

Abstract
Vitamin B6 metabolism influences the adaptive response of non-small lung carcinoma (NSCLC) cells to distinct, potentially lethal perturbations in homeostasis, encompassing nutrient deprivation, hyperthermia, hypoxia, irradiation as well as the exposure to cytotoxic chemicals, including the DNA-damaging agent cisplatin (CDDP). Thus, the siRNA-mediated downregulation of pyridoxal kinase (PDXK), the enzyme that generates the bioactive form of vitamin B6, protects NSCLC cells (as well as a large collection of human and murine malignant cells of distinct histological derivation) from the cytotoxic effects of CDDP. Accordingly, the administration of pyridoxine, one of the inactive precursors of vitamin B6, exacerbates cisplatin-induced cell death, in vitro and in vivo, but only when PDXK is expressed. Conversely, antioxidants such as non-oxidized glutathione (GSH) are known to protect cancer cells from CDDP toxicity. Pyridoxine increases the amount of CDDP-DNA adducts formed upon the exposure of NSCLC cells to CDDP and aggravates the consequent DNA damage response. On the contrary, in the presence of GSH, NSCLC cells exhibit near-to-undetectable levels of CDDP-DNA adducts and a small fraction of the cell population activates the DNA damage response. We therefore wondered whether vitamin B6 metabolism and GSH might interact with CDDP in a pharmacokinetic fashion. In this short communication, we demonstrate that GSH inhibits the intracellular accumulation of CDDP, while pyridoxine potentiates it in a PDXK-dependent fashion. Importantly, such pharmacokinetic effects do not involve plasma membrane transporters that mediate a prominent fraction of CDDP influx, i.e., solute carrier family 31, member 1 (SLC31A1, best known as copper transporter 1, CTR1) and efflux, i.e., ATPase, Cu2+ transporting, β polypeptide (ATP7B).
Keywords: A549, apoptosis, N-acetyl-cysteine, PDXP, reactive oxygen species, Wilson disease
Introduction
Patients affected by a large panel of solid tumors, including head and neck, lung, bladder, colorectal, prostate, ovarian and germ cell cancers, are routinely treated with cis-diammineplatinum (II) dichloride (CDDP), best known as cisplatin, a platinum-containing chemical first approved by the Food and Drug Administration for use in cancer patients in 1978.1 CDDP is highly efficient against testicular germ cell cancer, leading to long-term complete remission in > 80% of the patients.2 However, in other clinical settings, including colorectal, lung and prostate carcinoma, the therapeutic efficacy of CDDP is limited, owing to the facts that (1) a significant fraction of tumors is intrinsically resistant to therapy, and (2) a consistent percentage of initially sensitive neoplasms acquires chemoresistance, relapses and provokes the death of the patient.3,4 Thus, innate or acquired chemoresistance constitutes by far the most prominent obstacle to the clinical use of CDDP. In addition, the administration of CDDP has been associated with various adverse effects, including grade I–II nephrotoxicity (in > 30% of patients), neurotoxicity and ototoxicity (both in 10–30% of patients).1
Upon aquation, i.e., the incorporation of one molecule of water, intracellular CDDP mediates antineoplastic effects via at least two distinct mechanisms. First, CDDP causes the formation of intra- and inter-strand DNA adducts, resulting in the activation of a DNA damage response that ultimately promotes senescence or cell death, mostly via apoptosis.5-8 Second, CDDP can engage poorly characterized cytoplasmic pathways that promote apoptotic or necrotic cell death responses characterized by the overproduction of reactive oxygen species (ROS) and, at least in some instances, the activation of pro-apoptotic proteins, including the BCL-2 family members BAX and BAK as well as voltage-dependent anion channel 1 (VDAC1).9-13 The relative contribution of the nuclear and cytoplasmic signaling modules activated by CDDP to its cytotoxicity remains elusive and may be highly context-dependent.
Since the approval of CDDP as an antineoplastic agent for use in humans, the signaling pathways by which malignant cells elude the cytotoxic effects of CDDP have been extensively investigated. Although a comprehensive description of these mechanisms goes beyond the scope of this introduction, it should be noted that (1) malignant cells can elude CDDP cytotoxicity by at least four distinct types of mechanisms (i.e., pre-target, on-target, post-target and off-target resistance), and (2) in most, if not all, settings, CDDP resistance is multifactorial, meaning that it results from the concomitant activation of non-overlapping molecular mechanisms.3,14,15 The pleiotropic nature of the mechanisms that promote CDDP resistance complicates the development of clinically meaningful strategies of chemosensitization.
During the past few years, we have engaged in a relatively comprehensive system biology study to investigate CDDP-elicited signaling pathways in non-small cell lung carcinoma (NSCLC), a common and aggressive subtype of lung cancer that provokes more than 1 million deaths worldwide annually.16,17 This approach, which involved multiple in vitro and in vivo models, has allowed us to identify several CDDP response modifiers (CRMs), i.e., factors that inhibit or exacerbate the response of NSCLC cells to CDDP.18-21 Among these, a central position is occupied by the metabolism of vitamin B6, which appears to be involved not only in the cytotoxic response of a large collection of cancer cell lines to CDDP, but also in the adaptation of NSCLC cells to various perturbations in homeostasis, including nutrient deprivation, heat shock, hypoxia, irradiation as well as the exposure to multiple cytotoxic agents.19 Accordingly, high expression levels of pyridoxal kinase (PDXK), the enzyme that converts inactive precursors into the bioactive form of vitamin B6, i.e., pyridoxal-5-phosphate, have been associated with improved disease outcome in two independent cohorts of NSCLC patients.19
In this short communication, we demonstrate that vitamin B6 metabolism, as well as non-oxidized glutathione (GSH, a well-known inhibitor of CDDP-induced cell death) influence the cytotoxic response of NSCLC A549 cells to CDDP as they aggravate and reduce, respectively, its cellular uptake. Importantly, these pharmacokinetic effects do not involve the transmembrane proteins that, at least according to current knowledge, transport the largest fraction of CDDP across the plasma membrane.
Results and Discussion
Vitamin B6 metabolism and GSH influence the uptake of CDDP
We and others have previously shown that pyridoxine exacerbates the cytotoxic effects of CDDP in a PDXK-dependent fashion, whereas antioxidants such as GSH and N-acetyl-cysteine (NAC) mediate striking cytoprotective effects.19,22-24 Pyridoxine increases the number of CDDP-DNA adducts that form upon exposure of NSCLC A549 cells to CDDP and aggravates the consequent DNA damage response,19 manifesting with the activating phosphorylation of ataxia telangiectasia mutated (ATM) on Ser 1981,25 the phosphorylation of histone 2AX on Ser 139,26 as well as the activating phosphorylation of p53 on both Ser 15 and Ser 46.27 Conversely, when CDDP is administered to A549 cells together with GSH or NAC, CDDP-DNA adducts are barely detectable, irrespective of the presence of pyridoxine, and a limited fraction (< 25%) of the cell population exhibits markers of the DNA damage response.19
Based on these premises, we decided to investigate whether the effects of pyridoxine and antioxidants on the cytotoxic response of A549 to CDDP may be due, at least in part, to a pharmacokinetic interaction. To this aim, we pre-treated A549 cells with pyridoxine or GSH for 1 h, and then exposed them to a suboptimal dose of CDDP (25 μM) for additional 24 h. Then, intracellular CDDP accumulation was determined by flameless atomic absorption spectrometry, and results were normalized to total protein content. In the presence of pyridoxine, A549 cells accumulated ~30% more CDDP than in control conditions, whereas GSH reduced intracellular CDDP accumulation by ~60%, irrespective of the simultaneous presence of pyridoxine (Fig. 1A). In addition, A549 cells depleted of PDXK by means of a validated siRNA not only accumulated 15–20% less CDDP than A549 cells transfected with a control, non-targeting siRNA, but also were entirely insensitive to the pharmacokinetic effects of pyridoxine on intracellular CDDP concentrations (Fig. 1B).
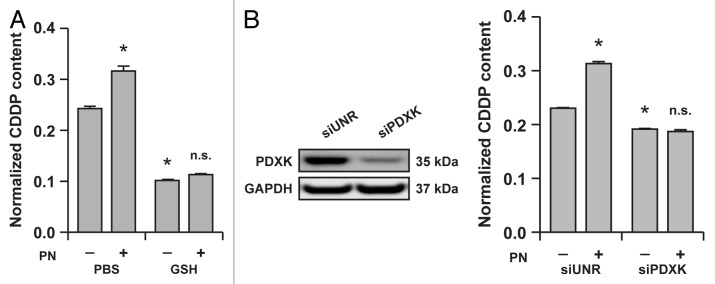
Figure 1. Reduced glutathione (GSH) and the vitamin B6 metabolism influence the intracellular accumulation of cisplatin (CDDP) in A549 cells. (A) Intracellular levels of CDDP (normalized to protein content) in lysates from A549 cells treated for 24 h with 25 μM CDDP alone (in the presence of a control amount of PBS) or combined with 5 mM GSH and/or 5 mM pyridoxine (PN). Means ± SEM (n = 6). *p < 0.05 (Student’s t-test), as compared to cells treated with CDDP only; n.s. = non-significant (Student’s t-test), as compared to cells treated with CDDP plus GSH. (B) Intracellular levels of CDDP (normalized to protein content) in lysates from A549 transfected with a control siRNA (siUNR) or with a siRNA specific for pyridoxal kinase (siPDXK) and then treated for 24 h with 25 μM CDDP alone (in the presence of a control amount of PBS) or combined with 5 mM PN. Means ± SEM (n = 6). *p < 0.05 (Student’s t-test), as compared to siUNR-transfected cells treated with CDDP only; n.s. = non-significant (Student’s t-test), as compared to siPDXK-transfected cells treated with CDDP only. Immunoblots in (B) depict the efficacy of siRNA-mediated PDXK downregulation. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels were monitored to ensure the equal loading of lanes.
Taken together, these findings demonstrate that GSH limits the intracellular accumulation of CDDP, while pyridoxine aggravates it in a PDXK-dependent fashion.
The pharmacokinetic effects of vitamin B6 and GSH on CDDP accumulation are not mediated by CTR1 and ATP7B
Until the late 1990s, CDDP was assumed to access the intracellular compartment mainly by passive diffusion across the plasma membrane, presumably because the uptake of CDDP (a highly polar chemical) is relatively slow as compared to that of chemically analogous antineoplastic agents that are actively transported.3,28 However, this notion has been progressively invalidated starting from the early 2000s, and it is now clear that a prominent fraction of CDDP is actively transported across the plasma membrane.3 In particular, it has been demonstrated that both the influx and efflux of CDDP are mediated, for a large fraction, by plasma membrane transporters that normally carry Cu2+ ions, namely solute carrier family 31, member 1 (SLC31A1, best known as copper transporter 1, CTR1)29-31 and ATPase, Cu2+ transporting, β polypeptide (ATP7B).32-34
Prompted by these observations, we investigated whether the capacity of GSH and pyridoxine to limit and augment, respectively, the intracellular accumulation of CDDP would be mediated by SLC31A1/CTR1 or ATP7B. To this aim, A549 cells were transfected with custom-designed siRNAs specific for SLC31A1/CTR1 and ATP7B for 48 h and then subjected to the same experimental maneuvers described above. In line with previous findings,29,31,33,35 the knockdown of SLC31A1/CTR1 by two non-overlapping siRNAs significantly inhibited the accumulation of CDDP by A549 cells. In addition, A549 cells transfected with two non-overlapping siRNAs targeting ATP7B accumulated approximately 20% more CDDP than cells of the same type receiving a control siRNA (Fig. 2A and B). Importantly, both GSH and pyridoxine preserved their capacity to modulate the intracellular accumulation of CDDP, even in A549 cells depleted of SLC31A1/CTR1 or ATP7B (Fig. 2A and B).
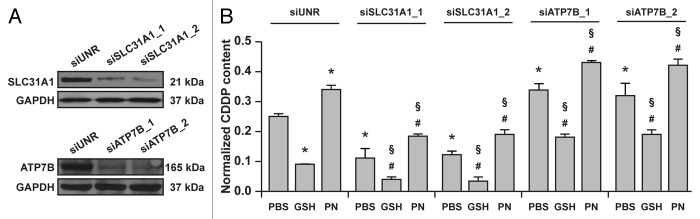
Figure 2. Vitamin B6 exacerbates the intracellular accumulation of cisplatin (CDDP) independent of ATP7B and SLC31A1. (A) Immunoblots depicting the efficacy of siRNA-mediated ATP7B and SLC31A1 downregulation. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels were checked to monitor equal lane loading. (B) Intracellular levels of CDDP (normalized to protein content) in lysates from A549 transfected with a control siRNA (siUNR) or with siRNAs specific for ATPase, Cu2+ transporting, β polypeptide (siATP7B) and solute carrier family 31, member 1 (siSLC31A1) then treated for 24 h with 25 μM CDDP alone (in the presence of a control amount of PBS) of combined with 5 mM reduced glutathione (GSH) or 5 mM pyridoxine (PN). Means ± SEM (n = 6). *p < 0.05 (Student’s t-test), as compared to siUNR-transfected cells treated with CDDP only; #p < 0.05 (Student’s t-test), as compared to cells transfected with the same siRNA and treated with CDDP only; §p < 0.05 (Student’s t-test), as compared to siUNR-transfected cells treated with the same compounds.
These results demonstrate that GSH as well as the vitamin B6 metabolism influence the intracellular levels of CDDP independent of the main CDDP transporters SLC31A1/CTR1 and ATP7B.
Concluding remarks
Here, we provide evidence in support of the hypothesis that the metabolism of vitamin B6 as well as GSH modulate the cytotoxic response of NSCLC cells to CDDP as they influence its intracellular accumulation. In addition, by RNA interference studies, we demonstrate that such pharmacokinetic effects do not involve the transmembrane proteins that are currently believed to account for the largest fraction of CDDP import (SLC31A1/CTR1) and export (ATP7B).
Our findings are in line with the previously documented capacity of pyridoxine to exacerbate CDDP cytotoxicity in a PDXK-dependent fashion19 as well as with the well-known ability of antioxidants, including GSH and NAC, to potently block CDDP-induced cell death.19,22-24 Moreover, our data are compatible with the observation that the amount of CDDP-DNA adducts that form in the presence of CDDP is increased by pyridoxine and decreased to nearly undetectable levels by antioxidants.19
Nevertheless, our observations cannot explain why pyridoxine and PDXK conjointly affect the adaptive response of A549 cells to several cisplatin-unrelated chemical and physical perturbations, including nutrient deprivation, hyperthermia, hypoxia, chemical inhibition of the respiratory chain, irradiation as well as the exposure to a large collection of cytotoxic agents.19 As pyridoxal-5-phosphate (the bioactive form of vitamin B6) operates as a prosthetic group for > 4% of the enzymatic activities of the cell,36 it is tempting to speculate, though remains to be formally demonstrated, that vitamin B6 metabolism is wired to a complex network of signaling pathways that influence adaptive responses in a general fashion.
High expression levels of PDXK have been associated with improved disease outcome in two distinct cohorts of NSCLC patients irrespective of whether patients received CDDP or not after surgery.19 These clinical findings strongly suggest that vitamin B6 metabolism influences the natural course of NSCLC, rather than the response to antineoplastic therapy, perhaps by affecting the adaptation of malignant cells to nutrient deprivation, hypoxia or other adverse conditions that accompany oncogenesis and tumor progression. The clinical relevance of vitamin B6 metabolism has been highlighted by large population studies in which the circulating levels of one or more B6 vitamers were correlated with a decreased risk of developing multiple malignancies, including NSCLC,37 colorectal cancer38 and ovarian carcinoma.39 Future studies will have to unveil whether parameters that reflect the proficiency of vitamin B6 metabolism may also constitute predictive, rather than merely prognostic, biomarkers in CDDP-treated cancer patients.
Materials and Methods
Chemicals and cell cultures
Unless otherwise specified, chemicals were obtained from Sigma-Aldrich, cell culture reagents and supplements from Gibco-Invitrogen and plasticware from Corning. Human wild-type (WT) non-small cell lung carcinoma (NSCLC) A549 cells were routinely maintained (at 37°C under 5% CO2) in Glutamax®-containing DMEM/F12 medium supplemented with 10% fetal calf serum (FCS), 10 mM HEPES buffer, 100 units/mL penicillin G sodium and 100 µg/mL streptomycin sulfate. Cells were seeded into appropriate supports and allowed to adapt for at least 24 h before experimental procedures.
Transfections
A549 cells at 30–40% confluence were transfected by means of the Oligofectamine™ transfection reagent (Invitrogen), following the manufacturer’s recommendations. The following siRNA duplexes specific for ATPase, Cu2+ transporting, β polypeptide (ATP7B), pyridoxal kinase (PDXK) and solute carrier family 31, member 1 (SLC31A1/CTR1) were used:19 siATP7B_1 (5′-CCAGGTTGGCATCAACAAAdTdT-3′), siATP7B_2 (5′-AATTGATATTGAGCGGTTAdTdT-3′), siPDXK (5′-GAGTGACTTTCTAACCCAAdTdT-3′), siSLC31A1_1 (5′-GCATGAACTTGCCAATCAAdTdT-3′) and siSLC31A1_2 (5′-GCCTGTTGTCTAAAGCCAAdTdT-3′). In addition, a siRNA duplex with a sequence unrelated to the murine and human genome (siUNR, sense 5′-GCCGGUAUGCCGGUUAAGUdTdT-3′)40,41 was employed to provide negative control conditions. Cells were used in experimental determinations 48 h after transfection, when target downregulation was monitored by immunoblotting (see below).
Immunoblotting
For the preparation of total protein extracts, A549 cells were washed and lysed according to standard procedures.42,43 Forty to 50 μg of proteins were then separated according to molecular weight on pre-cast 4–12% polyacrylamide NuPAGE® Novex® Bis-Tris gels (Invitrogen), electrotransferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad) and probed with primary antibodies specifically recognizing ATP7B (rabbit antiserum #AB41146, Abcam plc.), PDXK (rabbit antiserum #AV53615, Sigma-Aldrich) and SLC31A1/CTR1 (rabbit antiserum #AB108481, Abcam plc.). Equal-lane loading was monitored by probing membranes with an antibody specific for glyceraldehyde-3-phosphate dehydrogenase (GAPDH, mouse monoclonal IgG1 #MAB374, Millipore-Chemicon International). Finally, membranes were incubated with suitable secondary IgG conjugated to horseradish peroxidase (Southern Biotech), followed by chemiluminescence detection with the SuperSignal West Pico® reagent (Thermo Scientific-Pierce) and either CL-XPosure® X-ray films (Thermo Scientific-Pierce) or the ImageQuant LAS 4000 software-assisted imager (GE Healthcare).
Quantification of intracellular CDDP
Elemental platinum in cell lysates was determined by flameless atomic absorption spectrometry as previously described,44 with slight modifications. In brief, an AAnalyst 600 atomic absorption spectrometer (Perkin-Elmer Life Science) equipped with a graphite tube atomisator and a platinum hollow cathode lamp was used with the following thermal program: 120°C (20 s), 130°C (50 s), 350°C (20 s), 600°C (20 s), 1,300°C (20 s), 2,400°C (8 s, reading step), 2,450°C (4 s), 20°C (10 s) and 2,450°C (4 s). The technique was validated and met the international requirements on bioanalytical methods.
Quantification of protein content
Intracellular protein content was quantified by means of the DCTM Protein Assay (Bio-Rad), following the manufacturer’s instructions. Absorbance was recorded on a FLUOstar OPTIMA microplate based multi-detection reader (BMG Labtech).
Common statistical procedures
Unless otherwise indicated, experiments were performed in triplicate instances and repeated at least twice, yielding comparable results. Data, which are presented as means ± SEM, were analyzed with Microsoft Excel (Microsoft Co.). Statistical significance was determined by means of two-tailed unpaired Student’s t-tests. Unless otherwise indicated, p values < 0.05 were considered statistically significant.
Acknowledgments
The authors are supported by the European Commission (ArtForce); Agence National de la Recherche (ANR); Ligue contre le Cancer (Equipe labellisée); Fondation pour la Recherche Médicale (FRM); Institut National du Cancer (INCa); LabEx Immuno-Oncologie; Fondation de France; Fondation Bettencourt-Schueller; AXA Chair for Longevity Research; Cancéropôle Ile-de-France and Paris Alliance of Cancer Research Institutes (PACRI).
Glossary
Abbreviations:
- ATM
ataxia telangiectasia mutated
- ATP7B
ATPase, Cu2+ transporting, β polypeptide
- CDDP
cisplatin
- CRM
CDDP response modifier
- CTR1
copper transporter 1
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GSH
non-oxidized glutathione
- NAC
N-acetyl-cysteine
- NSCLC
non-small cell lung carcinoma
- PDXK
pyridoxal kinase
- PVDF
polyvinylidene fluoride
- SEM
standard error of the mean
- SLC31A1
solute carrier family 31, member 1
- VDAC1
voltage-dependent anion channel 1
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/23275
References
- 1.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–84. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 2.Winter C, Albers P. Testicular germ cell tumors: pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2011;7:43–53. doi: 10.1038/nrendo.2010.196. [DOI] [PubMed] [Google Scholar]
- 3.Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–83. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 4.Köberle B, Tomicic MT, Usanova S, Kaina B. Cisplatin resistance: preclinical findings and clinical implications. Biochim Biophys Acta. 2010;1806:172–82. doi: 10.1016/j.bbcan.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 5.Carrassa L, Broggini M, Erba E, Damia G. Chk1, but not Chk2, is involved in the cellular response to DNA damaging agents: differential activity in cells expressing or not p53. Cell Cycle. 2004;3:1177–81. doi: 10.4161/cc.3.9.1080. [DOI] [PubMed] [Google Scholar]
- 6.Nardella C, Clohessy JG, Alimonti A, Pandolfi PP. Pro-senescence therapy for cancer treatment. Nat Rev Cancer. 2011;11:503–11. doi: 10.1038/nrc3057. [DOI] [PubMed] [Google Scholar]
- 7.Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 8.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–20. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mandic A, Hansson J, Linder S, Shoshan MC. Cisplatin induces endoplasmic reticulum stress and nucleus-independent apoptotic signaling. J Biol Chem. 2003;278:9100–6. doi: 10.1074/jbc.M210284200. [DOI] [PubMed] [Google Scholar]
- 10.Berndtsson M, Hägg M, Panaretakis T, Havelka AM, Shoshan MC, Linder S. Acute apoptosis by cisplatin requires induction of reactive oxygen species but is not associated with damage to nuclear DNA. Int J Cancer. 2007;120:175–80. doi: 10.1002/ijc.22132. [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez VM, Fuertes MA, Alonso C, Perez JM. Is cisplatin-induced cell death always produced by apoptosis? Mol Pharmacol. 2001;59:657–63. doi: 10.1124/mol.59.4.657. [DOI] [PubMed] [Google Scholar]
- 12.Shulga N, Wilson-Smith R, Pastorino JG. Hexokinase II detachment from the mitochondria potentiates cisplatin induced cytotoxicity through a caspase-2 dependent mechanism. Cell Cycle. 2009;8:3355–64. doi: 10.4161/cc.8.20.9853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–14. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 14.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–79. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 15.Borst P, Rottenberg S, Jonkers J. How do real tumors become resistant to cisplatin? Cell Cycle. 2008;7:1353–9. doi: 10.4161/cc.7.10.5930. [DOI] [PubMed] [Google Scholar]
- 16.Beers MH. Lung carcinoma. In: Porter RS, Jones TV, eds. The Merck manual of diagnosis and therapy. Rahway: Merck & Co., Inc., 2008:2992. [Google Scholar]
- 17.Jemal A, Thun MJ, Ries LA, Howe HL, Weir HK, Center MM, et al. Annual report to the nation on the status of cancer, 1975-2005, featuring trends in lung cancer, tobacco use, and tobacco control. J Natl Cancer Inst. 2008;100:1672–94. doi: 10.1093/jnci/djn389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panaretakis T. Cisplatin-induced apoptosis and development of resistance are transcriptionally distinct processes. Cell Cycle. 2012;11:3723. doi: 10.4161/cc.22114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galluzzi L, Vitale I, Senovilla L, Olaussen KA, Pinna G, Eisenberg T, et al. Prognostic impact of vitamin B6 metabolism in lung cancer. Cell Rep. 2012;2:257–69. doi: 10.1016/j.celrep.2012.06.017. [DOI] [PubMed] [Google Scholar]
- 20.Galluzzi L, Vitale I, Senovilla L, Eisenberg T, Carmona-Gutierrez D, Vacchelli E, et al. Independent transcriptional reprogramming and apoptosis induction by cisplatin. Cell Cycle. 2012;11:3472–80. doi: 10.4161/cc.21789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yao Z, Szabadkai G. Transcriptional profiling of apoptosis: cell death classification moves toward the systems era. Cell Cycle. 2012;11:3721–2. doi: 10.4161/cc.22116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park SA, Choi KS, Bang JH, Huh K, Kim SU. Cisplatin-induced apoptotic cell death in mouse hybrid neurons is blocked by antioxidants through suppression of cisplatin-mediated accumulation of p53 but not of Fas/Fas ligand. J Neurochem. 2000;75:946–53. doi: 10.1046/j.1471-4159.2000.0750946.x. [DOI] [PubMed] [Google Scholar]
- 23.Previati M, Lanzoni I, Corbacella E, Magosso S, Guaran V, Martini A, et al. Cisplatin-induced apoptosis in human promyelocytic leukemia cells. Int J Mol Med. 2006;18:511–6. [PubMed] [Google Scholar]
- 24.Tajeddine N, Galluzzi L, Kepp O, Hangen E, Morselli E, Senovilla L, et al. Hierarchical involvement of Bak, VDAC1 and Bax in cisplatin-induced cell death. Oncogene. 2008;27:4221–32. doi: 10.1038/onc.2008.63. [DOI] [PubMed] [Google Scholar]
- 25.Lavin MF, Kozlov S. ATM activation and DNA damage response. Cell Cycle. 2007;6:931–42. doi: 10.4161/cc.6.8.4180. [DOI] [PubMed] [Google Scholar]
- 26.DeMicco A, Bassing CH. Deciphering the DNA damage histone code. Cell Cycle. 2010;9:3845. doi: 10.4161/cc.9.19.13381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.MacLaine NJ, Hupp TR. How phosphorylation controls p53. Cell Cycle. 2011;10:916–21. doi: 10.4161/cc.10.6.15076. [DOI] [PubMed] [Google Scholar]
- 28.Yoshida M, Khokhar AR, Siddik ZH. Biochemical pharmacology of homologous alicyclic mixed amine platinum(II) complexes in sensitive and resistant tumor cell lines. Cancer Res. 1994;54:3468–73. [PubMed] [Google Scholar]
- 29.Ishida S, McCormick F, Smith-McCune K, Hanahan D. Enhancing tumor-specific uptake of the anticancer drug cisplatin with a copper chelator. Cancer Cell. 2010;17:574–83. doi: 10.1016/j.ccr.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holzer AK, Howell SB. The internalization and degradation of human copper transporter 1 following cisplatin exposure. Cancer Res. 2006;66:10944–52. doi: 10.1158/0008-5472.CAN-06-1710. [DOI] [PubMed] [Google Scholar]
- 31.Ishida S, Lee J, Thiele DJ, Herskowitz I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc Natl Acad Sci USA. 2002;99:14298–302. doi: 10.1073/pnas.162491399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katano K, Kondo A, Safaei R, Holzer A, Samimi G, Mishima M, et al. Acquisition of resistance to cisplatin is accompanied by changes in the cellular pharmacology of copper. Cancer Res. 2002;62:6559–65. [PubMed] [Google Scholar]
- 33.Komatsu M, Sumizawa T, Mutoh M, Chen ZS, Terada K, Furukawa T, et al. Copper-transporting P-type adenosine triphosphatase (ATP7B) is associated with cisplatin resistance. Cancer Res. 2000;60:1312–6. [PubMed] [Google Scholar]
- 34.Zischka H, Lichtmannegger J, Schmitt S, Jägemann N, Schulz S, Wartini D, et al. Liver mitochondrial membrane crosslinking and destruction in a rat model of Wilson disease. J Clin Invest. 2011;121:1508–18. doi: 10.1172/JCI45401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Safaei R. Role of copper transporters in the uptake and efflux of platinum containing drugs. Cancer Lett. 2006;234:34–9. doi: 10.1016/j.canlet.2005.07.046. [DOI] [PubMed] [Google Scholar]
- 36.Amadasi A, Bertoldi M, Contestabile R, Bettati S, Cellini B, di Salvo ML, et al. Pyridoxal 5′-phosphate enzymes as targets for therapeutic agents. Curr Med Chem. 2007;14:1291–324. doi: 10.2174/092986707780597899. [DOI] [PubMed] [Google Scholar]
- 37.Johansson M, Relton C, Ueland PM, Vollset SE, Midttun O, Nygård O, et al. Serum B vitamin levels and risk of lung cancer. JAMA. 2010;303:2377–85. doi: 10.1001/jama.2010.808. [DOI] [PubMed] [Google Scholar]
- 38.Larsson SC, Orsini N, Wolk A. Vitamin B6 and risk of colorectal cancer: a meta-analysis of prospective studies. JAMA. 2010;303:1077–83. doi: 10.1001/jama.2010.263. [DOI] [PubMed] [Google Scholar]
- 39.Harris HR, Cramer DW, Vitonis AF, Depari M, Terry KL. Folate, vitamin B(6), vitamin B(12), methionine and alcohol intake in relation to ovarian cancer risk. Int J Cancer. 2011 doi: 10.1002/ijc.26455. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de La Motte Rouge T, Galluzzi L, Olaussen KA, Zermati Y, Tasdemir E, Robert T, et al. A novel epidermal growth factor receptor inhibitor promotes apoptosis in non-small cell lung cancer cells resistant to erlotinib. Cancer Res. 2007;67:6253–62. doi: 10.1158/0008-5472.CAN-07-0538. [DOI] [PubMed] [Google Scholar]
- 41.Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010;1:e10. doi: 10.1038/cddis.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G. Cell death assays for drug discovery. Nat Rev Drug Discov. 2011;10:221–37. doi: 10.1038/nrd3373. [DOI] [PubMed] [Google Scholar]
- 43.Galluzzi L, Morselli E, Vitale I, Kepp O, Senovilla L, Criollo A, et al. miR-181a and miR-630 regulate cisplatin-induced cancer cell death. Cancer Res. 2010;70:1793–803. doi: 10.1158/0008-5472.CAN-09-3112. [DOI] [PubMed] [Google Scholar]
- 44.LeRoy AF, Wehling ML, Sponseller HL, Friauf WS, Solomon RE, Dedrick RL, et al. Analysis of platinum in biological materials by flameless atomic absorption spectrophotometry. Biochem Med. 1977;18:184–91. doi: 10.1016/0006-2944(77)90089-8. [DOI] [PubMed] [Google Scholar]