

Abstract
Primary ovarian cancer is responsive to treatment, but chemoresistant recurrent disease ensues in majority of patients. Recent compelling evidence demonstrates that a specific population of cancer cells, the cancer stem cells, initiates and sustains tumors. It is therefore possible that this cell population is also responsible for recurrence. We have shown previously that CD44+/MyD88+ epithelial ovarian cancer stem cells (CD44+/MyD88+ EOC stem cells) are responsible for tumor initiation. In this study, we demonstrate that this population drives tumor repair following surgery- and chemotherapy-induced tumor injury. Using in vivo and in vitro models, we also demonstrate that during the process of tumor repair, CD44+/MyD88+ EOC stem cells undergo self-renewal as evidenced by upregulation of stemness-associated genes. More importantly, we show that a pro-inflammatory microenvironment created by the TLR2-MyD88-NFκB pathway supports EOC stem cell-driven repair and self-renewal. Overall, our findings point to a specific cancer cell population, the CD44+/MyD88+ EOC stem cells and a specific pro-inflammatory pathway, the TLR2-MyD88-NFκB pathway, as two of the required players promoting tumor repair, which is associated with enhanced cancer stem cell load. Identification of these key players is the first step in elucidating the steps necessary to prevent recurrence in EOC patients.
Keywords: ovarian cancer stem cells, recurrence, tumor repair, TLR2, self-renewal
Introduction
Epithelial ovarian cancer (EOC) continues to be the leading cause of gynecologic cancer deaths in the United States.1,2 Although 80% of patients with primary disease respond to surgery and chemotherapy, a majority of them eventually present with recurrent disease. These patients inevitably become refractory to chemotherapy, which results in disease progression and death. Therefore, identifying the source of recurrence and understanding the mechanisms involved would have a significant impact on patient survival.
The standard of treatment for EOC is cytoreductive surgery followed by taxane- and platinum-based combination chemotherapy.1,3 In patients diagnosed with stage I disease, complete surgical removal of tumor leads to remission. However, most patients are diagnosed with advanced-stage disease and maximal cytoreduction is limited by carcinomatosis.4 For these patients, optimal cytoreduction or de-bulking is performed followed by chemotherapy. Unfortunately, this mode of management is not optimal, and ~85% of patients recur and present with chemoresistant disease.5,6 Successful removal of macroscopic disease makes a major difference in the outcome of the disease. The median overall survival and progression-free survival increase from 35.0 to 81.1 mo with complete macroscopic surgical resection.5 The mechanisms by which residual disease achieves dissemination and aggressive growth are poorly understood.
Tumor recurrence following surgery and chemotherapy may parallel normal tissue repair. Normal tissue repair requires an inflammatory environment, which can enhance removal of dying cells, prevent death of the surrounding tissue, protect against pathogens and promote blood flow.7 Restoration of the lost cell population is sustained by a small group of long-lived cells with extraordinary expansion potential and pluripotency, including self-renewal, known as adult stem cells.8 In the tumor microenvironment, the repair process may be sustained by the cancer stem cells (CSCs).9-12 CSCs or tumor-initiating cells represent a unique cancer cell population with the capacity to initiate and expand the heterogeneous nature of the tumor.9,13 The CSC hypothesis implies that inherently chemoresistant CSCs can persist after chemotherapy and repopulate the tumor leading to recurrence. Contrary to the stochastic model of cancer (clonal expansion), the CSC model holds that tumors are hierarchically organized, and only some cells have the capacity to self-renew (i.e., maintain the stem cell pool) and differentiate to create tumor bulk.14
The ability of both normal and CSCs to divide and produce more stem cells is known as “self-renewal.” It is accomplished by symmetric cell division, which produces two daughter cells with equivalent stemness capacity, or by asymmetric cell division, with only one daughter cell maintaining the exact stemness phenotype and the other daughter acquiring a more differentiated phenotype. Stem cells can rely either completely on symmetric cell division or on a combination of symmetric and asymmetric divisions.15 The balance between these two modes is controlled by developmental and environmental signals, which will determine the appropriate number of stem cells and differentiated daughter cells. The ability to switch between asymmetric and symmetric modes of division and maintain the balance between stem cells and daughter cells is defective in some disease states such as cancer.
We and others have described the occurrence of CSCs in EOC tissue samples.16-24 In previous studies, we demonstrated that CD44+/MyD88+ EOC stem cells have tumor-initiating capacity.16 We isolated and established CD44+/MyD88+ EOC stem cell clones that can rebuild the original tumor in mice (tumor-initiation potential), express stemness markers, give origin to CD44-/MyD88- EOC cells (differentiation capacity), serve as tumor vascular progenitors (pleuripotency) and are chemoresistant.14,16,25-27 Our findings concur with other studies that have shown the existence of tumor-initiating cells in EOC through the use of different markers, suggestive of the heterogeneity of the disease.16,19,28-31
Evaluation of CD44+/MyD88+ EOC stem cells in tumor samples revealed that levels of these cells are associated with shorter progression-free survival in EOC patients.27 Moreover, CD44+/MyD88+ EOC stem cells have a unique phenotype that confers a high capacity for maintenance of a pro-inflammatory microenvironment. They possess a constitutively active NFκB and a functional Toll-like receptor 4 (TLR4) -MyD88- NFκB pathway,16,32 which are absent in CD44-/MyD88- EOC cells.
TLRs are a family of transmembrane proteins that recognize and respond to conserve pathogen-associated molecular patterns (PAMPs) that are expressed by microorganisms.33 To date, 10 human TLRs and their specific ligands have been identified. While individually TLRs respond to limited ligands, collectively as a family TLRs respond to a wide range of PAMPs. In addition, some TLRs, such as TLR-2, -3 and -4 respond to endogenous “stress” proteins, such as heat shock protein 60 (Hsp 60), hyaluronan and fibrinogen.34 These proteins are known to be released as cellular debris following cell injury or cell death.35 TLRs share a common signaling pathway via the adaptor molecule MyD88, leading to the activation of NFκB and the production of inflammatory cytokines.36 There is mounting evidence that TLR activation plays an important role in cancer progression and development.37-39 In EOC stem cells, ligation of TLR4 elicits increased NFκB activity and enhanced secretion of pro-inflammatory cytokines and chemokines.36,39 Interestingly, this occurs with the chemotherapeutic agent, Paclitaxel, which is a known TLR4 ligand.39
In this study we demonstrate that a functional TLR2 pathway in CD44+/MyD88+ EOC stem cells plays a relevant role in tumor repair following injury induced by surgery or chemotherapy. The repair process is associated with self-renewal and, hence, enrichment of these chemoresistant cancer cells. We demonstrate that the capacity to promote tumor repair is a specific property of the CD44+/MyD88+ EOC stem cells and is absent in CD44-/MyD88- EOC cells. Inhibition of the TLR2 pathway in EOC stem cells through consequent inhibition of both NFκB and release of downstream cytokines, inhibits the capacity of cancer stem cells to repair the wound. These data suggest that TLR2, expressed by EOC stem cells, plays a relevant role in promoting a pro-inflammatory microenvironment that supports EOC stem cell-associated tumor repair. This process provides a unique advantage for tumor renewal and its potential association with EOC recurrence.
Results
Tumor injury accelerates tumor growth and promotes self-renewal in cancer stem cells
Several studies have shown that CSCs represent the cell population that can survive chemotherapy and re-populate the tumor by differentiating into fast-dividing progeny.14,40 This implies the significant contribution of this cell population in the sustenance of the tumor and therefore tumor recurrence. This also suggests that CSCs should comprise only a very small percentage of the cancer cell population. Indeed, quantitation of the CSC load in various types of cancer has shown that CSCs represent a very small percentage of the tumor bulk (usually < 10%).12 In EOC, however, we previously reported that recurrent patients could have tumors with more than 20% CD44+ EOC stem cells.27Figure S1 shows representative sections of ovarian tumors, with almost 50% CD44+ EOC stem cells. This suggests that in some patients, recurrence may be brought about by CSC self-renewal instead of differentiation.
Since therapy has been associated with the enrichment of CSCs, we hypothesized that tumor injury, induced by either surgery or chemotherapy, may promote EOC stem cell self-renewal. To test this hypothesis, we first established a “surgery-induced” tumor injury/repair model using three different clones of CD44+/MyD88+ EOC stem cells isolated from patients with ovarian cancer, as previously reported.13,16 CD44+/MyD88+ EOC stem cells were injected s.c. into the right and left flanks of nude mice. Once the tumor reached an average size of 70 mm2, we surgically removed 50% of the right tumor (designated as “wound”), thus mimicking partial tumor debulking and residual disease. A skin incision was performed on the left tumor (designated as “control”), but the tumor was untouched (Fig. 1A). After 21 d, the “wounded” tumor (right side) was significantly bigger than the control (left side) (Fig. 1A and B). In addition, we observed faster growth kinetics on the “wounded” tumor compared with control (Fig. 1C). Flow cytometry analyses of dissociated cancer cells from the “wounded” tumors showed an increase in the number of CD44+ cells (Fig. 1D). The upregulation in CD44+ cells suggests that the repair process following tumor injury is associated with self-renewal.
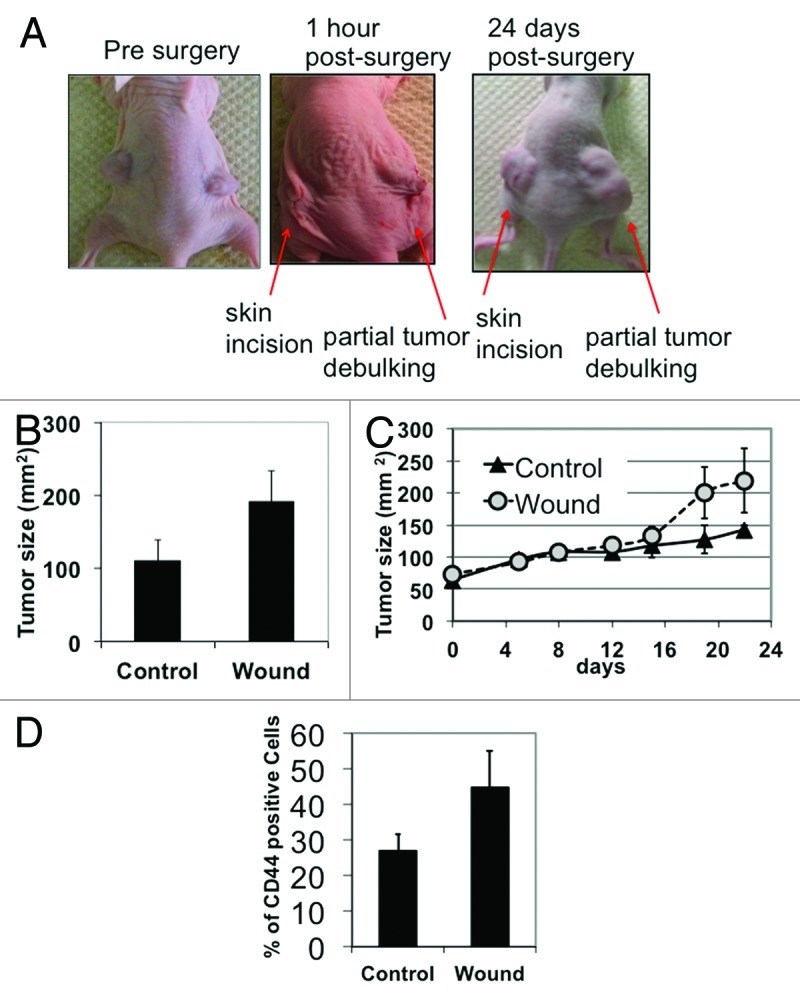
Figure 1. Tumor injury accelerates tumor growth in vivo. (A) Subcutaneous ovarian cancer xenografts were established on both sides of nude mice. Partial tumor debulking was performed on the right tumor, and skin was incised on the left side but the tumor was left untouched. (B) Size of tumor xenografts was measured ex vivo at day 24 post-surgery. Note that day 24 post-surgery, partially debulked or “wounded” tumor appears larger than the tumor that was not debulked. (C) Graph of tumor kinetics show accelerated growth in “wounded” tumors compared with non-debulked control. Day 0 indicates the measurement immediately preceding wounding. (D) Flow cytomety analysis for CD44 was performed on cancer cells dissociated from the excised tumor xenografts at day 24 post-surgery.
We then evaluated whether we will observe the same results with chemotherapy-induced tumor injury. Thus, we implanted in nude mice EOC tumors obtained from a patient who failed first-line chemotherapy. This tumor initially showed a significantly high number of CD44+ cells. Once the implant was established, mice were treated with Paclitaxel (20 mg/kg every 3 d) or vehicle as control. As shown in Figure 2A, tumors in treated mice grew faster compared with the control. Furthermore, similar to that observed with surgical wounding, evaluation of the CD44 content at the end of the treatment revealed expansion of the CD44+ population in the Paclitaxel-treated animals (Fig. 2B).
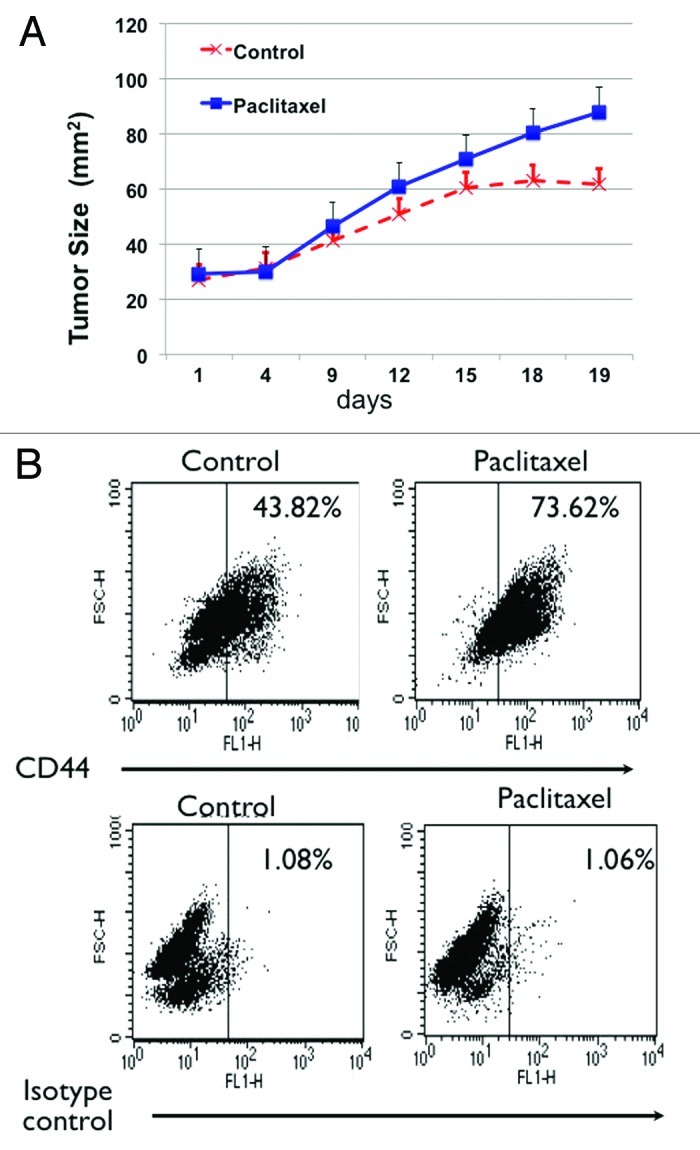
Figure 2. Chemotherapy-induced tumor injury enriches for CD44+/MyD88+ EOC stem cells. (A) Paclitaxel treatment (20 mg/kg q3d) enhances growth of tumor implants. (B) Enrichment of CD44 positive cells in tumors from mice treated with Paclitaxel. Tumor implants treated as in (A) were dissociated at the end of the treatment and the number of CD44+ cells was determined by flow cytometry. Note the significant increase in CD44+ cells in the Paclitaxel-treated group.
Taken together, these results show that tumor injury, as a result of either surgery or chemotherapy, is associated with acceleration of tumor growth and enrichment of CD44+/MyD88+ EOC stem cells.
In vitro wound/repair process is a unique property of the CD44+/MyD88+ EOC stem cells
To determine whether the tumor repair observed in vivo is an inherent characteristic of the CD44+/MyD88+ EOC stem cells, or if it is triggered by the tumor microenvironment, we established an in vitro wound/repair model using three clones of CD44+/MyD88+ EOC stem cells that were isolated and characterized as previously described.13,16,39,41 Following a mechanical scratch wound, CD44+/MyD88+ EOC stem cells are able to repair in vitro wound in a well-coordinated and organized process (Fig. 3A; Vid. S1). First, the cells formed a defined straight line to replace the irregular edge of the wound (Vid. S1). Afterwards, the cells proliferated toward the wound, “repairing” the wound until confluence was reached. Once the wound was repaired, the CD44+/MyD88+ EOC stem cells showed cell-cell contact growth inhibition (Fig. 3Ai). In contrast, CD44-/MyD88- EOC cells, were not able to repair the wound (Fig. 3Aii; Vid S1).
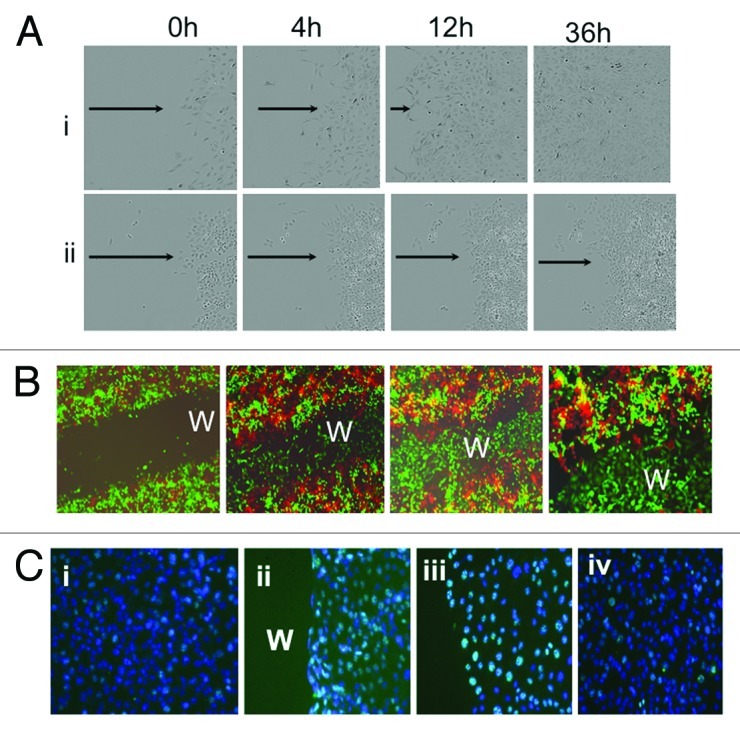
Figure 3. CD44+/MyD88+ EOC stem cells repair in vitro wound. (A) CD44+/MyD88+ EOC stem cells (i) and CD44-/MyD88- EOC cells (ii), were grown to confluence and “wounded” as described in the “Materials and Methods” section. Note that only CD44+/MyD88+ EOC stem cells repair in vitro wound. (B) GFP+/CD44+/MyD88+ EOC stem cells and RFP+/ CD44-/MyD88- EOC cells were grown to confluence and wounded (i). Wound healing was determined by fluorescent microscope at 24 h (ii), 48 h (iii) and 72 h (iv). Note that only GFP+/CD44+/MyD88+ EOC stem cells can repair the wound. (C) Immunohistochemistry for Ki67 (green staining) was determined in (i) no wound control, (ii) wound edge 4 h after wounding, (iii) wound edge 24 h after wounding and (iv) in wound back 24h after wounding. w, wound area. Note: for wound edge and wound back definitions, please see text.
To further support the concept that tumor repair is driven by CD44+/MyD88+ EOC stem cells, we developed an in vitro co-culture model that mimics tumor heterogeneity. We established clones of GFP labeled CD44+/MyD88+ EOC stem cells (GFP+ CD44+/MyD88+ EOC stem cells) and clones of RFP labeled CD44-/MyD88-cells (RFP+ CD44-/MyD88- EOC EOC cells). Cells were seeded to yield a confluent culture comprising of 50% GFP+ CD44/MyD88+ EOC stem cells and 50% RFP+CD44-/MyD88- EOC cells. Afterwards, a mechanical scratch wound was performed (Fig. 3Bi), and the repair process was monitored using fluorescence microscopy. As shown in Figure 3B, only GFP+/CD44+/MyD88+ EOC stem cells were observed to repair the wound. The “healed” wound is comprised of only GFP+/CD44+/MyD88+ EOC stem cells, while the RFP+/CD44-/MyD88- EOC cells remained in the margin and away from the wounded area (Fig. 3Bii−iv).
We then determined whether the repair process observed with CD44+/MyD88+ EOC stem cells is a result of cell proliferation or cell migration. Ki67 expression is only observed in the nuclei of cells undergoing cell proliferation, therefore it is an excellent marker to determine whether the cells are dividing or migrating. Growing cultures of CD44+/MyD88+ EOC stem cells show few Ki67-positive cells, which is a characteristic of its slow growth rate (Fig. 3Ci). However, in wounded CD44+/MyD88+ EOC stem cell cultures, we observed a significant increase in the number of Ki67-positive cells along the wound edge (Fig. 3Cii, nuclear green fluorescence). Further evaluation of the wound edge revealed that almost 90% of the cells were Ki67-positive (Fig. 3Ciii). Increase in Ki67 positivity was also observed in areas of the culture away from the wound (Fig. 3Civ) although significantly less compared with the wound edge.
Taken together, these results demonstrate that the capacity to repair in vitro wound is a specific and unique property of the CD44+/MyD88+ EOC stem cells and tumor repair is indeed driven by this cell population.
In vitro wound/repair process is associated with self-renewal
We previously demonstrated that under specific conditions, CD44+/MyD88+ EOC stem cells could differentiate in vitro and in vivo into CD44-/MyD88- EOC cells. During this process, the loss of CD44 is accompanied by loss of stemness-associated genes, such as β-catenin, Nanog, Oct-4 and Sox-2.42-44 Our next objective was to determine whether the repair process observed above is associated with maintenance of these stemness-associated genes. Thus, using the in vitro wound/repair model, CD44+/MyD88+ EOC stem cell cultures were wounded, and repair was allowed to occur. Once 50% of the wound was repaired, cells from the wound edge (WE; Fig. S3) were collected. Expression of CD44, Nanog, Oct-4 and Sox-2 were compared between cells from the WE and cells from a parallel control culture without wound (Fig. S3). qPCR results show that all genes tested were significantly upregulated in the WE compared with control (Fig. 4A). To validate these findings, we determined the levels of some of these markers at the protein level. In addition to the cells obtained from the control and WE cultures, we also included cells from an area distant to the wound (WB, wound back; Fig. S3). Western blot results show that compared with control (no wound), cells from WB and WE have higher levels of Oct-4 and β-catenin (Fig. 4B). CD44 expression was detected in all the samples (Fig. 4B). Maintenance of CD44 and upregulation of stemness-associated markers suggest that the repair process is associated with self-renewal.
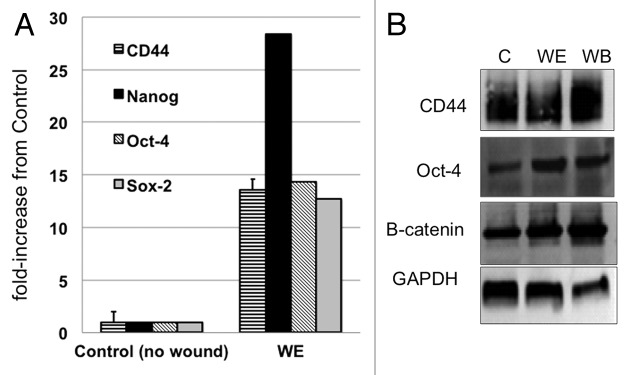
Figure 4. In vitro wound/repair process is associated with CD44+/MyD88+ EOC stem cell self-renewal. (A) Levels of CD44, Nanog, Sox2 and Oct4 were determined by qRT-PCR in control cultures (no wound) and in cells from wound edge (WE) 24h after wounding. (B) Levels of CD44, Oct-4, β-catenin and GAPDH were determined by western blot analysis from control cultures (no wound) and from cells isolated from WE and wound back areas. C, control no wound; WE, wound edge; WB, wound back.
TLR2 expression is upregulated during tumor injury in vivo
As mentioned above, TLRs can recognize cellular debris released following injury and can therefore contribute to the inflammatory process during repair.38 A unique property of CD44+/MyD88+ EOC stem cells is the ability to response to TLR ligands due to the presence of the adaptor protein, MyD88. Indeed, we previously showed that activation of the TLR4-MyD88-NFκB pathway in these cells by Paclitaxel treatment induce NFκB activation, increased secretion of pro-inflammatory cytokines, and enhanced cancer cell proliferation.39 Thus, we hypothesized that the TLR pathway may contribute to tumor repair. To test this hypothesis, we evaluated the expression of TLRs in the xenograft tumors described in Figure 1A. TLR2 was found to be highly expressed in the “wounded” tumors compared with control (Fig. 5A). Furthermore, when we determined the expression of TLR2 and MyD88 in the tumors that were treated with Paclitaxel (shown in Figure 2A and B), we found a significant increase in the expression of both genes in Paclitaxel-treated group compared with the control (Fig. 5B). These results suggest the possible involvement of the TLR2-MyD88 pathway in tumor repair.

Figure 5. TLR2 signaling enhances repair and self-renewal. (A) qRT-PCR analyses for TLR2 expression was performed on cancer cells dissociated from the excised tumor fragments at day 24 post-surgery. *p < 0.05. (B) qRT-PCR analyses for TLR2 expression was performed on cancer cells dissociated from tumors of animals treated with Paclitaxel as described in Figure 2A. (C) Levels of TLR2, TLR3 and TLR4 were determined by qRT-PCR from control cultures (no wound) and from cells isolated from wound edge (WE) area 8 h post-wounding. Data represent the average from three independent experiments, measured in triplicate and normalized to GAPDH mRNA. (D) Wounded CD44+/MyD88+ EOC stem cultures were left untreated or treated with 2 µg/ml of PGN or 10 μg/ml of LPS and effect on wound repair was determined. (E) Levels of CD44 and Nanog were determined by qRT-PCR from wounded CD44+/MyD88+ EOC stem cells in the presence or absence of PGN.
TLR2 activation enhances repair and self-renewal in EOC stem cells
As shown above, we observed a significant increase in TLR2 in wounded and Paclitaxel-treated xenograft tumors compared with controls. Thus, we hypothesize that TLR2 may affect wound repair and the self-renewal capacity of CD44+/MyD88+ EOC stem cells. To test this hypothesis, we used our in vitro wound/repair model. Similar to what was observed in vivo, the in vitro wound/repair process is also associated with a significant increase in the expression of TLR2. qPCR results show that compared with control (no wound), cells from WE have significantly higher levels of TLR2 (74-fold) and to a lesser extent, TLR3 (7-fold) and TLR4 (6-fold) (Fig. 5C).
To determine the functional significance of TLR2 upregulation, in vitro wounds were created, and the repair process was allowed to proceed in the presence or absence of peptidoglycan (PGN), a TLR2 agonist. We also used lipopolysaccharide (LPS), a TLR4 agonist, to determine specificity. We saw a significant acceleration in wound repair in cultures treated with PGN compared with no-treatment control (Fig. 5D). In contrast, no significant difference in wound repair was observed between no-treatment control and LPS-treated cultures, suggesting a specific role of TLR2 in tumor repair (Fig. 5D).
We also determined the effect of TLR2 ligation on the expression levels of CD44 and Nanog during the process of repair. As shown in Figure 5E, wounded cultures treated with PGN showed enhanced levels of these genes. CD44 was upregulated 40-fold, while Nanog was upregulated more than 2-fold with PGN compared with no-treatment control (wound alone). These changes were not observed in CD44-MyD88- EOC cells treated with similar concentration of PGN (data not shown). Taken together, these results suggest that TLR2 activation can enhance repair and self-renewal in CD44+/MyD88+ EOC stem cells during the process of tumor injury.
Inhibition of TLR2 or MyD88 prevents in vitro wound repair
To conclusively confirm the role of TLR2 in CD44+/MyD88+ EOC stem cell-associated repair, CD44+/MyD88+ EOC stem cells were transfected with a dominant-negative form of TLR2 (TLR2ΔTIR) prior to wounding. TLR2ΔTIR is able to bind TLR2 ligands but lacks the TIR domain required for downstream signaling.45 We observed a significant delay in in vitro wound repair in cultures stably expressing TLR2ΔTIR (Fig. 6). Since MyD88 is required for TLR2 signaling, we also determined the role of MyD88 in the repair process. EOC stem cell cultures stably expressing the dominant-negative form of MyD88 (MyD88-dn) also demonstrated a decreased capacity to repair in vitro wounds. Similarly, PGN was not able to accelerate the wound/repair process in cells expressing TLR2ΔTIR or MyD88-dn (data not shown). Taken together, these results suggest that the observed repair process is at least partially dependent on the TLR2-MyD88 pathway.
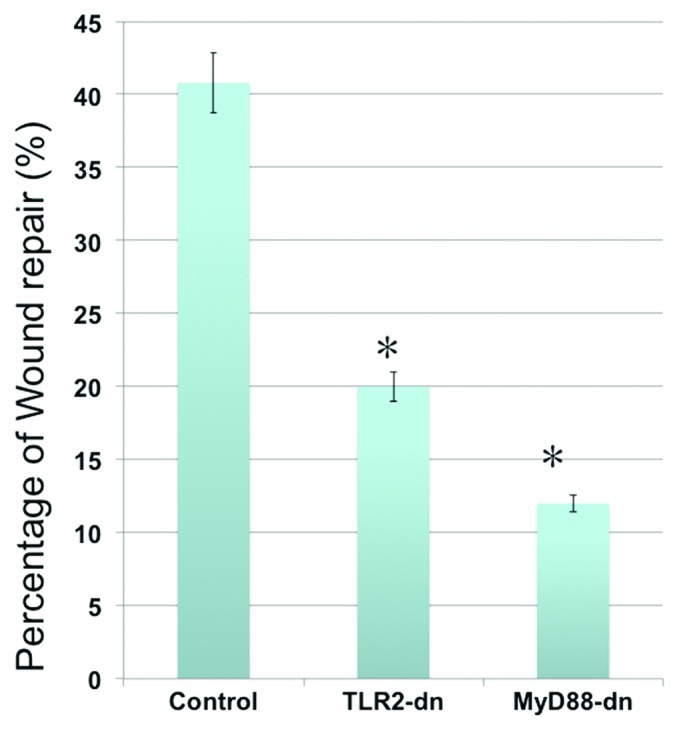
Figure 6. Inhibition of TLR2 or MyD88 activity inhibits repair. CD44+/MyD88+ EOC stem cells were transfected with a dominant-negative form of TLR2 or a dominant-negative form MyD88 prior to wounding. The effect of blocking TLR2 or MyD88 function on repair is then determined. Note the significant decrease on wound repair in cells transfected with TLR2 or MyD88 dominant-negative plasmids. TLR2-dn, TLR2-dominant-negative; MyD88-dn, MyD88-dominant-negative. *p < 0.005.
Tumor repair is associated with a pro-inflammatory microenvironment
Activation of TLR2 leads to signaling through MyD88 to NFκB (Fig. S4) and results in the upregulation of pro-inflammatory cytokines.46,47 To further show that the TLR2-MyD88-NFκB pathway is involved in the process of repair and self-renewal we measured the cytokine levels in EOC stem cell cultures during the process of repair. Cytokines were measured from cell lysates and not in culture supernatants to avoid detecting the cytokines that may be potentially released by dying cells. Cytokine levels were compared between cell lysates from CD44+/MyD88+ EOC stem cell cultures without wound (control) and from two sites of wounded cultures, the WE and WB areas. We observed a significant increase in IL-6, IL-8, MCP-1 and GROα in EOC stem cells as a result of wounding (Fig. 7). This increase was observed both at WE and WB sites. It should be noted that cells from WB had higher levels of cytokines than cells from WE suggesting that the release of soluble factors may promote a response from cells away from the wound.

Figure 7. In vitro wound/repair process upregulates NFκB-mediated pro-inflammatory cytokines. Levels of IL-6, IL-8, MCP-1 and GROa were determined from cell lysates obtained from control cultures (no wound) and from cells isolated from wound edge (WE) and wound back (WB) areas using xMAP technology.
Finally, to evaluate the role of NFκB-mediated inflammatory response on wound repair, in vitro wounds were created, and the repair process was allowed to proceed in the presence or absence of the NFκB inhibitor, BAY 11-7082. We observed a delay in wound repair in cultures treated with BAY 11-7082 (Fig. 8A). This delay was not associated due to cell death, since the dose used does not affect cell viability. More importantly, analysis of stemness-associated genes showed a parallel decrease in the levels of CD44, Nanog and Sox-2 in wounded cultures that were treated with BAY 11-7082 (Fig. 8B). Likewise, IL-6 was significantly decreased in the presence of BAY 11-7082 (Fig. S5). Taken together, these data suggest that the TLR2-MyD88-NFκB pathway promotes tumor repair and enhances self-renewal in CD44+/MyD88+ EOC stem cells.
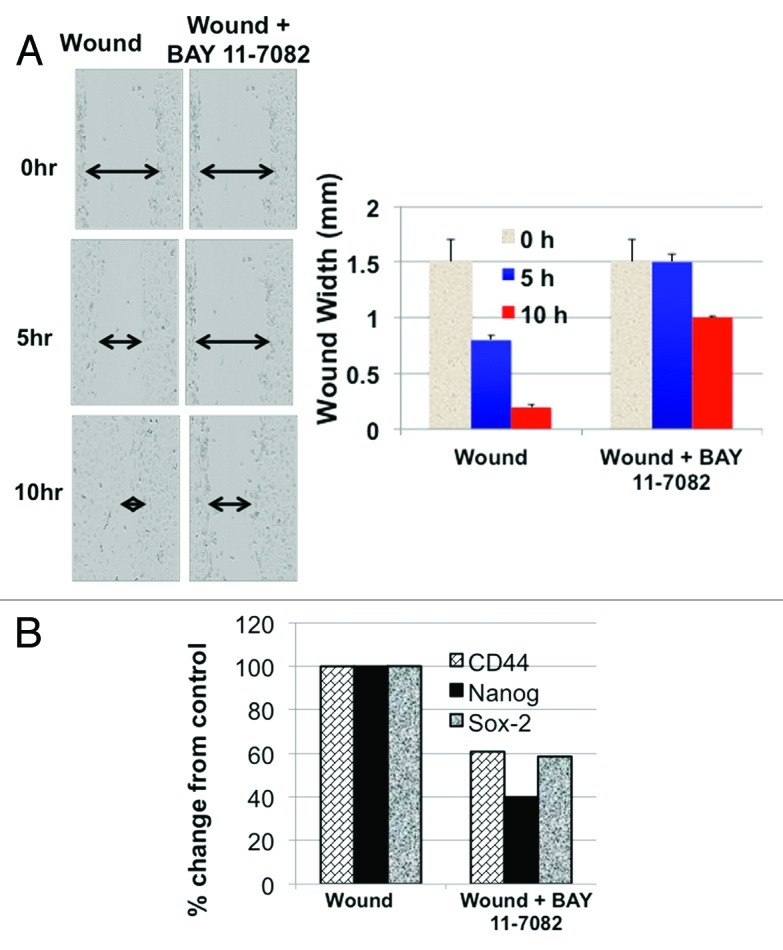
Figure 8. Inhibition of NFκB inhibits repair and self-renewal. (A) CD44+/MyD88+ EOC stem cells were wounded and repair process determined in the presence of absence of the NFκB inhibitor, BAY 11-7082 (2.5 μM). (B) CD44, Sox2, Nanog expression levels were determined using qRT-PCR from wounded CD44+/MyD88+ EOC stem cells in the presence or absence of BAY 11-7082. *p < 0.05.
Discussion
We show in this study that in EOC, tumor injury can accelerate tumor growth, and that the capacity to repair the tumor is a unique and inherent property of the CD44+/MyD88+ EOC stem cells. More importantly, we demonstrate the specific role of the TLR2-MyD88-NFκB pathway in enhancing EOC stem cell-driven repair and self-renewal during tumor injury. Promotion of self-renewal in the CSCs leads to enrichment of this chemo-resistant cell population and may partially confer acquired chemo-resistance in recurrent EOC patients. The pro-inflammatory microenvironment created as a result of the activation of the TLR2-MyD88-NFκB pathway is critical to support tumor repair and EOC stem cell self-renewal, and therefore, blocking this pathway by inhibiting TLR2, MyD88 and/or NFκB may prevent both repair and enrichment of the EOC stem cell population.
In normal conditions, tissues maintain their original mass and architecture through a tightly regulated process of tissue remodeling. During this process, new cells derived from adult stem cells replace aging or damaged cells. Stem cells have the capacity to self-renew and differentiate.15 Self-renewal involves the ability to form new identical stem cells and therefore maintenance of the stem cell pool. On the other hand, differentiation is the ability of a stem cell to give rise to a heterogeneous progeny of differentiated cells with specialized function.15 In normal stem cells, mechanisms that strictly regulate homeostatic control balance self-renewal and differentiation according to genetic or environmental stimuli. Tissue injury is accompanied by the expansion of tissue specific stem cells through asymmetric cell divisions in order to repair the injury. Once the wound is repaired, the stem cell compartment returns to quiescence. As such, in normal tissues, stem cells are a minority of the whole organ.48
Similar to normal stem cells, CSCs were also initially reported to comprise only a minor portion of the tumor mass. However, our work with EOC showed a wide range of variations in the percentage of CD44+/MyD88+ EOC stem cells. We have identified multiple cases wherein these cells comprise more than 50% of the tumor tissue.27 This suggests that the size of the CSC compartment can be highly variable, and indicates that the regulation of self-renewal and homeostatic control are altered in CSCs.
The heterogeneity of cancer cells that comprise a tumor mass has been described in most solid tumors. In EOC, our group has identified at least two types of EOC cells. CD44+/MyD88+ EOC stem cells possess stemness-associated markers,14,16,27 can initiate tumors in mice,16 can serve as tumor vascular progenitors,13 give origin to CD44-/MyD88- mature EOC16 and cells with metastatic potential41,49 and are inherently chemoresistant.14,16 In contrast, CD44-/MyD88- mature EOC represent the “classical” cancer cells. They have quick doubling time (16 h vs. 56 h for CD44+/MyD88+ EOC stem cells), are terminally differentiated and are more chemo-responsive.
During the process of tumor progression, tumor expansion is characterized by the generation of a high number of CD44-/MyD88- EOC cells originated from CD44+/MyD88+ EOC stem cells. This can be achieved by asymmetric cell division, which yields one daughter cell equivalent to the parent cell and one daughter cell with a more differentiated phenotype. However, this type of division cannot explain the increase in the number of CSCs that was observed in EOC patients. Therefore, it is possible that during the process of tumor recurrence, there is a unique microenvironment that may promote symmetric cell division or self-renewal in EOC stem cells, leading to the production of daughter cells with equivalent stemness potential as the parent CSC. By using in vitro and in vivo models of mechanical and chemical tumor injury, we have shown that such microenvironment is pro-inflammatory in nature and occurs as a result of activation of at least a specific pathway, the TLR2-MyD88-NFκB pathway, in a specific cancer cell population, the CD44+/MyD88+ EOC stem cells.
Inflammation is known to be critical for normal tissue repair and renewal.50 Cytokines produced at the site of injury can enhance tissue repair by promoting cell division, angiogenesis and cell migration.51 The major challenges in understanding the connection between inflammation and cancer is the identification of triggering events that lead to the inflammatory response, identification of the source and target of inflammatory signals and understanding how this can contribute to tumor progression. Thus, the identification of TLR2 as an important upstream component of the inflammatory response that occurs and is required during the process of repair and EOC stem cell self-renewal is critical in future endeavors to prevent EOC recurrence.
Our findings are in the same line with recent reports linking TLRs with tumor growth.46 Indeed, a recent study showed that TLR2 activation promotes breast cancer cell invasiveness and adhesiveness.52 Moreover, in a melanoma mouse model, TLR2 targeting attenuates pulmonary metastases of tumor, whereas combining an anti-TLR2 antibody and a cytotoxic agent, gemcitabine, provided a further improvement in the survival of tumor-bearing mice.53 Remarkably, TLR2 stimulation has been shown to sense oxidation-associated molecular patterns, providing a key link connecting inflammation, oxidative stress and innate immunity.46 Our data demonstrating the synergistic effect of wound-associated injury and PGN on TLR2 expression and activation provide a rationale for TLR2’s important role in maintaining EOC stem cell self-renewal.
The role of surgery and optimal debulking in EOC patients has been extensively evaluated.54 Prognosis has been correlated with the extent of residual disease after primary debulking surgery. It is clear that patients with the least tumor burden after surgery have the best prognosis, and that prognosis worsens as the diameter of the residual disease increases.55 Our data provides insights into the molecular mechanism by which residual disease may promote tumor recurrence and open the possibility to develop new approaches to prevent tumor repair in patients with residual disease.
Overall, our present findings point to a specific cancer cell population, the CD44+/MyD88+ EOC stem cells and a specific pro-inflammatory pathway, the TLR2-MyD88-NFκB pathway, as two of the required players to orchestrate tumor recurrence that is associated with enhanced CSC load. Given the inherent chemoresistant property of the CD44+/MyD88+ EOC stem cells, this may explain at least in part the acquisition of chemoresistance observed during EOC recurrence. Taken together, our results suggest that during surgical debulking or chemotherapy, cellular debris that are released during the process of injury and cell death may activate the TLR2-MyD88-NFκB pathway in a specific cancer cell population, the EOC stem cells, leading to tumor repair and hence disease recurrence. The identification of the specific cancer cells responsible and the associated signaling pathway that is required is a first step in elucidating the steps necessary to prevent recurrence in EOC patients. Understanding how these processes are regulated will impact the mode of management for ovarian cancer.
Materials and Methods
Cell cultures and culture conditions
Cells used in these studies were isolated from either ascites or cancer tissue from ovarian cancer patients and grown as previously described.41,56,57 The experiments described here were performed using three clones of CD44+/MyD88+ EOC stem cells and three clones of CD44-/MyD88- EOC cells.16,27 CD44-/MyD88- EOC cells correspond to the CD44- component of the cells derived from CD44+ EOC stem cells following in vitro and in vivo differentiation.27 CD44-/MyD88- EOC cells directly isolated from patient samples and those obtained by differentiation from CD44+/MyD88+ EOC cells share the same molecular phenotype.16 None of the cell cultures used in this study originated from commercially available cell lines. All patients signed consent forms, and the use of patient samples was approved under the Yale University’s Human Investigations Committee (HIC # 10425).
In vivo model
The Yale University Institutional Animal Care and Use Committee approved all in vivo studies described. Subcutaneous (s.c.) tumors were established in athymic nude mice as previously described.16 After 12 d, the tumor sizes were roughly equivalent, and wounding was performed. On a single mouse, on the right flank tumor, a small skin incision was made the length of the tumor, and a small section of the tumor (50%) was removed. On the left flank tumor, an equivalent skin incision was made, but the tumor was left intact to serve as a non-wounding control. At day 24 post-wounding, the tumors were excised, tumor size was measured ex vivo, and tumor tissue was taken for enzymatic tissue digestion to measure CD44 level by flow cytometry. All in vivo wounding experiments were performed three times with at least six animals per group.
To study in vivo chemotherapy response, we used tumor samples from patients with ovarian cancer, which received neoadjuvant chemotherapy. Subcutaneous tumors were established by implantation of human ovarian cancer tumor fragments in athymic nude mice. Specifically, the freshly obtained gross tumor tissue was placed in a dish containing cold 10% RPMI 1640 media. All gross normal and necrotic tissue was aseptically removed from the tumor surface prior to fragmenting. The tumor was dissected into 5 × 5 × 5 mm tumor fragments which were placed into a cryovial containing Matrigel (BD Biosciences) and allowed to incubate on ice for 10 min prior to implantation. Mice were anesthetized using an isoflurane vaporizer, and two small (1–1.5 cm) incisions were made in the right and left flanks of the mice. Using straight forceps, the tumor fragment was extracted from the Matrigel and inserted into the incision and located away from the wound. The incision site was elevated with forceps and closed using VetBond (3M). Therapy consisted of Paclitaxel, 20 mg/kg three times a week (q3) for 21 d. Tumor size was monitored daily.
Reagents
Mouse anti-human GAPDH antibody was purchased from Sungene Biotech, (Tianjin, China). Mouse anti-human MyD88, rabbit anti-human β-catenin, rabbit anti-human Oct4 antibodies were purchased from Cell Signaling Technology. Mouse-anti-human CD44 antibody was purchased from Novus. Lipopolysaccharides from Escherihia coli 0111:B4 (LPS) was purchased from Sigma. Peptidoglycan S. aureus (PGN) was purchased from InvivoGen. MyD88 siRNA was purchased from Invivogen.
Immunohistochemistry
Sections (5 μm) were deparaffinized in Histosolve and rehydrated. Antigen retrieval was performed using pre-warmed Target Retrieval Solution in a steamer for 30 min. Primary CD44 antibody (1:1,000) was applied to slides for 20 min at room temperature. CD44 sections were developed using DAB (Envision Double Stain System). Slides were counterstained with Hematoxylin and mounted with Aqueous Mounting Medium.
Wound/repair assay
Cells (80,000 cells/well) were plated in a 24-well Image Lock Plate (Essen Bioscience). After 24 h, the 100% confluent cells were wounded using a semi-manual wound maker tool. Wound width was calculated by imaging plates using the Incucyte system (Essen Instruments), during around-the-clock kinetic imaging (Fig. S1).
GFP and RFP lentiviral transduction
To create GFP-expressing CD44+/MyD88+ EOC cells and RFP-expressing CD44-/MyD88- cells, we used a DNA GFP or RFP lentiviral vector containing viral packaging signals and regulatory elements to package the DNA sequence into infectious virions.41 We transduced two clones of each cell type of ovarian cancer cells with lentiviral particles according to previously published protocols.36 Briefly, 24 h before transduction, cells were grown in 6-well plates up to 1.6 × 104 cells per well. One–5 μl of viral stock and 2 μl of 4 mgml−1 polybrene were added to the cells and incubated for 18–20 h at 37°C in 5% CO2-humidified incubator. Twenty-four hours after transduction, the cells were washed twice in 1 × PBS and maintained in complete growth medium. After expansion in culture for 48 h, the cells were maintained in a regular growth medium.
Quantitative RT-PCR
Total RNA was isolated using the high pure RNA isolation kit (Roche). One μg of total RNA isolated from each sample was then used as a template for cDNA synthesis, prepared with a first-strand cDNA synthesis kit (Thermo Scientific). The expression of various transcripts was assessed by real-time PCR amplification 50°C for 2 min; 95°C for 10 min; (95°C for 15 sec, 62°C for 45 sec; 40 cycles) with Kapa-Sybr Fast qPCR mix (Kapa Biosystems), using the CFX96-Real-Time System (Bio-Rad). All primers are designed as exon spanning primers; primer sequences are available upon request. All PCR reactions were performed in triplicate and validated by the presence of a single peak in the melt curve analysis. Changes in gene expression were calculated relative to GAPDH using the 2-ΔCt method. After the quantification procedure, the products were resolved by 2.5% agarose gel electrophoresis to confirm that the reaction had amplified DNA fragments of expected size.
Ki67 immunocytochemistry
Cells were fixed with 4% paraformaldehyde for 20 min at room temperature, washed twice with PBS and permeabilized with ice-cold 100% methanol for 10 min at -20°C. The following steps were performed at room temperature. Ki67 antibody (Vector Labs) was added at a dilution of 1:500 and incubated for 1 h. Cells were washed with PBS; secondary antibody (anti-rabbit FITC, Vector Labs) was applied at a 1:500 dilution and incubated for 1 h. Cells were washed twice with PBS then incubated with Hoechst (1:5,000) for 5 min. After one PBS wash, slides were mounted with Dako Fluorescent Mounting Medium (Dako), coverslip was applied and slides were imaged using Volocity software (PerkinElmer).
Transfection of EOC stem cells
Cells were transiently transfected with a dominant-negative form of MyD88 (pDeNy-MyD88) or dominant-negative form of TLR2 (TLR2ΔTIR) (InvivoGen). Briefly, 7 × 104 cells were seeded per well of a 24-well Image Lock Plate (Essen Bioscience) and 2 μg of plasmid DNA mixed with X-tremeGENE 9 transfection reagent (Roche Applied Science) was overlayed. Wells were wounded 24h post-transfection.
Flow cytometry
Flow cytometric analyses were performed as previously described.16 First, cells were dissociated from tumor tissue using collagenase as previously described.58 Next, pelleted cells were incubated with either FITC-anti CD44 or FITC-anti RatIgG2b isotype control (eBioscience). Data was acquired using BD FACSCalibur and analyzed using Cell Quest Pro (BD Bioscience).
Protein preparation
Protein extraction was performed as previously described.57 Briefly, cell pellets were lysed on ice in 1 × phosphate-buffered saline with 1% NP40, 0.1% SDS and freshly added protease inhibitor cocktail (Sigma Chemical) and phenylmethylsulfonyl fluoride (Sigma Chemical). Protein concentration was determined by BCA Protein Assay (Pierce Biotechnology), and proteins were stored at -80°C until further use.
SDS-PAGE and western blots
A quantity of 20 μg of each protein sample was denatured in sample buffer and subjected to 12% SDS-PAGE as previously described.57 The following antibody dilutions were used: rabbit anti-human β-catenin (1:1,000), rabbit anti-human Oct4 (1:1,000), mouse anti-human MyD88 (1:1,000), mouse anti-human CD44 (1:2,000) and mouse anti-human GAPDH (1:10,000). Specific protein bands were visualized using enhanced chemiluminescence (Pierce Biotechnology).
Cytokine profiling
Levels of cytokines and chemokines were measured in cell-free supernatants and cell lysates using the Bioplex Pro Cytokine Assay (Biorad). Data were acquired using the Bioplex system (Biorad), and analysis was performed using the Bioplex software as previously described.59
Statistical analysis
Data are presented as mean ± SD. Statistical significance (*p < 0.05, **p < 0.01, ***p < 0.005) was determined using one-way analysis of variance with the Bonferonni correction.
Supplementary Material
Acknowledgment
This study was supported in part by grants from NCI/NIH RO1CA127913, RO1CA118678, the Sands Family Foundation and the Discovery to Cure Research Program. I.C. is a Life Science Research Foundation Postdoctoral fellow. M.G. is a fellow from Fundacion Alfonso Martin Escudero.
Glossary
Abbreviations:
- EOC
epithelial ovarian cancer
- CSC
cancer stem cell
- TLR
Toll-like receptor
- WB
wound back
- WE
wound edge
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/23406
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/23406
References
- 1.Silasi DA, Illuzzi JL, Kelly MG, Rutherford TJ, Mor G, Azodi M, et al. Carcinosarcoma of the ovary. Int J Gynecol Cancer. 2008;18:22–9. doi: 10.1111/j.1525-1438.2007.00948.x. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.Baker TR, Piver MS, Hempling RE. Response to Taxol chemotherapy in resistant ovarian carcinoma. Eur J Gynaecol Oncol. 1993;14:449–54. [PubMed] [Google Scholar]
- 4.Armstrong DK. Relapsed ovarian cancer: challenges and management strategies for a chronic disease. Oncologist. 2002;7(Suppl 5):20–8. doi: 10.1634/theoncologist.7-suppl_5-20. [DOI] [PubMed] [Google Scholar]
- 5.Bast RC, Jr., Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9:415–28. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berchuck A, Elbendary A, Havrilesky L, Rodriguez GC, Bast RC., Jr. Pathogenesis of ovarian cancers. J Soc Gynecol Investig. 1994;1:181–90. doi: 10.1177/107155769400100302. [DOI] [PubMed] [Google Scholar]
- 7.Nickoloff BJ, Ben-Neriah Y, Pikarsky E. Inflammation and cancer: is the link as simple as we think? J Invest Dermatol. 2005;124:x–xiv. doi: 10.1111/j.0022-202X.2005.23724.x. [DOI] [PubMed] [Google Scholar]
- 8.Neth P, Ries C, Karow M, Egea V, Ilmer M, Jochum M. The Wnt signal transduction pathway in stem cells and cancer cells: influence on cellular invasion. Stem Cell Rev. 2007;3:18–29. doi: 10.1007/s12015-007-0001-y. [DOI] [PubMed] [Google Scholar]
- 9.Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells–perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- 10.Maitland NJ, Bryce SD, Stower MJ, Collins AT. Prostate cancer stem cells: a target for new therapies. Ernst Schering Found Symp Proc 2006; 5:155-79. [DOI] [PubMed] [Google Scholar]
- 11.Maitland NJ, Collins A. A tumour stem cell hypothesis for the origins of prostate cancer. BJU Int. 2005;96:1219–23. doi: 10.1111/j.1464-410X.2005.05744.x. [DOI] [PubMed] [Google Scholar]
- 12.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 13.Alvero AB, Fu HH, Holmberg J, Visintin I, Mor L, Marquina CC, et al. Stem-like ovarian cancer cells can serve as tumor vascular progenitors. Stem Cells. 2009;27:2405–13. doi: 10.1002/stem.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mor G, Yin G, Chefetz I, Yang Y, Alvero A. Ovarian cancer stem cells and inflammation. Cancer Biol Ther. 2011;11:708–13. doi: 10.4161/cbt.11.8.14967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441:1068–74. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 16.Alvero AB, Chen R, Fu HH, Montagna M, Schwartz PE, Rutherford T, et al. Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle. 2009;8:158–66. doi: 10.4161/cc.8.1.7533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alvero AB, Montagna MK, Craveiro V, Liu L, Mor G. Distinct subpopulations of epithelial ovarian cancer cells can differentially induce macrophages and T regulatory cells toward a pro-tumor phenotype. Am J Reprod Immunol. 2012;67:256–65. doi: 10.1111/j.1600-0897.2011.01068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alvero AB, Montagna MK, Holmberg JC, Craveiro V, Brown D, Mor G. Targeting the mitochondria activates two independent cell death pathways in ovarian cancer stem cells. Mol Cancer Ther. 2011;10:1385–93. doi: 10.1158/1535-7163.MCT-11-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bapat SA, Mali AM, Koppikar CB, Kurrey NK. Stem and progenitor-like cells contribute to the aggressive behavior of human epithelial ovarian cancer. Cancer Res. 2005;65:3025–9. doi: 10.1158/0008-5472.CAN-04-3931. [DOI] [PubMed] [Google Scholar]
- 20.Kurrey NK, K A, Bapat SA. Snail and Slug are major determinants of ovarian cancer invasiveness at the transcription level. Gynecol Oncol. 2005;97:155–65. doi: 10.1016/j.ygyno.2004.12.043. [DOI] [PubMed] [Google Scholar]
- 21.Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM, et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68:4311–20. doi: 10.1158/0008-5472.CAN-08-0364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deng S, Yang X, Lassus H, Liang S, Kaur S, Ye Q, et al. Distinct expression levels and patterns of stem cell marker, aldehyde dehydrogenase isoform 1 (ALDH1), in human epithelial cancers. PLoS One. 2010;5:e10277. doi: 10.1371/journal.pone.0010277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silva IA, Bai S, McLean K, Yang K, Griffith KA, Thomas D, et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res. 2011;71:3991–4001. doi: 10.1158/0008-5472.CAN-10-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dyall S, Gayther SA, Dafou D. Cancer stem cells and epithelial ovarian cancer. J Oncol. 2010;2010:105269. doi: 10.1155/2010/105269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alvero AB, Fu HH, Holmberg J, Visintin I, Mor L, Marquina CC, et al. Stem-like ovarian cancer cells can serve as tumor vascular progenitors. Stem Cells. 2009;27:2405–13. doi: 10.1002/stem.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leizer AL, Alvero AB, Fu HH, Holmberg JC, Cheng YC, Silasi DA, et al. Regulation of inflammation by the NF-κB pathway in ovarian cancer stem cells. Am J Reprod Immunol. 2011;65:438–47. doi: 10.1111/j.1600-0897.2010.00914.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steffensen KD, Alvero AB, Yang Y, Waldstrøm M, Hui P, Holmberg JC, et al. Prevalence of epithelial ovarian cancer stem cells correlates with recurrence in early-stage ovarian cancer. J Oncol. 2011;2011:620523. doi: 10.1155/2011/620523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao Q, Geng L, Kvalheim G, Gaudernack G, Suo Z. Identification of cancer stem-like side population cells in ovarian cancer cell line OVCAR-3. Ultrastruct Pathol. 2009;33:175–81. doi: 10.1080/01913120903086072. [DOI] [PubMed] [Google Scholar]
- 29.Moserle L, Indraccolo S, Ghisi M, Frasson C, Fortunato E, Canevari S, et al. The side population of ovarian cancer cells is a primary target of IFN-alpha antitumor effects. Cancer Res. 2008;68:5658–68. doi: 10.1158/0008-5472.CAN-07-6341. [DOI] [PubMed] [Google Scholar]
- 30.Szotek PP, Pieretti-Vanmarcke R, Masiakos PT, Dinulescu DM, Connolly D, Foster R, et al. Ovarian cancer side population defines cells with stem cell-like characteristics and Mullerian Inhibiting Substance responsiveness. Proc Natl Acad Sci U S A. 2006;103:11154–9. doi: 10.1073/pnas.0603672103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Curley MD, Therrien VA, Cummings CL, Sergent PA, Koulouris CR, Friel AM, et al. CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells. 2009;27:2875–83. doi: 10.1002/stem.236. [DOI] [PubMed] [Google Scholar]
- 32.Chen R, Alvero AB, Silasi DA, Mor G. Inflammation, cancer and chemoresistance: taking advantage of the toll-like receptor signaling pathway. Am J Reprod Immunol. 2007;57:93–107. doi: 10.1111/j.1600-0897.2006.00441.x. [DOI] [PubMed] [Google Scholar]
- 33.Uematsu S, Akira S. Toll-like receptors and innate immunity. J Mol Med (Berl) 2006;84:712–25. doi: 10.1007/s00109-006-0084-y. [DOI] [PubMed] [Google Scholar]
- 34.Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med. 2005;11:1173–9. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 35.Apetoh L, Ghiringhelli F, Tesniere A, Criollo A, Ortiz C, Lidereau R, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev. 2007;220:47–59. doi: 10.1111/j.1600-065X.2007.00573.x. [DOI] [PubMed] [Google Scholar]
- 36.Chen R, Alvero AB, Silasi DA, Kelly MG, Fest S, Visintin I, et al. Regulation of IKKbeta by miR-199a affects NF-kappaB activity in ovarian cancer cells. Oncogene. 2008;27:4712–23. doi: 10.1038/onc.2008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–9. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 38.Tsan MF. Toll-like receptors, inflammation and cancer. Semin Cancer Biol. 2006;16:32–7. doi: 10.1016/j.semcancer.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 39.Kelly MG, Alvero AB, Chen R, Silasi DA, Abrahams VM, Chan S, et al. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res. 2006;66:3859–68. doi: 10.1158/0008-5472.CAN-05-3948. [DOI] [PubMed] [Google Scholar]
- 40.Clarke MF, Fuller M. Stem cells and cancer: two faces of eve. Cell. 2006;124:1111–5. doi: 10.1016/j.cell.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 41.Yin G, Alvero AB, Craveiro V, Holmberg JC, Fu HH, Montagna MK, et al. Constitutive proteasomal degradation of TWIST-1 in epithelial-ovarian cancer stem cells impacts differentiation and metastatic potential. Oncogene. 2013;32:39–49. doi: 10.1038/onc.2012.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bourguignon LY, Earle C, Wong G, Spevak CC, Krueger K. Stem cell marker (Nanog) and Stat-3 signaling promote MicroRNA-21 expression and chemoresistance in hyaluronan/CD44-activated head and neck squamous cell carcinoma cells. Oncogene. 2012;31:149–60. doi: 10.1038/onc.2011.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–14. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- 44.Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, et al. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423:448–52. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- 45.Abrahams VM, Aldo PB, Murphy SP, Visintin I, Koga K, Wilson G, et al. TLR6 modulates first trimester trophoblast responses to peptidoglycan. J Immunol. 2008;180:6035–43. doi: 10.4049/jimmunol.180.9.6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim S, Karin M. Role of TLR2-dependent inflammation in metastatic progression. Ann N Y Acad Sci. 2011;1217:191–206. doi: 10.1111/j.1749-6632.2010.05882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Q, Dziarski R, Kirschning CJ, Muzio M, Gupta D. Micrococci and peptidoglycan activate TLR2-->MyD88-->IRAK-->TRAF-->NIK-->IKK-->NF-kappaB signal transduction pathway that induces transcription of interleukin-8. Infect Immun. 2001;69:2270–6. doi: 10.1128/IAI.69.4.2270-2276.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamashita YM, Fuller MT, Jones DL. Signaling in stem cell niches: lessons from the Drosophila germline. J Cell Sci. 2005;118:665–72. doi: 10.1242/jcs.01680. [DOI] [PubMed] [Google Scholar]
- 49.Yin G, Chen R, Alvero AB, Fu HH, Holmberg J, Glackin C, et al. TWISTing stemness, inflammation and proliferation of epithelial ovarian cancer cells through MIR199A2/214. Oncogene. 2010;29:3545–53. doi: 10.1038/onc.2010.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luo JL, Kamata H, Karin M. IKK/NF-kappaB signaling: balancing life and death--a new approach to cancer therapy. J Clin Invest. 2005;115:2625–32. doi: 10.1172/JCI26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–50. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 52.Wu S, Lu X, Zhang ZL, Lei P, Hu P, Wang M, et al. CC chemokine ligand 21 enhances the immunogenicity of the breast cancer cell line MCF-7 upon assistance of TLR2. Carcinogenesis. 2011;32:296–304. doi: 10.1093/carcin/bgq265. [DOI] [PubMed] [Google Scholar]
- 53.Yang HZ, Cui B, Liu HZ, Mi S, Yan J, Yan HM, et al. Blocking TLR2 activity attenuates pulmonary metastases of tumor. PLoS One. 2009;4:e6520. doi: 10.1371/journal.pone.0006520. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 54.Memarzadeh S, Holschneider CH, Bristow RE, Jones NL, Fu YS, Karlan BY, et al. FIGO stage III and IV uterine papillary serous carcinoma: impact of residual disease on survival. Int J Gynecol Cancer. 2002;12:454–8. doi: 10.1046/j.1525-1438.2002.01149.x. [DOI] [PubMed] [Google Scholar]
- 55.Jelovac D, Armstrong DK. Recent progress in the diagnosis and treatment of ovarian cancer. CA Cancer J Clin. 2011;61:183–203. doi: 10.3322/caac.20113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Flick MB, O’Malley D, Rutherford T, Rodov S, Kamsteeg M, Hao XY, et al. Apoptosis-based evaluation of chemosensitivity in ovarian cancer patients. J Soc Gynecol Investig. 2004;11:252–9. doi: 10.1016/j.jsgi.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 57.Kamsteeg M, Rutherford T, Sapi E, Hanczaruk B, Shahabi S, Flick M, et al. Phenoxodiol--an isoflavone analog--induces apoptosis in chemoresistant ovarian cancer cells. Oncogene. 2003;22:2611–20. doi: 10.1038/sj.onc.1206422. [DOI] [PubMed] [Google Scholar]
- 58.Green JM, Alvero AB, Kohen F, Mor G. 7-(O)-Carboxymethyl daidzein conjugated to N-t-Boc-hexylenediamine: a novel compound capable of inducing cell death in epithelial ovarian cancer stem cells. Cancer Biol Ther. 2009;8:1747–53. doi: 10.4161/cbt.8.18.9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Visintin I, Feng Z, Longton G, Ward DC, Alvero AB, Lai Y, et al. Diagnostic markers for early detection of ovarian cancer. Clin Cancer Res. 2008;14:1065–72. doi: 10.1158/1078-0432.CCR-07-1569. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.