

Abstract
Circumstantial evidence suggests that colon carcinogenesis can ensue the transient tetraploidization of (pre-)malignant cells. In line with this notion, the tumor suppressors APC and TP53, both of which are frequently inactivated in colon cancer, inhibit tetraploidization in vitro and in vivo. Here, we show that—contrarily to their wild-type counterparts—Tp53−/− colonocytes are susceptible to drug-induced or spontaneous tetraploidization in vitro. Colon organoids generated from tetraploid Tp53−/− cells exhibit a close-to-normal morphology as compared to their diploid Tp53−/− counterparts, yet the colonocytes constituting these organoids are characterized by an increased cell size and an elevated expression of the immunostimulatory protein calreticulin on the cell surface. The subcutaneous injection of tetraploid Tp53−/− colon organoids led to the generation of proliferating tumors in immunodeficient, but not immunocompetent, mice. Thus, tetraploid Tp53−/− colonocytes fail to survive in immunocompetent mice and develop neoplastic lesions in immunocompromised settings only. These results suggest that tetraploidy is particularly oncogenic in the context of deficient immunosurveillance.
Keywords: apoptosis, cell cycle, cytochalasin D, mitotic catastrophe, nocodazole, p53
Introduction
One prominent mechanism of tumorigenesis involves an initial event of illicit tetraploidization, corresponding to the acquisition by a single cell of two copies of the normal, diploid genome.
Tetraploidization can stem from (and proceed toward higher order polyploidization owing to) (1) unscheduled cell-to-cell fusion between somatic cells,1 (2) endomitosis, a process whereby cells with duplicated chromosomes fail to complete mitosis,2,3 or (3) endocycling, a cell cycle alteration whereby cells undergo two consecutive rounds of DNA replication (S phases) that are not separated by mitosis.4 This latter mechanism, which may occur following prolonged DNA damage or owing to telomere dysfunction, results in the generation of tetraploid cells containing one single set of polytenic chromosomes.4
Tetraploidy has been observed in the early stages of esophageal, colorectal, mammary and cervical cancer.5 According to currently accepted models, tetraploidy drives carcinogenesis by acting as a stochastic generator of genomic instability. Indeed, tetraploid cells experience intrinsic problems in maintaining their genome and often gain or lose chromosomes during aberrant rounds of bipolar mitosis3 and/or revert to an aneuploid (most often near-to-diploid) state upon one successful multipolar division. This process, which is known as “polyploidization/depolyploidization cascade,” is normally avoided by multiple oncosuppressive mechanisms, including mitotic catastrophe.1,2,5-8 In line with this notion, inactivating mutations of two crucial oncosuppressor genes, i.e., TP53 and RB1, have been correlated with the accumulation of tetraploid cells in a large spectrum of human neoplasms.9 Moreover, a plethora of additional oncogenes and tumor-suppressor genes has been shown to control the generation, survival and/or proliferation of tetraploid cells.1,2,5,6,8,10,11 It has recently been shown that primary epithelial cells that are previously rendered tetraploid in vitro, but not their parental diploid counterparts, can form tumors in vivo.12 These results provide strong evidence in favor of the direct implication of tetraploidy in oncogenesis.
We have recently demonstrated that the polyploidization/depolyploidization cascade is controlled not only by cell-intrinsic barriers, but also by cell-extrinsic mechanisms involving the immune system.13 Indeed, at odds with their parental near-to-diploid counterparts, near-to-tetraploid cancer cells injected into syngenic immunocompetent mice often fail to form tumors. When such tumors develop, they invariably exhibit signs of immunoselection, including a reduction in nuclear size and DNA content. In contrast, near-to-tetraploid cancer cells normally form tumors when injected into immunodeficient mice, and as these tumors progress-malignant cells do not exhibit significant alterations in ploidy, that is, they maintain a near-to-tetraploid state.13
Based on these results, we have postulated a functional link between tetraploidy and immunosurveillance. Indeed, similar to cancer cells that undergo immunogenic cell death, a functionally peculiar type of apoptosis that can elicit antitumor cognate immune responses,14 tetraploid cells (in steady-state conditions) experience high degrees of endoplasmic reticulum stress, leading to the exposure on the cell surface of an immunostimulatory protein, calreticulin (CRT).13 Driven by these premises and incognita, we investigated the possibility that tetraploidization would drive colon carcinogenesis. Using a method allowing for the isolation and culture of colon structures (hereafter referred to as colon organoids),15,16 we found that the absence of Tp53 is permissive for the generation and survival of tetraploid colonocytes that form morphologically near-to-normal colon organoids. Tetraploid Tp53−/− colonocytes, however, exhibit increased baseline levels of CRT on their surface, and the corresponding organoids are able to form tumors once injected in immunodeficient, but not immunocompetent, mice.
Results and Discussion
The absence of Tp53 is permissive for colonocyte tetraploidization
Small intestine or colon organoids were obtained from wild-type (WT) C57Bl/6 mice and their Tp53−/− counterparts following established protocols,15,16and they were cultured in the presence of three distinct tetraploidizing agents, nocodazole (which acts on microtubuli to perturb mitotic spindle dynamics), dihydrocytochalasin B (DCB) or cytochalasin D (both of which interfere with the actin cytoskeleton, hence preventing cytokinesis). Forty-eight hours later, organoids were dissociated to generate single-cell suspensions, and their DNA content was determined by cytofluorometry upon staining with the chromatinophylic dye Hoechst 33342.17 While WT colon organoids failed to generate a sizeable fraction of viable polyploid cells in these conditions, Tp53−/− colonocytes were susceptible to drug-induced tetraploidization (Fig. 1A and B). Similar to WT colon organoids, small intestine cells from both WT and Tp53−/− mice, were refractory to polyploidization (Fig. 1C). Diploid and tetraploid Tp53−/− cells were then purified from colon organoids by means of fluorescence-activated cell sorting (FACS), based on DNA content. As expected, tetraploid Tp53−/− colonocytes (which contain a 4n DNA content in the G1 phase of the cell cycle and an 8n DNA content in the G2/M phases) were larger than their diploid Tp53−/− counterparts (Fig. 1D). The cytofluorometric purification of diploid colonocytes from Tp53−/− colon organoids treated with cytochalasin D for 48 h yielded a population of initially diploid cells that tended to undergo spontaneous tetraploidization upon long-term culture (Fig. 2A and B). Of note, colonocytes from untreated Tp53−/− colon organoids also tended to spontaneously acquire a tetraploid state upon prolonged culture (Fig. 2C).

Figure 1. The absence of Tp53 facilitates the drug-induced tetraploidization of colonocytes. (A) DNA content analysis of cells obtained from Tp53−/− colon organoids that have been left untreated or treated with 0.6 μg/mL cytochalasin D for 48 h. (B and C) Percentage of polyploid cells (DNA content > 4n, means ± SEM, n = 1–3) developing in wild-type (WT) and Tp53−/− colon (B) or small intestine organoids (C) upon exposure to the indicated concentrations of nocodazole (Noco), dihydrocytochalasin B (DCB) and cytochalasin D (CytD). *p < 0.05, **p < 0.01, as compared to equally treated WT organoids. (D) Morphological aspects of freshly sorted diploid and tetraploid cells from Tp53−/− colon organoids, as determined by phase contrast microscopy. Representative microphotographs (scale bar = 10 μm) and quantitative data (means ± SEM, n = 10) are shown. ***p < 0.001.

Figure 2. Spontaneous generation of tetraploid cells from diploid Tp53−/− colon organoids. (A–C) DNA content of cells obtained from Tp53−/− colon organoids at various time points after isolation (day 0, D0). At D50, 2n cells were sorted upon transient exposure (48 h) to 0.6 μg/mL cytochalasin D (CytD), and ploidy was followed over time. Representative profiles and the sorting gate are indicated in panel (A). The DNA content of cells isolated from Tp53−/− diploid colon organoids as sorted on D50 upon CytD exposure is illustrated in (B). (C) depicts the DNA content of cells isolated from untreated Tp53−/− colon organoids over time.
Taken together, these results demonstrate that the absence of Tp53 renders colonocytes susceptible to drug-induced and spontaneous tetraploidization.
Normal development but increased calreticulin exposure on tetraploid colon organoids in vitro
Upon purification from Tp53−/− colon organoids by cytofluorometry, diploid and tetraploid cells were seeded in limiting dilution assays to generate new organoids that would arise in vitro from one or a few stem cells.16 In these conditions, diploid and tetraploid organoids developed with similar kinetics and exhibited similar morphologies (although the latter tended to adopt a less regular overall shape before the first dissociation) (Fig. 3A and B), as cells maintained a stable diploid or tetraploid DNA content (Fig. 3C and D). Along similar lines, 10 d after cell sorting, the size of organoids arising from diploid and tetraploid cells was comparable (Fig. 3E). Metaphase chromosome spreads confirmed that tetraploid cells contained twice as many chromosomes as their diploid counterparts (Fig. 3F and G). In addition, while we observed no difference in the proliferation rate of diploid and tetraploid colon organoids, as assessed by the percentage of cells incorporating the thymidine analog 5-ethynyl-2'-deoxyuridine (EdU, a marker of active DNA synthesis)17 (Fig. 4A and B), tetraploid colonocytes maintained a larger cell size than their diploid counterparts (Fig. 4C). As hyperploidy is closely linked to the translocation of CRT to the outer leaflet of the plasma membrane,13 we determined whether tetraploid Tp53−/− colonocytes would expose higher levels of CRT on the cell surface than their diploid counterparts. Hence, colon organoids were immunostained with an anti-CRT antibody in non-permeabilizing conditions, to avoid the detection of intracellular CRT. This revealed that tetraploid Tp53−/− colon organoids expose higher levels of CRT at the cell surface than their diploid Tp53−/− counterparts (Fig. 5A and B).

Figure 3. Tetraploid Tp53−/− colon organoids are viable and genetically stable. (A and B) Growth of organoids originating from a freshly sorted (D0) diploid (A) or tetraploid (B) Tp53−/− cell (scale bar = 20 μm). (C) DNA content of cells obtained from diploid or tetraploid Tp53−/− clonal colon organoids 18 d after sorting. (D) DNA content of cells from a tetraploid Tp53−/− clonal colon organoid at the indicated time point after cloning. (E) Mean diameter of diploid or tetraploid Tp53−/− colon organoids 10 d after sorting (mean ± SEM, n = 15). (F) Representative chromosomal spread of a tetraploid cell (number of chromosomes = 86, scale bar = 5 μm). (G) Number of chromosomes per cell in diploid and tetraploid Tp53−/− colon organoids (means ± SEM, n = 15, ***p < 0.001).
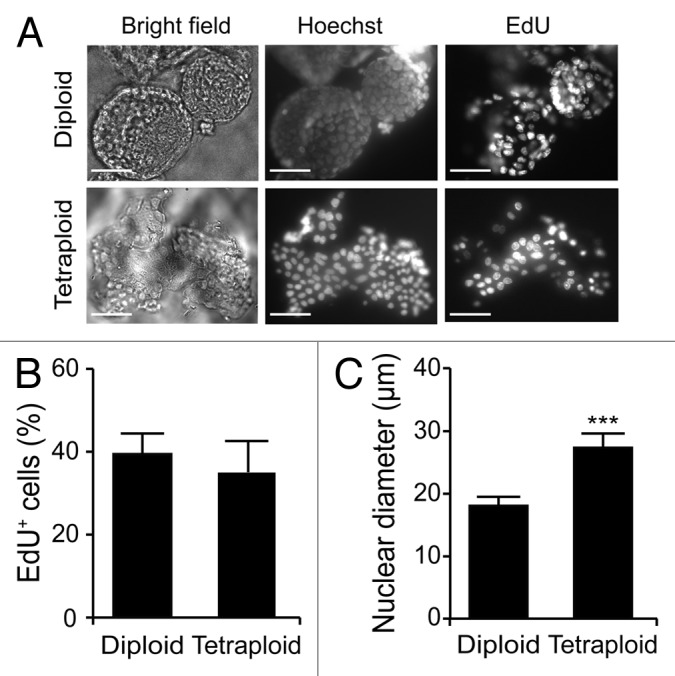
Figure 4. Proliferative rate of diploid and tetraploid organoids. (A–C) Diploid and tetraploid Tp53−/− colon organoids were cultured in the presence of the thymidine analog 5-ethynyl-2'-deoxyuridine (EdU) for 1 h and then processed for the fluorescence microscopy-assisted detection of EdU+ cells. Hoechst 33342 was employed for nuclear counterstaining. (A) reports representative images (scale bar = 100 μm), while in (B and C) quantitative data on the percentage of EdU+ cells and on nuclear diameter are illustrated (means ± SEM, n = 15, ***p < 0.001).
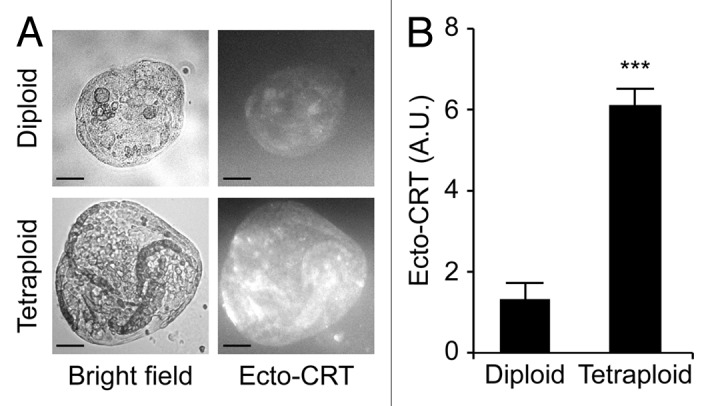
Figure 5. Increased calreticulin exposure on tetraploid Tp53−/− colon organoids. (A) Representative immunofluorescence microphotographs of diploid and tetraploid Tp53−/− colon organoids stained to visualize surface-exposed calreticulin (ecto-CRT, scale bar = 100 μm). (B) Quantification of ecto-CRT-dependent fluorescence of diploid and tetraploid Tp53−/− colon organoids (means ± SEM, n = 20, ***p < 0.001).
Altogether, these results indicate that tetraploid colonocytes can conserve the ability to generate colon organoids in vitro (hence maintaining stem cell features) as they manifest immune-related alterations that characterize other tetraploid cell types.
Tetraploid Tp53−/− colon organoids form tumors in immunodeficient mice
To investigate the tumorigenic potential of tetraploid Tp53−/− organoids, we embedded them in 20% matrigel and injected them s.c. into syngenic immunocompetent C57Bl/6 hosts or severely immunodeficient mice lacking the Rag2 recombinase and the common γ chain of cytokine receptors (Rag2−/− γc−/−, commonly known as Rag γ mice).18 In C57Bl/6 hosts, tetraploid Tp53−/− organoids formed small tumors that failed to expand after 3 wk and became entirely fibrotic, accompanied by the disappearance of all discernible epithelial elements (Fig. 6A and B). In stark contrast, tetraploid Tp53−/− organoids evolving in immunodeficient Rag γ mice proliferated for an extended period as tumors conserved epithelial structures. These epithelial cells formed tubular monolayers and occasional multilayers exhibiting multiple histological signs of malignancy, including visible metaplasia, cellular pleomorphy and local invasion. As compared to normal colonocytes from C57Bl/6 mice, tetraploid Tp53−/− colonocytes growing in immunodeficient mice contained larger nuclei and tended to exhibit a more heterogeneous shape (Fig. 6B and C). In this context, we observed a positive correlation between nuclear size and the degree of endoplasmic reticulum stress, as monitored by the phosphorylation status of the eukaryotic initiation translation factor 2α (eIF2α) (Fig. 6C). Altogether, these results indicate that tetraploid Tp53−/− colon stem cells can grow ectopically in immunodeficient, but not in immunocompetent, mice as they acquire a (pre-)malignant phenotype.

Figure 6. In vivo growth of tetraploid Tp53−/− colon organoids in immunocompetent and immunodeficient mice. (A) Growth of spontaneously tetraploid Tp53−/− colon organoids inoculated s.c. in immunocompetent (C57Bl/6) and immunodeficient (Rag γ) mice on day 0 (means ± SEM, n = 5, *p < 0.05, **p < 0.01). (B and C) Tumors were recovered at the end of the experiment shown in (A) and subjected to immunohistochemistry for the visualization of phosphorylated eukaryotic initiation factor 2α (P-eIF2α) or hematoxilin and eosin (HES) staining. Normal colons obtained from C57Bl/6 mice served as controls. (B) illustrates representative histologies of tumors generated by Tp53−/− colon organoids in C57Bl/6 and Rag γ mice. In (C), quantitative results on nuclear diameter and eIF2α phosphorylation levels are provided (means ± SEM, n = 100 cells from no less than five different tumors. Left panel: *,#p < 0.05; **,##p < 0.01, as compared to normal colonocytes and tumor cells recovered from C57Bl/6, respectively. Right panel: **p < 0.01).
Concluding Remarks
Previous work has established that defects in the TP53 system are permissive for the survival and proliferation of tetraploid mammary epithelial cells.2,19 Moreover, Tp53−/− mammary epithelial cells manipulated to become tetraploid in vitro (by transient exposure to cytochalasin D) have been shown to generate tumor cells in vivo upon transfer into immunodeficient nu/nu mice.2 However, in the latter study, no comparison was done to determine the tumorigenic potential of tetraploid mammary epithelial cells in immunocompetent vs. immunodeficient hosts. Here, we recapitulated and extended these findings, showing that the absence of Tp53 allows for the tetraploidization of colon stem cells. Conversely, in our hands, small intestinal epithelial stem cells were refractory to tetraploidization, independent from their Tp53 status. The reasons underlying this apparent discrepancy remain to be elucidated.
Tetraploid Tp53−/− colonocytes were able to form apparently normal colon organoids in vitro. As observed in cancer cell lines as well as in primary fibroblasts, the tetraploidization of Tp53−/− colonocytes resulted in the exposure of CRT on the cell surface. The mechanisms leading to this phenotypic alteration have not been entirely understood yet, although they have been linked to elevated degrees of endoplasmic reticulum stress.13,20 We have previously demonstrated that tetraploid cancer cells bearing high levels of CRT on their surface are way less efficient at inducing sizeable tumors in immunocompetent mice than in immunodeficient hosts.13 Along similar lines, we found that tetraploid Tp53−/− colon organoids fail to proliferate in immunocompetent mice, whereas they survive and proliferate in immunocompromised settings. Under these conditions, tetraploid Tp53−/− colon organoids form lesions exhibiting histopathological features of malignancy as well as immunohistochemical signs of endoplasmic reticulum stress.
Based on these findings, we surmise that a combination of tetraploidy and immunodeficiency may be particularly efficient at favoring colon carcinogenesis. Intriguingly, loss-of-function mutations of adenomatous polyposis coli (APC), one of the main oncosuppressor genes that limits colon carcinogenesis, promote the tetraploidization of intestinal epithelial cells.21-23 Thus, it is tempting to speculate that cell-autonomous events that favor or facilitate tetraploidization, including (but not limited to) the inactivation of APC or TP53, may contribute to colon carcinogenesis via this mechanism. An abundant amount of literature indicates the existence of an immunosurveillance system that dictates the long-term fate of colorectal carcinoma patients by virtue of its ability to control colon carcinogenesis.24-27 Future studies on patient specimens are required to explore the anatomical and functional relationship between tetraploid colonocytes, their malignant derivatives and immune effectors.
Materials and Methods
Reagents
Unless specified otherwise, reagents were purchased from Sigma-Aldrich. Matrigel was obtained from BD Biosciences, culture supplements including recombinant mouse epidermal growth factor (EGF) from Invitrogen, recombinant mouse noggin from Peprotech and A-83-01 from Tocris Bioscience. Human recombinant R-spondin 1-conditioned medium was obtained with 293T cells transfected with a plasmid encoding R-spondin 1 (kind gift of Dr. Wim De Lau), and this medium was diluted 1:10 in Advanced DMEM (Invitrogen). Mouse recombinant Wnt3a-conditioned medium28 was obtained from the L-Wnt3a cell line29 and diluted 1:2 in advanced DMEM.
Organoid culture
Organoids were established from small intestines and colons freshly isolated from WT and Tp53−/− C57Bl/6 mice. The isolation and maintenance of small intestine and colon organoids have been previously described.15,16
Cytofluorometry
For the assessment of cell cycle distribution, organoids were harvested with cold TrypLE express (Invitrogen), kept for 10 min at 37°C and then subjected to mechanical disruption to obtain single-cell suspensions. Thereafter, cells were washed twice with cold fetal bovine serum (10% in PBS) and incubated for 30 min at 37°C with 10 μg/mL Hoechst 33342 (Molecular Probes-Invitrogen).30,31 Samples were analyzed on a Gallios cytometer (Beckman Coulter), and first-line statistical analyses were performed by means of the Kaluza software (Beckman Coulter). Single viable cells were discriminated from doublets upon gating on forward scatter, side scatter and pulse width.
Generation of tetraploid cells
Parental diploid organoids were treated for 48 h with 0.3 or 0.6 µg/mL cytochalasin D, 2 or 4 µM DCB or 25, 50 or 100 nM nocodazole, and then either analyzed by cytofluorometry for DNA content or cultured for 1 wk in drug-free culture medium. To generate diploid and tetraploid clonal organoids, organoids were treated for 48 h with 0.6 µg/mL cytochalasin D, and cells with a 2n or 8n DNA content, respectively, were sorted by means of a FACSVantage cell sorter (BD Biosciences), as previously described.32 Diploid and tetraploid cells were then collected and embedded in Matrigel supplemented with 10 μM Y-27632.16 One week later, growing organoids were manually isolated with a micropipette and cultured separately.
Immunofluorescence microscopy
For cell surface staining, organoids were placed on ice, washed once with PBS and incubated with CRT-specific rabbit polyclonal antibodies (#ab2907, from Abcam) in cold blocking buffer (5% bovine serum albumin, w/v in PBS) for 30 min. Organoids were then washed twice in cold PBS, incubated for 30 min with anti-rabbit secondary antibodies (diluted 1:500 in cold blocking buffer), washed again with PBS and mounted on slides. Fluorescence microscopy was performed on a DM IRE2-inverted fluorescence microscope (Leica Microsystems) equipped with a Cool Snap Ez camera (Roper Scientific, Princeton Instruments).
Chromosomal spreads
Chromosomal spreads were realized according to standard protocols.33 Briefly, organoids were treated with 1 μM nocodazole for 6 h to enrich mitotic cells, collected and subjected to hypotonic lysis by incubation in 75 mM KCl for 12 min at 37°C. Upon removal of the hypotonic solution, cells were fixed in freshly prepared Carnoy solution (3:1 ethanol:acetic acid) and stored at -20°C. Fixed cells were then dropped onto glass slides, air-dried and mounted with 4',6-diamidino-2-phenylindole (DAPI)-containing Vectashield® mounting medium (Vector Labs). Chromosomal spreads were evaluated on a DM IRE2 inverted fluorescence microscope equipped with a Cool Snap Ez camera.
EdU staining
EdU staining was performed following the manufacturer’s recommendations (Click-IT kit; Invitrogen).17,34
Mice and genotyping
Mice were kept in pathogen-free conditions, and experiments respected both the Federation of European Laboratory Animal Science Association (FELASA) guidelines and the EU Directive 63/2010. In addition, all experimental protocols were approved by the Ethical Committee of the Institut Gustave Roussy (CEEA IRCIV/IGR n° 26, registered at the French Ministry of Research). WT C57Bl/6 mice were obtained from Harlan France, while Rag γ and Tp53−/− C57Bl/6 animals from CNRS UMR6218. To obtain primary Tp53−/− cells, C57Bl/6 mice bearing a heterozygous deletion of Tp53 (CNRS UMR6218) were cross-bred to obtain Tp53+/+ (WT) and Tp53−/− animals. F1 mice were genotyped by standard PCR procedures, as previously described.13
Tumorigenicity assays
For tumorigenicity assays, 500 tetraploid organoids were injected s.c. into C57Bl/6 or Rag γ mice (both exhibiting a MHC class I haplotype H-2b) in 20% Matrigel. Tumor size was routinely evaluated using a common caliper, and after 50 d, tumors were aseptically harvested for histological and immunohistochemical assays.
Histology and immunohistochemistry
Samples from tumors were cut in two parts, one fixed in 10% neutral buffered formalin and embedded in paraffin, the other fixed in 10% neutral buffered formalin for 4 h, dehydrated in 30% saccharose (overnight, 4°C) and embedded in Tissue-Tek® cryomold (Sakura Finetek). Sections of 10 μm were then stained with hematoxylin and eosin following standard protocols.35 For the detection of phosphorylated eIF2α, tissue sections were stained with monoclonal anti-phospho-eIF2α(S51) antibodies (#3597, Cell Signaling Technology Inc.) following previously established protocols.13
Statistical procedures
Unless otherwise specified, experiments were performed at least three times, yielding similar results. Data were analyzed by means of Microsoft Excel (Microsoft Co.), and statistical significance was assessed with unpaired Student’s t-tests. p values were considered significant when < 0.05.
Acknowledgments
Authors are supported by the European Commission (ArtForce); Agence National de la Recherche (ANR); Ligue contre le Cancer (Equipe labellisée); Fondation pour la Recherche Médicale (FRM); Institut National du Cancer (INCa); LabEx Immuno-Oncologie; Fondation de France; Fondation Bettencourt-Schueller; AXA Chair for Longevity Research; Cancéropole Ile-de-France and Paris Alliance of Cancer Research Institutes (PACRI).
Glossary
Abbreviations:
- APC
adenomatous polyposis coli
- CRT
calreticulin
- DCB
dihydrocytochalasin B
- EGF
epidermal growth factor
- eIF2α
eukaryotic initiation factor 2α
- EdU
5-ethynyl-2'-deoxyuridine
- FELASA
Federation of European Laboratory Animal Science Association
- WT
wild-type
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/23369
References
- 1.Duelli D, Lazebnik Y. Cell-to-cell fusion as a link between viruses and cancer. Nat Rev Cancer. 2007;7:968–76. doi: 10.1038/nrc2272. [DOI] [PubMed] [Google Scholar]
- 2.Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043–7. doi: 10.1038/nature04217. [DOI] [PubMed] [Google Scholar]
- 3.Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev. 2007;17:157–62. doi: 10.1016/j.gde.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 4.Davoli T, Denchi EL, de Lange T. Persistent telomere damage induces bypass of mitosis and tetraploidy. Cell. 2010;141:81–93. doi: 10.1016/j.cell.2010.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vitale I, Galluzzi L, Senovilla L, Criollo A, Jemaa M, Castedo M, et al. Illicit survival of cancer cells during polyploidization and depolyploidization. Cell Death Differ. 2011;18:1403–13. doi: 10.1038/cdd.2010.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ganem NJ, Pellman D. Limiting the proliferation of polyploid cells. Cell. 2007;131:437–40. doi: 10.1016/j.cell.2007.10.024. [DOI] [PubMed] [Google Scholar]
- 7.Margolis RL. Tetraploidy and tumor development. Cancer Cell. 2005;8:353–4. doi: 10.1016/j.ccr.2005.10.017. [DOI] [PubMed] [Google Scholar]
- 8.Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12:385–92. doi: 10.1038/nrm3115. [DOI] [PubMed] [Google Scholar]
- 9.Davoli T, de Lange T. The causes and consequences of polyploidy in normal development and cancer. Annu Rev Cell Dev Biol. 2011;27:585–610. doi: 10.1146/annurev-cellbio-092910-154234. [DOI] [PubMed] [Google Scholar]
- 10.Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, et al. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008;22:2189–203. doi: 10.1101/gad.1700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vitale I, Senovilla L, Jemaa M, Michaud M, Galluzzi L, Kepp O, et al. Multipolar mitosis of tetraploid cells: inhibition by p53 and dependency on Mos. EMBO J. 2010;29:1272–84. doi: 10.1038/emboj.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Padilla-Nash HM, Hathcock K, McNeil NE, Mack D, Hoeppner D, Ravin R, et al. Spontaneous transformation of murine epithelial cells requires the early acquisition of specific chromosomal aneuploidies and genomic imbalances. Genes Chromosomes Cancer. 2012;51:353–74. doi: 10.1002/gcc.21921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, et al. An immunosurveillance mechanism controls cancer cell ploidy. Science. 2012;337:1678–84. doi: 10.1126/science.1224922. [DOI] [PubMed] [Google Scholar]
- 14.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–20. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett's epithelium. Gastroenterology. 2011;141:1762–72. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 16.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–5. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 17.Jemaa M, Vitale I, Kepp O, Berardinelli F, Galluzzi L, Senovilla L, et al. Selective killing of p53-deficient cancer cells by SP600125. EMBO Mol Med. 2012;4:500–14. doi: 10.1002/emmm.201200228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68:855–67. doi: 10.1016/0092-8674(92)90029-C. [DOI] [PubMed] [Google Scholar]
- 19.Senovilla L, Vitale I, Galluzzi L, Vivet S, Joza N, Younes AB, et al. p53 represses the polyploidization of primary mammary epithelial cells by activating apoptosis. Cell Cycle. 2009;8:1380–5. doi: 10.4161/cc.8.9.8305. [DOI] [PubMed] [Google Scholar]
- 20.Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2012 doi: 10.1146/annurev-immunol-032712-100008. In press. [DOI] [PubMed] [Google Scholar]
- 21.Baker DJ, van Deursen JM. Chromosome missegregation causes colon cancer by APC loss of heterozygosity. Cell Cycle. 2010;9:1711–6. doi: 10.4161/cc.9.9.11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caldwell CM, Green RA, Kaplan KB. APC mutations lead to cytokinetic failures in vitro and tetraploid genotypes in Min mice. J Cell Biol. 2007;178:1109–20. doi: 10.1083/jcb.200703186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dikovskaya D, Schiffmann D, Newton IP, Oakley A, Kroboth K, Sansom O, et al. Loss of APC induces polyploidy as a result of a combination of defects in mitosis and apoptosis. J Cell Biol. 2007;176:183–95. doi: 10.1083/jcb.200610099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 25.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 26.Senovilla L, Vacchelli E, Galon J, Adjemian S, Eggermont A, Fridman WH, et al. Trial Watch: Prognostic and predictive value of the immune infiltrate in cancer. OncoImmunology. 2012;1:1323–44. doi: 10.4161/onci.22009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zitvogel L, Kepp O, Kroemer G. Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol. 2011;8:151–60. doi: 10.1038/nrclinonc.2010.223. [DOI] [PubMed] [Google Scholar]
- 28.Hoffman J, Kuhnert F, Davis CR, Kuo CJ. Wnts as essential growth factors for the adult small intestine and colon. Cell Cycle. 2004;3:554–7. doi: 10.4161/cc.3.5.858. [DOI] [PubMed] [Google Scholar]
- 29.Jung P, Sato T, Merlos-Suarez A, Barriga FM, Iglesias M, Rossell D, et al. Isolation and in vitro expansion of human colonic stem cells. Nat Med. 2011;17:1225–7. doi: 10.1038/nm.2470. [DOI] [PubMed] [Google Scholar]
- 30.Galluzzi L, Vitale I, Senovilla L, Eisenberg T, Carmona-Gutierrez D, Vacchelli E, et al. Independent transcriptional reprogramming and apoptosis induction by cisplatin. Cell Cycle. 2012;11:3472–80. doi: 10.4161/cc.21789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lainey E, Sebert M, Thepot S, Scoazec M, Bouteloup C, Leroy C, et al. Erlotinib antagonizes ABC transporters in acute myeloid leukemia. Cell Cycle. 2012;11:4079–92. doi: 10.4161/cc.22382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Castedo M, Coquelle A, Vivet S, Vitale I, Kauffmann A, Dessen P, et al. Apoptosis regulation in tetraploid cancer cells. EMBO J. 2006;25:2584–95. doi: 10.1038/sj.emboj.7601127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mouhamad S, Galluzzi L, Zermati Y, Castedo M, Kroemer G. Apaf-1 deficiency causes chromosomal instability. Cell Cycle. 2007;6:3103–7. doi: 10.4161/cc.6.24.5046. [DOI] [PubMed] [Google Scholar]
- 34.Jemaa M, Galluzzi L, Kepp O, Boileve A, Lissa D, Senovilla L, et al. Preferential killing of p53-deficient cancer cells by reversine. Cell Cycle. 2012;11:2149–58. doi: 10.4161/cc.20621. [DOI] [PubMed] [Google Scholar]
- 35.Lainey E, Wolfromm A, Marie N, Enot D, Scoazec M, Bouteloup C, et al. Azacytidine and erlotinib exert synergistic effects against acute myeloid leukemia. Oncogene. 2012 doi: 10.1038/onc.2012.469. In press. [DOI] [PubMed] [Google Scholar]