

Abstract
Here, we provide the necessary proof of concept, that it is possible to metabolically create a non-permissive or “hostile” stromal microenvironment, which actively prevents tumor engraftment in vivo. We developed a novel genetically engineered fibroblast cell line that completely prevents tumor formation in mice, with a 100% protection rate. No host side effects were apparent. This could represent a viable cellular strategy for preventing and treating a variety of human cancers. More specifically, we examined the autocrine and paracrine effects of the cellular delivery of TNFα on breast cancer tumor growth and cancer metabolism. For this purpose, we recombinantly overexpressed TNFα in human breast cancer cells (MDA-MB-231) or human immortalized fibroblasts (hTERT-BJ1). Our results directly show that TNFα functions as a potent tumor suppressor. Remarkably, TNFα-expressing breast cancer cells were viable, without any significant increases in their basal apoptotic rate. However, after 4 weeks post-implantation, TNFα-expressing breast cancer cells failed to form any tumors in xenografted mice (0 tumors/10 injections), ultimately conferring 100% protection against tumorigenesis. Similarly, TNFα-overexpressing fibroblasts were also viable, without any increases in apoptosis. Significantly, complete tumor suppression was obtained by co-injecting TNFα expressing stromal fibroblasts with human breast cancer cells, indicating that paracrine cell-mediated delivery of TNFα can also prevent tumor engraftment and growth (0 tumors/10 injections). Mechanistically, TNFα induced autophagy and mitochondrial dysfunction in both epithelial cancer cells and stromal fibroblasts, preventing energy transfer from the tumor microenvironment, likely “starving” the cancer cells to death. In addition, via qRT-PCR analysis of MDA-MB-231 cells, we observed that TNFα mediated the upregulation of gene transcripts associated with inflammation and senescence [IL-1-β, IL-6, IL-8, MCP-1, COX-2, p21(WAF1/CIP1)] and downregulated known tumor-promoting genes (collagen VI and MMP2). Recombinant overexpression of TNFα receptor(s) in MDA-MB-231 cells also significantly reduced tumor growth, but was not as effective as the TNFα ligand itself in preventing tumor growth. Thus, we propose that stromal cell-mediated delivery of TNFα to human tumors [using transfected fibroblasts or mesenchymal stem cells (hMSCs)] may be a novel and effective strategy for the prevention and treatment of human cancers.
Keywords: tumor necrosis factor (TNF), cancer prevention, cellular therapy, fibroblast mediated delivery, mitochondrial dysfunction, breast cancer, tumor growth, tumor cell engraftment, autophagy, apoptosis
Introduction
Tumor necrosis factor α (TNFα) is an inflammatory cytokine that is key part of the innate immune response, in both infectious disease and cancer(s).1,2 It has also been implicated in the pathogenesis of chronic inflammatory diseases (rheumatoid arthritis, inflammatory bowel disease, psoriasis, refractory asthma), as well as Alzheimer disease.3,4 TNFα is able to induce acute inflammatory events [fever (pyrogen) and sepsis (via IL-1 and IL-6)], as well as chronic inflammation.5,6 It behaves as a catabolic cytokine that drives ROS production, oxidative stress, autophagy and mitochondrial dysfunction, as well as programmed cell death (apoptosis).7-12 In accordance with these metabolic findings, for many years TNFα was also known as the hormone cachexin or cachectin, a mediator of tumor-induced cachexia (“wasting”), which results in negative energy balance.7-12 TNFα is named for its ability to kill tumor cells, but this activity has not yet been successfully exploited for anticancer therapy.
Here, we assessed the compartment-specific functional effects of the cellular expression of TNFα on tumor engraftment and cancer metabolism. Remarkably, cellular expression of TNFα in epithelial cancer cells (MDA-MB-231) was not cytotoxic in vitro, but completely prevented tumor growth in vivo. Similarly, cellular expression of TNFα in stromal fibroblasts also completely prevented tumor formation in vivo, via paracrine mechanism(s). We propose that the tumor suppressor effects of TNFα are due to its ability to interrupt symbiotic metabolic coupling between epithelial cancer cells and their host stromal microenvironment. Thus, cellular delivery of TNFα to human tumors in vivo may represent a viable new strategy for inducing primary tumor regression and preventing metastatic cancer progression.
As such, TNFα expressing stromal cells [either fibroblasts or mesenchymal stem cells (MSCs)] may represent the missing “magic bullets” that we need for successful targeted anticancer therapies. In fact, TNFα pre-treatment of MSCs increases their ability to “home” or accumulate within tumors in vivo.13
Finally, our results also suggest that successful tumor-host engraftment requires metabolic co-operation or symbiosis with the host, as normally occurs during a parasite-host infection. Thus, we should consider treating cancer as an “infectious disease,” with “antibiotics” that prevent or interrupt metabolic co-operation. Our current findings with the cellular delivery of TNFα provide the required proof-of-principal that that this simple “antibiotic” or “metabolic uncoupling” strategy can effectively prevent tumor growth in vivo.
Results
MDA-MB-231 cancer cells overexpressing TNFα display increased autophagy and decreased mitochondrial activity, without any changes in apoptosis
MDA-MB-231 cancer cells were transduced with a TNFα lentiviral vector or an LV empty vector control. After selection, stable cell lines were subjected to immunoblotting and immunofluorescence analysis to validate the TNFα overexpression. Figure 1A and B show, respectively, the results of this analysis, demonstrating successful TNFα overexpression.
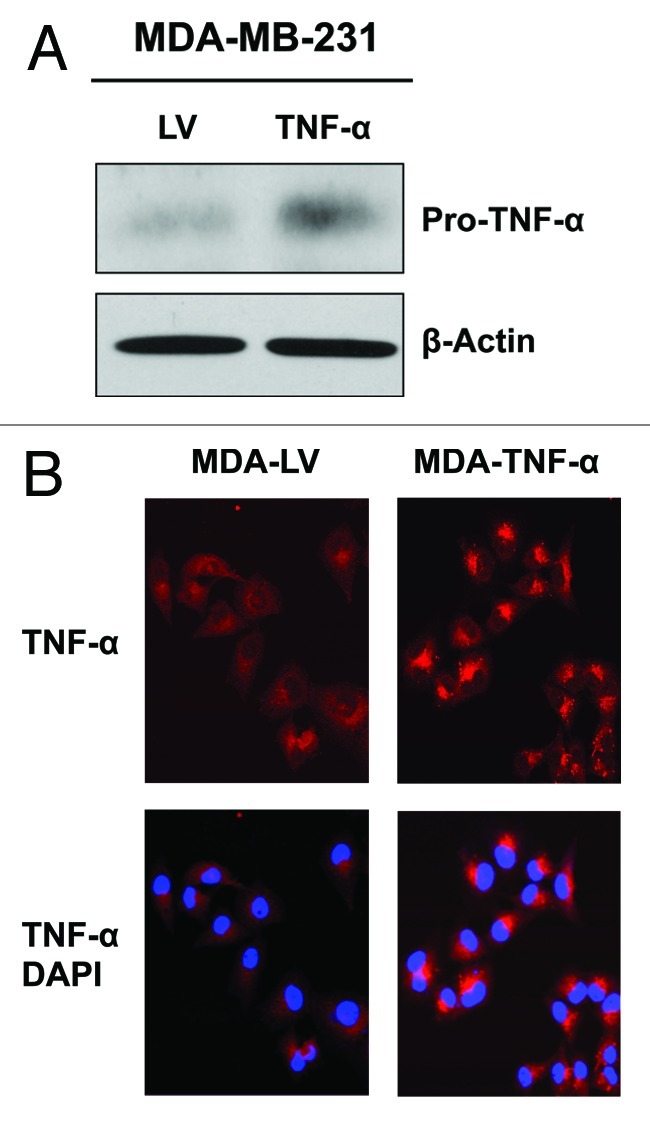
Figure 1. Stable TNFα overexpression in human epithelial breast cancer cells. MDA-MB-231 human breast cancer cells were transduced with a TNFα lentiviral vector or the LV empty vector control, and selected with puromycin to generate stable cell lines. Then, transduced cells were subjected to immunoblotting and immunofluorescence analysis to validate TNFα overexpression. (A) Immunoblotting shows a band at ~24 KDa, corresponding to the pro-TNFα precursor form. β-actin is shown as a control for equal protein loading. (B) Immunofluorescence with anti-TNFα antibodies demonstrates TNFα overexpression in cancer cells. Red, TNFα specific staining; blue, DAPI nuclear staining.
We first assessed if TNFα overexpression promotes the apoptosis of MDA-MB-231 cells by immunoblotting with specific antibodies against apoptotic markers. Figure 2A shows that caspase-3 is significantly upregulated and activated in MDA-MB-231 cells overexpressing TNFα relative to control cells. However, MDA-MB-231 cells overexpressing TNFα display the downregulation of the pro-apoptotic marker Bax, and this is consistent with the upregulation of the anti-apoptotic molecule BCL-2. These results suggest that TNFα induces the upregulation of certain components of the apoptosis machinery and may prime cancer cells toward apoptosis.
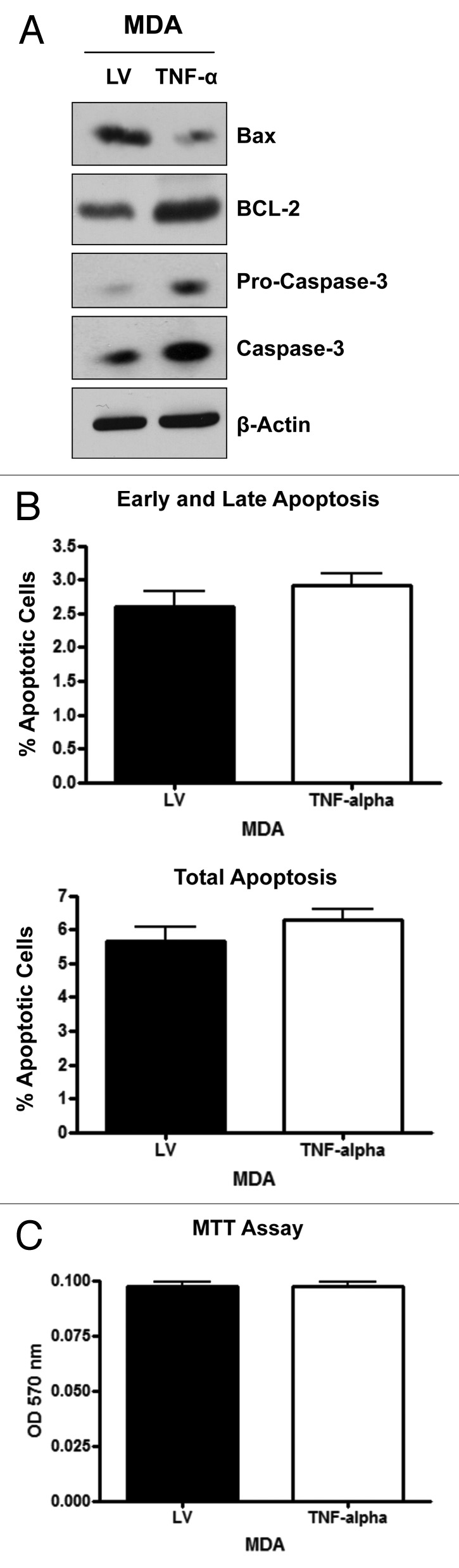
Figure 2. MDA-MB-231 cells overexpressing TNFα show the upregulation of apoptotic machinery, without a steady-state induction of increased apoptosis. (A) MDA-MB-231 cells overexpressing TNFα or the LV empty vector were subjected to immunoblotting with specific antibodies directed against apoptotic markers. Note that caspase-3 is significantly upregulated and activated in TNFα-expressing MDA-MB-231 cells, while Bax is downregulated consistent with BCL-2 upregulation. β-actin is shown as a control for equal loading. (B) Apoptosis was directly measured with Annexin-V and PI probes by flow cytometry (FACS analysis). MDA-MB-231 cells overexpressing TNFα did not show any significant increases in apoptotic rates relative to the empty vector control. Early and late apoptosis, represents Annexin-V-positive/PI-negative cells; Total apoptosis, represents Annexin-V-positive and/or PI-positive cells. (C) MTT assay demonstrates that MDA-MB-231 cells overexpressing TNFα show no differences in cell viability/proliferation, relative to empty vector control cells. These results indicate that TNFα does not increase the steady-state apoptotic rate in epithelial cancer cells.
To directly evaluate steady-state apoptosis levels, MDA-MB-231 cells overexpressing TNFα or control cells were analyzed by flow cytometry using Annexin-V and PI probes. Figure 2B shows that MDA-MB-231 cells overexpressing TNFα did not show any significant differences in apoptotic rates relative to the empty vector control cells. To independently validate these results, MDA-MB-231 cells overexpressing TNFα or control cells were subjected to an MTT assay. Figure 2C demonstrates that MDA-MB-231 cells overexpressing TNFα show no differences in cell viability/proliferation relative to control cells. These results indicate that TNFα does not induce non-specific cytotoxicity in MDA-MB-231 cells.
We next assessed if TNFα overexpression promotes autophagy in MDA-MB-231 cells using immunoblotting with specific antibodies directed against autophagic markers. Figure 3A shows that several autophagy markers (Beclin-1, BNIP3 and Lamp-1) are upregulated in TNFα overexpressing MDA-MB-231 cells relative to control cells. Since the mitophagy marker BNIP3 is upregulated in TNFα-overexpressing MDA-MB-231 cancer cells, we postulated that mitochondrial activity might also be decreased. To this end, we investigated the status of OXPHOS complexes (I–V) by immunoblotting. Figure 3 shows the downregulation of the key subunits of the OXPHOS complexes (namely components III and V) in MDA-MB-231 cells overexpressing TNFα, relative to LV control cells, providing an indication of impaired mitochondrial activity.
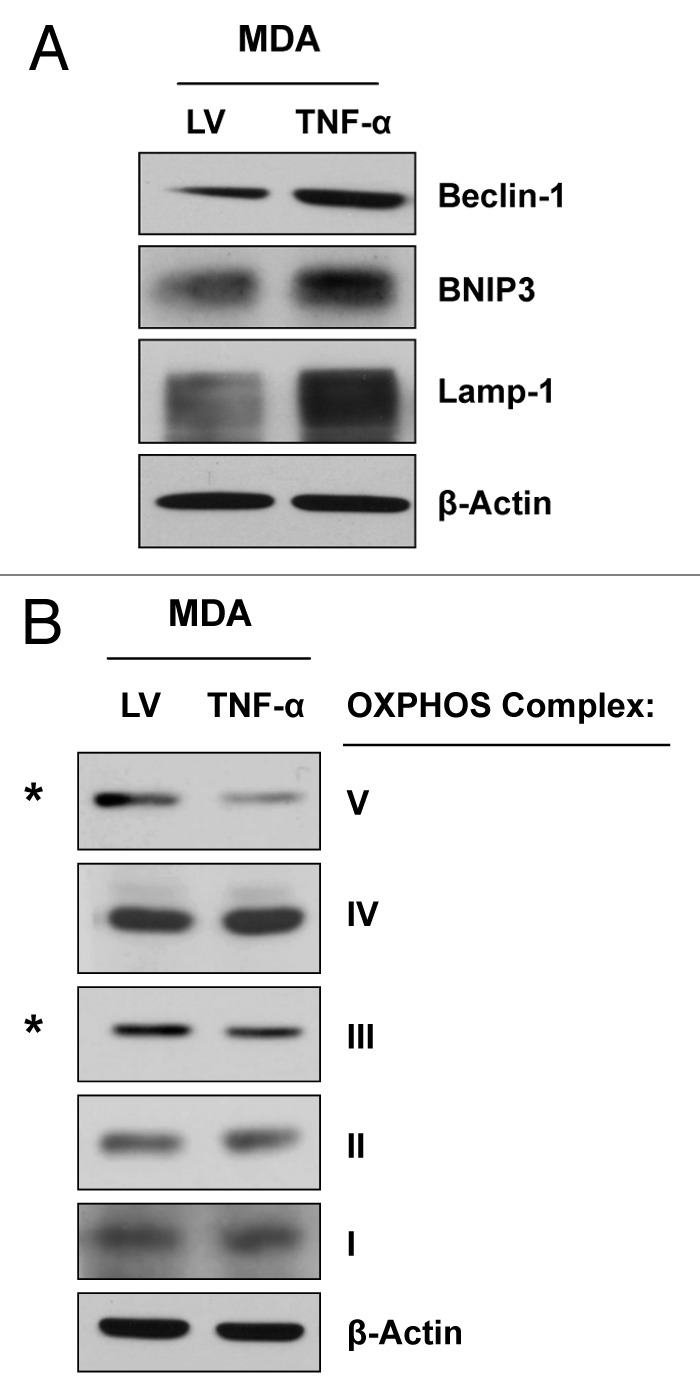
Figure 3. MDA-MB-231 cells overexpressing TNFα display the induction of autophagy and a reduction in mitochondrial OXPHOS. MDA-MB-231 cells overexpressing TNFα or LV empty vector were subjected to immunoblotting with specific antibodies directed against autophagic markers and OXPHOS complex components. (A) Note that several autophagy markers (Beclin-1, BNIP3 and Lamp-1) are upregulated in TNFα-overexpressing MDA-MB-231 cells relative to control cells. (B) MDA-MB-231 cells overexpressing TNFα display the downregulation of the OXPHOS complexes III and V, relative to control cells. β-actin is shown as an equal loading control.
TNFα overexpression in epithelial cancer cells completely abolishes breast cancer tumor growth in vivo
To evaluate the effects of TNFα overexpression in MDA-MB-231 cells on tumor growth in vivo, we employed a murine xenograft model. To this end, MDA-MB-231 cells carrying TNFα or the LV empty control vector were injected into the flanks of nude mice, and tumors were measured using calipers for a period of 30 d. Figure 4A−C shows the tumor growth curve as well as the tumor mass and volume of the dissected tumors, clearly demonstrating that MDA-MB-231 cells overexpressing TNFα did not develop tumors in any of the injected mice (0/10; 0 tumors out of 10 injections). However, MDA-MB-231 vector alone control cells formed robust tumor xenografts, as expected (10/10; 10 tumors out of 10 injections). These results demonstrate that TNFα overexpression completely inhibits the in vivo growth of MDA-MB-231 cells.
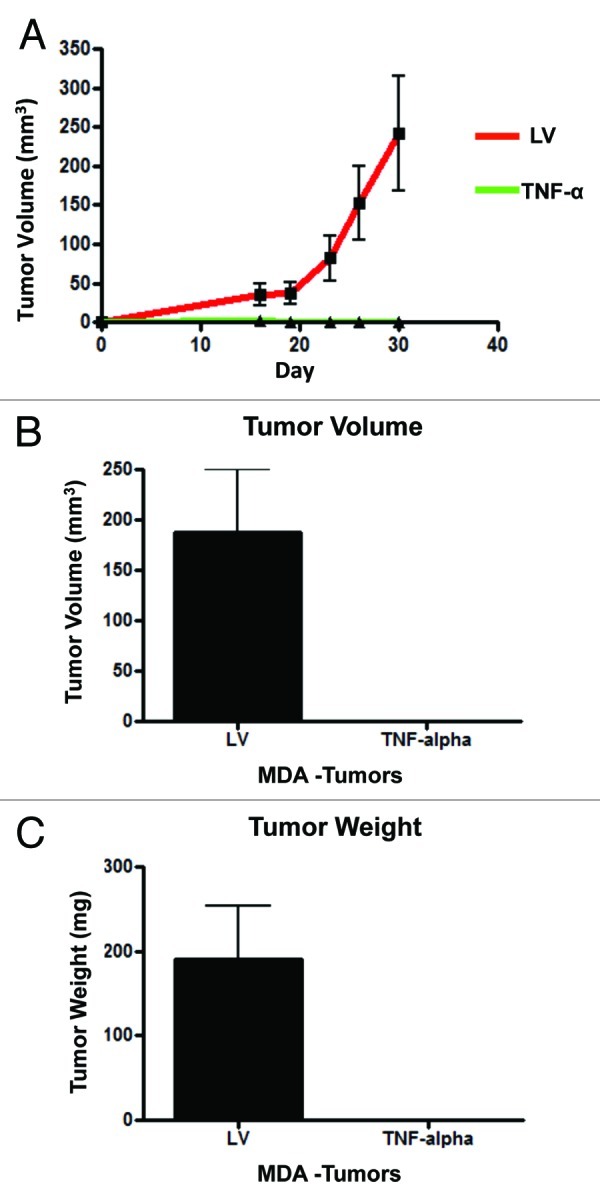
Figure 4. TNFα overexpression in cancer cells completely abolishes tumor growth in vivo. MDA-MB-231 cells (carrying TNFα or the LV empty vector control) were injected into the flanks of nude mice. Tumors growth was serially followed using calipers for 30 d post-injection. (A) Note that the growth curves clearly show that MDA-MB-231 cells overexpressing TNFα did not develop any tumors. After mice were sacrificed, tumor volume (B) and weight (C) were measured, demonstrating no growth of TNFα-MDA-MB-231 cells in all xenografted mice (0/10; 0 tumors grew from 10 injections). Thus, expression of TNFα confers 100% protection against tumor formation.
TNFα overexpression in fibroblasts induces autophagy and decreases mitochondrial activity, without causing apoptosis
To evaluate if the effects of TNFα are cell type-specific, we overexpressed TNFα in the hTERT-BJ1 fibroblast cell line. LV empty vector control fibroblasts were generated in parallel. Immunoblotting (Fig. 5A) and immunofluorescence analysis (Fig. 5B) validated the overexpression of TNFα in hTERT-BJ1 cells. We next checked if fibroblasts overexpressing TNFα display increased levels of apoptosis by immunoblotting with antibodies against apoptotic markers. Figure 6A shows no changes in the levels of caspase-3, Bax and BCL-2. To independently assess apoptotic rates, hTERT-BJ1 cells overexpressing TNFα or LV control cells were analyzed by flow cytometry (FACS) using Annexin-V and PI probes. Figure 6B demonstrates that hTERT-BJ1 cells overexpressing TNFα did not show any significant differences in their apoptotic rates relative to the empty vector controls. These results indicate that TNFα does not induce apoptosis in non-transformed fibroblasts.
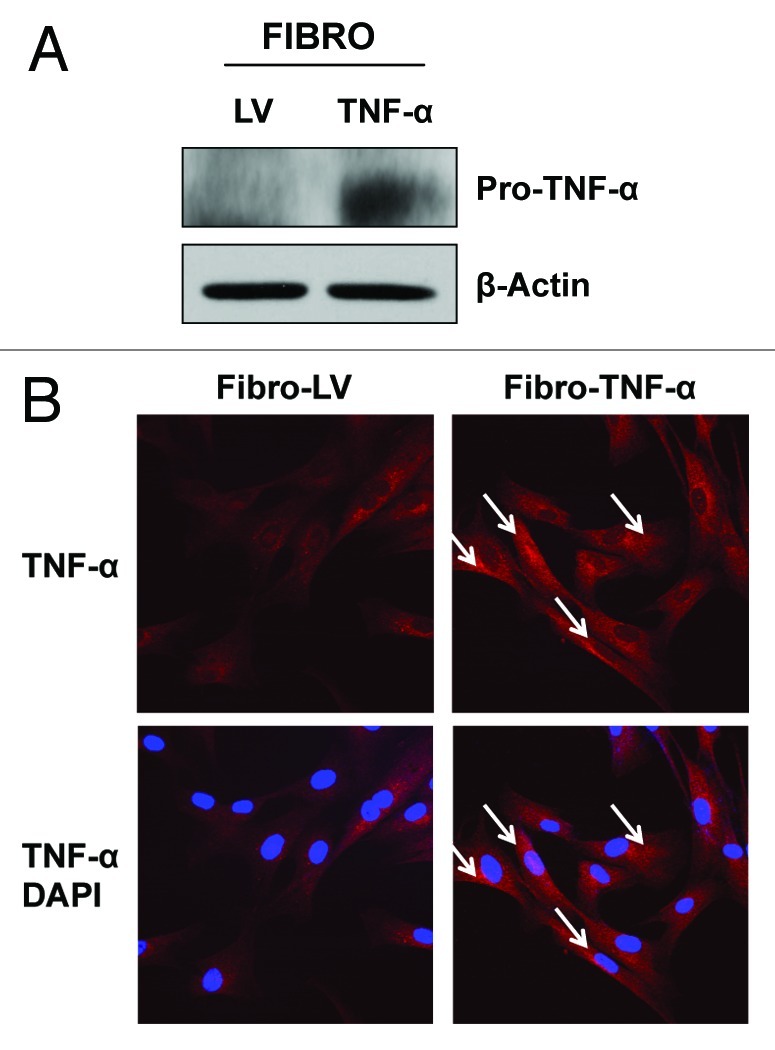
Figure 5. Stable TNFα overexpression in immortalized human fibroblasts. hTERT-BJ1 fibroblasts were transduced with a TNFα expressing vector or LV empty vector control. Then, the resulting transduced fibroblasts were subjected to immunoblotting or immunofluorescence analysis to validate TNFα overexpression. (A) Immunoblotting shows a band at ~24 KDa, corresponding to pro-TNFα, the precursor form. β-actin demonstrates equal protein loading. (B) Immunofluorescence with TNFα specific antibodies clearly demonstrates TNFα expression (see white arrows) in hTERT-BJ1 cells. Red, TNFα specific staining; blue, DAPI nuclear staining.

Figure 6. TNFα overexpression induces autophagy and deregulates mitochondrial activity in fibroblasts, without inducing apoptosis. (A) Immunoblotting shows that fibroblasts overexpressing TNFα have similar levels of the apoptotic markers (caspase-3, Bax and BCL-2), as control cells. (B) Apoptosis was measured with Annexin-V and PI probes by flow cytometry (FACS analysis). hTERT-BJ1 fibroblasts overexpressing TNFα did not show any significant increases in apoptosis, relative to empty vector controls. Early and late apoptosis, represents Annexin-V-positive/PI-negative cells; total apoptosis, represents Annexin-V-positive and/or PI-positive cells. (C) TNFα overexpression in hTERT-BJ1 cells induces autophagy, as assessed by the elevated expression and/or activation of cathepsin B, Lamp-1 and LC3B (LC3B-II is the cleaved active form). (D) Immunoblotting shows that TNFα overexpression in hTERT-BJ1 cells induces the downregulation of key subunits of the OXPHOS complex, namely components III and V. β-actin indicates equal protein loading.
We next evaluated the activation status of the autophagy program by immunoblotting with specific antibodies directed against autophagic markers. Figure 6C shows that TNFα overexpression induces autophagy in hTERT-BJ1 cells, as judged by elevated expression and activation of cathepsin B, Lamp-1 and LC3B. Finally, we asked if TNFα overexpression in hTERT-BJ1 cells results in decreased mitochondrial activity, similarly to what we observed in MDA-MB-231 cells. Immunoblot analysis shows that TNFα overexpression in hTERT-BJ1 cells induces the downregulation of key subunits of the OXPHOS complexes (namely subunits III and V; Fig. 6D), providing an indication of decreased mitochondrial respiration.
Fibroblasts overexpressing TNFα completely prevent breast cancer tumor growth in vivo
To evaluate if fibroblasts overexpressing TNFα inhibit breast cancer tumor growth in vivo, hTERT-BJ1 cells (carrying TNFα or LV empty vector) were co-injected with MDA-MB-231 cells into the flanks of nude mice. Tumor growth was monitored for over a 4-week period post-implantation. The tumor growth curves show that MDA-MB-231 cells co-injected with TNFα-overexpressing fibroblasts did not form any tumors (Fig. 7A). Measurements of the weight and volume (Fig. 7A and B) of tumors shows that TNFα-overexpressing fibroblasts completely prevented the growth of co-injected MDA-MB-231 cells, relative to control cells (0/10; 0 tumors out of 10 injections). These results indicate that fibroblast-mediated delivery of TNFα to the tumor site may be an effective and feasible strategy for tumor inhibition and regression.
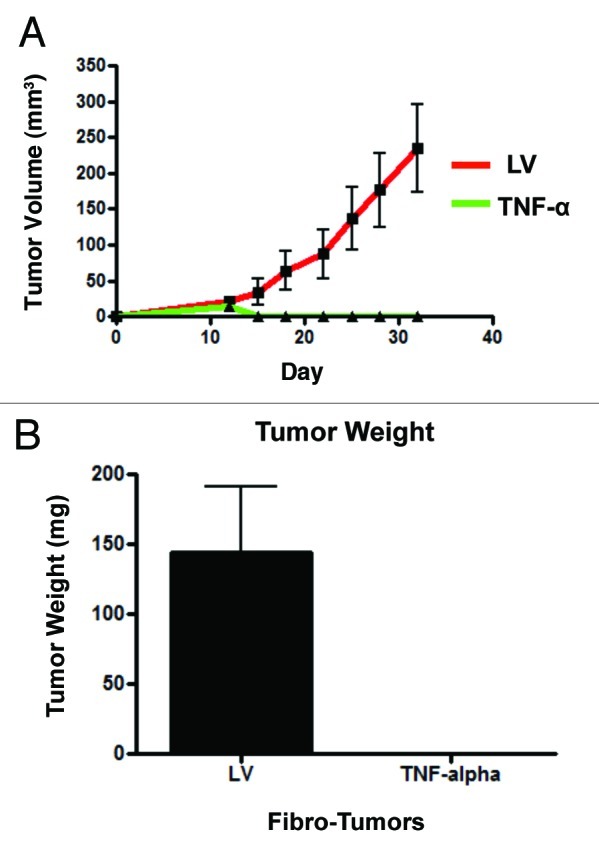
Figure 7. Fibroblasts harboring TNFα completely prevent breast cancer tumor growth in vivo. MDA-MB-231 cells were co-injected with hTERT-BJ1 cells (carrying TNFα or the LV empty vector control) into the flanks of nude mice. Tumors growth was serially measured using calipers for 31 d. (A) Note that the growth curves clearly show that MDA-MB-231 cells co-injected with TNFα-overexpressing fibroblasts did not develop any tumors. After 31 d, the mice were sacrificed. Measurements of tumor weight (B) show that TNFα-overexpressing fibroblasts completely prevented tumor growth of co-injected MDA-MB-231 cells, relative to control cells.
MDA-MB-231 cancer cells overexpressing TNFα receptors show the upregulation of apoptotic markers in vitro, and show significantly reduced tumor growth in vivo
To further evaluate the cell-autonomous tumor inhibition properties of TNFα, MDA-MB-231 cells were transduced with vectors encoding TNFα receptors (TNFR-1A and TNFR-1B) or a LV empty vector. Cells were first subjected to immunoblotting with antibodies directed against apoptotic markers. Figure 8 shows that MDA-MB-231 cells overexpressing TNFR-1A and TNFR-1B display the upregulation and activation of caspase-3 relative to empty vector control cells. Conversely, the levels of the apoptotic activator Bax were slightly decreased.
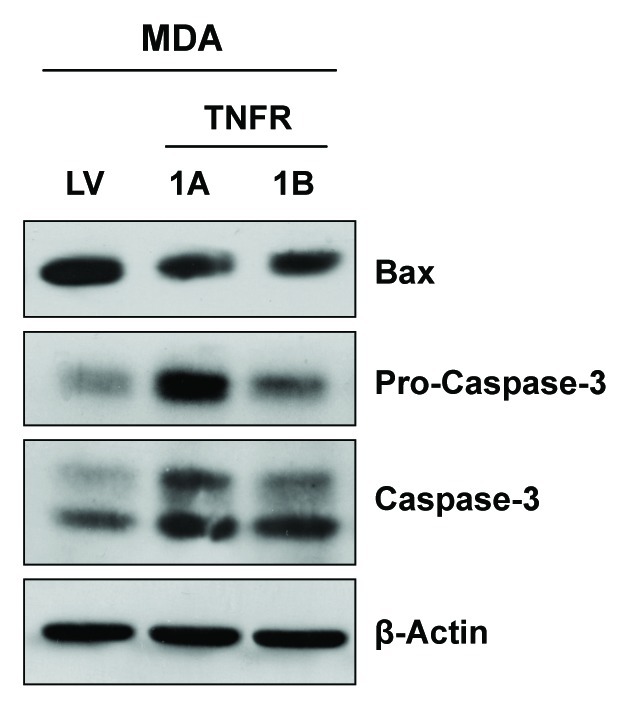
Figure 8. Overexpression of TNFα receptors induces the upregulation of apoptotic markers in cancer cells. MDA-MB-231 cells were transduced with TNFα receptor (TNFR-1A and TNFR-1B) vectors or an LV empty vector control and subjected to immunoblotting with apoptotic markers. Note the upregulation and activation of caspase-3 in MDA-MB-231 harboring TNFR-1A and TNFR-1B, relative to empty vector control cells. β-actin is shown as a control for equal protein loading.
We next employed a tumor xenograft model to establish the behavior of MDA-MB-231 cells expressing TNFR-1A and TNFR-1B in vivo. To this end, MDA-MB-231 cancer cells overexpressing TNFR-1A or TNFR-1B or LV empty vector were injected into the flanks of mice, and tumor growth was monitored for 30 d. The tumor growth curves (Fig. 9A), tumor volume (Fig. 9B) and tumor weight (Fig. 9C) all directly show that MDA-MB-231 cells overexpressing TNFR-1A and TNFR-1B display significantly reduced tumor growth rates, relative to control cells. However, recombinant expression of TNFR-1A had the most robust tumor suppressor activity.
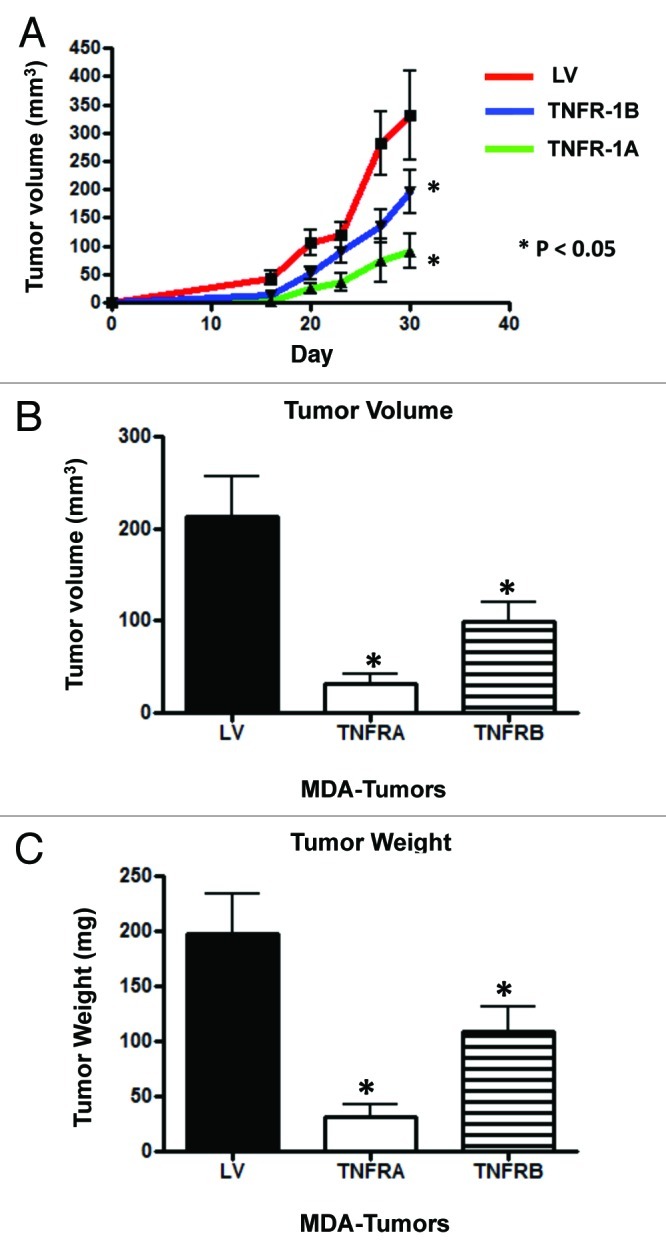
Figure 9. Cancer cells overexpressing TNFR-1A or TNFR-1B show significant tumor growth inhibition in vivo. MDA-MB-231 cells (carrying TNFR-1A, TNFR-1B or the LV empty vector control) were injected into the flanks of nude mice. Tumors were measured with calipers for 30 d. Note that the serial growth curve (A), final tumor volume (B) and final tumor weight (C) all clearly show that TNFR-1A and TNFR-1B transduced MDA-MB-231 cells display significantly reduced tumor growth relative to control cells.
To verify if TNFR-1A and TNFR-1B expression is maintained in the tumor xenografts, tumor xenograft sections derived from TNFR-1A, TNFR-1B or LV-control MDA-MB-231 cells were subjected to immunohistochemistry with anti-TNFR-1A or anti-TNFR-1B antibodies. Figure 10A and B shows that the expression of TNFR-1A and TNFR-1B (brown staining) is preserved in the epithelial compartments of the tumor xenografts.
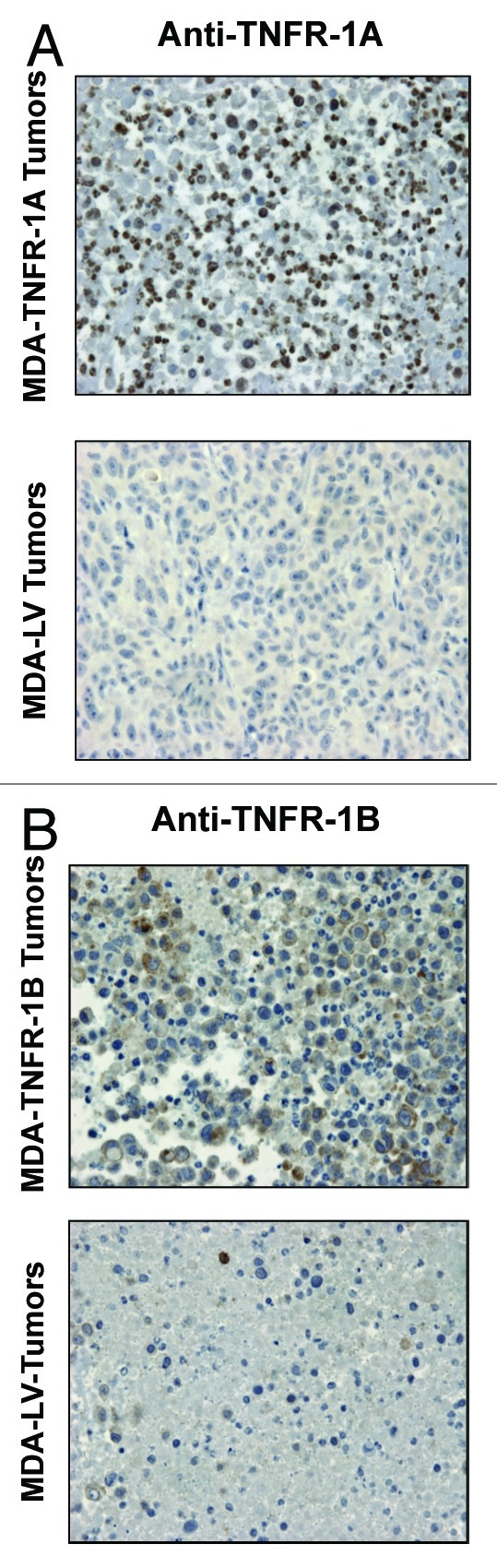
Figure 10. TNFR-1A and TNFR-1B overexpression in MDA-MB-231 cell tumor xenografts. TNFR-1A and TNFR-1B overexpression in MDA-MB-231 cells was validated on the tumor xenografts derived from TNFR-1A, TNFR-1B or LV-control MDA-MB-231 cells. Sections were subjected to immunohistochemistry with anti-TNFR-1A (A) or anti-TNFR-1B (B) antibodies. The positive-staining (brown color) represents TNFR-1A or TNFR-1B overexpression. Original magnification, 60×.
MDA-MB-231 cells overexpressing TNFα show the transcriptional elevation of genes functionally associated with cellular inflammation and senescence and decreased expression of genes associated with extracellular matrix remodeling
To further understand the mechanism(s) by which TNFα suppresses tumor growth, MDA-MB-231 cells overexpressing TNFα or vector alone control cells were analyzed by quantitative real time PCR (RT-PCR) for markers of inflammation, and extracellular matrix remodeling. Figure 11A shows that TNFα induces the upregulation of several transcripts associated with the inflammatory response, including IL-1-β, IL-8, IL-6, MCP-1 and COX-2. TNFα was also found greatly upregulated, as expected. In addition, several markers associated with extracellular matrix remodeling, such as collagen VI and MMP2 were selectively downregulated in TNFα-expressing cancer cells, relative to controls. Previous studies have shown that collagen VI deposition favors tumor growth.14 Similarly, it is well known that increased MMP2 levels are associated with tumor aggressiveness.15

Figure 11. MDA-MB-231 cancer cells overexpressing TNFα display elevated transcripts associated with inflammation and senescence and decreased transcripts associated with extracellular matrix remodeling. MDA-MB-231 cells overexpressing TNFα or vector alone were analyzed by quantitative real time PCR (qRT-PCR) for markers of inflammation, senescence and extracellular matrix remodeling. (A) Note that TNFα induces the upregulation of several transcripts associated with an inflammatory response, including IL-1-β, IL-8, IL-6, MCP-1 and COX-2. TNFα was also found to be elevated, as expected. (B) In addition, several markers associated with extracellular matrix remodeling, including collagen VI and MMP2, were selectively downregulated in TNFα-expressing cancer cells, relative to controls. (C) Finally, a marker of hypoxia, HIF2a, was decreased and a marker of senescence, p21 (Cip1/Waf1) was found to be elevated in TNFα-expressing cancer cells, relative to controls. We have previously shown that increased HIF2a expression in MDA-MB-231 cancer cells promotes tumor growth, and that increased p21 expression in MDA-MB-231 cancer cells inhibits tumor growth.16,17
Interestingly, TNFα-expressing cancer cells display the downregulation of a marker of hypoxia, HIF-2, relative to controls (Fig. 11C). We have previously shown that increased HIF-2a expression in MDA-MB-231 cancer cells promotes tumor growth.16 Finally, a marker of senescence, p21 (WAF1/CIP1) was found to be elevated in TNFα-expressing MDA-MB-231 cancer cells, relative to controls (Fig. 11C). We have previously shown increased p21 expression in MDA-MB-231 cancer cells inhibits tumor growth, by inducing senescence and autophagy.17
Discussion
Here, we determined the specific effects of the stable recombinant expression of TNFα in human breast cancer cells (MDA-MB-231) and human immortalized fibroblasts (hTERT-BJ1 cells). Interestingly, stable expression was achieved via lentiviral transduction, but did not lead to any significant increases in basal apoptotic rates, indicating that the cells remained viable. As expected, TNFα expression resulted in the induction of multiple autophagy markers as well as the onset of mitochondrial dysfunction, as evidenced by a loss of expression of specific OXPHOS components, especially Complex V, the mitochondrial ATP synthase.
Most importantly, MDA-MB-231 cells expressing TNFα failed to form any tumors in vivo, resulting in 100% protection against tumorigenesis. These results suggest that TNFα induces metabolic dysfunction in cancer cells, preventing their ability to form viable tumors in vivo. These findings are also consistent with the idea that autophagy and mitochondrial dysfunction in cancer cells has a potent tumor-suppressor effect.18-25
Similarly, the tumor suppressor effects of TNFα could be achieved by co-injecting TNFα-expressing human fibroblasts with human breast cancer cells. As such, we propose that generalized metabolic dysfunction in both tumor cells and their surrounding microenvironment will prevent the onset of metabolic symbiosis, effectively “starving” the cancer cells to death18-25 (Fig. 12).
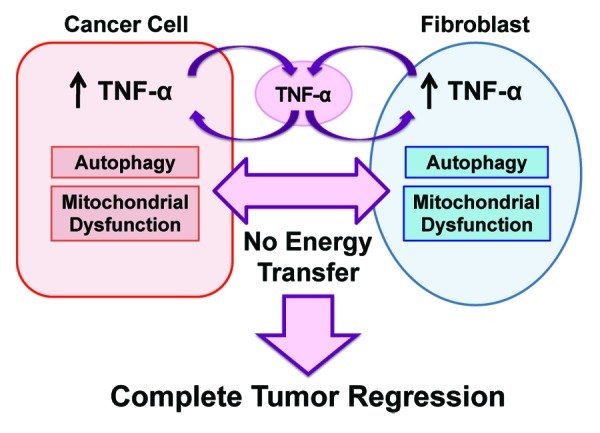
Figure 12. Proposed metabolic mechanism(s) underlying the potent tumor suppressor effects of TNFα in the tumor microenvironment. TNFα can be release either by tumor cells or cancer associated fibroblasts (CAFs). Then, TNFα induces autophagy both in the fibroblast cell compartment and in the epithelial cancer cell compartment. In addition, TNFα induces mitochondrial dysfunction in both compartents. The resulting lack of energy transfer between the two metabolic comparments (epithelia vs. stroma) promotes complete tumor regression and/or inhibits tumor growth.
This has important therapeutic implications, because mesenchymal stem cells (MSCs) and fibroblasts are known to migrate or “home” to human tumors in vivo, where they contribute significantly to the mass of the tumor microenvironment. So, fibroblasts or MSCs transfected with TNFα could be used for the direct cellular delivery of this inflammatory cytokine in vivo, thereby promoting tumor regression.
While this manuscript was being finalized for publication, several other papers were published suggesting that TNFα has potent tumor suppressor activity. For example, Munich et al., showed that dendritic cell exosomes, which contain TNFα, can kill melanoma (B16), squamous cell carcinoma (KLN205) and colon carcinoma cells (MC38).26 In addition, skeletal muscle myoblasts, which naturally secrete TNFα, were effective in preventing the tumor growth and metastasis of prostate cancer cells.27 Finally, Prockop and colleagues most recently showed that pre-treatment of hMSCs with TNFα can be used to inhibit the progression of lung metastatic foci in vivo.28 However, none of these papers directly demonstrated that recombinant expression and cellular delivery of TNFα was indeed sufficient to prevent tumor growth in vivo.
Interestingly, pre-treatment of hMSCs with TNFα increases their ability to home to and accumulate within tumors, and this phenomenon is dependent upon NFkB activation.13 As such, cellular delivery of TNFα via the injection of non-tumorigenic cells (human fibroblasts or hMSCs) could be an attractive new approach for anticancer therapy.
Materials and Methods
Materials
Antibodies were purchased from the following commercial sources: Beclin-1 (NBP1–00085, Novus), Lamp-1 (E-5, sc-17768, Santa Cruz Biotech), BNIP3 (ab10433, Abcam), Cathepsin B (FL-339, sc-13985, Santa Cruz Biotech), OXPHOS (MS601, Mitosciences), β-actin (A5441, Sigma), caspase-3 (9662, Cell Signaling), Bax (06–499, Millipore), TNFα (ab9739, Abcam), TNFR-1A (ab107860, Abcam), TNFR-1B (ab15563, Abcam), BCL-2 (ab7973, Abcam). Other reagents were purchased as follows: DAPI nuclear stain (Sigma), anti-fade mounting reagent (S2828, Invitrogen), biotinylated anti-rabbit antibody (BA-1000, Vector), normal goat serum (S-1000, Vector), streptavidin-HRP (K0690, Dako), Avidin (AB972H-A, Biocare Medical), Biotin (AB972H-B, Biocare Medical), DAB (K3468, Dako).
Cell cultures
MDA-MB-231 cells (a human triple negative breast cancer cell line) and hTERT-BJ1 cells (an hTERT-immortalized fibroblast cell line) were cultured in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum in a 37°C humidified atmosphere containing 5% CO2.
Cell line derivation
Lentiviral plasmids (from GeneCopoeia) were as follows: EX-NEG-Lv105 (empty vector), TNFα (EX-A0276-Lv105), TNFR-1A (EX-Z2785-Lv105), TNFR-1B (EX-A0254-Lv105). Lentiviral particles were generated by transfecting the GeneCopoeia 293Ta packaging cells with lentiviral plasmids, using the Lenti-Pac HIV Expression Packing Kit (GeneCopoeia), according to manufacturer’s recommendations. Cells were transduced with lentiviral particles in the presence of 5 μg/ml Polybrene (Santa Cruz Biotech). Transduced cells were selected with 2 μg/ml (MDA-MB-231 cells) or 1.5 µg/ml (hTERT-BJ1 cells) puromycin.
Immunoblotting
Protein lysates were obtained by cell scraping into lysis buffer (10 mM Tris pH 7.5, 150 mM NaCl, 1% Triton X-100 and 60 mM n-octylglucoside) containing protease and phosphatase inhibitors. Samples were incubated on a rotating platform at 4°C, and then centrifuged at 12,000 g for 10 min at 4°C to remove insoluble debris. Protein concentrations were determined using the BCA reagent (Pierce). Samples were separated by SDS-PAGE (10% acrylamide) and transferred to nitrocellulose. All subsequent wash buffers contained 10 mM Tris pH 8.0, 150 mM NaCl, 0.1% Tween 20, which was supplemented with 5% nonfat dry milk for the blocking solution and 1% BSA for the antibody diluent. Horseradish peroxidase-conjugated secondary antibodies were used to visualize bound primary antibodies with an ECL detection kit (Pierce).
Immunofluorescence
Cells were plated onto coverslips in 12-well plates for 48 h in complete media. Then, cells were rinsed with PBS containing 0.1 mM CaCl2 and 1 mM MgCl2 (PBS/CM) and fixed with 2% paraformaldehyde in PBS/CM for 30 min. After fixation, cells were washed three times with PBS/CM and permeabilized with IF buffer (PBS/CM with 0.1% Triton-X100 and 0.2% BSA) for 10 min. Then, fixed cells were quenched with 50 mM NH4Cl in PBS/CM for 10 min, rinsed and incubated with anti-TNFα antibodies for 24 h at 4°C. Cells were washed with IF buffer and incubated with fluorescent secondary antibodies (Molecular Probes) for 30 min. After washing, cells were incubated with DAPI nuclear stain, rinsed and mounted with ProLong Gold anti-fade reagent (Molecular Probes). Images were acquired with a Zeiss LSM510 Meta confocal microscope system and analyzed with Zeiss LSM Browser.
Apoptosis assay by Annexin V and PI staining (flow cytometry)
Cells were seeded in a 12-well plate (50,000 cells/well). The day after, the medium was collected into a 15-ml tube, and cells were rinsed with PBS and trypsinized (100 μl of 0.05% trypsin-EDTA per well). Cells were placed into the same tube as the collected medium. Cells were spin down at 1,000 rpm for 5 min and resuspended in 500 μl of 1x Annexin binding buffer (556454, BD Biosciences), with 4 μl of Annexin V-APC conjugate (550474, BD Biosciences) and 0.5 μl propidium iodide (PI) (71–04–00, KPL). Samples were incubated in the dark for 5–15 min, and then analyzed by flow cytometry under the following parameters: voltage settings-FSC (250), SSC (245), APC (500), PE Texas Red (PI, 400).
MTT assay
The MTT assay (M5655, Sigma) was performed according to manufacturer’s recommendations. Cells were seeded in 48-well plate (9 wells for each cell line) and incubated at 37°C for 48–72 h. Then, the MTT solution (5 mg/ml in phenol-free medium) was added to each well to equal one-tenth of the original culture volume, and incubated for 3–4 h. At the end of the incubation period, the medium was removed and the converted dye was solubilized with 200 μL of DMSO. Absorbance was measured in a plate reader at a wavelength of 570 nm.
Pre-clinical animal studies
Tumor cells (MDA-MB-231; 1 × 106 cells) were injected alone or co-injected with hTERT-BJ1 fibroblasts (3 × 105 cells) in 100 µl of sterile PBS into the flanks of athymic NCr nude mice (NCRNU; Taconic Farms; 6–8 weeks of age). Mice were sacrificed at 4–5 weeks post-injection; tumors were excised to determine their weight and size, using calipers. Tumor volumes were calculated using the formula V = (x2 y)/2, where V is the tumor volume, x is the length of the short axis, and y is the length of the long axis.
Immunohistochemistry
Formalin-fixed paraffin embedded sections (6 µm) were deparaffinized by heating for 30 min at 55°C, followed by xylene incubation and rehydration to water. Antigen retrieval was performed with citrate buffer (pH 6.0) using a pressure cooker. Endogenous peroxidase was blocked with 3% H2O2 in PBS for 15 min, followed by avidin/biotin blocking. Then, slides were blocked with 10% goat serum for 60 min and incubated overnight with anti-TNFR-1A (1:200) or anti-TNFR-1B (1:100) at 4°C. Slides were then incubated with biotinylated secondary antibodies (1:500) for 30 min at room temperature, followed by streptavidin-HRP incubation for 30 min at room temperature. Bound antibodies were visualized with DAB solution. Slides were counterstained with hematoxylin and mounted.
Real-time PCR
RNA extraction was performed using Trizol-Phenol-Isoprobanol. cDNA was prepared using the First-Strand cDNA Synthesis Kit for Real-Time PCR (75780 50 RX) from Affymetrix. All primers were from Invitrogen (Taqman Gene Expression Assay, GEx, 4331182), TaqMan Gene Expression Assay consist of a pair of unlabeled PCR primers and a TaqMan probe with a FAM for all genes and VIC for the GAPDH housekeeping gene (20 X). Primers were diluted 1:20 in 2X master mix (Hot Start-IT Master Mix (2X), usb 75766). Q-PCR was performed by mixing 1:1 the primer master mix with the cDNA and run under the following conditions: 95°C for 2 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min.
Statistical analysis
Statistical significance was examined using the Student t-test. Values of p < 0.05 were considered significant.
Acknowledgments
F.S. was the recipient of a Young Investigator Award from the Breast Cancer Alliance. U.E.M. was supported by a Young Investigator Award from the Margaret Q. Landenberger Research Foundation. Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.).
This work was also supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the UK and an Advanced ERC Grant from the European Research Council.
Also, these developments were made possible through the resources of Thomas Jefferson University.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/23370
References
- 1.Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood. 2012;119:651–65. doi: 10.1182/blood-2011-04-325225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts NJ, Zhou S, Diaz LA, Jr., Holdhoff M. Systemic use of tumor necrosis factor alpha as an anticancer agent. Oncotarget. 2011;2:739–51. doi: 10.18632/oncotarget.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Solomon DH, Mercer E, Kavanaugh A. Observational studies on the risk of cancer associated with tumor necrosis factor inhibitors in rheumatoid arthritis: a review of their methodologies and results. Arthritis Rheum. 2012;64:21–32. doi: 10.1002/art.30653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cassaday RD, Malik JT, Chang JE. Regression of Hodgkin lymphoma after discontinuation of a tumor necrosis factor inhibitor for Crohn’s disease: a case report and review of the literature. Clin Lymphoma Myeloma Leuk. 2011;11:289–92. doi: 10.1016/j.clml.2011.03.018. [DOI] [PubMed] [Google Scholar]
- 5.Keystone EC. Does anti-tumor necrosis factor-α therapy affect risk of serious infection and cancer in patients with rheumatoid arthritis?: a review of longterm data. J Rheumatol. 2011;38:1552–62. doi: 10.3899/jrheum.100995. [DOI] [PubMed] [Google Scholar]
- 6.Dommasch ED, Abuabara K, Shin DB, Nguyen J, Troxel AB, Gelfand JM. The risk of infection and malignancy with tumor necrosis factor antagonists in adults with psoriatic disease: a systematic review and meta-analysis of randomized controlled trials. J Am Acad Dermatol. 2011;64:1035–50. doi: 10.1016/j.jaad.2010.09.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashkenazi A, Holland P, Eckhardt SG. Ligand-based targeting of apoptosis in cancer: the potential of recombinant human apoptosis ligand 2/Tumor necrosis factor-related apoptosis-inducing ligand (rhApo2L/TRAIL) J Clin Oncol. 2008;26:3621–30. doi: 10.1200/JCO.2007.15.7198. [DOI] [PubMed] [Google Scholar]
- 8.Daniel D, Wilson NS. Tumor necrosis factor: renaissance as a cancer therapeutic? Curr Cancer Drug Targets. 2008;8:124–31. doi: 10.2174/156800908783769346. [DOI] [PubMed] [Google Scholar]
- 9.Grosse-Wilde A, Kemp CJ. Metastasis suppressor function of tumor necrosis factor-related apoptosis-inducing ligand-R in mice: implications for TRAIL-based therapy in humans? Cancer Res. 2008;68:6035–7. doi: 10.1158/0008-5472.CAN-08-0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Secchiero P, Zauli G. Tumor-necrosis-factor-related apoptosis-inducing ligand and the regulation of hematopoiesis. Curr Opin Hematol. 2008;15:42–8. doi: 10.1097/MOH.0b013e3282f15fa6. [DOI] [PubMed] [Google Scholar]
- 11.ten Hagen TL, Seynhaeve AL, Eggermont AM. Tumor necrosis factor-mediated interactions between inflammatory response and tumor vascular bed. Immunol Rev. 2008;222:299–315. doi: 10.1111/j.1600-065X.2008.00619.x. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Lin Y. Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol Sin. 2008;29:1275–88. doi: 10.1111/j.1745-7254.2008.00889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uchibori R, Tsukahara T, Mizuguchi H, Saga Y, Urabe M, Mizukami H, et al. NFκB Activity Regulates Mesenchymal Stem Cell Accumulation at Tumor Sites. Cancer Res. 2013;73:364–72. doi: 10.1158/0008-5472.CAN-12-0088. [DOI] [PubMed] [Google Scholar]
- 14.Iyengar P, Espina V, Williams TW, Lin Y, Berry D, Jelicks LA, et al. Adipocyte-derived collagen VI affects early mammary tumor progression in vivo, demonstrating a critical interaction in the tumor/stroma microenvironment. J Clin Invest. 2005;115:1163–76. doi: 10.1172/JCI23424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Williams TM, Medina F, Badano I, Hazan RB, Hutchinson J, Muller WJ, et al. Caveolin-1 gene disruption promotes mammary tumorigenesis and dramatically enhances lung metastasis in vivo. Role of Cav-1 in cell invasiveness and matrix metalloproteinase (MMP-2/9) secretion. J Biol Chem. 2004;279:51630–46. doi: 10.1074/jbc.M409214200. [DOI] [PubMed] [Google Scholar]
- 16.Chiavarina B, Martinez-Outschoorn UE, Whitaker-Menezes D, Howell A, Tanowitz HB, Pestell RG, et al. Metabolic reprogramming and two-compartment tumor metabolism: opposing role(s) of HIF1α and HIF2α in tumor-associated fibroblasts and human breast cancer cells. Cell Cycle. 2012;11:3280–9. doi: 10.4161/cc.21643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Capparelli C, Chiavarina B, Whitaker-Menezes D, Pestell TG, Pestell RG, Hulit J, et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts, “fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle. 2012;11:3599–610. doi: 10.4161/cc.21884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez-Outschoorn UE, Pestell RG, Howell A, Tykocinski ML, Nagajyothi F, Machado FS, et al. Energy transfer in “parasitic” cancer metabolism: mitochondria are the powerhouse and Achilles’ heel of tumor cells. Cell Cycle. 2011;10:4208–16. doi: 10.4161/cc.10.24.18487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Power surge: supporting cells “fuel” cancer cell mitochondria. Cell Metab. 2012;15:4–5. doi: 10.1016/j.cmet.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 20.Sotgia F, Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Lisanti MP. Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment. Breast Cancer Res. 2011;13:213. doi: 10.1186/bcr2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathol. 2012;7:423–67. doi: 10.1146/annurev-pathol-011811-120856. [DOI] [PubMed] [Google Scholar]
- 22.Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I, et al. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal. 2012;16:1264–84. doi: 10.1089/ars.2011.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lisanti MP, Martinez-Outschoorn UE, Chiavarina B, Pavlides S, Whitaker-Menezes D, Tsirigos A, et al. Understanding the “lethal” drivers of tumor-stroma co-evolution: emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor micro-environment. Cancer Biol Ther. 2010;10:537–42. doi: 10.4161/cbt.10.6.13370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lisanti MP, Martinez-Outschoorn UE, Lin Z, Pavlides S, Whitaker-Menezes D, Pestell RG, et al. Hydrogen peroxide fuels aging, inflammation, cancer metabolism and metastasis: the seed and soil also needs “fertilizer”. Cell Cycle. 2011;10:2440–9. doi: 10.4161/cc.10.15.16870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lisanti MP, Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Pestell RG, Howell A, et al. Accelerated aging in the tumor microenvironment: connecting aging, inflammation and cancer metabolism with personalized medicine. Cell Cycle. 2011;10:2059–63. doi: 10.4161/cc.10.13.16233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munich S, Sobo-Vujanovic A, Buchser WJ, Beer-Stolz D, Vujanovic NL. Dendritic cell exosomes directly kill tumor cells and activate natural killer cells via TNF superfamily ligands. Oncoimmunology. 2012;1:1074–83. doi: 10.4161/onci.20897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stölting MN, Ferrari S, Handschin C, Becskei A, Provenzano M, Sulser T, et al. Myoblasts inhibit prostate cancer growth by paracrine secretion of TNF alpha. J Urol. 2012 doi: 10.1016/j.juro.2012.10.071. In press. [DOI] [PubMed] [Google Scholar]
- 28.Lee RH, Yoon N, Reneau JC, Prockop DJ. Preactivation of Human MSCs with TNFα Enhances Tumor-Suppressive Activity. Cell Stem Cell. 2012;11:825–35. doi: 10.1016/j.stem.2012.10.001. [DOI] [PubMed] [Google Scholar]