

Abstract
Mutant K-Ras and survivin both contribute to oncogenesis, but little is known about K-Ras requirement for the maintenance of the high levels of survivin in human tumors. Here we demonstrate that K-Ras depletion significantly decreases survivin levels in human cancer cells that harbor mutant but not wild type K-Ras. K-Ras depletion attenuates both basal and drug-induced survivin levels. The mechanism by which K-Ras depletion decreases survivin levels is through ubiquitination and proteasomal degradation of survivin and is independent of survivin-Thr-34 phosphorylation. Depletion of RalA and RalB, but not Raf-1, Akt1 and Akt2, decreases survivin levels, suggesting that K-Ras may regulate survivin stability through its RalGDS/Ral but not PI3K/Akt and Raf-1/Mek effector pathways. Furthermore, the ability of mutant K-Ras to induce anchorage-independent growth, invasion and survival is compromised by depletion of survivin. These studies suggest that mutant K-Ras contributes to the maintenance of the aberrantly high levels of survivin in tumors by regulating its stability, and that the ability of mutant K-Ras to induce malignant transformation is, at least in part, dependent on these high levels of survivin.
Keywords: K-Ras, Survivin, apoptosis, cancer, proteasome, protein degradation
Introduction
One of the critical requirements for normal cells to become cancerous is to acquire the ability to evade programmed cell death (apoptosis) even when challenged with unfavorable conditions such as cytotoxic agent or radiation exposure.1 The Bcl-2 family as well as the inhibitors of apoptosis (IAP) family of proteins play essential roles in the regulation of apoptosis. While the Bcl-2 family proteins monitor cell death by controlling the release of cytochrome c from mitochondria, the IAP family proteins prevent cell death by inhibiting the activation of caspases.2,3 Survivin is one of the members of the IAP family known to play critical roles in promoting cell cycle progression4 and in preventing apoptosis.5 During cell cycle progression, survivin mediates proper loading of the chromosomal passenger complex, chromosomal segregation, spindle formation and microtubule stabilization.6,7 The role of survivin as an anti-apoptotic protein has also been investigated thoroughly, and depending on the cell type, survivin inhibits either spontaneous apoptosis or drug-induced apoptosis.8 Despite its prominent role in apoptosis, the mechanism by which survivin blocks apoptosis remains elusive. Some studies suggested that survivin protects against apoptosis through direct binding to caspases,5 whereas other studies demonstrated that XIAP (another IAP member) but not survivin directly binds caspases.9 More recent studies demonstrated that survivin binds and cooperates with XIAP to efficiently block caspase activation.10
Unlike other IAPs, little to no survivin is expressed in normal cells. In contrast, virtually all cancer cells maintain very high levels of survivin protein.11 The fact that survivin protein levels are much higher in cancer cells as compared to normal cells indicates that some oncogene may be responsible for the maintenance of these high levels. Indeed, the induction of survivin expression in cancer cells has been attributed to some mutated or deregulated oncogenes/proto-oncogenes in cancer. For example, aberrant activation of STAT3, NFkB, Notch and Wnt as well as inactivation of the tumor suppressors p53 and Rb all have been shown to increase survivin expression by regulating its transcription.12
Mutations in the small GTPase Ras and high levels of the survivin protein are prevalent in human tumors, but the role of mutant Ras in regulating survivin protein levels has not been thoroughly studied, and the very few studies reported investigated mainly the role of ectopically expressed H-Ras but not K-Ras, the most frequently mutated isoform of Ras in human cancer. Previous studies showed that, ectopic overexpression of c-H-Ras in Rat cells as well as in human keratinocytes induces survivin expression.13,14 Furthermore, Fukuad et al. showed that exogenous H-Ras is required for interleukin induced survivin in Baf-3 cells.15 However, other studies showed that mutant H-Ras is not able to increase survivin.16 Furthermore, the requirements of endogenous Ras for the maintenance of the high levels of survivin in cancer cells have not been investigated. For example, the effects of depleting Ras on survivin levels are not known. Finally, several studies have documented both in vitro and in vivo the importance of survivin to malignant transformation17,18 but little is known about the contributions of survivin to mutant K-Ras-driven transformation.
In this manuscript, we demonstrated that depletion of K-Ras decreases survivin levels in human cancer cells that harbor mutant K-Ras but not wild type Ras, and that this decrease in the survivin protein levels is due to ubiquitination and proteasome degradation, suggesting that mutant K-Ras regulates survivin stability. Our results also demonstrated that depletion of RalA and/or RalB but not Akt1, Akt2 and Raf-1 decrease the levels of survivin. Furthermore, we also demonstrated that depletion of survivin significantly reduces mutant K-Ras-driven anchorage-independent growth and invasion, suggesting that survivin contributes to K-Ras driven malignant transformation.
Results
Depletion of K-Ras decreases survivin levels in human cancer cells that harbor mutant, but not wild type K-Ras
Both mutant K-Ras and aberrantly-high levels of survivin contribute to malignant transformation and are associated with poor cancer patient prognosis. However, whether mutant K-Ras is required for the maintenance of high survivin levels in tumors is not known. To address this important question we first depleted K-Ras from several human cancer cell lines that either harbor mutant K-Ras or wild type Ras and determined the effect of this depletion on the levels of survivin. Human cancer cell lines from various origins, including pancreatic (Panc-1, MiaPaca-2, SW1990, ASPC-1, BXPC-3), lung (A-549, Calu-1, H460, HOP-92) and ovarian (SKOV-3), were transfected with non-targeting (NT) or K-Ras siRNA and processed for western blotting as described under “Materials and Methods.” Figure 1A shows that siRNA to K-Ras depleted K-Ras, and that this resulted in a significant decrease in the protein levels of survivin in cell lines that harbor mutant K-Ras (Panc-1, AsPC-1, A-549, H460, MiaPaca-2, Calu-1 and SW1990) but not in those cancer cell lines that express only wild type Ras (BXPC3, HOP-92 and SKOV-3).

Figure 1. Depletion of K-Ras decreases survivin levels in human cancer cells that harbor mutant K-Ras. Effects on endogenous, exogenous and drug-induced survivin. (A) Human cancer cell lines that harbor mutant K-Ras (Panc-1, AsPC-1, A549, H460, MiaPaCa-2, SW1990 and Calu-1) or wild type Ras (PXPC3, HOP-92, SKOV3) were transfected either with non-targeting siRNA (NT) or K-Ras siRNA and the cells were processed for western blot analysis as described in Materials and Methods. (B) Panc-1 cells were transfected with siRNA#2 that targets different regions of K-Ras mRNA and processed for western blot analysis as described above. (C) Depletion of K-Ras reduces both exogenous and endogenous survivin. Panc-1 cells that stably express either vector or MYC-DDK-Survivin were transfected with siRNA and the cells processed for western blot analysis as above. (D) Panc-1 cells were treated with siRNA for 36 h followed by VP-16 (D) treatment for 24 h. Cells were processed for western blot analysis as above. Data are representative of at least two independent experiments.
Next we used another K-Ras siRNA that targets a different region of K-Ras mRNA to confirm the results. Figure 1A and B show that depletion of K-Ras by two distinct K-Ras siRNAs reduces survivin levels. Whether depleting K-Ras also reduces the levels of ectopically expressed survivin was investigated next. To this end, cells were stably transfected with Myc-DDK-tagged survivin, and survivin protein levels were determined following siRNA to NT or K-Ras transfection as described under “Materials and Methods.” Figure 1C shows that depletion of K-Ras reduces the levels of endogenous as well as exogenous survivin. The fact that depletion of K-Ras reduces the levels of exogenous survivin, the expression of which was driven by a foreign promoter, suggests that K-Ras may affect survivin levels by regulating its stability (see more below).
Depletion of K-Ras compromises the ability of VP16 to increase survivin levels
Previous studies have shown that treatment with chemotherapeutic drugs such as VP16 results in survivin induction, and that this is an intrinsic mechanism by which tumor cells develop resistance to chemotherapy. Since we found depletion of K-Ras to decrease basal survivin protein levels, we reasoned that depletion of K-Ras may also prevent induction of survivin by anticancer drugs in cancer cells that harbor mutant K-Ras. Therefore, we next investigated whether the VP16-mediated increase in survivin levels requires mutant K-Ras. To this end, Panc-1 cells were transfected with siRNA to K-Ras or NT siRNA and treated with vehicle or VP16 and processed for western blotting as described under “Materials and Methods.” Figure 1D shows that in the absence of siRNA to K-Ras VP16 treatment of Panc-1 cells resulted in a robust increase in survivin levels. In contrast, in Panc-1 cells where K-Ras was depleted, the ability of VP16 to induce survivin was compromised (Fig. 1D). These results suggest that K-Ras expression is required for the ability of VP16 to increase survivin protein levels, further confirming the role of mutant K-Ras in maintaining high levels of survivin in human cancer cells.
Ectopic expression of mutant K-Ras is not sufficient to induce survivin
Figure 1 demonstrated that depletion of K-Ras resulted in decreased levels of endogenous and exogenous survivin as well as drug-induced survivin. Next we determined if ectopic expression of mutant, constitutive active K-Ras (CA-K-Ras) increases survivin levels. Human embryonic kidney HEK293 cells that ectopically express CA-K-Ras and their parental counterparts, which were transfected with an empty vector construct, were used. Figure 2A shows that forced expression of CA-K-Ras did not affect the protein levels of survivin. Similar results were also obtained with NIH-3T3 cells as well as two primary pancreatic ductal cell lines HPNE and C7 that stably express either CA-K-Ras or empty vector (Fig. 2B−D), suggesting that K-Ras is not sufficient to drive the expression of survivin. Taken together, the results from Figures 1 and 2 suggest that mutant K-Ras is required but not sufficient for the maintenance of the high survivin levels in human cancer cells.
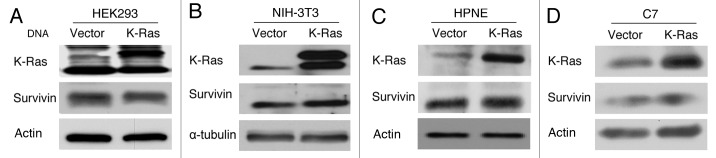
Figure 2. Ectopic expression of CA-K-Ras is not sufficient to induce survivin. HEK293 (A), NIH3T3 (B), HPNE (C) and C7 (D) cells that stably express CA-K-Ras or vector were plated and grown for 48 h, and western blot analysis was performed to determine the survivin levels.
Depletion of K-Ras decreases the stability of survivin by promoting its ubiquitination and proteasome degradation
Previous studies have shown that survivin protein undergoes proteasomal degradation. To determine if depletion of K-Ras leads to decreased survivin protein levels by inducing its proteasomal degradation, we used the proteasome inhibitor bortezomib. First, we demonstrated that treatment of Panc-1 cells with bortezomib resulted in the accumulation of survivin in a time-dependent manner starting as early as 1 h and reaching a maximum at 4 h (Fig. 3A). Next, whether bortezomib can rescue from the K-Ras depletion-mediated decrease of survivin levels was investigated. To this end, Panc-1 cells that stably express Myc-DDK tagged survivin were transfected with either NT or K-Ras siRNA for 48 h prior to incubation with bortezomib for 1 h, and the cells were processed for western blotting as described under “Materials and Methods.” Figure 3B shows that vector cells express only endogenous survivin, whereas Myc-survivin transfected cells express both endogenous and exogenous survivin (compare lanes 1 and 2). In the absence of bortezomib, depleting K-Ras resulted in a significant decrease of both endogenous and exogenous survivin levels (compare lanes 3 and 5). In contrast, treatment of cells with bortezomib inhibited the ability of K-Ras siRNA to decrease the levels of both endogenous and ectopically expressed survivin (compare lanes 4 and 6 to lanes 3 and 5). Figure 3C shows that bortezomib inhibited the CT-L activity of the proteasome by 83% and 88% in Panc-1 cells transfected with NT siRNA and K-Ras siRNA, respectively.

Figure 3. Depletion of K-Ras decreases survivin protein levels in a proteasome-dependent manner and a survivin-T34 phosphorylation-independent manner. (A) Panc-1 cells were treated with the indicated dose of bortezomib for 1 h. The media was then replaced with drug-free media and the cells were allowed to grow for the indicated length of time and processed for western blotting as described in “Materials and Methods.” (B and C) Panc-1 cells stably expressing Myc-DDK survivin were treated with siRNA to NT or K-Ras, followed by vehicle or bortezomib treatment. Cells were then harvested for western blot analysis (B) or CT-L proteasome activity assay (C) as described in “Materials and Methods.” (D) A549 cells were transfected with NT or K-Ras (K) siRNA for 48 h followed by bortezomib treatment for 30 min. Survivin was immunoprecipitated and processed for western blot analysis for either ubiquitin or survivin. (E) Panc-1 cells that stably express vector, wt, T34A mutant or T34D mutant survivin were transfected with NT or K-Ras siRNA and the cells processed for western blotting as described in “Materials and Methods.” Data are representative of at least two independent experiments.
We next determined whether K-Ras siRNA induces the degradation of survivin by increasing its ubiquitination. To this end, cells were transfected with siRNA to NT or K-Ras for 48 h followed by bortezomib treatment for 30 min. The cells were then processed for immunoprecipitation with survivin antibody and the immune-precipitates separated by SDS-PAGE and immunoblotted with ubiquitin antibody as described under “Materials and Methods.” Figure 3D shows that, in the presence of bortezomib, depletion of K-Ras did not decrease survivin levels, consistent with the results of Figure 3B. Figure 3D also shows that depletion of K-Ras increased the levels of ubiquitinated survivin. Therefore, bortezomib inhibited the degradation of survivin, which, in the absence of K-Ras, accumulated in its ubiquitinated state. Taken together, the results from Figure 3B and D suggest that K-Ras depletion leads to ubiquitination and proteasome-mediated degradation of survivin.
Phosphorylation of survivin at T34 has been shown to increase the stability of survivin.22,23 One possible mechanism by which depletion of K-Ras could decrease the stability of survivin is by inhibiting the phosphorylation of survivin at T34. We therefore reasoned that the phospho mimic T34D survivin mutant should be resistant to K-Ras depletion. To this end, we generated the T34D survivin mutant as well as the non-phosphorylatable T34A mutant. Panc-1 cells that stably express either wild type (WT), T34D mutant or T34A mutant were transfected with siRNA to NT or K-Ras for 48 h, and the expression of survivin was followed by western blotting as described under “Materials and Methods.” Figure 3E shows that depletion of K-Ras reduces the expression of WT survivin as well as T34A and T34D survivin mutants, suggesting that the ability of K-Ras siRNA to decrease survivin protein levels is not dependent on the phosphorylation status of survivin at T34.
Depletion of K-Ras decreases selectively survivin over the anti-apoptotic proteins Bcl-2, Bcl-XL and Mcl-1 and does not increase the levels of the pro-apoptotic proteins Bax and Bad
The ability of cancer cells to survive depends not only on IAP family members, but also on a balance between anti-apoptotic and pro-apoptotic Bcl-2 family members (see “Introduction” section). We therefore determined whether K-Ras depletion selectively affects survivin over other proteins that regulate cancer cell survival. To this end, in addition to survivin, we evaluated the effects of K-Ras depletion on anti-apoptotic (Bcl-2, Bcl-XL and Mcl-1) and pro-apoptotic (Bad and Bax) proteins. Figure 4 shows that siRNA to K-Ras decreased the levels of survivin but did not affect the levels of the Bcl-2 family proteins in Panc-1 and MiaPaca-2 cells. These results suggest that the effects of depleting K-Ras on survivin are specific.
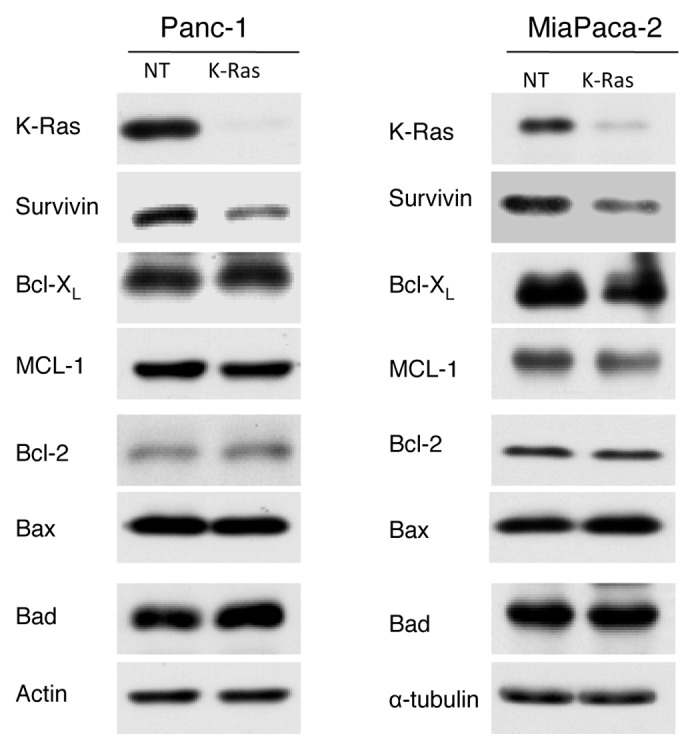
Figure 4. Depletion of K-Ras reduces survivin levels but does not affect the levels of Bcl-2 family proteins. Panc-1 and MiaPaCa-2 cells were transfected with siRNA to K-Ras or NT for 48 h and the expression of different members of BCL-2 family proteins as well as expression of survivin were detected by western blot analysis. Data are representative of two (MiaPaCa) or three (Panc-1) independent experiments.
Depletion of RalA and/or RalB but not Akt1, Akt2 and c-Raf-1 decreases survivin levels
Ras mediates its biological effects by triggering the activation of several downstream signaling pathways, among which RalGDS/Ral, PI3K/Akt and Raf/Mek play pivotal roles. Therefore, we next determined whether depletion of RalA, RalB, Akt1, Akt2 and/or Raf-1 phenocopies the effect of K-Ras depletion on survivin levels. To this end, A549 and H460 cells were transfected with siRNAs to RalA, RalB, Akt1/2 or Raf-1 and processed the cells for western blot analysis as described in “Materials and Methods.” Figure 5 shows that, in A-549 cells, depletion of K-Ras, RalA and RalB decreased the survivin levels, with RalB being more potent than K-Ras and RalA. In H460 cells, depletion of K-Ras and RalA was much more potent than depletion of RalB at decreasing the levels of survivin. Furthermore, depletion of Raf-1 and Akt1/2 did not result in decreased survivin levels in both cell lines (Fig. 5). These results demonstrate that depletion of RalA and RalB but not that of Raf-1 or Akt1/2 phenocopies K-Ras depletion effects on survivin, and suggest that the RalGSD/RalA-RalB pathway may mediate the effects of K-Ras in regulating the stability of survivin
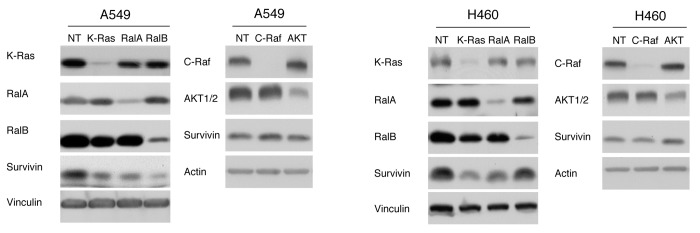
Figure 5. Depletion of RalA and/or RalB but not Akt1/2 and Raf-1 decreases survivin levels. A549 and H460 cells were transfected with siRNA to NT, K-Ras, RalA, RalB, Akt1/2 or c-Raf-1 siRNA for 48 hrs. Samples were processed for western blot analysis and immunoblotted with the indicated antibody as described in “Materials and Methods.” Data are representative of two independent experiments.
Survivin is required for mutant K-Ras-driven malignant transformation
Both survivin and mutant K-Ras contribute to malignant transformation, but whether survivin is required for K-Ras-driven malignant transformation is not known. In an attempt to address this question, we next determined if depletion of survivin induces apoptosis in HNPE cells where malignant transformation was induced by mutant K-Ras. Figure 6 shows that depletion of survivin induces apoptosis in HPNE-K-Ras cells, as measured by caspase 3 activation and PARP cleavage, suggesting that survivin mediates at least in part the anti-apoptotic function of K-Ras. To further confirm this result, we used different cell lines that harbor mutant K-Ras and determined if depletion of survivin induces apoptosis. Our result shows that depletion of survivin induces caspase 3 activation and PARP cleavage in A549 and H460 but not in Panc-1 cells.

Figure 6. Effects of depleting survivin on apoptosis. HPNE-K-Ras, A549, Panc-1 and H460 cells were transfected with either control (NT) or survivin siRNA for 96 h, as described under “Materials and Methods.” Top panel: western blot analysis showing knockdown of survivin and cleaved PARP. Bottom panel: caspase-3 activity following depletion of survivin. Data are representative of two independent experiments.
In addition to the well-studied function of survivin in apoptosis, recent studies have also demonstrated that survivin contributes to invasion and metastasis, and this role is independent of its anti-apoptotic activity.24 The ability of cancer cells to grow in an anchorage-independent manner is a prerequisite to metastasis. We therefore determined if mutant K-Ras-induced colony formation in soft agar of HPNE pancreatic cells depends on survivin. Figure 7 shows that HPNE cells transfected with vector grew 62 colonies in soft agar. Transfection with mutant K-Ras enhanced the ability of HPNE cells to grow soft agar colonies by 6.6-fold from 62 (vector cells) to 412 (K-Ras cells) colonies. Figure 7 shows that depletion of survivin potently inhibited this K-Ras-dependent increase in colony formation in soft agar. This suggests that the ability of K-Ras to promote anchorage-independent growth partly depends on survivin. Furthermore, the ability of H-460 and Panc-1 cells, both of which harbor mutant K-Ras, to grow soft agar colonies was also inhibited by depletion of survivin

Figure 7. Depletion of survivin inhibits K-Ras-driven anchorage-independent growth on soft agar. HPNE-Vector, HPNE-K-Ras, Panc-1 and H460 cells were transfected with the indicated siRNA for 48 h and were plated for soft agar analysis as described under “Materials and Methods.” Data are representative of two (H460) or three (HPNE and Panc-1) independent experiments.
While anchorage-independent growth is a pre-requisite for metastasis, another close hallmark of metastasis is invasion. We therefore determined if mutant K-Ras-driven invasion requires survivin. Figure 8 shows that mutant K-Ras increased the ability of HPNE cells to invade by ~3.7-fold from 18 in vector cells to 67 in K-Ras transformed cells. Figure 8 also shows that depletion of survivin inhibited the ability of K-Ras to induce invasion. Similarly, in A-549, H-460 and Panc-1 cells, all of which harbor mutant K-Ras, depletion of survivin significantly compromised their ability to invade (Fig. 8).

Figure 8. Depletion of survivin inhibits K-Ras-driven invasion. HPNE-Vector, HPNE-K-Ras, A549, Panc-1 and H460 were transfected as described for Figure 7 and were analyzed for invasion through matrigel coated transwell inserts after 24 h (HPNE cells), 48 h (Panc-1 and H460) or 72 h (A549) as described under “Materials and Methods.” Data are representative of two (HPNE and A549) or three (Panc-1 and H460) independent experiments.
Discussion
The contributions of mutant K-Ras to several hallmarks of cancer, including deregulation of anchorage-dependent and -independent cell growth, invasion, metastasis and apoptosis evasion, have been well documented. In contrast, until recently, survivin has only been implicated in the prevention of apoptosis and the promotion of mitosis. Recent studies have demonstrated, as is the case for K-Ras, that survivin is also involved in other hallmarks of cancer such as invasion and metastasis.24 However, it is not known whether K-Ras contributes to the maintenance of the high levels of survivin in human tumors, and whether this contributes to mutant K-Ras-mediated malignant transformation
In this manuscript, we used siRNA to knockdown K-Ras expression in several human cancer cell lines, and demonstrated that depletion of K-Ras reduces survivin protein levels in cancer cells that harbor mutant K-Ras but not wild type Ras. In addition to decreasing the basal levels of survivin, depletion of K-Ras also inhibited the ability of VP-16 to induce survivin, further confirming the importance of K-Ras in the maintenance of survivin. Furthermore, the K-Ras depletion-mediated reduction in survivin levels was demonstrated in cancer cell lines from different tissue of origin. Therefore, our results suggest that the requirement of K-Ras expression for the maintenance of aberrantly high levels of survivin in human cancer cells depends on the mutation status of K-Ras but is not tissue-specific.
Our studies also demonstrated that while mutant K-Ras is required for the maintenance of survivin levels (Fig. 1), on its own, it is not sufficient to induce survivin in several cell models, including NIH-3T3 murine fibroblasts and human kidney embryonic HEK293 cells as well as two human pancreatic cell lines C7 and HPNE (Fig. 2). Consistent with our results, expression of mutant K-Ras in mouse kidneys in the Wilms tumor mouse model was not able to induce survivin.25 Interestingly, in this model, expression of both mutant K-Ras and β-catenine was able to induce survivin.25 This study, coupled with our K-Ras depletion results, suggests that while mutant K-Ras and other genetic aberrations are required for increasing the levels of survivin, depletion of only K-Ras is able to decrease survivin. Therefore, K-Ras is necessary for the maintenance of survivin but not sufficient to increase survivin levels. Whether ectopic expression of CA-H-Ras can increase survivin levels is not clear, with some studies showing that it can14,15 and another showing that it cannot.16
In addition to depleting the endogenous levels of survivin, our studies also demonstrated that depletion of K-Ras was able to decrease the levels of exogenous survivin whose ectopic expression was under the control of a foreign promoter. This suggested that depletion of K-Ras may affect the stability of the survivin protein, and this was further confirmed by the demonstration that depletion of K-Ras increased survivin ubiquitination, and that treatment with the proteasome inhibitor Bortezomib compromised the ability of K-Ras siRNA to decrease the levels of the survivin protein. Therefore, our studies indicate that depletion of K-Ras leads to decreased survivin levels through a proteasome degradation mechanism. Survivin has a fairly short half-life of 30 min,26 and its stability has been suggested to depend on phosphorylation of survivin at T34 by CDK1.22,23 We have made the phosphomimic T34D as well as the non-phosphorylatable T34A survivin mutants, and have demonstrated that depletion of K-Ras is as effective at decreasing the levels of these mutants as it is at decreasing the levels of wild type survivin, suggesting that the ability of K-Ras depletion to promote the proteasome degradation of survivin is independent of T34 phosphorylation. While these results exclude the phosphorylation of survivin at T34 as the sole determinant of K-Ras regulated survivin stability, it doesn’t rule out possible involvement of this phosphorylation site in combination with other plausible K-Ras-regulated mechanisms that are involved in the stability of survivin.
Another important finding of our study is that depletion of RalA and/or RalB but not that of Raf-1 and Akt1/2 phenocopies the effects of depleting K-Ras on the survivin levels. This suggests that the ability of K-Ras to regulate the stability of survivin may be mediated by RalA and/or RalB but not Raf-1 or Akt1/2. The fact that not all effectors of Ras mediate the same biological effects of Ras is consistent with previous studies that showed, for example, that the Raf/Mek pathway predominantly mediates proliferation whereas the PI3K/Akt drives survival.27-32 Furthermore, our results are consistent with other reports that demonstrated that the RalGDS/RalA-RalB pathway downstream of Ras may be more important at mediating Ras-driven malignant transformation than the PI3K/Akt and the Raf-1/Mek pathways in some but not all tumors.27 Interestingly, we have found that depletion of RalA and RalB in some tumors, particularly those of pancreatic origin, such as Panc-1 and MiaPacca-2 cells, does not result in decreasing survivin levels. Whether this difference is due to tissue of origin (lung vs. pancreas) is not known. However, it is clear that, while in the panel of human cancer cells that we have investigated, the ability of K-Ras depletion to decrease survivin appears to be dependent only on the K-Ras mutation status, this is not the case for RalA and RalB. Indeed depletion of RalA and/or RalB decreases the survivin levels in A-549 and H460 but not in Panc-1 and MiaPacca-2 cells, yet all four cell lines harbor mutant K-Ras.
Depletion of K-Ras induces apoptosis in many human cancer cell lines that depend on mutant K-Ras for survival.33,34 However, whether this depends on the K-Ras depletion-mediated decrease in survivin levels is not known. Using isogenic HCT-116 colon cell lines, Sarthy et al.35 have found that survivin depletion induces apoptosis preferentially in cancer cells that harbor mutant K-Ras. However, several studies using RNAi screens have not all identified that depletion of survivin induces cell death in cancer cells with K-Ras mutation.33,34 Consistent with this, our results also show that mutation of K-Ras alone may not predict sensitivity to survivin depletion. For example, depleting survivin in H460 and A549 but not in Panc-1cells (Fig. 6) and MiaPaca 2 cells (data not shown) induces apoptosis, yet all four cell lines harbor mutant K-Ras. Depletion of K-Ras may induce apoptosis through other mechanisms; however, apoptosis following depletion of K-Ras in these two cell lines does not involve anti-apoptotic (Bcl-xL, Bcl-2 and Mcl-1) or pro-apoptotic (Bax and Bad) Bcl2 family members (Fig. 4). Future studies that use a large number of cell lines that harbor mutant and wild type K-Ras are warranted to identify the effect of survivin depletion in the context of K-Ras mutation.
Survivin has been associated with malignant transformation as its expression increases at early stages of tumor development and its targeted expression in skin predispose mice to UV-induced tumors.36 Our studies show that, mutant K-Ras also depends on survivin for its ability to transform cells as well as to induce invasion. Using a genetically defined pancreatic cancer cell model (HPNE), we have shown that the ability of mutant K-Ras to induce anchorage-independent cell growth on soft agar was inhibited by depletion of survivin. Depletion of survivin also inhibited the anchorage-independent growth of human cancer cells that harbor mutant K-Ras such as Panc-1 and H-460 cells. Furthermore, the ability of mutant K-Ras to induce invasion in HPNE pancreatic cells was also inhibited by depletion of survivin. The ability of A-549, H-460 and Panc-1 cancer cells, all of which harbor mutant K-Ras, to invade was also inhibited when survivin expression was knocked down. These results suggest that survivin may be critical to the ability of mutant K-Ras to induce metastasis. This is consistent with recent studies that implicated survivin in invasion and metastasis.24
In summary, this manuscript demonstrates that in the absence of K-Ras, survivin ubiquitination levels are increased, leading to its degradation by the proteasome, and suggests that this important biochemical link between mutant K-Ras, and the stability of survivin contributes to K-Ras-driven malignant transformation.
Materials and Methods
Cell culture
The human tumor cell lines were grown in their respective media: A549 in Kaighn's modification medium (F12K) (Gibco Laboratories), Calu-1 and SKOV-3 in McCoy’s 5a medium modified (Sigma-Aldrich), H460, AsPC-1 and BxPC-3 in RPMI, (Invitrogen), MiaPaCa-2, Panc-1, SW1990, and HOP-92 in Dulbecco’s modified minima essential medium (DMEM) (Invitrogen). All cells were grown in their respective media containing 10% FBS and 1% penicillin streptomycin at 37°C in a humidified incubator at 5% CO2. Human Pancreatic Nestin Expressing (HPNE) cells were the kind gift of Dr. Channing Der and Dr. Paul Campbell (University of North Carolina) and were maintained in media containing 3:1 mixture of DMEM and M3F base media (INCELL). NIH-3T3 cells that stably express mutant K-Ras and C7 cells that stably express mutant K-Ras were grown as described by us,19,20 respectively.
SiRNAs and antibodies
Non-targeting (NT), K-Ras, RalA, RalB, c-Raf and survivin siRNA were purchased from Dharmacon and Ambion, respectively; Akt1/2 siRNA was purchased from Cell Signaling. Survivin antibodies were purchased from Novus Biologicals, (used for immunoprecipitation) and Abcam (used for western blotting). Bax, MCL-1, Bad, Akt1/2, c-Raf and ubiquitin antibodies were purchased from SantaCruz Biotechnology. Cleaved PARP antibody was purchased from Cell Signaling. β-actin, vinculin, α-tubulin and GAPDH antibodies were purchased from Sigma. K-Ras (OP-24), Bcl-2 and Bcl-XL antibodies were purchased from Calbiochem. RalA antibody was purchased from BD Biosciences. RalB antibody was purchased from (Millipore) Peroxidase conjugated Goat anti-mouse IgG, Rabbit anti-Goat IgG, Mouse anti-Rabbit IgG antibodies were purchased from Jackson ImmunoResearch Laboratories. TrueBlot mouse secondary was purchased from e-Bioscience.
DNA constructs and site directed mutagenesis
Survivin T34 mutants were generated using QuickChange II XL site-directed mutagenesis kit (Agilent Biotechnologies). Survivin ORF plasmid was purchased from Origene and used as a template for site directed mutagenesis according to the manufacturer’s recommendation. Briefly, primers were designed using an online primer designing tool from Agilient Biotechnology, (www.agilent.com/genomics).
The following primers were purchased from Integrated DNA Technology (IDT). Survivin T34D: Sense primer: 5′-GGGCTGCGCCTGCGACCCGGAGCGGATG-3′; Antisense primer: 5′-CATCCGCTCCGGGTCGCAGGCGCAGCCC-3′. Survivin T34A: Sense primer: 5′-GCTGCGCCTGCGCCCCGGAGCGG-3′; Antisense primer:- 5′-CCGCTCCGGGGCGCAGGCGCAGC-3′.
PCR conditions
The following components were added into a PCR tube-reaction buffer to 1×, 200 ng DNA, 125 ng of each sense and antisense primers, 1 ul of dNTP mix, 3 μl of quicksolution and distilled water to a final volume of 50 μl and DNA polymerase (all reagents from Agilent Biotechnologies). PCR cycle conditions: 95°C, 2 min; 95°C, 1 min; 60°C, 50 sec; 68°C, 14 min; 68°C, 7 min, repeat 18 times then end. Following temperature cycling, the reaction tubes were placed on ice for 2 min, followed by the addition of 2 μl DPN1 restriction enzyme to each sample. This reaction was allowed to go for at least 4 h at 37°C. After generation of the plasmid, bacteria (XL10- Gold Ultra Competent Cells) were transformed per the manufacturer’s instructions. After overnight incubation, 10 colonies were picked for each sample. DNA was purified to determine mutation status.
For cloning myc-tagged CA-K-Ras12V, we used our previously described K-Ras-Q12V pBABE expression construct21 as template to clone into myc-tagged pCMV-Tag3B vector. The following primers were used to shuttle the construct:
Forward primer: 5′-CGC GGA TCC AAG CTT ATG ACT GAA TAT AAA CTT GTG GTA-3′,
Reverse primer: 5′-TGC TCT AGA CTC GAG TTA CAT AAT TAC ACA CTT TGT CTT-3′
SiRNA transfection
Cells were transfected with siRNA using Lipofectamin RNAiMAX transfection reagent (Invitrogen) according to the manufacturer’s instructions. Briefly, cells were plated overnight to reach ~40–50% confluence at the time of transfection. Lipofecatmine RNAiMAX and Optimum were mixed in one tube, and a mixture of siRNA and Optimum was prepared in another tube, and the content of the two tubes were mixed. The mixture was incubated for 20 min at room temperature, after which the siRNA-RNAiMax mixture was added to cells drop-wise. After 24 h of transfection, appropriate media that contain 10% serum only was added until the desired time of transfection.
DNA transfection and generation of stable cell lines
Cells were plated overnight to reach 70–80% confluence and were then transfected with DNA using Lipofectamine 2,000 transfection reagent according to the manufacturer’s instructions. Briefly, Lipofectamine 2,000 reagent (1.5 μl/μg of DNA) was diluted in OPTI-MEM medium (Invitrogen) and allowed to equilibrate for 5 min at room temperature. Two μg of DNA (vector or Myc-DDK-survivin construct) (Origene) was diluted in OPTI-MEM in a separate tube. The contents of the two tubes were mixed and allowed to complex for 20 min at room temperature then added drop wise to cells that contain 10% FBS but no 1% PS. The cells were transfected for 6 h before changing the medium to medium containing 10% FBS only and incubated for an additional 18 h. The medium was then replaced with complete growth medium containing 1.2 mg/mL of G418 (Fisher Scientific). The cells were kept in selection medium for 2 weeks, after which the resistant cells were pooled and grown for further experiments.
Western blotting
Cells were rinsed with ice-cold PBS twice and lysed on the plate in lysis buffer containing 20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerolphosphate, 1 mM sodium orthovanadate, 1ug/mL leupeptin, and 1 mM PMSF. The lysate was collected by scraping and centrifuging at 13,000 g for 13 min to remove the debris. The protein content was measured using Bradford protein assay. Samples were resolved using 12.5% SDS-PAGE and transferred to PVDF membranes (Millipore). The membranes were blocked with 5% milk in Tris-buffered saline and Tween 20 (TBST) and then probed with the desired antibodies. The enhanced chemiluminescence blotting system (PerkinElmer Inc.) was used for antibody reaction.
Immunoprecipitation
Cells were rinsed with ice cold PBS twice and lysed on the plate in NP-40 lysis buffer containing 20 mM Tris pH 7.5, 0.05% Igepal, 250 mM NaCl, 3 mM EGTA, 3 mM EDTA, 2 mM NaVO4, 2 mM PMSF and 7.4 mg/ml PNPP, 0.5 uM bortezomib, Ub-aldehyde 4 μM, 20 mM n-ethylmaleimide. After collecting the lysate, the lysate was sonicated (4 pulses), using Branson Sonifier, VWR Scientific, at output level 3, duty cycle (%) 30. After clearing the debris by centrifuging at 13,000 g for 13 min, the protein amount was measured. 600–1,000 μg of protein was incubated with 30 μl of 50/50 mix of protein A/ protein G agarose beads (Millipore) and 2 μg/ml of survivin antibody (Novus) at 4°C overnight on an orbital shaker. After overnight incubation, the sample was centrifuged for 5 sec to collect the beads. The supernatant was discarded, and the beads were washed two times with 500 μl lysis buffer. The agarose beads were re-suspended in 30 ul of sample buffer. The agarose beads were boiled for 5 min to dissociate the immunocomplex from the beads. The beads were collected by centrifuging and the proteins in the supernatant were run on a 12.5% SDS-PAGE.
In vivo ubiquitination assay
Panc-1 cells were first transfected with siRNA for 48 hr. The cells were then treated with 0.5 uM of bortezomib (Velcade) for 30 min. The cells were then lysed with NP-40 lysis buffer. Survivin was immunoprecipitated as described above and ubiquitinated survivin was detected using ubiquitin antibody from SantaCruz Biotechnology and TrueBlot mouse secondary (eBioscience).
Caspase activation assay
Caspase-3 activity was measured in cell lysates using a fluorogenic substrate. The assay measures the cleavage of AMC groups from a Caspase-3 specific substrate (Ac-DEVD-AMC) (Biomol Research Labs, Inc.). Liberated AMC groups were quantified on fluorometer at excitation max.: 365–380 nm and emission max.: 430–460 nm. Briefly, cells were lysed in a protease inhibitor free lysis buffer (50 mM Tris, pH 8, 5 mM EDTA, 150 mM NaCl, 0.5% NP-40). The lysate was collected by scraping. After clearing the lysate by centrifugation, protein was measured. 20 μg of protein and 20 μM caspase-3 substrate were added into 50 mM Tris, pH 7.5 reaction buffer in triplicate in a 96-well plate. The reactions were incubated for 1h at 37°C and the plate was read by a fluorometer, WALLAC Victor 1420 Multilabel counter with 355 nm excitation and 460 nm emission filters (Perkin Elmer Life Sciences).
Assay for the chymotrypsin-like activity (CT-L) of the proteasome
The (CT-L of the proteasome was measured using fluorogenic peptide substrate as described elsewhere.20 Briefly, 20 μg of cell lysate protein was incubated with 20 μM of Suc-Leu-Leu-Val-AMC substrate for CT-L activity in 100 μl of assay buffer (50 mM TRIS-HCl, pH 7.6) for 1 hr at 37°C. After incubation, production of 7-hydrolyzed 7-amido-4-methyl-coumarin (AMC) groups were measured using WALLAC Victor 1420 Multilabel counter with 355 nm excitation and 460 nm emission filters (Perkin Elmer Life Sciences).
Soft agar colony-formation assay
After 48 h of siRNA treatment, the cells were trypsinized, counted and seeded at a cell density of 2,000/well in triplicate in 12-well culture plates in 0.3% agar over a 0.6% bottom agar layer as previously described.21 Cultures were incubated until colonies formed (approximately 2–3 weeks). Plates were scanned after overnight incubation with 1 mg/ml MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Calbiochem], in cell growth medium. The colony numbers were visually determined and quantified.
Cell invasion assay
Analysis of cell invasion was performed using artificial extracellular matrix (Matrigel, 8 uM pore size, BD Biosciences) according to the manufacturer’s instructions. Briefly, the inserts were rehydrated by adding warm serum-free medium for 2 h at 37°C, 5% CO2 atmosphere. The medium was then removed from the inserts, and 750 ul of medium containing FBS was added to each well of the plate and the inserts were placed on top of the medium. The transfected A549, Panc-1, and H460 (40,000 cells in 500 ul of serum free-media) and HPNE cells (20,000) were added into the inserts. The cells were incubated for 48 h at 37°C. Cell invasion was determined by staining the bottom parts of the insert with crystal violet (0.5% crystal violet in 25% methanol). The porous membrane was removed and placed on the slide. The number of cells that invaded were counted in three high-power microscope (40×) fields and averaged as the mean number of invaded cells per field for each membrane.
Acknowledgments
The authors would like to thank the molecular biology core facility at the Moffitt Cancer Center for sequencing the DNA constructs used in this manuscript. We also would like to thank Maria E. Balasis for her careful reading of the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/23407
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Salvesen GS, Duckett CS. IAP proteins: blocking the road to death’s door. Nat Rev Mol Cell Biol. 2002;3:401–10. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- 3.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 4.Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, et al. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580–4. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 5.Shin S, Sung BJ, Cho YS, Kim HJ, Ha NC, Hwang JI, et al. An anti-apoptotic protein human survivin is a direct inhibitor of caspase-3 and -7. Biochemistry. 2001;40:1117–23. doi: 10.1021/bi001603q. [DOI] [PubMed] [Google Scholar]
- 6.Lens SM, Vader G, Medema RH. The case for Survivin as mitotic regulator. Curr Opin Cell Biol. 2006;18:616–22. doi: 10.1016/j.ceb.2006.08.016. [DOI] [PubMed] [Google Scholar]
- 7.Lens SM, Wolthuis RM, Klompmaker R, Kauw J, Agami R, Brummelkamp T, et al. Survivin is required for a sustained spindle checkpoint arrest in response to lack of tension. EMBO J. 2003;22:2934–47. doi: 10.1093/emboj/cdg307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen XM, Luan XY, Lei DP, Ma XJ, Liu XX, Liu J, et al. Suppression of survivin expression by short hairpin RNA induces apoptosis in human laryngeal carcinoma cells. ORL J Otorhinolaryngol Relat Spec. 2008;70:168–75. doi: 10.1159/000124290. [DOI] [PubMed] [Google Scholar]
- 9.Li C, Wu Z, Liu M, Pazgier M, Lu W. Chemically synthesized human survivin does not inhibit caspase-3. Protein Sci. 2008;17:1624–9. doi: 10.1110/ps.036145.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dohi T, Okada K, Xia F, Wilford CE, Samuel T, Welsh K, et al. An IAP-IAP complex inhibits apoptosis. J Biol Chem. 2004;279:34087–90. doi: 10.1074/jbc.C400236200. [DOI] [PubMed] [Google Scholar]
- 11.Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–21. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 12.Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer. 2008;8:61–70. doi: 10.1038/nrc2293. [DOI] [PubMed] [Google Scholar]
- 13.Sommer KW, Schamberger CJ, Schmidt GE, Sasgary S, Cerni C. Inhibitor of apoptosis protein (IAP) survivin is upregulated by oncogenic c-H-Ras. Oncogene. 2003;22:4266–80. doi: 10.1038/sj.onc.1206509. [DOI] [PubMed] [Google Scholar]
- 14.Sommer KW, Rodgarkia-Dara CJ, Schreiner C, Holzmann K, Krupitza G, Cerni C. Oncogenic c-H-ras deregulates survivin expression: an improvement for survival. FEBS Lett. 2007;581:4921–6. doi: 10.1016/j.febslet.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 15.Fukuda S, Pelus LM. Activated H-Ras regulates hematopoietic cell survival by modulating Survivin. Biochem Biophys Res Commun. 2004;323:636–44. doi: 10.1016/j.bbrc.2004.08.149. [DOI] [PubMed] [Google Scholar]
- 16.Raj D, Liu T, Samadashwily G, Li F, Grossman D. Survivin repression by p53, Rb and E2F2 in normal human melanocytes. Carcinogenesis. 2008;29:194–201. doi: 10.1093/carcin/bgm219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grossman D, Kim PJ, Blanc-Brude OP, Brash DE, Tognin S, Marchisio PC, et al. Transgenic expression of survivin in keratinocytes counteracts UVB-induced apoptosis and cooperates with loss of p53. J Clin Invest. 2001;108:991–9. doi: 10.1172/JCI13345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin Q, Menter DG, Mao L, Hong WK, Lee HY. Survivin expression in normal human bronchial epithelial cells: an early and critical step in tumorigenesis induced by tobacco exposure. Carcinogenesis. 2008;29:1614–22. doi: 10.1093/carcin/bgm234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lerner EC, Zhang TT, Knowles DB, Qian Y, Hamilton AD, Sebti SM. Inhibition of the prenylation of K-Ras, but not H- or N-Ras, is highly resistant to CAAX peptidomimetics and requires both a farnesyltransferase and a geranylgeranyltransferase I inhibitor in human tumor cell lines. Oncogene. 1997;15:1283–8. doi: 10.1038/sj.onc.1201296. [DOI] [PubMed] [Google Scholar]
- 20.Kazi A, Lawrence H, Guida WC, McLaughlin ML, Springett GM, Berndt N, et al. Discovery of a novel proteasome inhibitor selective for cancer cells over non-transformed cells. Cell Cycle. 2009;8:1940–51. doi: 10.4161/cc.8.12.8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Falsetti SC, Wang DA, Peng H, Carrico D, Cox AD, Der CJ, et al. Geranylgeranyltransferase I inhibitors target RalB to inhibit anchorage-dependent growth and induce apoptosis and RalA to inhibit anchorage-independent growth. Mol Cell Biol. 2007;27:8003–14. doi: 10.1128/MCB.00057-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.O’Connor DS, Grossman D, Plescia J, Li F, Zhang H, Villa A, et al. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci U S A. 2000;97:13103–7. doi: 10.1073/pnas.240390697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wall NR, O’Connor DS, Plescia J, Pommier Y, Altieri DC. Suppression of survivin phosphorylation on Thr34 by flavopiridol enhances tumor cell apoptosis. Cancer Res. 2003;63:230–5. [PubMed] [Google Scholar]
- 24.Mehrotra S, Languino LR, Raskett CM, Mercurio AM, Dohi T, Altieri DC. IAP regulation of metastasis. Cancer Cell. 2010;17:53–64. doi: 10.1016/j.ccr.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clark PE, Polosukhina D, Love H, Correa H, Coffin C, Perlman EJ, et al. β-Catenin and K-RAS synergize to form primitive renal epithelial tumors with features of epithelial Wilms’ tumors. Am J Pathol. 2011;179:3045–55. doi: 10.1016/j.ajpath.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao J, Tenev T, Martins LM, Downward J, Lemoine NR. The ubiquitin-proteasome pathway regulates survivin degradation in a cell cycle-dependent manner. J Cell Sci. 2000;113:4363–71. doi: 10.1242/jcs.113.23.4363. [DOI] [PubMed] [Google Scholar]
- 27.Hamad NM, Elconin JH, Karnoub AE, Bai W, Rich JN, Abraham RT, et al. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 2002;16:2045–57. doi: 10.1101/gad.993902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pagès G, Lenormand P, L’Allemain G, Chambard JC, Meloche S, Pouysségur J. Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc Natl Acad Sci U S A. 1993;90:8319–23. doi: 10.1073/pnas.90.18.8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moodie SA, Wolfman A. The 3Rs of life: Ras, Raf and growth regulation. Trends Genet. 1994;10:44–8. doi: 10.1016/0168-9525(94)90147-3. [DOI] [PubMed] [Google Scholar]
- 30.McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE, et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul. 2006;46:249–79. doi: 10.1016/j.advenzreg.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 31.Drosten M, Dhawahir A, Sum EY, Urosevic J, Lechuga CG, Esteban LM, et al. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J. 2010;29:1091–104. doi: 10.1038/emboj.2010.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wee S, Jagani Z, Xiang KX, Loo A, Dorsch M, Yao YM, et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69:4286–93. doi: 10.1158/0008-5472.CAN-08-4765. [DOI] [PubMed] [Google Scholar]
- 33.Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–12. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, et al. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell. 2009;15:489–500. doi: 10.1016/j.ccr.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sarthy AV, Morgan-Lappe SE, Zakula D, Vernetti L, Schurdak M, Packer JC, et al. Survivin depletion preferentially reduces the survival of activated K-Ras-transformed cells. Mol Cancer Ther. 2007;6:269–76. doi: 10.1158/1535-7163.MCT-06-0560. [DOI] [PubMed] [Google Scholar]
- 36.Thomas J, Liu T, Cotter MA, Florell SR, Robinette K, Hanks AN, et al. Melanocyte expression of survivin promotes development and metastasis of UV-induced melanoma in HGF-transgenic mice. Cancer Res. 2007;67:5172–8. doi: 10.1158/0008-5472.CAN-06-3669. [DOI] [PMC free article] [PubMed] [Google Scholar]