

Myofibroblasts play a critical role in pathogenesis of liver fibrosis. Hepatic fibrosis results from many chronic liver diseases, including hepatitis B virus (HBV), hepatitis C virus (HCV), alcoholic liver disease and non-alcoholic steatohepatitis (NASH).1 Hepatic fibrosis is the scar caused by deregulation of physiological wound healing and results in excessive production of extracellular matrix (ECM), mostly collagen type I. Myofibroblasts, which are not present in normal liver, are the major source of the ECM during fibrogenesis. Activation and proliferation of hepatic myofibroblasts are required for the development of fibrosis.1 Myofibroblasts are characterized by stellate shape, expression of abundant intracellular filaments [α-smooth muscle actin (α-SMA), vimentin], high contractility and secretion of ECM (fibronectin, collagen type I and III).2 Hepatic stellate cells (HSCs) are the major source of myofibroblasts in hepatotoxic liver fibrosis.1,3 Under physiological conditions, HSCs reside in the space of Disse and exhibit a quiescent phenotype (qHSCs). They express neural markers, such as GFAP, synemin, synaptophysin and nerve growth factor receptor p75, and store vitamin A in lipid droplets.4 In response to injury, qHSCs decrease vitamin A storage and peroxisome proliferator-activated receptor gamma (PPARγ) expression and activate into collagen type I- and α-SMA-expressing myofibroblasts.1 Although the mechanism of HSC activation has been comprehensively studied, insights into the fate of HSCs during regression of liver fibrosis are new.
Reversibility of liver fibrosis. Many studies have clearly demonstrated that hepatic fibrosis is reversible in patients (e.g., HBV, HCV, biliary obstruction or alcohol) and in experimental rodent models (alcohol feeding, CCl4 or bile duct ligation).1 Upon removal of the etiological source of the chronic injury, regression of liver fibrosis is associated with decreased cytokines and ECM production, increased collagenase activity, disappearance of myofibroblasts and dissolution of the fibrous scar.1,5 Only recently has the fate of these myofibroblasts been revealed. The previous concept was that the myofibroblasts undergo apoptosis on the basis of the documented senescence and apoptosis of some aHSCs during reversal of fibrosis. We,6 and subsequently others,7 have used genetic marking to demonstrate an alternative pathway in which myofibroblasts revert to a quiescent-like phenotype in CCl4-induced liver injury and experimental alcoholic liver disease. The fate of these genetically marked myofibroblasts is quantitated in experimental models of fibrosis and its reversal.
Inactivation of HSCs (iHSCs). Using Cre-LoxP-based genetic labeling of myofibroblasts, we elucidated the fate of activated (a)HSCs/myofibroblasts during recovery from CCl4-induced liver fibrosis. Genetic labeling of aHSC/myofibroblasts results from crossing mice expressing Cre under control of the collagen-α1(I) enhancer/promoter (Colα1(I)Cre mice) with reporter mice (Rosa26f/f-YFP mice) ubiquitously expressing a yfp gene in which transcription is blocked by a floxed Stop cassette.6 In the offspring (Colα1(I)Cre-YFP mice), genetic Cre-lox recombination causes excision of the floxP-Stop-floxP sequence from genomic DNA and activation of YFP transcription in cells expressing type I collagen. Following induction of liver injury in these mice, aHSCs and their progeny are permanently labeled by YFP expression even if the cells undergo a phenotypic change.6 Phenotypical changes of aHSCs and the mechanism of their inactivation can now be studied during regression of liver fibrosis. Using two models of hepatotoxic-induced liver fibrosis (such as carbon-tertrachloride (CCl4) or intragastric alcohol feeding), we demonstrated that half of the myofibroblasts escape apoptosis during regression of liver fibrosis, downregulate fibrogenic genes (Col1a1, Col1a2, αSMA, TIMP1, TGFβ-RI) and acquire a phenotype similar to, but distinct from, quiescent HSCs.6
Inactivated HSCs acquire a novel phenotype which has not been previously described and is now referred as iHSCs. In particular, iHSCs more rapidly reactivate into myofibroblasts in response to fibrogenic stimuli and more effectively contribute to liver fibrosis. Inactivation of HSCs is associated with re-expression of lipogenic genes PPAR-γ, Insig1 and CREBP.8 Our findings in mice in vivo support previous in vitro studies demonstrating the importance of PPAR-γ for maintaining and re-establishing the quiescent phenotype (qHSCs).8,9
While PPAR-γ drives HSC inactivation during reversal from liver fibrosis, Hspa1a/b, two members of Hsp70 family of heat shock proteins implicated in protection from apoptosis,10 appear to regulate HSC survival. Genetic ablation of Hspa1a/b renders aHSCs more susceptible to TNF-α and glyotoxin-induced apoptosis in culture, while deletion of Hspa1a/b in mice results in increased disappearance of α-SMA+Desmin+ HSCs and accelerates regression of liver fibrosis.
Future prospectives. Inactivation of HSCs is a newly described phenomenon6 that now requires mechanistic investigation. Inactivation of aHSCs is associated with regression of liver fibrosis and is characterized by inhibition of fibrogenic gene (Col1a1, αSMA) expression and upregulation of adipogenic (PPARγ, Insig1, CEBPd) genes. Understanding of the mechanism of HSC inactivation upon cessation of fibrogenic stimuli may identify new approaches to revert already existing aHSCs/myofibroblasts into a quiescent-like state. Since reversibility of fibrosis has been reported in lungs and kidneys, these concepts and approaches may be applicable to study fibrosis of other organs and provide new targets for anti-fibrotic therapy.
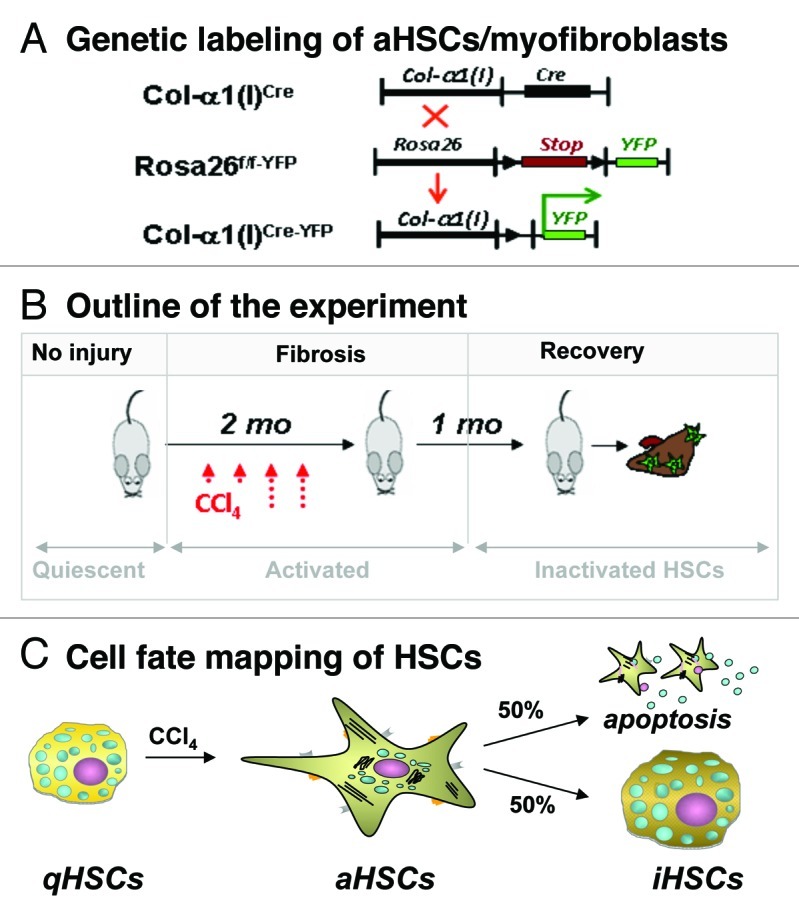
Figure 1. Study design to determine the cell fate of aHSCs during regression of CCl4-induced liver fibrosis. (A) Cre-loxP-based genetic labeling marks the fate of Col-α1(I)-expressing aHSCs/ myofibroblasts in Col-α1(I)Cre-YFP mice generated by crossing Col-α1(I)Cre and Rosa26f/f-YFP mice. (B) Col-α1(I)Cre-YFP mice were subjected to CCl4-induced liver injury (1.5 mo) then recuperated upon cessation of injuring agent (for 1 mo). Mice were sacrificed, and livers were analyzed for the presence of Vitamin A+ YFP+ and Vitamin A+ YFP- HSCs. (C) CCl4 induces qHSC activation into aHSCs/myofibroblasts in Col-α1(I)Cre-YFP mice. After CCl4 withdrawal, some aHSCs apoptose, while some inactivate (YFP+ iHSCs number < 100% of aHSCs).6
Glossary
Abbreviations:
- HSCs
hepatic stellate cells
- qHSCs
quiescent HSCs
- aHSCs
activated HSCs
- iHSCs
inactivated HSCs
- CCl4
carbon tetrachloride
- α-SMA
α-smooth muscle actin
- Col-α2(I)
Collagen-α2(I)
- Col-α1(I)
Collagen-α1(I)
- Col-α1(1)Cre-YFP mice
(Col-α1(I)Cre mice x Rosa26flox-Stop-flox-YFP mice)
- Hspa1a/b
heat shock proteins 1a/b
- PPARγ
peroxisome proliferator-activated receptor gamma
- Adfp
adipose differentiation relate protein
- Adfp
adipose differentiation relate protein
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/23549
References
- 1.Bataller R, et al. J Clin Invest. 2005;115:209–18. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parola M, et al. Mol Aspects Med. 2008;29:58–66. doi: 10.1016/j.mam.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 3.Friedman SL, et al. Proc Natl Acad Sci USA. 1985;82:8681–5. doi: 10.1073/pnas.82.24.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kisseleva T, et al. J Gastroenterol Hepatol. 2006;21(Suppl 3):S84–7. doi: 10.1111/j.1440-1746.2006.04584.x. [DOI] [PubMed] [Google Scholar]
- 5.Iredale JP, et al. J Clin Invest. 1998;102:538–49. doi: 10.1172/JCI1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kisseleva T, et al. Proc Natl Acad Sci U S A. 2012;109:9448–53. doi: 10.1073/pnas.1201840109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Troeger JS, et al. Gastroenterology. 2012;143:1073–83, e22. doi: 10.1053/j.gastro.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.She H, et al. J Biol Chem. 2005;280:4959–67. doi: 10.1074/jbc.M410078200. [DOI] [PubMed] [Google Scholar]
- 9.Tsukamoto H. Alcohol Clin Exp Res. 2005;29(Suppl):132S–3S. doi: 10.1097/01.alc.0000189279.92602.f0. [DOI] [PubMed] [Google Scholar]
- 10.Yenari MA, et al. Antiapoptotic and anti-inflammatory mechanisms of heat-shock protein protection. Ann NY Acad Sci. 2005;1053:74–83. doi: 10.1196/annals.1344.007. [DOI] [PubMed] [Google Scholar]