapid HPLC-EC method for the isocratic separation of amino acid neurotransmitters from brain tissue and microdialysis perfusates. Life Sci. 1988;43:913–922. doi: 10.1016/0024-3205(88)90267-6. [DOI] [PubMed] [Google Scholar]
- Farfel GM, Seiden LS. Role of hypothermia in the mechanism of protection against serotonergic toxicity. I. experiments using 3,4-methylenedioxymethamphetamine, dizocilpine, CGS 19755 and NBQX. J Pharmacol Exp Ther. 1995;272:860–867. [PubMed] [Google Scholar]
- Garcia MC, Kim HY. Mobilization of arachidonate and docosahexaenoate by stimulation of the 5-HT2A receptor in rat C6 glioma cells. Brain Res. 1997;768:43–48. doi: 10.1016/s0006-8993(97)00583-0. [DOI] [PubMed] [Google Scholar]
- Green AR, Mechan AO, Elliott JM, O’Shea E, Colado MI. The pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA, “ecstasy”) Pharmacol Rev. 2003;55:463–508. doi: 10.1124/pr.55.3.3. [DOI] [PubMed] [Google Scholar]
- Gudelsky GA, Yamamoto BK. Neuropharmacology and neurotoxicity of 3,4-methylenedioxymethamphetamine. Methods Mol Med. 2003;79:55–73. doi: 10.1385/1-59259-358-5:55. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen EB, Kieu K, Nielsen J. The efficiency of systematic sampling in stereology--reconsidered. J Microsc. 1999;193:199–211. doi: 10.1046/j.1365-2818.1999.00457.x. [DOI] [PubMed] [Google Scholar]
- Jacobsen LK, Mencl WE, Pugh KR, Skudlarski P, Krystal JH. Preliminary evidence of hippocampal dysfunction in adolescent MDMA (“ecstasy”) users: Possible relationship to neurotoxic effects. Psychopharmacology (Berl) 2004;173:383–390. doi: 10.1007/s00213-003-1679-4. [DOI] [PubMed] [Google Scholar]
- Kay C, Harper DN, Hunt M. The effects of binge MDMA on acquisition and reversal learning in a radial-arm maze task. Neurobiol Learn Mem. 2011;95:473–483. doi: 10.1016/j.nlm.2011.02.010. [DOI] [PubMed] [Google Scholar]
- Kelley KA, Ho L, Winger D, Freire-Moar J, Borelli CB, Aisen PS, Pasinetti GM. Potentiation of excitotoxicity in transgenic mice overexpressing neuronal cyclooxygenase-2. Am J Pathol. 1999;155:995–1004. doi: 10.1016/S0002-9440(10)65199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kermanian F, Mehdizadeh M, Soleimani M, Ebrahimzadeh Bideskan AR, Asadi-Shekaari M, Kheradmand H, Haghir H. The role of adenosine receptor agonist and antagonist on hippocampal MDMA detrimental effects; a structural and behavioral study. Metab Brain Dis. 2012 doi: 10.1007/s11011-012-9334-6. [DOI] [PubMed] [Google Scholar]
- Kerner JA, Standaert DG, Penney JB, Jr, Young AB, Landwehrmeyer GB. Expression of group one metabotropic glutamate receptor subunit mRNAs in neurochemically identified neurons in the rat neostriatum, neocortex, and hippocampus. Brain Res Mol Brain Res. 1997;48:259–269. doi: 10.1016/s0169-328x(97)00102-2. [DOI] [PubMed] [Google Scholar]
- Lister JP, Tonkiss J, Blatt GJ, Kemper TL, DeBassio WA, Galler JR, Rosene DL. Asymmetry of neuron numbers in the hippocampal formation of prenatally malnourished and normally nourished rats: A stereological investigation. Hippocampus. 2006;16:946–958. doi: 10.1002/hipo.20221. [DOI] [PubMed] [Google Scholar]
- Mackowiak M, Chocyk A, Sanak M, Czyrak A, Fijal K, Wedzony K. DOI, an agonist of 5-HT2A/2C serotonin receptor, alters the expression of cyclooxygenase-2 in the rat parietal cortex. J Physiol Pharmacol. 2002;53:395–407. [PubMed] [Google Scholar]
- Meller R, Harrison PJ, Elliott JM, Sharp T. In vitro evidence that 5-hydroxytryptamine increases efflux of glial glutamate via 5-HT(2A) receptor activation. J Neurosci Res. 2002;67:399–405. doi: 10.1002/jnr.10126. [DOI] [PubMed] [Google Scholar]
- Meyer JS, Grande M, Johnson K, Ali SF. Neurotoxic effects of MDMA (“ecstasy”) administration to neonatal rats. Int J Dev Neurosci. 2004;22:261–271. doi: 10.1016/j.ijdevneu.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Moga DE, Janssen WG, Vissavajjhala P, Czelusniak SM, Moran TM, Hof PR, Morrison JH. Glutamate receptor subunit 3 (GluR3) immunoreactivity delineates a subpopulation of parvalbumin-containing interneurons in the rat hippocampus. J Comp Neurol. 2003;462:15–28. doi: 10.1002/cne.10710. [DOI] [PubMed] [Google Scholar]
- Morris AM, Churchwell JC, Kesner RP, Gilbert PE. Selective lesions of the dentate gyrus produce disruptions in place learning for adjacent spatial locations. Neurobiol Learn Mem. 2012;97:326–331. doi: 10.1016/j.nlm.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller GJ, Moller A, Johansen FF. Stereological cell counts of GABAergic neurons in rat dentate hilus following transient cerebral ischemia. Exp Brain Res. 2001;141:380–388. doi: 10.1007/s002210100879. [DOI] [PubMed] [Google Scholar]
- Okada K, Okaichi H. Functional differentiation and cooperation among the hippocampal subregions in rats to effect spatial memory processes. Behav Brain Res. 2009;200:181–191. doi: 10.1016/j.bbr.2009.01.011. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 4th Ed Academic Press; San Diego, CA: 1998. [Google Scholar]
- Perrine SA, Ghoddoussi F, Michaels MS, Hyde EM, Kuhn DM, Galloway MP. MDMA administration decreases serotonin but not N-acetylaspartate in the rat brain. Neurotoxicology. 2010;31:654–661. doi: 10.1016/j.neuro.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj V, Liang HC, Woodward ND, Bauernfeind AL, Lee J, Dietrich MS, Park S, Cowan RL. MDMA (ecstasy) use is associated with reduced BOLD signal change during semantic recognition in abstinent human polydrug users: A preliminary fMRI study. J Psychopharmacol. 2010;24:187–201. doi: 10.1177/0269881109103203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riezzo I, Cerretani D, Fiore C, Bello S, Centini F, D’Errico S, Fiaschi AI, Giorgi G, Neri M, Pomara C, Turillazzi E, Fineschi V. Enzymatic-nonenzymatic cellular antioxidant defense systems response and immunohistochemical detection of MDMA, VMAT2, HSP70, and apoptosis as biomarkers for MDMA (ecstasy) neurotoxicity. J Neurosci Res. 2010;88:905–916. doi: 10.1002/jnr.22245. [DOI] [PubMed] [Google Scholar]
- Sang N, Yun Y, Yao GY, Li HY, Guo L, Li GK. SO(2)-induced neurotoxicity is mediated by cyclooxygenases-2-derived prostaglandin E(2) and its downstream signaling pathway in rat hippocampal neurons. Toxicol Sci. 2011;124:400–413. doi: 10.1093/toxsci/kfr224. [DOI] [PubMed] [Google Scholar]
- Sanon N, Carmant L, Emond M, Congar P, Lacaille JC. Short-term effects of kainic acid on CA1 hippocampal interneurons differentially vulnerable to excitotoxicity. Epilepsia. 2005;46:837–848. doi: 10.1111/j.1528-1167.2005.21404.x. [DOI] [PubMed] [Google Scholar]
- Sanzgiri RP, Araque A, Haydon PG. Prostaglandin E(2) stimulates glutamate receptor-dependent astrocyte neuromodulation in cultured hippocampal cells. J Neurobiol. 1999;41:221–229. [PubMed] [Google Scholar]
- Schmued LC. Demonstration and localization of neuronal degeneration in the rat forebrain following a single exposure to MDMA. Brain Res. 2003;974:127–133. doi: 10.1016/s0006-8993(03)02563-0. [DOI] [PubMed] [Google Scholar]
- Shankaran M, Yamamoto BK, Gudelsky GA. Ascorbic acid prevents 3,4-methylenedioxymethamphetamine (MDMA)-induced hydroxyl radical formation and the behavioral and neurochemical consequences of the depletion of brain 5-HT. Synapse. 2001;40:55–64. doi: 10.1002/1098-2396(200104)40:1<55::AID-SYN1026>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Skelton MR, Williams MT, Vorhees CV. Treatment with MDMA from P11-20 disrupts spatial learning and path integration learning in adolescent rats but only spatial learning in older rats. Psychopharmacology (Berl) 2006;189:307–318. doi: 10.1007/s00213-006-0563-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terao A, Matsumura H, Saito M. Interleukin-1 induces slow-wave sleep at the prostaglandin D2-sensitive sleep-promoting zone in the rat brain. J Neurosci. 1998;18:6599–6607. doi: 10.1523/JNEUROSCI.18-16-06599.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhu SP, Kuang WH, Li J, Sun X, Huang MS, Sun XL. Neuron apoptosis induced by 3,4-methylenedioxy methamphetamine and expression of apoptosis-related factors in rat brain. Sichuan Da Xue Xue Bao Yi Xue Ban. 2009;40:1000–2. 1037. [PubMed] [Google Scholar]
- Warren MW, Larner SF, Kobeissy FH, Brezing CA, Jeung JA, Hayes RL, Gold MS, Wang KK. Calpain and caspase proteolytic markers co-localize with rat cortical neurons after exposure to methamphetamine and MDMA. Acta Neuropathol. 2007;114:277–286. doi: 10.1007/s00401-007-0259-9. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Xie T, Tong L, McLane MW, Hatzidimitriou G, Yuan J, McCann U, Ricaurte G. Loss of serotonin transporter protein after MDMA and other ring-substituted amphetamines. Neuropsychopharmacology. 2006;31:2639–2651. doi: 10.1038/sj.npp.1301031. [DOI] [PubMed] [Google Scholar]