

Abstract
Phenotypic switching of vascular smooth muscle cells (VSMCs) is known to play a key role in the development of atherosclerosis. However, the mechanisms that mediate VSMC phenotypic switching are unclear. We report here that TIPE2, the tumor necrosis factor (TNF) α-induced protein 8-like 2 (TNFAIP8L2), plays an atheroprotective role by regulating phenotypic switching of VSMCs in response to oxidized low-density lipoprotein (ox-LDL) stimuli. TIPE2-deficient VSMCs treated with ox-LDL expressed lower levels of contractile proteins such as SMαA, SM-MHC and calponin, whereas the proliferation, migration and the synthetic capacity for growth factors and cytokines were increased remarkably. Furthermore, TIPE2 inhibited VSMCs proliferation by preventing G1/S phase transition. Interestingly, these effects of TIPE2 on VSMCs were dependent on P38 and ERK1/2 kinase signals. As a result, neointima formation was accelerated in the carotid arteries of TIPE2-deficient mice. These results indicate that TIPE2 is a potential inhibitor of atherosclerosis.
Keywords: TNFAIP8L2 (TIPE2), vascular smooth muscle cell, phenotypic switching, cell cycle, atherosclerosis
Introduction
Vascular smooth muscle cell (VSMC) migratory and proliferatory responses to inflammation of the vessel wall are considered to play a critical role in the development of atherosclerosis.1 The earliest recognizable lesion of atherogenesis is the so-called “fatty streak” of intima. Studies show that the transition from the fatty streak to the more complex lesion is characterized by the immigration of smooth muscle cells (SMCs) from the medial layer of the artery wall into the intimal or subendothelial space.2 Thus, intimal smooth muscle cells undergo phenotypic switching, synthesize extracellular matrix proteins and express a variety of adhesion molecules, all of which contribute to the initiation of atherogenesis.
“Phenotypic switching” or “phenotypic modulation” means that SMCs in lesions have changed from a contractile to a synthetic phenotype, which can result in an increase in the rate of cell proliferation and migration and a decrease in the expression of SMC-specific marker genes,3 including smooth muscle actin (SMαA),4 smooth muscle myosin heavy chain (SM-MHC),5 SM-22 (SM22)6 and calponin (CN).7 A high concentration of circulating ox-LDL is a major risk factor for atherosclerosis, not only because it can influence the functions of macrophages, but also stimulate MAP kinase activation in VSMC.2,8 Recent data have indicated that numerous transcription regulators mediate responses to the proliferation and migration of VSMCs.9,10 However, the exact mechanisms are not well-understood.
TIPE2, the tumor necrosis factor (TNF)-α-induced protein 8-like 2 (TNFAIP8L2), is newly identified as a negative regulator for inflammation through inhibiting immune receptor signaling.11-13 TIPE2 deficiency in mice causes fetal inflammatory diseases, and abnormal expression in humans is associated with infectious disease and autoimmune disorders.11,14-16 Early studies showed that TIPE2 expressed in macrophages plays an atheroprotective role by negatively regulating inflammation (unpublished data). However, as the major effector cell in atherosclerosis, it is unknown whether TIPE2 takes a part in VSMCs functions. This study will focus on the roles of TIPE2 in VSMCs functions during atherogenesis. We hypothesize that TIPE2 expressed in VSMCs prevents the development of atherosclerosis by negatively regulating phenotypic switching. Therefore, VSMCs isolated from TIPE2−/− mice and WT controls were used to determine the expression of contractile proteins, the ability of migration and proliferation and the synthetic capacity following ox-LDL stimuli in this study.
Results
TIPE2 expression in cultured VSMCs was upregulated by ox-LDL stimulation
Murine VSMCs were obtained as described previously. When cultured in regular medium, SMαA protein was detected both in VSMCs from TIPE2-deficient mice and WT controls, but the staining in WT cells was much stronger than that in TIPE2−/− cells (Fig. 1A). To evaluate the effect of ox-LDL, a major atherogenic factor,2 on TIPE2 expression, VSMCs from WT mice were grown in growth medium containing 10% FBS in the absence or presence of different concentrations of ox-LDL. As shown in Figure 1C and D, both the mRNA and protein levels of TIPE2 in VSMCs were increased significantly following ox-LDL stimulation in a dose-dependent manner. We found that 20 μg/mL was the most effective concentration. Figure 1B illustrates that ox-LDL could upregulate the mRNA levels of TIPE2 in a time-dependent manner. These data suggest that TIPE2 may regulate VSMCs functions after exposure to ox-LDL stimulation.

Figure 1. Roles of TIPE2 on ox-LDL-induced VSMCs phenotypic switching. (A) SMαA was detected in cultured VSMCs from C57BL/6 (WT) and TIPE2−/− mice by immunostaining (×200). (B) Levels of TIPE2 mRNA in WT VSMCs treated with 20 μg/mL ox-LDL for different times determined by real-time RT-PCR. (C and D) Both the mRNA and protein levels of TIPE2 in WT VSMCs treated with ox-LDL stimuli were upregulated in a time- and dose-dependent manner. β-actin served as control for equal quality and protein loading. One representative result of three independent experiments is shown (n = 3). *p < 0.05, **p < 0.005, ***p < 0.001. (E) The mRNA levels of differentiated marker genes, such as SMαA, SM-MHC and calponin in TIPE2−/− VSMCs treated with 20 μg/mL ox-LDL were significantly decreased compared with WT controls. (F) Protein levels of SMαA and TIPE2 in TIPE2−/− VSMCs and WT controls were determined by western blot at 24, 48 h after 20 μg/mL ox-LDL stimulation. (G) mRNA levels of contractile protein were upregulated in TIPE2-overexpressed VSMCs. (H) The overexpression of TIPE2 significantly upregulated SMαA protein levels. β-actin served as control for equal qualify and protein loading. Three independent experiments were performed and one representative result is shown. *p < 0.05, **p < 0.005, ***p < 0.001.
TIPE2 was required for contractile protein expression in cultured mice aortic VSMCs
The process of transition from contractile phenotype to synthetic phenotype in VSMCs contributes to many human vascular diseases, including atherosclerosis.3,17,18 The modulation from contractile to synthetic phenotype is associated with a marked decrease in expression of contractile proteins.19 In this study, TIPE2-deficient and overexpressed VSMCs were used to determine whether TIPE2 could modify phenotypic switching. We found that the mRNA levels of concerned contractile genes, such as SMαA, SM-MHC and calponin, were decreased significantly in TIPE2-deficient VSMCs compared with WT controls (the first column from left of each figure in Fig. 1E, TIPE2−/−vs. WT, 0.645 ± 0.011 vs. 1.001 ± 0.027 for SMαA, p = 0.0002; 0.661 ± 0.030 vs. 1.000 ± 0.013 for SM-MHC, p = 0.0005; 0.636 ± 0.049 vs. 1.000 ± 0.012 for calponin, p = 0.0019). The downregulations of marker gene mRNA levels were remarkably enlarged by ox-LDL stimulation in TIPE2-deficient VSMCs (Fig. 1E, TIPE2−/−vs. WT at 6 h, 0.134 ± 0.024 vs. 0.611 ± 0.023 for SMαA, p = 0.0001; 0.080 ± 0.022 vs. 0.545 ± 0.065 for SM-MHC, p = 0.0025; 0.114 ± 0.025 vs. 0.600 ± 0.038 for calponin, p = 0.0004). We also examined the expression levels of SMαA protein by western blot. As expected, SMαA protein decreased significantly in TIPE2-deficient VSMCs compared with WT controls, especially in cells exposed to 20 μg/mL ox-LDL stimulation (Fig. 1F), whereas both the mRNA and protein levels of contractile genes expression were significantly increased in VSMCs transfected with TIPE2 expression vectors (Fig. 1G and H). These results indicate that TIPE2 deficiency in VSMCs could promote the transition from contractile to synthetic phenotype response to ox-LDL stimulation, while TIPE2 overexpression could upregulate contractile genes expression.
TIPE2 prevents migration of cultured VSMCs
It has been reported that TIPE2 could inhibit cells migration.20 Migration of VSMCs from media to intima is pivotal to atherogenesis.21 In this study, Boyden chamber migration assays were used to determine whether TIPE2 could inhibit the migration of cultured VSMCs. As shown in Figure 2A and B, the migrated cells in TIPE2−/− VSMCs were significantly increased compared with WT controls (TIPE2−/−vs. WT, 206 ± 18 vs. 96 ± 11, p = 0.0009), especially when exposed to ox-LDL stimulation, the migrated cells were remarkably enhanced (TIPE2−/−vs. WT, 504 ± 15 vs. 170 ± 19, p < 0.0001), whereas overexpressed TIPE2 in cultured VSMCs could prevent the migration. As shown in Figure 2C−E, VSMC/pEGFP-mTIPE2 expressed higher levels of TIPE2 protein and migrated cells were obviously decreased compared with VSMC/pEGFP control cells. Taken together, results of these studies provide evidence that TIPE2 can prevent migration in cultured VSMC.

Figure 2. TIPE2 prevents migration of cultured murine VSMCs. (A and B) TIPE2 deficiency promotes cell migration, especially exposed to ox-LDL stimulation (×100). (C) Enhanced expression of TIPE2 protein in VSMC/pEGFP-mTIPE2 at 24 h. (D and E) TIPE 2 overexpression prevents VSMC migration detected by Boyden Chamber analysis. Five fields of migrated cells in the lower side were counted with a microscope at ×100. (F) mRNA levels of MMP-9 in WT and TIPE2−/− VSMCs determined by real-time RT-PCR at 2, 4, 6 h after ox-LDL stimulation at a concentration of 20 μg/mL. Data represent means ± SEM of three independent experiments. *p < 0.05, **p < 0.005, ***p < 0.001.
Matrix metalloproteinase-9 (MMP-9) are known to play important roles during changes in vascular structure associated with atherosclerotic progression. The stimulation with ox-LDL leads to increased activity of MMP-9, and this upregulation is linked to increased cell migration.22 Therefore, we further tested the effect of TIPE2 on MMP-9 expression in cultured VSMCs. As shown in Figure 2F, without ox-LDL treatment, although both TIPE2−/− VSMCs and WT controls expressed very low levels of MMP-9, the mRNA levels of MMP-9 detected in TIPE2−/− VSMCs were much higher than that in WT controls (TIPE2−/−vs. WT, 1.327 ± 0.021 vs. 1.001 ± 0.028, p = 0.0008). Ox-LDL treatment strongly induced MMP-9 expression, especially in TIPE2 deficient cells. These data suggest that MMP-9 may be involved in TIPE2-inhibited migration of cultured SMCs during atherosclerosis.
TIPE2 affects synthetic functions of VSMCs
Secretory functions of VSMCs play an important role in the initiation and progression of AS.23 In this study, we found that either in presence or absence of ox-LDL, the mRNA levels of PDGF-A and its receptor PDGFR-α, were significantly increased in TIPE2-deficient VSMCs compared with WT controls (Fig. 3A and B). Whereas, the mRNA levels of pro-inflammatory cytokines, such as IL-1, IL-6, IL-8 and TNFα, were not different between TIPE2-deficient VSMCs and WT controls cultured in normal medium. When the cells were exposed to ox-LDL stimulation, the expression of these cytokines was strongly upregulated, while the levels in TIPE2-deficient cells were significant higher than that in WT controls (Fig. 3C−F, p < 0.05). We also found that the mRNA levels of MCP-1, a chemokine for macrophages, were increased significantly in TIPE2-deficient cells, especially incubated with ox-LDL (Fig. 3G). Furthermore, the mRNA levels of TGF-β, an atheroprotective factor that can suppress effector leukocytes and promote VSMCs differentiation,24 were decreased significantly in cultured TIPE2-deficient VSMCs compared with WT controls (Fig. 3H). These results indicate that TIPE2 could regulate VSMCs synthetic functions and subsequently attenuate AS development.

Figure 3. TIPE2 affects synthetic functions of VSMCs. The mRNA levels of PDGF-A (A), PDGFR-α (B) and IL-1β (C), IL-6 (D), IL-8 (E), TNF-α (F), MCP-1 (G) and TGF-β (H) in TIPE2−/− VSMCs and WT controls were determined by real-time PCR at 2, 4, 6 h after 20 μg/mL ox-LDL stimulation. One representative result of three independent experiments is shown.*p < 0.05, **p < 0.005, ***p < 0.001.
TIPE2 suppresses proliferation through preventing cell cycle G1/S phase transition
It is well known that AS development could be partly explained by the proliferation of VSMCs. To determine whether TIPE2 plays roles in proliferation, VSMCs from TIPE2−/− mice and WT controls were cultured to detect their proliferative capacity in this study. As shown in Figure 4A, the 450 nm absorbance of TIPE2−/− VSMCs with or without ox-LDL stimulation was obviously higher than WT cells, indicating that TIPE2 can inhibit VSMCs proliferation. Furthermore, the expression of PCNA (proliferating cell nuclear antigen), a protein involved in cell cycle progression,25 was remarkably increased in cultured TIPE2-deficient VSMCs, neither in absence nor presence of ox-LDL stimulation (Fig. 4D).

Figure 4. TIPE2 inhibits VSMC proliferation and prevents cell cycle entry/progression in the G0/G1 phase. (A) Proliferation ability is significantly increased in TIPE2−/− VSMCs with or without ox-LDL treatment compared with WT controls. (B and C) TIPE2 prevents cell cycle entry/progression in G0/G1 phase in cultured VSMCs. (D) SMαA, PCNA and cell cycle proteins such as Cyclin D1, Cyclin E, P21 and P27 in VSMCs treated with PD/SB plus ox-LDL were determined by western blot. β-actin served as a loading control. Data shown are means ± SEM (n = 5) and are representative of three independent experiments. *p < 0.05, **p < 0.005, ***p < 0.001.
To elucidate the mechanisms responsible for proliferation effect of TIPE2, we determine the cell cycle progression and the expression of cell cycle-regulating proteins in cultured TIPE2−/− VSMCs and WT cells. As shown in Figure 4B and C, the baseline proportion of TIPE2−/− VSMCs in S+G2/M phase was much higher than that of WT control cells (35.15 ± 2.73% vs. 20.97 ± 3.55%, p = 0.0124). While treated with 20 μg/mL ox-LDL, the proportion of TIPE2-deficient VSMCs in S+G2/M phase was upregulated remarkably compared with WT cells (54.30 ± 2.92% vs. 31.58 ± 2.32%, p = 0.0002). These results indicate that TIPE2 deficiency prevents cell cycle entry/progression in G0/G1 phase in cultured VSMCs. Further studies showed that TIPE2 prevents the downregulation of the cyclin-dependent kinase inhibitors p21cip1 and p27kip1, as well as upregulation of cyclin D1 and cyclin E, which are essential for the progression through the G0/G1 phase (Fig. 4D). However, there was no specific regulation of one of these molecules, suggesting that TIPE2 may not affect the expression or function of a single cell cycle regulatory protein, but rather may interfere with upstream signaling mechanisms that regulate early cell cycle entry. Taken together, TIPE2 can prevent VSMCs proliferation via interfering with upstream signaling that regulates cell cycle entry.
The effects of TIPE2 on phenotypic switching, migration and proliferation of VSMCs is dependent on ERK and P38 signaling pathways
Previous reports pointed out that ERK1/2 and P38 signal transduction pathways played important roles in VSMCs function.26-29 The mitogen-activated protein kinase pathway is an essential trigger for early cell cycle events, such as the expression of the cyclin-dependent kinase inhibitors p21cip/waf and p27Kip1, or the expression of cyclins.30 Therefore, in this study we evaluated the effect of TIPE2 on the activation of ERK1/2 and P38 pathways in cultured VSMCs. As shown in Figure 5A, the basal levels of phosphorylation of ERK1/2 and P38 were increased in TIPE2-deficient VSMCs, especially when exposed to ox-LDL stimulation. Phosphorylated ERK1/2 and P38 were upregulated remarkably in TIPE2-deficient VSMCs than that in WT controls, suggesting that TIPE2 can negatively regulate the activation of these signaling pathways.

Figure 5. The effects of TIPE2 on phenotypic switching, migration and proliferation of cultured VSMCs are dependent on ERK and P38 signaling pathways. (A) TIPE2 can inhibit the activation of ERK1/2 and P38 pathways. (B–D) The effects of TIPE2 on phenotypic switching, cell migration and proliferation of VSMCs are ERK1/2 and P38 dependent. Data are means ± SEM of one representative result obtained from three independent experiments. ***p < 0.001.
To further determine whether the enhanced signal transduction was due to TIPE2 deficiency, PD98059 plus SB203580 were used to block ERK1/2 and P38 signaling in cultured VSMCs. As expected, both the decrease of contractile marker genes expression and the increase of migratory cells were prevented by PD98059 and SB203580 in ox-LDL stimulated TIPE2-deficient VSMCs (Figs. 4D and 5B−D). Similarly, treatment with both inhibitors suppressed the upregulation of PCNA, Cyclin D1 and Cyclin E, as well as the downregualtion of P21 and P27 (Fig. 4D). These data suggest that TIPE2 deficiency can activate ERK1/2 and P38 signaling transduction; as a result, the capacity of phenotypic switching, migration and proliferation of cultured TIPE2-deficient VSMCs was enhanced.
TIPE2 deficiency accelerates neointima formation in vivo
VSMCs dysfunction plays important role in neointima formation, which is one of major events in early atherosclerosis plague development. To determine the potential role of TIPE2 in atherosclerosis (AS) development, we generated rapid atherogenesis modeled by perivascular carotid collar placement in TIPE2−/− mice, while age- and sex-matched ApoE−/− mice were used as controls. Remarkably, we found that TIPE2−/− mice were significantly prone to early AS lesion formation. Specifically, 3 wk after HFD feeding, all TIPE2−/− mice generated visible lesions in the proximal carotid site, but not ApoE−/− mice (Fig. 6A). Furthermore, we also observed increased VSMCs in the lesions of TIPE2−/− mice (Fig. 6B). These data suggest that TIPE2 may prevent neointima formation by negatively regulating VSMCs functions and subsequently delay the early plague formation during AS development in vivo.
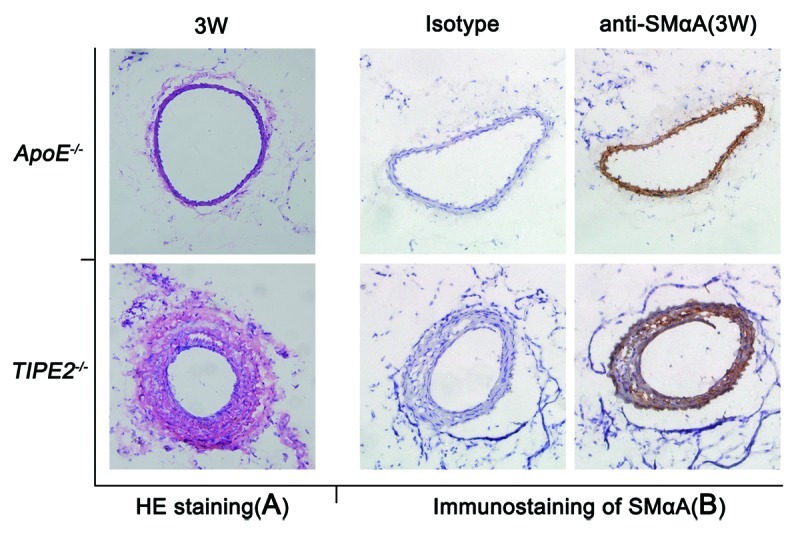
Figure 6. TIPE2 deficiency accelerates neointima formation in vivo. (A) Representative microphotographs of HE-stained neointima obtained from ApoE−/− and TIPE2−/− mice 3 weeks after high-fat diet feeding. (B) The expression of SMαA in neointima lesions of ApoE−/− and TIPE2−/− mice three weeks after perivascular carotid collar placement was detected by immunostaining (×200).
Discussion
Phenotypic switching characterized by loss of contractility and abnormal proliferation, migration and matrix synthesis of vascular smooth muscle cells (VSMCs) is involved in the development of atherosclerosis, which is considered as a vascular proliferation disease.31-33 In this study, we first report that TIPE2 can negatively regulate phenotypic switching of cultured VSMCs following ox-LDL stimulation. As a result, the expression of contractile genes or differentiation markers was significantly decreased in TIPE2-deficient VSMCs, while the capacity of migration, matrix synthesis and cellular proliferation were increased. In addition, we demonstrated that TIPE2 represses SMC proliferation by preventing cell cycle G1/S phase transition, and this effect is dependent on ERK1/2 and P38 signaling transduction. As a result, neointima formation is accelerated in the carotid arteries of TIPE2-deficient mice that are fed with a high fat diet in vivo. As such, results of the present studies indicate that TIPE2 may be a new potential therapeutic target for the prevention of vascular proliferation disease, such as atherosclerosis, by negatively regulating VSMCs phenotypic switching.
Mouse SMCs can switch between the “contractile” and “synthetic” phenotypic states in response to a variety of atherogenic stimuli.3,17,34 In a contractile phenotype, SMCs predominantly express proteins involved in the contractile function of the cell, such as SM-MHC or SMαA, while those SMCs found in lesions express lower levels of these proteins.35 Consistent with recent findings,19 in this study we found that all cultured vascular smooth muscle cells from TIPE2−/− mice and C57BL/6 controls were positive for SMαA, whereas the intensity of the staining in TIPE2-deficient cells was much lower than that in normal controls. Moreover, when exposed to ox-LDL stimuli, the levels of contractile proteins, including SMαA, SM-MHC and calponin, are significantly decreased in TIPE2-deficient cells, while the synthetic capacity for some cytokines and growth factors are increased, indicating that a TIPE2-deficient cell is a synthetic predominant phenotype. Consequently, TIPE2 may be a new atheroprotective factor by preventing the transition from a contractile phenotype to synthetic phenotype.
Furthermore, we found that TIPE2 can inhibit VSMC migration and synthetic capacity. This is in accordance with previous reports that TIPE2 deficiency led to increased migration and dysregulation of exocyst complex formation in macrophages.20 Migration ability is another important component of vascular lesion formation. Current evidence suggests that intimal SMCs have unique atherogenic properties that make them migrate more readily in response to ox-LDL stimuli, and this can contribute to the initiation and propagation of atherosclerosis.36 Our results showed that TIPE2 deficiency can promote cultured VSMCs migration, while cells overexpressing TIPE2 showed decreased migration ability. This effect was also observed in vivo, because TIPE2 deficiency significantly accelerates neointima formation in a murine model of collar-induced atherosclerosis.
Previous studies showed that TIPE2 interferes with Ras downstream signaling.20 In the present study we demonstrate that TIPE2 can prevent VSMC proliferation by inhibiting the activation of P38 and ERK1/2 pathways in response to ox-LDL stimuli. ERK pathway is known to be essential for the induction of cell cycle entry.30 Thereby the cyclin-dependent kinase inhibitors p21cip/waf1 and p27Kip1 were decreased in TIPE2-deficient VSMCs treated with ox-LDL, while the transcriptional activation of G1 phase cyclins was increased. Subsequently, TIPE2 can prevent G1/S phase transition and proliferation of VSMC, suggesting that TIPE2 can interfere with upstream signaling mechanisms that regulate early cycle entry. Furthermore, the inhibition ability of proliferation represents a potentially interesting target for antiproliferative strategies.
On the other hand, TIPE2 could regulate cell death by promoting Fas-induced apoptosis.11 VSMCs apoptosis contributes to plaque destabilization. They are considered as the only vascular cells that express stabilizing matrix to hinder plaque rupture.41 In this study, we found that TIPE2−/− VSMCs could express a higher level of MMP-9 than wild type VSMCs, which is considered a maker for unstable plaque, indicating that TIPE2 may play roles in plaque stabilization by regulating VSMCs apoptosis. Otherwise, autophagy is related to cell death. Studies showed that the inhibition of autophagic capacity with aging generates the inflammaging condition via the activation of inflammasomes.42 Our findings are consistent with the view that age-related diseases such as atherosclerosis are active processes driven by increased cellular functions and signal transduction pathways.43 TIPE2 may inhibit this process by downregulating the P38 and ERK1/2 signaling pathways.
TIPE2 is a new negative regulator mainly expressed in immune tissues.11 Many cell types, such as macrophage, T lymphocyte and endothelia, are involved in atherogenesis. Although we discovered that the SMC contractile proteins or differentiation markers were downregulated in TIPE2-deficient VSMCs, in the present study, the migration ability and proliferation capacity were increased; it is possible that the loss of TIPE2 in other cell types may also contribute to neointima formation observed in TIPE2−/− mice. Indeed, our previous studies showed that TIPE2-deficient macrophages treated with ox-LDL produced more oxidative stress and proinflammatory cytokines, and exacerbated atherosclerosis development (our unpublished data). Therefore, in this study we cannot rule out the possibility that TIPE2 deficiency may alter the functions of these cells and thereby alter VSMC phenotypic switching and neointima formation. Perhaps, the loss of TIPE2 in endothelial cells also can contribute to enhanced neointima formation. Further studies are needed to determine the role of TIPE2 in endothelial cells during the development of atherosclerosis.
Overall, in the present study we provide evidence that TIPE2 plays a critical role in the regulation of VSMC phenotypic switching and neointima formation in response to ox-LDL stimulation. Indeed, TIPE2 is an inhibitor of VSMC proliferation by preventing cell cycle G1/S phase transition. These effects of TIPE2 were dependent on p38 and ERK1/2 activation. These results suggest that TIPE2 may be a new target for the atherosclerosis therapy. Further studies are needed to determine the role of TIPE2 in the development of other vascular disease in experimental animal models as well as in humans.
Material and Methods
Mice
All experiments with animals were performed according to the guidelines of the Animal Management Rules of the Chinese Ministry of Health (document No. 55, 2001) and were approved by the Animal Ethical Committee of Shandong University. Male C57BL/6J mice were purchased from Shanghai Laboratory Animal Center of Chinese Academy of Science and were 8–12-weeks-old at the time of entry into the study. The male TIPE2-deficient (TIPE2−/−) mice (C57BL/6J background, 8–12-weeks-old) have been described previously.11 All mice were housed in the Animal Facilities of Shandong University, under pathogen-free conditions.
Cell culture and plasmids
Primary murine VSMCs used in this study were obtained from the aorta ventralis of 8-week-old male C57BL/6J (wide type, WT) or TIPE2−/− mice by enzymatic digestion (type I collagenase and type III elastase, Sigma). Cells were grown in high-glucose version of DMEM medium (GIBCO-BRL) supplemented with 10% fetal bovine serum (FBS, Gibco-BRL), 100 μg/mL penicillin (Sigma) and 100 μg/mL streptomycin (Sigma), in 24-well culture dishes (Corning incorporated) at 37°C in a humidified atmosphere containing 5% CO2.37,38 Immunostaining was performed with anti-SMαA to determine the vascular smooth muscle cells as described previously.12 Then these cells of third to tenth passages were used to do experiments.
VSMC cells of WT mice were transfected with a TIPE2 expression vector (pEGFP-mTIPE2) or pEGFP alone, named as VSMC/pEGFP-mTIPE2 or VSMC/pEGFP respectively, or transfected with pRK5-mTIPE2 or pRK5 vectors, named as VSMC/pRK5-mTIPE2 or VSMC/pRK5 respectively, using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. The cells were then used for determining the differentiation maker genes or performing migration assay.
Ox-LDL stimulation
Ox-LDL was purchased from Peking Union Medical College. WT VSMCs at a concentration of 1 × 105 cells/mL were cultured in 12-well plates (Corning) or 6-well plates (Corning) overnight, then treated with various concentrations of ox-LDL (0, 5, 10, 20, 50 μg/mL, respectively). Four h later, cells in 12-well plates were collected for RNA extraction, while those in 6-well plates were collected 24 h later for TIPE2 protein detection. From this experiment, we found that 20 μg/mL was the most effective concentration. Therefore, cells were treated with 20 μg/mL ox-LDL for different time intervals (0, 2, 4, 6 h for RNA extraction and 0, 24, 48 h for western blot, respectively) and collected to determine TIPE2 expression both at mRNA and protein levels.
Quantitative real-time PCR
Total RNA was extracted according to the protocol for the TRIZOL kit (Invitrogen). Single-strand cDNA was synthesized using hexamer primers (Promega). cDNA was used as templates for the amplification of genes concerned. Quantitative PCR (qPCR) was performed as described previously.12 Each sample was run in triplicate. The primers used are shown in Table 1 and were synthesized by Invitrogen Corporation.
Table 1. The primers used for real-time RT-PCR.
| Species | Gene | Primer sequences |
|---|---|---|
| Mus | β-actin |
F: 5′-TGCGTGACATCAAAGAGAAG-3′ |
| R: 5′-TCCATACCCAAGAAGGAAGG-3′ | ||
| TIPE2 |
F: 5′-TCAGAAACATCCAAGGCCAGAC-3′ |
|
| R: 5′-CGGACCGACCAGCCATTTTAC-3′ | ||
| SMαA |
F: 5′-GAGAAGCCCAGCCAGTCG-3′ |
|
| R: 5′-CTCTTGCTCTGGGCTTCA-3′ | ||
| SM-MHC |
F: 5′-GAGGTGGTCGTGGAGTTGGT-3′ |
|
| R: 5′-GTATCGCTCCCTCAGGTTGT-3′ | ||
| Calponin |
F: 5′-CACCAACAAGTTTGCCAG-3′ |
|
| R: 5′-TGTGTCGCAGTGTTCCAT-3′ | ||
| MMP-9 |
F: 5′-CTGTCCAGACCAAGGGTACAGCCT-3′ |
|
| R: 5′-GTGGTATAGTGGGACACATAGTGG-3′ | ||
| PDGF-A |
F: 5′-GAAGGTCACGCAACTGGATCT-3′ |
|
| R: 5′-TGCATCTTCATGTAGCGTTCTT-3′ | ||
| PDGFR-α |
F: 5′-CCTGGCGCAAGGAAAAATTGT-3′ |
|
| R: 5′-CCAGAGCAGAATGCCATAGGA-3′ | ||
| IL-lβ |
F: 5′-TCGCTCAGGGTCACAAGAAA-3′ |
|
| R: 5′-ATCAGAGGCAAGGAGGAAA-3′ | ||
| IL-6 |
F: 5′-CAGGATACCACTCCCAACAGACC-3′ |
|
| R: 5′-AAGTGCATCATCGTTGTTCATACA-3′ | ||
| IL-8 |
F: 5′-CTCAAGAATGGTCGCGAGGCT-3′ |
|
| R: 5′-AGAGCAGTCTGTCTTCTTTCTCCGTT-3′ | ||
| TNF-α |
F: 5′AACTTCGGGGTGATCGGTCC-3′ |
|
| R: 5′ CAAATCGGCTGACGGTGTGGG-3′ | ||
| TGF-β1 |
F: 5′-CCGCAACAACGCCATCTATG-3′ |
|
| R: 5′-CTCTGCACGGGACAGCAAT-3′ | ||
| MCP-1 | F: 5′-CAGCCAGATGCAGTTAACGC -3′ |
|
| R: 5′-GCCTACTCATTGGGATCATCTTG-3′ |
Note: F, forward primer; R, reverse primer.
Migration assay
WT and TIPE2−/− VSMCs, or VSMC/pEGFP-mTIPE2 and VSMC/pEGFP, (1 × 105 cells per chamber) were cultured in serum-free-medium. Then the cells were seeded into the top chamber (24-well insert, 8 μm; Corning). After 6 h, the experimental groups were added with ox-LDL into the top chamber at a final concentration of 20 g/mL, while control groups were added with equivoluminal serum-free-medium.39 VSMC/pEGFP-mTIPE2 and VSMC/pEGFP could be observed in fluorescence microscopy directly. The migration ability was determined by calculating the number of migrated cells in fove visual fields per well by microscopy with 100× magnification. The experiments were repeated at least three times.
Proliferation
WT or TIPE2−/− VSMCs (2 × 103 cells/well) were cultured in 96-well dishes overnight and then allowed to proliferate in the absence or presence of 20 g/mL ox-LDL in DMEM medium containing 10% FBS. Cell proliferation was measured at different time intervals over a period of 5 d using Cell Counting Kit-8 reagent (CCK-8, Dojindo) as described by the manufacturer. At each time point, 10 μL of CCK-8 reagent was added to each well 1 h before the end of incubation. The optical density (OD) value of each sample was measured at a wavelength of 450 nm on a microplate reader (Model 680, BIO-RAD).
Flow cytometry
About 5 × 104 VSMCs from WT or TIPE2−/− mice were cultured in 6-well dishes for 24 h. Then cells were made quiescent by incubation with serum-free DMEM for 24 h and allowed to grow for another 24 h in the absence or presence of 20 μg/mL ox-LDL. Cells were collected to fix in methanol, then treated with 100 μg/mL RNase A solution in 37°C for 30 min., cells were then washed and incubated with 50 μg/mL Propidium iodide (PI) containing 0.2% Triton X-100, away from light for 30 min. PI incorporation was determined by flow cytometer (FC-500 MCL/MPL, BECKMAN) according to the instructions of the supplier.
Western blot
Cells were lysed on ice in the presence of a protease inhibitor and/or phosphatase inhibitor cocktails (Roche) and protein concentration was determined using BCA protein assay reagent (Thermo Scientific). 50 μg protein was separated by 12% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membrane (Millipore). The blocked membranes were then incubated with primary antibodies overnight. TIPE2 was detected using a specific antibody as previously described.12 The following primary antibodies were used: anti-SMA (Millipore, 1:1000); anti-ERK and p-ERK, anti-P38 and p-P38 (Cell Signaling Technology, 1:1000); anti-PCNA, anti-Cyclin D1 and anti-Cyclin E, anti-P21 and anti-P27 (Bioworlde, 1:500); anti-β-actin (1:1000). Immunoblotting was conducted by incubating the membranes with primary antibodies at 4°C overnight followed by secondary antibodies (goat anti-rabbit Ig G or goat anti-mouse Ig G, 1:1000) conjugated with peroxidase for 1 h at room temperature. After washing, bound peroxidase activity was detected by the ECL detection system (ECL, F-cheiBIsi.6pro, DNR) using the SuperSignal West Pico kit (Pierce Biotechnology).
Collar-induced atherosclerosis study
TIPE2−/− (n = 20) or ApoE−/− mice (n = 20, 8 to 10 wk old) were subjected to carotid collar placement surgery with a high-fat diet (HFD, 0.25% cholesterol and 15% cocoa butter). Carotid atherosclerotic lesions were induced by a perivascular silica collar of 3 mm length and 0.3 mm internal diameter placed on the left common carotid artery with an average adventitial diameter of 0.5 mm.40 Three weeks after surgery, animals were anesthetized by intraperitoneal pentobarbital injection and exanguinated by femoral artery transection. In situ perfusion fixation through the left cardiac ventricle was performed by normal saline instillation for 15 min, followed by infusion of 4% paraformaldehyde (Sigma-Aldrich) for 30 min. The aorta was dissected and fixed in 4% paraformaldehyde overnight, then embedded in optimal cutting temperature (OCT) compound (Tissue-Tek; Sakura Finetek). Sections about 7 min were stained with hematoxylin (Sigma Diagnostics) and eosin, (Merck Diagnostica) or with anti-SMαA (smooth muscle α-actin, dilution 1:200, Millepore) for smooth muscle cells.
Statistical analysis
All analyses were performed using SPSS 16.0 (SPSS Inc.). Data were expressed as mean ± SE. An independent-samples t test was used to compare continuous data for between-group differences and comparisons among groups involved in the use of ANOVA with LSD post hoc test used for multiple comparisons. p < 0.05 was considered statistically significant. All experiments were repeated at least three times.
Acknowledgments
This work was supported by the grants from the National Natural Science Foundation of China (No.81171578, No.81100205), the Key Grant from the Health Department of Shandong Province (2009HD009) and the Award Funds for Excellent Young and Middle-Aged Scientists of Shandong Province (BS2009YY007).
Glossary
Abbreviations:
- TIPE2
tumor necrosis factor (TNF)-alpha-induced protein 8-like 2 (TNFAIP8L2)
- VSMC
vascular smooth muscle cell
- AS
atherosclerosis
- ox-LDL
oxidized low-density lipoprotein
- SMαA
smooth muscle α-actin
- SM-MHC
smooth muscle myosin heavy chain
- MMP-9
matrix metalloproteinase-9
- FBS
fetal bovine serum
- PBS
phosphate-buffered saline
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/23325
References
- 1.Dzau VJ, Braun-Dullaeus RC, Sedding DG. Vascular proliferation and atherosclerosis: new perspectives and therapeutic strategies. Nat Med. 2002;8:1249–56. doi: 10.1038/nm1102-1249. [DOI] [PubMed] [Google Scholar]
- 2.Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell. 2001;104:503–16. doi: 10.1016/S0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 3.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 4.Gabbiani G, Schmid E, Winter S, Chaponnier C, de Ckhastonay C, Vandekerckhove J, et al. Vascular smooth muscle cells differ from other smooth muscle cells: predominance of vimentin filaments and a specific alpha-type actin. Proc Natl Acad Sci USA. 1981;78:298–302. doi: 10.1073/pnas.78.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nagai R, Kuro-o M, Babij P, Periasamy M. Identification of two types of smooth muscle myosin heavy chain isoforms by cDNA cloning and immunoblot analysis. J Biol Chem. 1989;264:9734–7. [PubMed] [Google Scholar]
- 6.Solway J, Seltzer J, Samaha FF, Kim S, Alger LE, Niu Q, et al. Structure and expression of a smooth muscle cell-specific gene, SM22 alpha. J Biol Chem. 1995;270:13460–9. doi: 10.1074/jbc.270.22.13460. [DOI] [PubMed] [Google Scholar]
- 7.Gimona M, Sparrow MP, Strasser P, Herzog M, Small JV. Calponin and SM 22 isoforms in avian and mammalian smooth muscle. Absence of phosphorylation in vivo. Eur J Biochem. 1992;205:1067–75. doi: 10.1111/j.1432-1033.1992.tb16875.x. [DOI] [PubMed] [Google Scholar]
- 8.Kusuhara M, Chait A, Cader A, Berk BC. Oxidized LDL stimulates mitogen-activated protein kinases in smooth muscle cells and macrophages. Arterioscler Thromb Vasc Biol. 1997;17:141–8. doi: 10.1161/01.ATV.17.1.141. [DOI] [PubMed] [Google Scholar]
- 9.Cherepanova OA, Pidkovka NA, Sarmento OF, Yoshida T, Gan Q, Adiguzel E, et al. Oxidized phospholipids induce type VIII collagen expression and vascular smooth muscle cell migration. Circ Res. 2009;104:609–18. doi: 10.1161/CIRCRESAHA.108.186064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar MS, Owens GK. Combinatorial control of smooth muscle-specific gene expression. Arterioscler Thromb Vasc Biol. 2003;23:737–47. doi: 10.1161/01.ATV.0000065197.07635.BA. [DOI] [PubMed] [Google Scholar]
- 11.Sun H, Gong S, Carmody RJ, Hilliard A, Li L, Sun J, et al. TIPE2, a negative regulator of innate and adaptive immunity that maintains immune homeostasis. Cell. 2008;133:415–26. doi: 10.1016/j.cell.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang G, Hao C, Lou Y, Xi W, Wang X, Wang Y, et al. Tissue-specific expression of TIPE2 provides insights into its function. Mol Immunol. 2010;47:2435–42. doi: 10.1016/j.molimm.2010.06.016. [DOI] [PubMed] [Google Scholar]
- 13.Zhang X, Wang J, Fan C, Li H, Sun H, Gong S, et al. Crystal structure of TIPE2 provides insights into immune homeostasis. Nat Struct Mol Biol. 2009;16:89–90. doi: 10.1038/nsmb.1522. [DOI] [PubMed] [Google Scholar]
- 14.Zhang S, Zhang Y, Wei X, Zhen J, Wang Z, Li M, et al. Expression and regulation of a novel identified TNFAIP8 family is associated with diabetic nephropathy. Biochim Biophys Acta. 2010;1802:1078–86. doi: 10.1016/j.bbadis.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 15.Xi W, Hu Y, Liu Y, Zhang J, Wang L, Lou Y, et al. Roles of TIPE2 in hepatitis B virus-induced hepatic inflammation in humans and mice. Mol Immunol. 2011;48:1203–8. doi: 10.1016/j.molimm.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Wei X, Liu L, Liu S, Wang Z, Zhang B, et al. TIPE2, a novel regulator of immunity, protects against experimental stroke. J Biol Chem. 2012;287:32546–55. doi: 10.1074/jbc.M112.348755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–9. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 18.Yoshida T, Owens GK. Molecular determinants of vascular smooth muscle cell diversity. Circ Res. 2005;96:280–91. doi: 10.1161/01.RES.0000155951.62152.2e. [DOI] [PubMed] [Google Scholar]
- 19.Worth NF, Rolfe BE, Song J, Campbell GR. Vascular smooth muscle cell phenotypic modulation in culture is associated with reorganisation of contractile and cytoskeletal proteins. Cell Motil Cytoskeleton. 2001;49:130–45. doi: 10.1002/cm.1027. [DOI] [PubMed] [Google Scholar]
- 20.Gus-Brautbar Y, Johnson D, Zhang L, Sun H, Wang P, Zhang S, et al. The anti-inflammatory TIPE2 is an inhibitor of the oncogenic Ras. Mol Cell. 2012;45:610–8. doi: 10.1016/j.molcel.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newby AC, Zaltsman AB. Molecular mechanisms in intimal hyperplasia. J Pathol. 2000;190:300–9. doi: 10.1002/(SICI)1096-9896(200002)190:3<300::AID-PATH596>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 22.Chen KC, Wang YS, Hu CY, Chang WC, Liao YC, Dai CY, et al. OxLDL up-regulates microRNA-29b, leading to epigenetic modifications of MMP-2/MMP-9 genes: a novel mechanism for cardiovascular diseases. FASEB J. 2011;25:1718–28. doi: 10.1096/fj.10-174904. [DOI] [PubMed] [Google Scholar]
- 23.Doran AC, Meller N, McNamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:812–9. doi: 10.1161/ATVBAHA.107.159327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lebastchi AH, Qin L, Khan SF, Zhou J, Geirsson A, Kim RW, et al. Activation of human vascular cells decreases their expression of transforming growth factor-beta. Atherosclerosis. 2011;219:417–24. doi: 10.1016/j.atherosclerosis.2011.07.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strzalka W, Ziemienowicz A. Proliferating cell nuclear antigen (PCNA): a key factor in DNA replication and cell cycle regulation. Ann Bot. 2011;107:1127–40. doi: 10.1093/aob/mcq243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayashi K, Shibata K, Morita T, Iwasaki K, Watanabe M, Sobue K. Insulin receptor substrate-1/SHP-2 interaction, a phenotype-dependent switching machinery of insulin-like growth factor-I signaling in vascular smooth muscle cells. J Biol Chem. 2004;279:40807–18. doi: 10.1074/jbc.M405100200. [DOI] [PubMed] [Google Scholar]
- 27.Hayashi K, Takahashi M, Kimura K, Nishida W, Saga H, Sobue K. Changes in the balance of phosphoinositide 3-kinase/protein kinase B (Akt) and the mitogen-activated protein kinases (ERK/p38MAPK) determine a phenotype of visceral and vascular smooth muscle cells. J Cell Biol. 1999;145:727–40. doi: 10.1083/jcb.145.4.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayashi K, Takahashi M, Nishida W, Yoshida K, Ohkawa Y, Kitabatake A, et al. Phenotypic modulation of vascular smooth muscle cells induced by unsaturated lysophosphatidic acids. Circ Res. 2001;89:251–8. doi: 10.1161/hh1501.094265. [DOI] [PubMed] [Google Scholar]
- 29.Yoshida K, Nishida W, Hayashi K, Ohkawa Y, Ogawa A, Aoki J, et al. Vascular remodeling induced by naturally occurring unsaturated lysophosphatidic acid in vivo. Circulation. 2003;108:1746–52. doi: 10.1161/01.CIR.0000089374.35455.F3. [DOI] [PubMed] [Google Scholar]
- 30.Sedding DG, Tröbs M, Reich F, Walker G, Fink L, Haberbosch W, et al. 3-Deazaadenosine prevents smooth muscle cell proliferation and neointima formation by interfering with Ras signaling. Circ Res. 2009;104:1192–200. doi: 10.1161/CIRCRESAHA.109.194357. [DOI] [PubMed] [Google Scholar]
- 31.Lagna G, Ku MM, Nguyen PH, Neuman NA, Davis BN, Hata A. Control of phenotypic plasticity of smooth muscle cells by bone morphogenetic protein signaling through the myocardin-related transcription factors. J Biol Chem. 2007;282:37244–55. doi: 10.1074/jbc.M708137200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pagano PJ, Gutterman DD. The adventitia: the outs and ins of vascular disease. Cardiovasc Res. 2007;75:636–9. doi: 10.1016/j.cardiores.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andrés V. Control of vascular cell proliferation and migration by cyclin-dependent kinase signalling: new perspectives and therapeutic potential. Cardiovasc Res. 2004;63:11–21. doi: 10.1016/j.cardiores.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 34.Su B, Mitra S, Gregg H, Flavahan S, Chotani MA, Clark KR, et al. Redox regulation of vascular smooth muscle cell differentiation. Circ Res. 2001;89:39–46. doi: 10.1161/hh1301.093615. [DOI] [PubMed] [Google Scholar]
- 35.Pidkovka NA, Cherepanova OA, Yoshida T, Alexander MR, Deaton RA, Thomas JA, et al. Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circ Res. 2007;101:792–801. doi: 10.1161/CIRCRESAHA.107.152736. [DOI] [PubMed] [Google Scholar]
- 36.Mosse PR, Campbell GR, Wang ZL, Campbell JH. Smooth muscle phenotypic expression in human carotid arteries. I. Comparison of cells from diffuse intimal thickenings adjacent to atheromatous plaques with those of the media. Lab Invest. 1985;53:556–62. [PubMed] [Google Scholar]
- 37.Golovina VA, Blaustein MP. Preparation of primary cultured mesenteric artery smooth muscle cells for fluorescent imaging and physiological studies. Nat Protoc. 2006;1:2681–7. doi: 10.1038/nprot.2006.425. [DOI] [PubMed] [Google Scholar]
- 38.Ray JL, Leach R, Herbert JM, Benson M. Isolation of vascular smooth muscle cells from a single murine aorta. Methods Cell Sci. 2001;23:185–8. doi: 10.1023/A:1016357510143. [DOI] [PubMed] [Google Scholar]
- 39.Goncharova EA, Goncharov DA, Krymskaya VP. Assays for in vitro monitoring of human airway smooth muscle (ASM) and human pulmonary arterial vascular smooth muscle (VSM) cell migration. Nat Protoc. 2006;1:2933–9. doi: 10.1038/nprot.2006.434. [DOI] [PubMed] [Google Scholar]
- 40.von der Thüsen JH, van Berkel TJ, Biessen EA. Induction of rapid atherogenesis by perivascular carotid collar placement in apolipoprotein E-deficient and low-density lipoprotein receptor-deficient mice. Circulation. 2001;103:1164–70. doi: 10.1161/01.CIR.103.8.1164. [DOI] [PubMed] [Google Scholar]
- 41.Stein S, Matter CM. Protective roles of SIRT1 in atherosclerosis. Cell Cycle. 2011;10:640–7. doi: 10.4161/cc.10.4.14863. [DOI] [PubMed] [Google Scholar]
- 42.Salminen A, Kaarniranta K, Kauppinen A. Inflammaging: disturbed interplay between autophagy and inflammasomes. Aging (Albany NY) 2012;4:166–75. doi: 10.18632/aging.100444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blagosklonny MV. Prospective treatment of age-related diseases by slowing down aging. Am J Pathol. 2012;181:1142–6. doi: 10.1016/j.ajpath.2012.06.024. [DOI] [PubMed] [Google Scholar]