

Abstract
Recent studies have highlighted an apparently paradoxical link between self-renewal and senescence triggered by DNA damage in certain cell types. In addition, the finding that TP53 can suppress senescence has caused a re-evaluation of its functional role in regulating these outcomes. To investigate these phenomena and their relationship to pluripotency and senescence, we examined the response of the TP53-competent embryonal carcinoma (EC) cell line PA-1 to etoposide-induced DNA damage. Nuclear POU5F1/OCT4A and P21CIP1 were upregulated in the same cells following etoposide-induced G2M arrest. However, while accumulating in the karyosol, the amount of OCT4A was reduced in the chromatin fraction. Phosphorylated CHK2 and RAD51/γH2AX-positive nuclear foci, overexpression of AURORA B kinase and moderate macroautophagy were evident. Upon release from G2M arrest, cells with repaired DNA entered mitoses, while the cells with persisting DNA damage remained at this checkpoint or underwent mitotic slippage and gradually senesced. Reduction of TP53 using sh- or si-RNA prevented the upregulation of OCT4A and P21CIP1 and increased DNA damage. Subsequently, mitoses, micronucleation and senescence were all enhanced after TP53 reduction with senescence confirmed by upregulation of CDKN2A/P16INK4A and increased sa-β-galactosidase positivity. Those mitoses enhanced by TP53 silencing were shown to be multicentrosomal and multi-polar, containing fragmented and highly deranged chromosomes, indicating a loss of genome integrity. Together, these data suggest that TP53-dependent coupling of self-renewal and senescence pathways through the DNA damage checkpoint provides a mechanism for how embryonal stem cell-like EC cells safeguard DNA integrity, genome stability and ultimately the fidelity of self-renewal.
Keywords: TP53, OCT4A/POU5F1, self-renewal, tumor cells, DNA damage, pluripotency, senescence
Introduction
Aggressive somatic tumors possess a gene expression profile similar to embryonic stem (ES) cells, suggesting that key signaling pathways and biological links exist between them.1 Importantly, both appear to display characteristics of self-renewal and differentiation potential, with current data implying that these properties underlie the resistance of cancer to genotoxic treatment modalities and help explain disease relapse.2,3 The characteristic self-renewal, extensive proliferation and differentiation potential of ES cells is also evident in cancer stem cells, which are highly proliferative and phenotypically plastic.4 The ability of cancer cells to utilize equivalent stem cell transcription networks is of great interest for the subsequent understanding and treatment of cancer.
Recent data shows that even normal somatic cells can be reprogrammed to become induced pluripotent stem (iPS) cells, and that upregulation of only a very small set of genes is sufficient to initiate de-differentiation to provide them with stem-like properties. OCT4A (POU5F1), SOX2 and NANOG are master transcription factors responsible for the maintenance and tight coordination of pluripotency and self-renewal in ES cells.5 The first human iPS cells were created from somatic fibroblasts ectopically expressing two of these factors (OCT4A and SOX2) along with KLF4 and c-MYC.6 Since then, expression of OCT4A and SOX2 alone has been shown to be sufficient to convert human somatic cells to iPS cells.7 These results suggest that cancer cells may be able to access ES cell properties through the upregulation of these genes alone. In keeping with this proposition, the prognostic significance of these two markers was recently reported for several kinds of cancer (lung, squamous cell carcinoma, colorectal and breast).8-10
We have previously reported the upregulation of OCT4A/SOX2/NANOG in TP53 mutant lymphoma cell lines as a response to DNA damage and highlighted the potential role of this ectopically upregulated embryonic self-renewal program as a survival strategy in TP53-deficient cells following genotoxic damage.11
Self-renewal and pluripotency factors have paradoxically also been linked to accelerated cellular senescence (termed senescense hereon). When Banito and colleagues transfected IMR90 human fibroblasts with the Yamanaka transcription factors (OCT4A, SOX2, c-MYC and KLF4), they found senescence was induced rather than pluripotency, which was associated with DNA damage.12 A study of normal IMR90 fibroblasts showed that at pre-senescence, cells signaling DNA damage in the ≥ 4C compartment simultaneously express cyclin-dependent kinase inhibitors P16INKA4A and P21CIP1, as well as the self-renewal transcription factor NANOG,13 further indicating that the properties of pluripotency, self-renewal and senescence are tightly coordinated by DNA damage.
The relationship between mitogenic activation of proliferation and cell senescence is also complex. For example, the concept of hyper-mitogenic arrest intimates that simultaneous stimulation of mitogen-activated pathways (integrated by mTOR) and downstream inhibition of cyclin-dependent kinases promotes senescence.14 Similarly, DNA damage-induced senescence occurs in immortalized WI38 fibroblasts only when mitogenic growth factors are present.15 In this context, the role of TP53 in regulating proliferation and senescence is critical, with evidence of its ability to both block and promote senescence. On one hand, TP53 signals to prevent cellular senescence, but conversely it can cause cell cycle arrest, thereby providing the cellular context from which the senescence program can start.16 These data imply that senescence is not in fact a barrier to cancer but almost a prerequisite for it.17 This concept and observations showing the complexity of the inter-regulation between these various cellular outcomes may help to understand how self-renewing (cancer) stem cells respond to DNA damage.
Together, these data indicate that intrinsic links exist between self-renewal, senescence and the DNA damage checkpoints of the cell. Further complexity arises from the fact that the regulation of the cell cycle in stem cells is evidently different from that in normal somatic cells and not yet fully understood. Current reports indicate that the cell cycle in stem cells lacks G0, has a short G1, absent G1-S checkpoint18 and a preferential activation of the G2M DNA damage checkpoint, which is responsible for the augmented resistance of stem cells to genotoxic damage.19 Presumably, these differences in cell cycle regulation are associated with the particular biological properties of ES cells, such as self-renewal (immortality).
EC cell lines, which are derived from germ cell tumors, can serve, to a certain extent, as a model of ES cells and express the key cassette of ES cell transcription factors, OCT4A, SOX2 and NANOG.20,21 At the same time, these tumors are highly malignant and so may be used to closely model the features of aggressive tumors that show ES cell transcription profiles.1,8-10 Therefore, in this current study, we chose to investigate the relationship between senescence and self-renewal regulation following DNA damage in the ovarian teratocarcinoma PA1 cell-line, which possesses functional TP53. For inducing DNA damage, we used the topoisomerase II inhibitor etoposide (ETO), which was previously used for studies of senescence in tumor and immortalized cells lines,15,22,23 and which is also in clinical use for the treatment of various cancer types.
Results
Characterization of PA-1 cells
The PA-1 cell line used in this study was established from the metastases of a malignant ovarian teratocarcinoma. It possesses the features of embryonal carcinoma (EC), as testified by its ability to form OCT4A/NANOG-positive spheres in serum-free medium (Fig. S1).24 It has a stable, near-diploid karyotype with a single chromosome translocation [t(15;20)(p11.2;q11.2)]25 and functional (although heterozygotic) TP53.26 Several sources show that PA1 expresses the key self-renewal genes OCT4A, SOX2 and NANOG.11,24,27
Response of PA-1 cells to DNA damage
To determine the response of PA-1 cells to DNA damage, the cells were assessed following ETO treatment over time. Phosphorylated CHK2 (pCHK2) and γH2AX staining were used to report on the presence of DNA damage, and the cell cycle response of surviving cells was monitored using DNA cytometry. In response to ETO treatment, virtually all cells became arrested in G2 within 2 days and displayed pCHK2 and γH2AX-positive nuclear foci as evidence of DNA damage and subsequently attempted DNA repair as judged by the presence of RAD51/γH2AX foci (Fig. 1A and B). The cells remained in G2 for up to 4 days. Simultaneously, small G1, 2C and polyploid (> 4C) cell fractions then appeared, increased for a few days before the polyploid fraction was overcome by the mitotic cycling fraction and disappeared by day 7 (Fig. 1C). Substantial apoptosis appeared from day 3, reached a maximum on day 5 and disappeared with the recovery of clonogenic growth after day 7 (data not shown and Fig. 5A).

Figure 1. DNA damage response of PA-1 cells to ETO treatment. PA-1 cells were treated with 8 µM ETO for 20 h, then washed and assessed at the indicated time point. Cells were pelleted, cytospun, fixed and stained for (A) pCHK2 (red) or (B) γH2AX (red) and RAD51 (green) in combination with DAPI (blue). Bar = 10 µM. (A) NT PA-1 cells show very faint background pCHK2 staining in non-dividing cells and increased staining of centrosomes in dividing cells (white arrowheads). On day 2 post-ETO, small pCHK2-positive foci accumulate in the nuclei of all cells. By day 4, some small nuclei have lost pCHK2 staining (yellow arrowheads), while large cells remain pCHK2 positive. (B) NT PA-1 cells show no positive staining for γH2AX or RAD51. On day 2, the majority of cells show accumulation of γH2AX or RAD51 positive foci in the nuclei. (C) Cells were cytospun, fixed and stained for DNA image cytometry. DNA content was determined for at least 200 cells in each condition and represented as a percentage. Profound G2 arrest on day 2 was observed followed by the simultaneous emergence of a polyploid (> 4C) and G1 fraction on day 5 before the recovery of the normal cell cycle profile by day 7. (D) The proportion of pCHK2-positive cells was examined in the context of DNA content with cells sub-divided into small or large cells. In the NT control sample, all were pCHK2-negative with an expected nuclei size distribution (2C 80%; ≥ 4C 20%). On day 2, all cells were pCHK2-positive and the vast majority of nuclei were large (≥ 4C). By day 5, cells with small nuclei appear, all of which are pCHK2-negative. Data are representative of > three independent experiments.

Figure 5. Effect of silencing TP53 on the cell cycle response of PA-1 cells after ETO treatment. PA-1 cells were treated with 8 µM ETO for 20 h, then washed and examined at the indicated time points by (A and B) flow cytometry, (C) DNA image cytometry and (D) mitotic counts. (A) Both control plasmid and shRNA p53-silenced cells undergo a G2M arrest after ETO treatment, which is, however, more profound from day 3 in TP53-silenced cells. The TP53-silenced cells also show a larger increase in the ≥ 4C cell fraction in response to ETO treatment. Conversely, the recovering G1-2C fraction is reduced. By day 5, the relative proportion of the G2 fraction is diminished in both samples, due to the increase in apoptosis. (B) The proportion of cells in G2 was enumerated over the time course in three independent experiments with the mean and SEM plotted (* p < 0.05). (C) DNA image cytometry analysis was also performed on day 4 after ETO treatment. Apoptotic cells were excluded from the measurements, and highlighted more clearly the relationship between the increased 4C fraction and relative delay in recovery of the proliferating G1-2C fraction (reduced 2.5-fold) after TP53-silencing. The corresponding mitotic index (MI) at this time point is also shown. For full cell cycle dynamics over the time course in this experiment, please see Fig. S6. Data representative of two similar experiments. (D) Mitotic counts were performed on control plasmid or shRNA p53-silenced cells over time after ETO treatment. The results from three independent experiments show a significant increase in the proportion of mitoses after TP53 silencing (* p < 0.05).
In addition, the G2-arrested cells showed enhanced nuclear staining of AURORA B kinase (Fig. S2), indicating the mitotic potential of these cells.
From day 3 onwards, pCHK2 nuclear staining disappeared in some cells, while persisting and even becoming enhanced in others (Fig. 1A). To elucidate which cells entered mitosis and which polyploidized, we examined them on day 5 using DAPI-integrated fluorescence to assess their DNA content and size as well as pCHK2 nuclear staining levels. Two distinct cell types were observed: those with small nuclei (G1, 2C-sized) and those with large (G2 and polyploid, 4C and > 4C) nuclei. As seen in Figure 1D, the majority of large cells (≥ 4C) were pCHK2-positive, and all pCHK2-positive cells were large cells. In contrast, all cells with small nuclei (G1) cells were pCHK2-negative. These results indicate that ~30% of cells have repaired DNA and are returning to the mitotic cell cycle, while the remainder display persistent DNA damage and are either maintained in the 4C fraction or become polyploid.
During this period, macroautophagy was observed in response to ETO treatment as seen by LC3B and P62 lysosomal staining being compatible with the enhanced OCT4A staining (Fig. S3A–D) and gradually increasing from day 1 to 5. Macroautophagy returned to resting levels in cells as they re-entered mitotic cycling, but remained elevated in cells that persisted in G2 and in most polyploid cells. TERT staining revealed nuclear foci in NT cells (not shown) and in the small cells re-entering the mitotic cycle following ETO treatment. Most large cells lacked nuclear foci and showed elimination of TERT in perinuclear LC3B-lined vacuoles; however, in parallel, large amounts of TERT also appeared in their cytoplasm (Fig. S3E and F), possibly due to the stress-protecting binding of TERT to mitochondria.28 Most PA-1 cells displayed low NANOG expression, which was more evident around chromosomes in recovering mitotic cells and clones (Fig. S4A). In contrast, rare cells expressed high levels of NANOG in both control and ETO-treated conditions (Fig. S4B). The NANOG-overexpressing cells observed after ETO treatment were large and presumably polyploid. We hypothesize that they arose by overcoming the G1-4N checkpoint and avoiding senescence. These cells have the potential to reach 8-16C and then de-polyploidize. Such a response is typical of mutant TP53 lymphoma cells after DNA damage,11,29 but is a rare event with PA-1.
Upregulation of OCT4A and P21CIP1 in response to DNA damage
We next assessed key molecular regulators of the DNA damage response by immunofluorescence (IF). As expected, TP53 and, subsequently, P21CIP1 (CDKN1A), were upregulated in response to ETO treatment. However, the pluripotency self-renewal transcription factor OCT4A was also upregulated (Fig. 2A–C). In fact, paradoxically, OCT4A and P21CIP1 were shown by IF to be both expressed in the same cells (Fig. 2B and C). Expression of high levels of both proteins displayed a strong positive correlation (Fig. 2D) in the first 3 d post-treatment in the G2 (4C by DNA content) and polyploid (> 4C) fractions described above (Fig. 2E; Fig. S5A). However, individual cells were shown to vary greatly in their expression of both of these factors after treatment as indicated by their mean expression levels, standard deviations and extended outliers (Fig. 2D; Fig. S5B).

Figure 2. Regulation of TP53, P21CIP1 and OCT4A in PA-1 cells after ETO treatment. PA-1 cells were treated with 8 µM ETO for 20 h, then washed and assessed at the indicated time point. Cells were pelleted, cytospun, fixed and stained for (A) TP53 (red) (B) P21CIP1 (red) or (C) P21CIP1 (red) and OCT4A (green) in combination with DAPI (blue); Bar = 10 µM. Results representative of nine separate experiments. (A) NT cells showed very faint background staining for TP53. On day 3 post-ETO treatment, TP53 accumulated in the majority of nuclei. (B and C) NT cells had faint background staining for P21CIP1 and weak staining of OCT4A. On day 3 post-ETO treatment, P21CIP1 and OCT4A accumulated in the majority of cell nuclei. (D) Using image analysis on fixed cytospins, the fluorescence intensity of P21CIP1 and OCT4A during the first 3 d post-ETO treatment was determined and plotted for 500 cells in four independent experiments. There was substantial increase and a clear positive correlation between the fluorescent intensity of staining for OCT4A and P21CIP1 after ETO treatment. At the same time, the amplitude of the expression of both factors greatly increased: Non-treated OCT4A expression mean = 2,448, SD = 1,236; OCT4A expression on day 3 mean = 12,641, SD = 6,189; non-treated P21CIP1 expression mean = 2,875, SD = 1,594; P21CIP1 expression on day 3 mean = 10,291, SD = 5,795. (E) Two-channel flow cytometry was used to measure DNA content (PI fluorescence) and OCT4A expression. In NT samples, the OCT4A expression was similar in the 2C and 4C fractions. After ETO treatment, OCT4A was highly elevated in the 4C and 8C fractions and to a lesser extent in the G1-2C fraction. These data indicate that OCT4A accumulation was mostly induced in the DNA damage checkpoint and persisted in the polyploid cells.
The TP53 dependency of the OCT4A response
To further investigate the link between increased OCT4A expression and TP53 induction after DNA damage in PA-1 cells, siRNA directed against TP53 was used. Western analysis of whole-cell lysates confirmed that ETO treatment strongly upregulated TP53, OCT4A and P21CIP1 (CDKN1A), confirming our IF results (Fig. 3). In the post-G2 period, from day 4 after ETO treatment, both TP53 and OCT4A levels partially decreased, while P21CIP1 continued to increase. Transfection of PA-1 cells with TP53-siRNA successfully silenced TP53 expression throughout the time-course and, as expected, inhibited the increase in P21CIP1 in response to ETO treatment. However, unexpectedly, silencing of TP53 also largely inhibited the upregulation of OCT4A. Thus, the increase in OCT4A in response to ETO is largely TP53-dependent. To confirm this surprising finding, we repeated these experiments using PA-1 cells stably transfected with a shRNA vector directed against a different region of TP53 and saw the same results (Fig. S6) with diminished OCT4A upregulation.
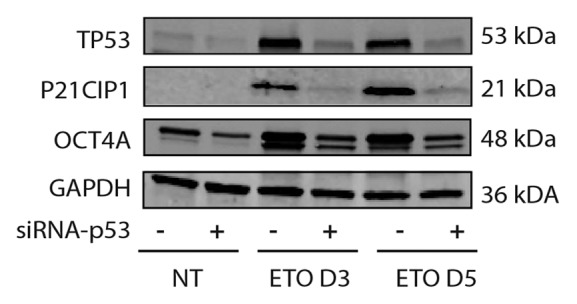
Figure 3. Immunoblot analysis of TP53, OCT4A and P21CIP1 in PA-1 cells after ETO treatment. PA-1 cells were treated with non-target (ntg) siRNA (-) or siRNA-Tp53 (+) for 24 h before treatment with 8 µM ETO, washing after 20 h and cell lysates made and assessed by immunoblotting for Tp53, P21CIP1, OCT4A or GAPDH as a loading control at the indicated time points (day 3 and day 5). Tp53 was upregulated in response to ETO treatment and suppressed by siRNA-TP53. P21CIP1 and OCT4A were also upregulated by ETO treatment, and the upregulation was restricted by treatment with siRNA-TP53. Data are representative of three independent experiments.
Previous studies have indicated that the expression of OCT4A in cancer cell lines may have been misreported due to the presence of other OCT4 isoforms (OCT4 B, B1 and/or transcribable pseudogenes-1, -3 and -4). To address this issue here, we performed RT-PCR and revealed that OCT4A transcription was present but not particularly enhanced after ETO treatment (Fig. 4A), whereas isoform B was entirely absent. The splicing isoform OCT4 B1 (characteristic for ES/EC cells and poorly differentiated cancer)30,31 was induced after ETO treatment (Fig. 4A). In addition, our results indicated that pseudogene-1 was transcribed, whereas pseudogenes-3 and -4 were not (not shown). The OCT4B1 splicing form is appreciably smaller in size in comparison to OCT4A, and was not detected in our immunoblotting analysis with either OCT4A or OCT4A/B antibodies and so can be disregarded. However, to evaluate our IF and immunoblotting findings in relation to pseudogene-1, expression vectors encoding HA-tagged forms of OCT4A or OCT4-pseudogene 1 were produced, transfected into HEK-293 cells and the ability of OCT4 antibodies to detect them examined. It was revealed that although both proteins were equivalently expressed as judged by detection with anti-HA antibodies, only the OCT4A form was detected with the anti-OCT4A monoclonal antibody reagent (Fig. S7). Together, these data confirm that the increase in OCT4A described here was attributed to bona fide OCT4A and not alternative splicing or pseudogene forms. However, it should be noted that an additional smaller protein species of OCT4A was also observed in our experiments, which increased in intensity in response to ETO treatment. As discussed above, the size of this protein does not correspond to OCT4B1, nor does this antibody detect other isoforms of OCT4. It therefore may reflect a post-translational change in OCT4A that increases in response to DNA damage.

Figure 4. Analysis of OCT4 regulation in PA-1 cells after ETO treatment. PA-1 cells were treated with 8 µM ETO for 20 h, then washed and examined at the indicated time points by (A) semi-quantitative PCR or (B) cell fractionation and immunoblotting using antibodies against OCT4A or OCT4A and B, actin or histone H3 (H3), the latter two as loading controls. (A) OCT4A transcription was shown to be modestly upregulated in response to ETO after 3 d (upper panel). OCT4B was not substantially detected (middle panel). OCT4B1 was upregulated in response to ETO. OCT4-PG-1 was also upregulated in response to ETO (lower panel). Controls were NTC, non-template control; PBL, peripheral blood lymphocytes. Equivalent amplification of actin after 30 cycles was used as a control for cDNA input (bottom panel). Data representative of three independent experiments. (B) OCT4A protein was upregulated in response to ETO treatment in the cytosol, membrane and nuclear fractions. OCT4A was downregulated in the chromatin-bound fraction. No additional bands were detected with the OCT4A/B dual specificity (C-terminal) antibody compared with the mono-specific OCT4A antibody (N-terminal), indicating only OCT4A species are expressed. Data representative of two independent experiments.
OCT4A does not bind chromatin during the DNA damage response
Subcellular fractionation was subsequently used to establish the location of the OCT4A within the cell. Cytoplasm, membrane-bound, nuclear (karyosolic) and chromatin-bound fractions were obtained, and OCT4A presence was assessed by immunoblotting. This analysis revealed a considerable increase of OCT4A in response to ETO treatment in the nuclear (karyosolic) fraction as attested to by two different antibodies, directed to the N or C terminus of OCT4. However, OCT4A was shown to be depleted after ETO treatment from the chromatin-bound fraction as compared with non-treated cells (Fig. 4B), indicating that although it is elevated, it may lack the ability to bind its own promoter or the promoters of its cooperative partners (SOX2 and NANOG) and influence the downstream transactivation targets.
Effect of TP53 on the cell cycle response
We next sought to examine the effect of TP53 on the cell cycle response of PA-1 cells after ETO treatment using flow cytometry and DNA image analysis (Fig. 5A–C). As indicated above using DNA image cytometry, ETO treatment of TP53-competent PA-1 cells resulted in a pronounced G2 arrest by day 1–2, followed by its release from day 3–4 and a degree of polyploidy and apoptosis. When TP53 was silenced by shRNA in transfected cell lines, a larger proportion of cells accumulated in the 4C and polyploid fractions. Subsequently, less cells recovered in the G1-2C fraction (Fig. 5A and B). These relationships were more evident upon DNA image analysis that omitted apoptotic cells (Fig. 5C; Fig. S8). Surprisingly, despite an apparent delay in proliferation, mitotic counts revealed a significant increase in the mitotic index (MI) of TP53-silenced samples compared with TP53-sufficient controls. (Fig. 5D; and shown for an individual experiment in Fig. 5C). To address the nature of this response further, pCHK2 and α-tubulin staining was performed on adherent cells on day 4 after ETO treatment. Using this approach, bipolar and bicentrosomal mitotic cells and aberrant multipolar mitotic cells were distinguished and enumerated (for examples of normal and abnormal patterns, see Fig. S9). The results of this analysis showed that all mitoses in TP53-silenced samples were multicentrosomal and most of them multipolar, with a smaller proportion showing coalescence of multiple centrosomes at two poles, indicating a high degree of chromosome and centrosome instability (Fig. 6A). Conversely, the mitoses in control cells after ETO treatment were mostly bipolar and bicentrosomal, with a smaller proportion of multicentriolar multipolarity. Inspection of metaphases by DNA staining after TP53-silencing revealed that they were highly aberrant with deranged plates, partly uninemic, fragmented and clumped chromosomes and no anaphases, while only rare aberrant mitoses were seen in the control sample (characteristic patterns are shown in Fig. S10A and B). Some of these aberrant metaphase plates in ETO-treated controls had chromosomes separated from the mitotic spindle and, when stained for γH2AX, displayed DNA damage (Fig. S10C). Intense DNA damage was also observed in the polyploid cells with micronuclei, while cells with small nuclei were free of γH2AX staining or contained only residual damage (Fig. S10D). Presumably, these observations explain the link between damaged DNA, aberrant mitoses, spindle checkpoint arrest and the induction of polyploidy by mitotic slippage. Therefore, we conclude that in spite of increased entrance into mitosis after Tp53 silencing, these cells are arrested in metaphase and delay proliferation, enhancing their propensity to undergo mitotic slippage, and become tetraploid. However, the eventual recovery of the cell population from small clones re-entering mitosis was comparable in both TP53-silenced and control cells, suggesting that TP53-silenced cells compensate for their initial delay in proliferation.
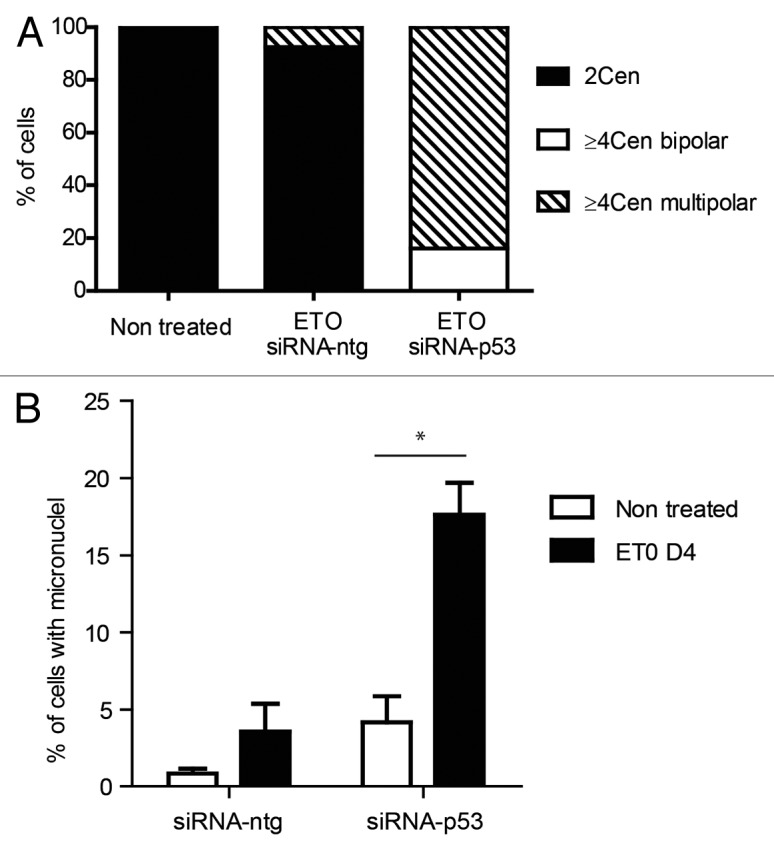
Figure 6. Effect of silencing TP53 on genome instability in PA-1 cells after ETO treatment. PA-1 cells were treated with 8 µM ETO for 20 h, then washed and examined at the indicated time points for the (A) the proportion of normal (2 centrosomal; 2 cen), ≥ 4 cen bipolar and ≥ 4 cen multipolar mitoses and (B) micronucleation. (A) Cells were treated as before and then examined by IF staining for pCHK2 and α-tubulin. Silencing of TP53 caused multi-centrosomal mitoses (≥ 4 cen) that were mostly multi-polar or showed coalescence of centrosomes in two poles. (B) Cells were treated as before and then assessed for the extent of micronucleation following DNA in situ staining. An increase in the proportion of interphase cells with micronuclei was observed in response to ETO treatment. Silencing of TP53 significantly increased the amount of micronucleation, which was further enhanced in response to ETO (p < 0.05). Data are representative of three independent experiments.
Effect of TP53-silencing on micronucleation
Next, we examined the effect of TP53-silencing on chromosome stability using the presence of micronuclei as a marker (Fig. 6B). As expected, treatment with ETO caused an increase in micronucleation and, thus, nuclear instability. Chromosome instability was substantially increased by silencing of TP53 as judged by the ~4-fold increase in micronucleation index.
Effect of TP53-silencing on senescence
Subsequently, we addressed how TP53 affected the induction of senescence after ETO treatment, initially by assessing the expression of p16inka4a. We found that an increase in P16INKA4A expression was detected from days 4–5 post-treatment in TP53-silenced cells (Fig. 7A), corresponding with the partial release from G2 arrest and increase in (aberrant) mitoses. In contrast, mock-transfected control cells had only marginal expression of P16INKA4A during this time. To confirm whether the increase in P16INKA4A expression corresponded with other indicators of senescence, senescence-associated β-galactosidase (sa-β-gal) staining and morphological analysis were performed. In control cells, ETO treatment evoked a modest but measurable increase in cell size (hypertrophy) and weak sa-β-gal staining at later time points. However, this was greatly accelerated by the silencing of TP53 with strong sa-β-gal staining in a significantly (p < 0.05, n = 3) higher proportion of cells in TP53-silenced (mean = 70.7%) vs. control (mean = 38.7%) samples at day 5 post-treatment, corresponding with the accelerated increase of DNA damage marked by pCHK2 (Fig. S6) and the senescence marker P16INKA4A (Fig. 7A) seen above.
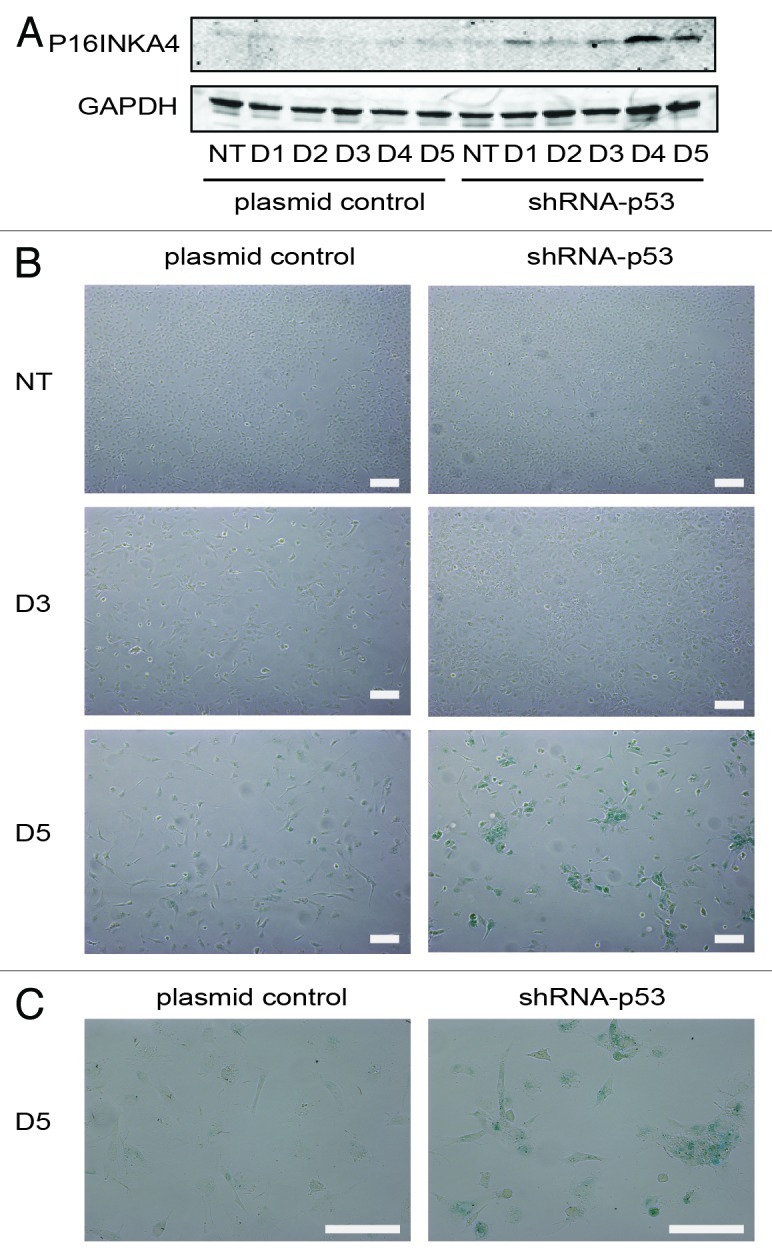
Figure 7. Senescence response of PA-1 cells to ETO treatment. PA-1 cells stably transfected with shRNA-p53 or plasmid control were treated with 8 µM ETO for 20 h, then washed and assessed at the indicated time point by (A) immunoblotting or (B) for sa-β-gal expression and cellular morphology. (A) Cell lysates were made and assessed by immunoblotting for P16INKA4 and GAPDH as a loading control. An accumulation of P16INKA4 was detected in TP53-silenced cells after ETO treatment. (B) sa-β-gal staining was used to detect sa-β-gal activity in response to ETO treatment at indicated time points. Detection of sa-β-gal activity was increased by TP53-silencing and largely mirrored the induction of P16INKA4 seen above. (C) Higher power image of the cells seen on day 5. Bar = 20 µM. Data are representative of > three independent experiments.
Discussion
In this study, we investigated the DNA damage response of the ovarian teratocarcinoma cell line PA-1, with particular focus on the relationship between self-renewal and senescence and the role of TP53 in regulating these different cell fates. Despite expressing functional TP53, PA-1 cells showed a preferential G2M arrest following ETO-induced DNA damage, a characteristic feature and cause of genotoxic resistance of ES cells.19 We also observed equivalent results with other forms of DNA damage such as X-ray irradiation (data not shown).
This arrest was typically characterized by extensive DNA damage signaling (indicated by the upregulation pCHK2 and γH2AX) and by attempted repair through homologous recombination (as shown by the induction of RAD51/γH2AX foci). The subsequent cellular response varied: while most cells died, some cells successfully repaired, halted DNA damage signaling and returned to mitotic cycling, whereas others displayed persistent DNA damage, remained in the 4C fraction or became polyploid. These latter cells underwent upregulated macroautophagy, indicative of a move toward senescence. However, these cells were also OCT4A and AURORA B kinase-positive, contained a large amount of cytoplasmic TERT and were therefore potentially able to enter mitosis, so retained a clear plasticity of response, not yet terminally committed to senescence.
The DNA damage response in the PA-1 cells appears to couple the upregulation of the pluripotency and self-renewal-promoting master molecule, OCT4A, with cell cycle arrest and senescence-promoting molecules (TP53 and P21CIP1) in the same cells, providing them with bipotentiality in the DNA damage checkpoint. Furthermore, the expression levels of these proteins in individual cells after treatment was highly varied. Although heterogeneous expression of NANOG within ES cell cultures has been observed,32 and this, in turn, has been attributed to fluctuation of NANOG levels,33 it is generally accepted that OCT4A expression is tightly regulated and expressed relatively uniformly in ES cell populations and does not fluctuate.34 In the context of TP53-dependent upregulation of OCT4A, the heterogeneity observed here may be due to fluctuation in TP53 expression itself, which is well-documented in cell stress responses.35,36
Artificially reducing the level of TP53 using si- or shRNA resulted in precocious mitosis and, subsequently, an increase in chromosome and centrosome instability in response to ETO treatment. In addition, TP53 silencing caused an increase in DNA damage and enhanced senescence. This is in agreement with previous studies that demonstrated TP53 can suppress cellular senescence.16 We therefore propose that unrepaired DNA damage and the increase in chromosome instability may link TP53 suppression to senescence.
The question then becomes how. Precocious aberrant mitoses containing DNA damage readily undergo mitotic slippage (to become G1-4N cells with micronuclei), which may undergo apoptosis or alternatively trigger terminal senescence through TP53-independent P21CIP1 and P16INKA4A targeting of the cyclin kinases governing the G1-S transition. Weaver and Cleveland also suggested adaptation of the spindle checkpoint as a pathway to cell survival vs. senescence through tetraploidy.37 Based upon our observations, we suggest that coupling the expression of self-renewal and senescence regulators through TP53 at the DNA damage checkpoint favors DNA damage repair, reduces chromosome and centrosome instability and, thus, safeguards the fidelity of self-renewal in the damage-resistant EC cells.
The question again follows, how are these opposing processes coordinated at the molecular level? The explanation may be provided by the observation that the OCT4A induced by TP53 at the DNA damage checkpoint is unable to bind chromatin. Presumably then, OCT4A is unable to execute its transactivation role in binding to the NANOG promoter regulatory element essential for the pluripotency function.34,38 Conversely, the NANOG promoter should be directly blocked by activated TP53.39 However, importantly, in ETO-treated PA-1 cells, OCT4A accumulates in the cell nucleus, and this should be a pre-requisite for its effective upregulation of NANOG through a positive feedback loop, when the NANOG promoter becomes free from inhibition by TP53 after DNA damage repair. Therefore, accumulation of OCT4A can be considered as a precursor for self-renewal potential. Simultaneously, the cell is primed for senescence through the accumulation of P21CIP1, again in a TP53-dependent manner, but is prevented from its full execution, possibly by the pre-mitotic molecular environment of the G2 cell. It follows that the PA-1 cells in the G2M damage checkpoint are in a state of bi-potential metastability between self-renewal and senescence, akin to the situation of bi-potentiality between self-renewal and differentiation in stem cells.
This interpretation fits well with the extensive heterogeneity in OCT4A and P21CIP1 levels observed in the individual cells held at the G2 arrest, potentially suggesting that their expression levels are dynamically fluctuating.40 To our knowledge, this notion of bi-potential metastability has not previously been considered as a key result of DNA damage, and we believe our data support further study of this area.
In conclusion, our studies have described two previously unknown phenomena relating to the intrinsic link between self-renewal and senescence in DNA damaged cells expressing stem cell-like transcriptional networks and revealed the role of TP53 in this process. This knowledge may be of use in improving our understanding of normal stem cells and also in designing better strategies to eliminate cancer stem cells.
Material and Methods
Cell culture and ETO treatment
PA-1 (ATCC) and HEK-293 cells were cultured in Dulbecco’s modified Eagle’s media (DMEM) supplemented with 10% fetal calf serum. Cells were grown without antibiotics in 5% CO2 incubators at 37°C. For the sphere formation assay, PA1 cells were inoculated in DMEM/F12 media supplemented with 20 ng/ml EGF (236-EG, R&D Systems), 10 ng/ml FGF (233-FB-025/CF, R&D Systems), 0.5 ng/ml hydrocortisone (H0888, Sigma), 10 ng/ml insulin (I9278, Sigma), 1x antibiotics/antimycotics (15240-062, Invitrogen), 1% L-glutamine (25030024, Invitrogen) and 20% methylcellulose (high viscosity, 4,000 cP; M0512, Sigma), and grown in 5% CO2 at 37°C. Exponentially growing PA-1 cells were incubated with an 8 µM dose of ETO for 20 h.
Small interfering RNA (siRNA)
TP53 protein Hs_TP53_9 FlexiTube siRNA (Qiagen) was used to silence TP53 expression and ON-TARGET plus non-targetting siRNA #1 (Dharmacon) was used as a negative control. PA-1 cells were transfected with siRNA using HiPerfect (Qiagen) according to the manufacturer’s protocol.
Small hairpin RNA (shRNA)
The following plasmids were purchased from Addgene: pMKO.1 puro TP53 plasmid (Addgene plasmid 19119) deposited by Bob Weinberg;41 and pMKO.1 puro (Addgene plasmid 8452) deposited by Bob Weinberg.42 PA-1 cells were transfected with plasmid using Fugene 6 (Promega) according to the manufacturer’s protocol and then selected using puromycin (0.3 µg/ml). After 2 wk, growing colonies were sub-cloned, stable cell lines established and maintained in media containing puromycin. Selection was removed 3 d prior to experiments.
Expression of HA-tagged forms of OCT4A and OCT4A-pseudogene-1
Expression vectors encoding HA-tagged forms of OCT4A or OCT4A-pseudogene-1 (OCT4A-PG1), a kind gift from Professor Aijun Hao,43 were transfected into HEK-293 cells using Fugene 6 (Promega) according to the manufacturer’s protocol.
DNA image cytometry
Cytospins were prepared and fixed in ethanol:acetone (1:1) for > 30 min at 4°C and air-dried. Slides were then treated with 5N HCl for 20 min at room temperature, washed in distilled water (5 × 1 min) and stained for 10 min with 0.05% toluidine blue in 50% citrate-phosphate McIlvain buffer pH 4. Slides were rinsed with distilled water, blotted dry and dehydrated by incubating twice in butanol for 3 min each at 37°C. Samples were then incubated twice in xylene for 3 min each at room temperature before being embedded in DPX. Slides were then evaluated using a Leitz Ergolux L03-10 microscope equipped with a calibrated Sony DXC 390P color video camera. DNA content was measured as the integral optical density in the green channel, using Image-Pro Plus 4.1 software (Media Cybernetics). Apoptotic cells were omitted from measurements. The stoichiometry of DNA staining was verified using the values obtained for metaphases compared with anaphases and telophases (ratio 2.0); the method error was estimated to be less than 5%.
DNA flow cytometry
Cells were trypsinized, harvested at indicated time points, washed in PBS and suspended in hypotonic fluorochrome solution [50 μg/ml propidium iodide (PI), 0.1% (w/v) sodium citrate, 0.1% (v/v) Triton X-100] and stored for at least 1 h in the dark at 4°C. Flow cytometry was performed using a FACScan (BD Biosciences) using Cell Quest Pro Software.
Two-channel flow cytometry
Cells were harvested at relevant time points, washed in cold PBS and fixed with 70% ethanol for 20 min at room temperature. After two washes in TBS, cells were permeabilized with TBS/4% bovine serum albumin (BSA)/0.1% Triton X-100 for 10 min at room temperature. Samples were then incubated with rabbit polyclonal anti-OCT4 antibody solution (ab19857, Abcam) (5 μg/ml) in TBS/4% BSA/0.1% Triton X-100 for 1 h at room temperature. Following two washes in TBS, cells were incubated with goat anti-rabbit Alexa Fluor 488 solution (1:200) in TBS/4%BSA/0.1% Triton X100, for 30 min in the dark. DNA was counterstained with 10 μg/ml PI solution in PBS, containing 200 μg/ml RNase (Sigma) and assessed by flow cytometry using a FACSCalibur (BD Biosciences) using Cell Quest Pro Software.
Immunofluorescent (IF) staining
IF was performed as described previously.44 Cytospins were fixed in methanol for 7 min at -20°C and dipped 10 times in ice-cold acetone. Slides were then washed three times in TBS 0.01% Tween 20 (TBST) for 5 min. Slides were subsequently blocked for 15 min in TBS, 0.05% Tween 20%, 1% BSA at room temperature. Samples were covered with 50 µL of TBS, 0.025% Tween 20%, 1% BSA containing primary antibody and incubated overnight at 4°C in a humidified chamber. Samples were then washed three times in TBST and covered with 50 µL of TBST containing the appropriate secondary antibody before incubation for 40 min at room temperature in the dark. Slides were washed three times for 5 min with PBST and once for 2 min in PBS. Samples were then counterstained with 0.25 µg/ml DAPI for 1 min, mounted using ProLong Gold antifade reagent (Invitrogen) and evaluated using a Leitz Ergolux L03-10 microscope equipped with a Sony DXC 390P color video camera. Antibodies and their source are listed in Tables 1 and 2. Note: when staining for tubulin, the fixation step using acetone was omitted and detergent was absent from all buffers. For TERT staining alone, the fixed cells were pre-treated with 2N HCl for 20 min at room temperature.
Table 1. Primary antibodies.
| Target | Description | Specifity/Immunogen | Product nr and manufacturer | Use* |
|---|---|---|---|---|
| TP53 |
Rabbit polyclonal |
Against full-length human p53 fusion protein |
#9282 Cell Signalling Technology |
W, IF |
| OCT |
Mouse mAb |
Peptide raised against amino acids 1–134 of Oct-3/4 of human origin non-cross-reactive with Oct-3/4 isoforms B and B1. |
sc-5279, Santa Cruz |
W, IF, F |
| OCT4A/B |
Rabbit polyclonal, ChIP grade |
Peptide derived from within residues 300 to the C terminus of human Oct4. |
ab19857, Abcam |
W, IF, F |
| NANOG |
Mouse mAb, clone NNG-811 |
Against human Nanog. |
N3038, Sigma |
IF |
| P16INK4A |
Rabbit polyclonal |
Human p16, C-terminal. |
ab7962, Abcam |
IF |
| P16INK4A |
Rabbit polyclonal |
Raised against a peptide mapping at the N terminus of p16 of human origin. |
sc-467, Santa Cruz |
W, IF |
| Aurora B kinase |
Rabbit polyclonal |
Peptide derived from within residues 1–100 of human Aurora B. |
ab2254, Abcam |
IF |
| Telomerase reverse transcriptase (TERT) |
Mouse mAb |
Against human TERT. |
ab5181, Abcam |
IF |
| P62 (SQSTM1) |
Rabbit polyclonal |
Epitope corresponding to amino acids 151–440 of SQSTM1 of human origin. |
sc-25575, Santa Cruz |
IF |
| pCHK2 (phosphorylated T68) |
Rabbit polyclonal |
Epitope around the phosphorylation site of Threonine 68 (VST pQE) of human Chk2. |
ab38461, Abcam |
W, IF |
| α-tubulin |
Mouse mAb |
Recognizes an epitope located at the C-terminal end of the α-tubulin isoform in a variety of organisms. |
T5168, Sigma |
IF |
| γ-H2AX |
Rabbit polyclonal |
Recognizes mammalian, yeast, D. melanogaster and X. laevis γ-H2AX. |
4411-PC-100, Trevigen |
IF |
| RAD51 |
Mouse mAb |
Targeted to amino acids 1–138 of human Rad51. |
ab213, Abcam |
IF |
| LC3B |
Rabbit polyclonal |
Peptide derived from within residues 1–100 of human LC3B. |
ab63817, Abcam |
IF |
| P21CIP1 |
Rabbit polyclonal |
Raised against a peptide mapping at the C terminus of p21 of human origin. |
sc-397, Santa Cruz |
W, IF |
| GAPDH |
Mouse mAb, clone 6C5 |
Rabbit muscle GAPDH. |
ab8245, Abcam |
W |
| β-actin | Rabbit polyclonal | Synthetic peptide derived from within residues 1–100 of human β-actin. | ab8227, Abcam | W |
W, western; IF, immunofluorescent staining; f, flow cytometry.
Table 2. Secondary antibodies.
| Antibody | Conjugate | Product nr and manufacturer | Use* |
|---|---|---|---|
| Goat anti-mouse IgG |
Alexa Fluor 488 |
A31619, Invitrogen |
IF, F |
| Goat anti-rabbit- IgG |
Alexa Fluor 594 |
A31631, Invitrogen |
IF |
| Goat anti-rabbit IgG |
HRP |
32460,Thermo Fisher Scientific |
W |
| Rabbit anti-mouse IgG |
HRP |
61-6520, Invitrogen |
W |
| Goat anti-rabbit IgG |
IRDye 800CW |
926-32211, IRDye Antibodies |
W |
| Goat anti-mouse IgG |
IRDye 800CW |
926-32210, IRDye Antibodies |
W |
| Goat anti-rabbit IgG | IRDye 680LT | 926-68021, IRDye Antibodies | W |
Detection of sa-β-galactosidase activity
The senescence β-galactosidase (sa-β-gal) staining kit (Cell Signaling, 9860) was used to detect sa-β-gal activity in cultured cells at indicated time points according to the manufacturer’s protocol.
Western blotting (whole-cell lysate)
Cells were harvested using trypsin digestion and lysed using RIPA buffer with protease inhibitor cocktail (Sigma P8340). Total protein was quantified using BCA protein assay kit (Pierce) and equal quantities of denatured protein were subjected to electrophoresis on SDS-polyacrylamide gels, blotted onto Immobilon-FC transfer membrane and probed with specific primary antibodies listed in Table 1 and secondary antibodies listed in Table 2. The signal was visualized using a LICOR Odyssey imaging system.
Subcellular fractionation
For cellular fractionation, the Subcellular Protein Fractionation Kit for Cultured Cells (Thermo Scientific) was used according to the manufacturer’s instructions. Cytoplasmic, membrane, nuclear soluble, karyosol and chromatin-bound protein extracts were obtained. Protein concentrations were determined by Bio-Rad (Bio-Rad Inc.) protein assay, using a BSA standard set (Fermentas MBI) for quantitation. Proteins (10 or 15 μg) were separated on 10, 12, 5 or 20% SDS PAGE gels, followed by electrophoretic transfer onto BA85 nitro-cellulose membranes (Schleicher and Schuell GmbH) overnight. Equal protein loading in each lane was verified by Ponceau S staining. Blots were probed with specific primary antibodies listed in Table 1 and secondary antibodies listed in Table 2. The signal was visualized using the Chemiluminescent Nucleic Acid Detection Module (Thermo Scientific).
RT-PCR analysis of Oct4-splicing forms
Total RNA was extracted from cells using TRIZOL (Invitrogen). cDNA was synthesized using First Strand cDNA Synthesis Kit (Fermentas MBI) according to the manufacturer’s protocol and then diluted 10 times. The absence of contamination with genomic DNA was verified by PCR using actin primers as described.11 cDNA from peripheral blood lymphocytes (PBL) as a control of somatic cells was kindly provided by Dr. Inta Vasiljeva. Amplification was performed with 1–4 μl of diluted cDNA and the following primers, β-actin F/R; Oct4A AF/AR; Oct4B/B1 BF1/BR2 under conditions previously described.11 Amplified PCR products were analyzed on an agarose gel after various PCR cycles and their identity determined by direct sequencing after ExoI/SAP treatment (Fermentas, MBI) using the fluorescent Big DyeTerminator v. 3.1 Cycle Sequencing protocol on a 3130xl Genetic Analyzer (Applied Biosystems).
Methods of statistical analysis
Statistical analysis was performed in Minitab. A paired t-test was used to calculate the statistical significance of difference of means where appropriate. Statistical significance was accepted when p < 0.05. Graphs were plotted in GraphPad Prism 5 and Statistica 6.
Supplementary Material
Acknowledgments
The authors would like to thank Professor Hao and Dr. Zhao, Dept. of Histology and Embryology, Shandong University School of Medicine for the kind gift of the OCT4A and OCT4A-Psg-1 vector constructs and Dr. Pawel Zayakin (Latvian Biomedical Centre) for insightful discussions. T.R.J. was funded through an MRC PhD studentship and the Gerald Kerkut Charitable Trust awarded to P.A.T. and M.S.C.; J.E. was funded by the Latvian National Research Programme 2010–2013 “BIOMEDICINE” and AH by European Social Fund within the project “Support for Doctoral Studies at University of Latvia.” Exchange visits between Riga and Southampton were supported by the Royal Society of London. The publishing costs associated with this article are in part provided by the ERDF project no. 2DP/2.1.1.2.0/10/ APIA/VIAA/004.
Glossary
Abbreviations:
- EC
embryonal carcinoma
- ES
embryonic stem
- ETO
etoposide
- IF
immunofluorescent
- iPS
induced pluripotent stem
- LC3
microtubule-associated protein 1 light chain 3
- MI
mitotic index
- NT
non-treated
- ntg
non-target
- pCHK2
phosphorylated CHK2
- PI
propidium iodide
- siRNA
small interfering RNA
- shRNA
small hairpin RNA
- Sa-β-gal
senescence-associated beta-galactosidase
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/23285
References
- 1.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–84. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 3.Jordan CT, Guzman ML, Noble M. Cancer stem cells. N Engl J Med. 2006;355:1253–61. doi: 10.1056/NEJMra061808. [DOI] [PubMed] [Google Scholar]
- 4.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 5.Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–56. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 7.Huangfu DW, Osafune K, Maehr R, Guo W, Eijkelenboom A, Chen S, et al. Induction of pluripotent stem cells from primary human fibroblasts with only Oct4 and Sox2. Nat Biotechnol. 2008;26:1269–75. doi: 10.1038/nbt.1502. [DOI] [PubMed] [Google Scholar]
- 8.Ge N, Lin HX, Xiao XS, Guo L, Xu HM, Wang X, et al. Prognostic significance of Oct4 and Sox2 expression in hypopharyngeal squamous cell carcinoma. J Transl Med. 2010;8:94. doi: 10.1186/1479-5876-8-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saigusa S, Tanaka K, Toiyama Y, Yokoe T, Okugawa Y, Ioue Y, et al. Correlation of CD133, OCT4, and SOX2 in rectal cancer and their association with distant recurrence after chemoradiotherapy. Ann Surg Oncol. 2009;16:3488–98. doi: 10.1245/s10434-009-0617-z. [DOI] [PubMed] [Google Scholar]
- 10.Xiang R, Liao D, Cheng T, Zhou H, Shi Q, Chuang TS, et al. Downregulation of transcription factor SOX2 in cancer stem cells suppresses growth and metastasis of lung cancer. Br J Cancer. 2011;104:1410–7. doi: 10.1038/bjc.2011.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salmina K, Jankevics E, Huna A, Perminov D, Radovica I, Klymenko T, et al. Up-regulation of the embryonic self-renewal network through reversible polyploidy in irradiated p53-mutant tumour cells. Exp Cell Res. 2010;316:2099–112. doi: 10.1016/j.yexcr.2010.04.030. [DOI] [PubMed] [Google Scholar]
- 12.Banito A, Rashid ST, Acosta JC, Li S, Pereira CF, Geti I, et al. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009;23:2134–9. doi: 10.1101/gad.1811609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huna A, Salmina K, Jascenko E, Duburs G, Inashkina I, Erenpreisa J. Self-Renewal signalling in presenescent tetraploid IMR90 cells. J Aging Res. 2011;2011:103253. doi: 10.4061/2011/103253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blagosklonny MV. Cell senescence and hypermitogenic arrest. EMBO Rep. 2003;4:358–62. doi: 10.1038/sj.embor.embor806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leontieva OV, Blagosklonny MV. DNA damaging agents and p53 do not cause senescence in quiescent cells, while consecutive re-activation of mTOR is associated with conversion to senescence. Aging (Albany NY) 2010;2:924–35. doi: 10.18632/aging.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci USA. 2010;107:9660–4. doi: 10.1073/pnas.1002298107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blagosklonny MV. Cell cycle arrest is not senescence. Aging (Albany NY) 2011;3:94–101. doi: 10.18632/aging.100281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boheler KR. Stem cell pluripotency: a cellular trait that depends on transcription factors, chromatin state and a checkpoint deficient cell cycle. J Cell Physiol. 2009;221:10–7. doi: 10.1002/jcp.21866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rich JN. Cancer stem cells in radiation resistance. Cancer Res. 2007;67:8980–4. doi: 10.1158/0008-5472.CAN-07-0895. [DOI] [PubMed] [Google Scholar]
- 20.Sperger JM, Chen X, Draper JS, Antosiewicz JE, Chon CH, Jones SB, et al. Gene expression patterns in human embryonic stem cells and human pluripotent germ cell tumors. Proc Natl Acad Sci USA. 2003;100:13350–5. doi: 10.1073/pnas.2235735100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Josephson R, Ording CJ, Liu Y, Shin S, Lakshmipathy U, Toumadje A, et al. Qualification of embryonal carcinoma 2102Ep as a reference for human embryonic stem cell research. Stem Cells. 2007;25:437–46. doi: 10.1634/stemcells.2006-0236. [DOI] [PubMed] [Google Scholar]
- 22.te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002;62:1876–83. [PubMed] [Google Scholar]
- 23.Sabisz M, Skladanowski A. Cancer stem cells and escape from drug-induced premature senescence in human lung tumor cells: implications for drug resistance and in vitro drug screening models. Cell Cycle. 2009;8:3208–17. doi: 10.4161/cc.8.19.9758. [DOI] [PubMed] [Google Scholar]
- 24.Lifantseva N, Koltsova A, Krylova T, Yakovleva T, Poljanskaya G, Gordeeva O. Expression patterns of cancer-testis antigens in human embryonic stem cells and their cell derivatives indicate lineage tracks. Stem Cells Int. 2011;2011:795239. doi: 10.4061/2011/795239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarraf S, Tejada R, Abawi M, Oberst M, Dennis T, Simon KC, et al. The human ovarian teratocarcinoma cell line PA-1 demonstrates a single translocation: analysis with fluorescence in situ hybridization, spectral karyotyping, and bacterial artificial chromosome microarray. Cancer Genet Cytogenet. 2005;161:63–9. doi: 10.1016/j.cancergencyto.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 26.Gao C, Miyazaki M, Li JW, Tsuji T, Inoue Y, Namba M. Cytogenetic characteristics and p53 gene status of human teratocarcinoma PA-1 cells in 407-445 passages. Int J Mol Med. 1999;4:597–600. doi: 10.3892/ijmm.4.6.597. [DOI] [PubMed] [Google Scholar]
- 27.Zhang XN, Jaramillo M, Singh S, Kumta P, Banerjee I. Analysis of regulatory network involved in mechanical induction of embryonic stem cell differentiation. PLoS One. 2012;7:e35700. doi: 10.1371/journal.pone.0035700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chiodi I, Mondello C. Telomere-independent functions of telomerase in nuclei, cytoplasm, and mitochondria. Front Oncol. 2012;2:133. doi: 10.3389/fonc.2012.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Erenpreisa J, Salmina K, Huna A, Kosmacek EA, Cragg MS, Ianzini F, et al. Polyploid tumour cells elicit paradiploid progeny through depolyploidizing divisions and regulated autophagic degradation. Cell Biol Int. 2011;35:687–95. doi: 10.1042/CBI20100762. [DOI] [PubMed] [Google Scholar]
- 30.Atlasi Y, Mowla SJ, Ziaee SAM, Gokhale PJ, Andrews PW. OCT4 spliced variants are differentially expressed in human pluripotent and nonpluripotent cells. Stem Cells. 2008;26:3068–74. doi: 10.1634/stemcells.2008-0530. [DOI] [PubMed] [Google Scholar]
- 31.Asadi MH, Mowla SJ, Fathi F, Aleyasin A, Asadzadeh J, Atlasi Y. OCT4B1, a novel spliced variant of OCT4, is highly expressed in gastric cancer and acts as an antiapoptotic factor. Int J Cancer. 2011;128:2645–52. doi: 10.1002/ijc.25643. [DOI] [PubMed] [Google Scholar]
- 32.Singh AM, Hamazaki T, Hankowski KE, Terada N. A heterogeneous expression pattern for NANOG in embryonic stem cells. Stem Cells. 2007;25:2534–42. doi: 10.1634/stemcells.2007-0126. [DOI] [PubMed] [Google Scholar]
- 33.Kalmar T, Lim C, Hayward P, Muñoz-Descalzo S, Nichols J, Garcia-Ojalvo J, et al. Regulated fluctuations in NANOG expression mediate cell fate decisions in embryonic stem cells. PLoS Biol. 2009;7:e1000149. doi: 10.1371/journal.pbio.1000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chambers I, Silva J, Colby D, Nichols J, Nijmeijer B, Robertson M, et al. Nanog safeguards pluripotency and mediates germline development. Nature. 2007;450:1230–4. doi: 10.1038/nature06403. [DOI] [PubMed] [Google Scholar]
- 35.Lev Bar-Or R, Maya R, Segel LA, Alon U, Levine AJ, Oren M. Generation of oscillations by the p53-Mdm2 feedback loop: a theoretical and experimental study. Proc Natl Acad Sci USA. 2000;97:11250–5. doi: 10.1073/pnas.210171597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lahav G, Rosenfeld N, Sigal A, Geva-Zatorsky N, Levine AJ, Elowitz MB, et al. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet. 2004;36:147–50. doi: 10.1038/ng1293. [DOI] [PubMed] [Google Scholar]
- 37.Weaver BAA, Cleveland DW. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell. 2005;8:7–12. doi: 10.1016/j.ccr.2005.06.011. [DOI] [PubMed] [Google Scholar]
- 38.Levasseur DN, Wang J, Dorschner MO, Stamatoyannopoulos JA, Orkin SH. Oct4 dependence of chromatin structure within the extended Nanog locus in ES cells. Genes Dev. 2008;22:575–80. doi: 10.1101/gad.1606308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E, et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol. 2005;7:165–71. doi: 10.1038/ncb1211. [DOI] [PubMed] [Google Scholar]
- 40.Huang S. Non-genetic heterogeneity of cells in development: more than just noise. Development. 2009;136:3853–62. doi: 10.1242/dev.035139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Godar S, Ince TA, Bell GW, Feldser D, Donaher JL, Bergh J, et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62–73. doi: 10.1016/j.cell.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003;9:493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao SD, Yuan QH, Hao HB, Guo YJ, Liu SM, Zhang YM, et al. Expression of OCT4 pseudogenes in human tumours: lessons from glioma and breast carcinoma. J Pathol. 2011;223:672–82. doi: 10.1002/path.2827. [DOI] [PubMed] [Google Scholar]
- 44.Erenpreisa J, Cragg MS, Salmina K, Hausmann M, Scherthan H. The role of meiotic cohesin REC8 in chromosome segregation in gamma irradiation-induced endopolyploid tumour cells. Exp Cell Res. 2009;315:2593–603. doi: 10.1016/j.yexcr.2009.05.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.